

Abstract
Deregulation of c-Myc plays a central role in the tumorigenesis of many human cancers. Yet, the development of drugs regulating c-Myc activity has been challenging. To facilitate the identification of c-Myc inhibitors, we developed a molecular imaging sensor based high throughput-screening (HTS) system. This system uses a cell-based assay to detect c-Myc activation in a HTS format, which is established from a pure clone of a stable breast cancer cell line that constitutively expresses a c-Myc activation sensor. Optimization of the assay performance in the HTS format resulted in uniform and robust signals at the baseline. Using this system, we performed a quantitative HTS against approximately 5,000 existing bioactive compounds from five different libraries. Thirty-nine potential hits were identified, including currently known c-Myc inhibitors. There are a few among the top potent hits that are not known for anti-c-Myc activity. One of these hits is nitazoxanide (NTZ), a thiazolide for treating human protozoal infections. Validation of NTZ in different cancer cell lines revealed a high potency for c-Myc inhibition with IC50 ranging between 10 - 500nM. Oral administration of NTZ in breast cancer xenograft mouse models significantly suppressed tumor growth by inhibition of c-Myc and induction of apoptosis. These findings suggest a potential of NTZ to be repurposed as a new anti-tumor agent for inhibition of c-Myc associated neoplasia. Our work also demonstrated the unique advantage of molecular imaging in accelerating discovery of drugs for c-Myc targeted cancer therapy.
Keywords: c-Myc activation sensor, molecular imaging, high throughput screening, c-Myc inhibitors, nitazoxanide
INTRODUCTION
MYC was the first oncogene linked to human cancer biology and among the most de-regulated genes in the cancer genome (1). Sustained activation of the c-Myc protein up-regulates a cohort of target genes involved in cell cycle, proliferation, growth, metabolism and apoptosis, which contribute to tumorigenesis in a majority of human cancers (2). Intense research on modulation of c-Myc function has demonstrated the feasibility and benefits of c-Myc inhibition as an anti-cancer therapeutic strategy (3, 4).
Yet the development of an effective therapeutic approach to target c-Myc has been challenging (5). Many attempts have been made to target c-Myc at the transcriptional level, either by inhibiting myc promoter activation (6-8) or destabilizing myc transcripts, to down-regulate c-Myc. Most of these approaches have involved using anti-sense oligonucleotides and siRNA, which showed certain efficacy in inhibiting tumor growth but were often unstable and difficult to deliver in vivo(9). Most recently, bromodomain proteins were found to regulate myc expression through a chromatin-dependent signal transduction. Inhibition of these proteins by de novo small molecules resulted in down-regulation of c-Myc expression and showed efficacy in animal multiple myeloma (10) and leukemia models (11). This may point to a new route to target myc expression using small molecule inhibitors, yet clinical development of new drug-like molecules is often challenging. Other attempts were made to repress c-Myc activity at the protein level, either by disrupting c-Myc interaction with other factors (12, 13) or suppressing its upstream activation signaling pathway, such as MAP kinase (MAPK), to inhibit c-Myc activation. MAPK inhibitors have shown great results in many preclinical models (14), however most of them have failed in clinical trials, either due to lack of therapeutic efficacy, such as for PD98059 and U0126, or poor bioavailability and high toxicity, such as for CI1040 and PD0325901 (15). Recently, Atorvastatin, a family member of the Statins, was shown to reduce phosphorylation-mediated c-Myc activation by inhibiting HMG-coA reductase, and to have efficacy in different preclinical tumor models, such as lymphoma and hepatocellular carcinoma (16, 17). However, its clinical efficacy in tumor inhibition remains to be determined. To date, despite ample efforts, there have been no effective approaches to target c-Myc for cancer therapy, underscoring the continuing need for new therapeutic agents, perhaps also new approaches to accelerate development of effective c-Myc drugs.
Multimodality molecular imaging, a spectrum of imaging technologies and strategies, provides a rapid way to detect and quantify tumor response to drugs in a noninvasive and repetitive manner, and has become a key approach to accelerate drug development in both preclinical and clinical settings (18, 19). Recent development of a c-Myc activation sensor provides a way to noninvasively monitor c-Myc activity in cells and living animals (16). This sensor is based on a split luciferase complementation system, where N-terminal and C-terminal Firefly luciferase fragments (NFL and CFL) were fused with a c-Myc activation motif and a GSK3β phospho-site binding domain respectively (16). Phosphorylation-mediated c-Myc activation induces recognition and binding of the c-Myc motif by the GSK3β domain, which brings two split FL fragments close together to complement. The complementation of the N-terminal and C-terminal FL fragments results in reconstitution of FL activity, thus c-Myc activation is detected upon the substrate addition of D-Luciferin (Supplementary Fig. S1).
In this study, we utilized the c-Myc sensor to establish a cell assay and optimized it to a high throughput-screening (HTS) format. Using this assay in a quantitative HTS (qHTS) platform, we screened about 5,000 existing bioactive compounds for potential c-Myc inhibitors. Among the most potent hits was nitazoxanide (NTZ), a human anti-protozoal drug. We further validated its inhibitory effect on c-Myc in different tumor cell lines and the efficacy of tumor inhibition in mouse xenograft models. Our HTS approach revealed for the first time the anti-c-Myc effect of NTZ and its potential role as an anti-neoplastic agent. This study also demonstrates the unique role of molecular imaging in accelerating drug development for c-Myc targeted cancer therapy.
MATERIALS AND METHODS
Chemicals and Drugs
PD98059 and U0126 were purchased from Cell Signaling. Purified Atorvastatin was purchased from Sequoia Research Products, Pangbourne, United Kingdom. The pure compound of nitazoxanide was purchased from Sigma. The 500mg tablet of nitazoxanide (Generic Alinia) was purchased from Buy-pharma.com. The purity and quantity of the drug was validated using LC-MS. D-Luciferin was purchased from BIOSYNTH.
Cell Culture and Cell Lines
All cell culture media were purchased from Invitrogen and supplemented with 10% Fetal Bovine Serum and 1% penicillin/streptomycin solution. SKBR3 (human breast adenocarcinoma) cells were cultured in McCoy’s 5a medium. Osteosarcoma-derived cell line 1325 was cultured in DMEM medium. Lymphoma cells were cultured in RPMI medium supplemented with extra 3.96×10-4% of 2-mercaptoethanol (Sigma). All cells were incubated at 37°C with 5% CO2. The SK-ST and SK-FST stable cells were established by transient transfection of the sensor system or the full-length firefly luciferase genes on a single vector, and selected with 3μg/ml puromycin (Invitrogen). SKBR3 cell line was originally purchased from ATCC and passaged in house. 1325 and Lymphoma cells were provided by the laboratory of Dr. Dean Felsher from the Department of Hematology and Oncology, Stanford University. Although cell lines were not subjected to further genetic authentication, their characteristic morphologies and certain biochemical features were specifically verified. For example, overexpression of c-Myc in SKBR3 was always observed. In addition, all cell lines were tested negative for PCR evaluation for Mycoplasma pathogen using MycoAlert Kit (Lonza).
Western Blotting
Total cell lysates were collected using 1× cell lysis buffer, as directed by the manufacturer (Cell Signaling). The protein concentration was determined using the BioRad DC protein assay. SDS-PAGE analysis of 50μg of protein was transferred to Immobilon-P membrane (Millipore). Anti-phospho-c-Myc (Thr58/Ser62) and anti-c-Myc antibody (Cell Signaling) were used to visualize the phospho c-Myc and c-Myc protein respectively, as directed by the manufacturer. Anti-FLuc-HRP antibody was used to detect the Firefly luciferase fragments, as directed by the manufacturer (Sigma). β-actin protein was used as loading control and detected by anti-β-actin antibody (Sigma). Immunoblots were developed using ECL kit (Pierce), as directed by the manufacturer. The protein level was determined by quantitation of the band intensities on western blots using the ROI manager of ImageJ software (Open Source).
Luciferase Activity Assay
Bioluminescence imaging of luciferase activity in intact cells was performed as described (5). Briefly, after indicated time of treatment, culture medium was added with PBS (pH 7.0) solution of 45μg/ml D-Luciferin for FL imaging in IVIS 50 (Xenogen). Images were acquired in 2mins interval until reaching the peak signal. The photon output of FL was normalized to the total protein of the cell lysates, measured by BioRad DC protein assay as directed by the manufacturer (Bio-rad).
Inhibition Assay
For measuring the inhibitory effect on the sensor signal, SKBR3 stable cells, constitutively expressing the sensor system (SK ST) or the full-length FL (SK FST), were treated with NTZ for 16hrs, PD98059 for 2hrs, U0126 for 1hr and Atorvastatin for 16hrs at the indicated concentration and subjected to Luciferase activity assay. For analyzing the effect on protein level, SKBR3, lymphoma and osteosarcoma cells were treated as above and lysed for western blotting of phospho c-Myc, c-Myc and β-actin proteins. The IC50 of protein inhibition was determined using non-linear regression based curve fitting and a variable model of four parameters in Prism. All treatments were repeated at least three times and the error bar indicates the plus and minus of the standard deviation from the mean of all the repeats.
Bioluminescence Imaging of Living Mice
Healthy female NU/NU mice age 8 to 12 weeks were used (Charles River). The Animal handling was performed in accordance with Stanford University Animal Research Committee guidelines. Bioluminescence imaging of mouse xenografts were performed in IVIS Spectrum (Xenogen) as described (20) with some modifications. FL imaging was acquired with D-Luciferin PBS solution (45μg/μl, 100μl) via i.p. injection. 5×106 of SK ST or SK FST cells were s.c. implanted into nude mice (nu/nu) (N=12 each type) as indicated to make two xenograft models. Each model was repeated three times. The mean of the three repeats and the standard error of the mean were graphed. FL imaging started one day after the implantation and continued on every other day thereafter. Three days after tumor implantation, half of the mice were gavaged twice a day with NTZ 0.5% CMC solution (50mg/ml, 100μl) or half with 0.5% CMC solution only (100μl). Mice were sacrificed after 27 days of treatment. Each xenograft tumor mass was completely dissected and tissue samples were collected for western blotting of phospho c-Myc, c-Myc and α-tubulin proteins. Three samples from each treatment and control group were collected for H&E and IHC staining for Ki67 and caspase 3 (Histo-tec laboratory, CA). At least three fields of view were examined for each stained sample. The mean of the average positive stained cells in each sample was calculated and the standard error mean between each sample was recorded.
Quantitative HTS of Compound Libraries
High throughput screening was performed at Stanford High throughput Bioscience Center (HTBC). Cells were dispensed using the Matrix Wellmate (Thermo Scientific, Hudson, NH, USA) in 100μl culture medium into each well of the 384 white bottom plate from columns 1–22 (E & K Scientific, Santa Clara, CA, USA) and incubated for 24hrs at 5% CO2 and 37°C before compound addition. 100μl of medium alone was added to column 23–24 to control for background signals. Total of 4,846 compounds from five different libraries, NIH Clinical Collection (NCC), Sigma LOPAC, Microsource Spectrum (MS), Biomol_ICB and Biomol_FDA library, were screened. All compounds are in 100% dimethylsulfoxide (DMSO) solution and the stock concentration is 10mM in NCC and seven two-fold titration series, 0.3125, 0.625, 1.25, 2.5, 5, 10 and 20mM, in the rest of the libraries. Approximately 100 nl of each compound at each concentration was added directly to each well and repeat one time in columns 3–22 using a PinTool (V&P Scientific, San Diego, CA, USA) on a Sciclone ALH3000 (Caliper Sciences, Hopkinton, MA, USA) to achieve 1000 fold dilution. Cells in columns 1–2 that were not treated with compounds were used as control for 100% activity. 24hrs after compound addition, the D-Luc substrate was added using multi-drop liquid dispenser and imaged using the Analyst GT (Molecular Device, Sunnyvale, CA, USA), with an acquisition time of 0.2 s per well. The coefficient of variation (CV) is calculated by σ/μ and the Z factor is calculated by 1-(3 σmax+3 σmin)/| μ max- μ min| (μ : mean, σ: standard deviation).
Data Analysis
Curve fitting and Student’s t test were performed using Excel (Microsoft 2008) and Prism. Data are presented as described in each experimental method. Generally, for in cellulo experiments, the data point on the graph is the mean between repeated measurements, and the error bar is the standard deviation from the mean. For in vivo experiments, the data point is the mean between repeated treatment experiments and the error bar is the standard error of that mean. P<0.05 was used as cutoff point for statistical significance.
RESULTS
Development of a HTS system using a c-Myc activation sensor based cell assay
A recent bioluminescence imaging sensor that can noninvasively detect the phosphorylation-mediated activation of c-Myc (Supplementary Fig. S1), provides a rapid and quantitative approach to measure c-Myc activity upon drug inhibition in an intact biological system (21). To facilitate the drug development against c-Myc, we constructed a stable cell line constitutively expressing this c-Myc sensor, developed it into a cell assay and optimized it in a HTS format. The sensor was first transfected into SKBR3 cells, a breast cancer cell line with overexpression of the c-Myc protein. A single pure clone of SKBR3 cells with stable and relatively high sensor signal was selected for the development of a cell assay that can detect c-Myc activation in a HTS format. To determine the optimal cell concentration for the detection assay, six thousand to 18,000 sensor stable cells (SK-ST), in increments of 2,000, were each plated in five consecutive columns of 384 well plates in 100μl/well and incubated for 24hrs before imaging. To measure the performance of the assay, the percentage of coefficient of variation (CV%) for comparing variation between concentrations and the Z factor for assessing the quality of entire screen were calculated. Although the sensor signals were correlated well with different cell concentrations (R2=0.99, Supplementary Fig. S2A and S2B), the CV of the sensor signals decreased from 32% at 6,000 cells/well to 7% at 12,000 cells/well and increased back to about 26% at 14,000 cells/well and above (Supplementary Fig. S2C). Since lower CV% indicates less intra-group variation, these results suggest that a cell concentration of 12,000 cells/100μl/well in a 384-well plate were optimal to achieve low signal variation between wells.
To obtain a higher signal to noise ratio, we evaluated the substrate concentration effect on the intensity and variability of the assay signal. 45mg/ml stock substrate, D-Luciferin (D-Luc), in 0.02, 0.01, 0.005, 0.002 and 0.001 dilution ratios, was added directly to each well of 12,000 cells. 0.02 (50-fold) dilution ratio, which results in 900μg/ml D-Luc, yielded the highest signal, 2.5E+05 p/s/cm2/sr, and the lowest CV%, 6.2% (Fig. S3A and S3B). Dilution ratios higher than 0.02 resulted in cell toxicity and dramatic color change of the medium. We also found, due to the kinetics of the enzyme-substrate reaction, the assay signal was unstable at the early time points after substrate addition. The average signal intensity increased and reached a plateau at 10mins after D-Luc addition and the CV% decreased from about 30% at 2mins to less than 10% at 10mins after D-Luc addition in three separate experiments (Fig. S3C and S3D). These results suggested that the optimal substrate concentration is 900μg/ml D-Luc and the optimal assay time is 10mins after substrate addition.
To validate the quality of the assay under optimal conditions at the baseline, six 384-well plates were seeded with 12,000 cells/100μl/well in the first to the 22nd columns and 100μl medium alone in the last two columns as the blank control. The assay signal was measured ten minutes after addition of 900μg/ml D-Luc. The CV% and Z factor (22) ranged between 11.4% to 9.3% and 0.67 to 0.72 respectively (Supplementary Fig. S4). These data demonstrated the uniformity and robustness of the assay in a HTS format.
A quantitative HTS for inhibitors of c-Myc activation
Using the optimized sensor-based cell assay, we performed a quantitative high throughput screening against 4,846 existing bioactive compounds from five different libraries, the NIH Clinical Collection (NCC), Sigma LOPAC, Microsource Spectrum (MS), Biomol_ICB (BMI) and Biomol_FDA (BMF) (Fig. 1). The NCC library has over 400 drugs that have been used in clinic or clinical trials. The LOPAC library from Sigma-Aldrich has close to 1,300 compounds with proven pharmacological activities in a variety of cell signaling and neuroscience fields (23). The MS, BMI and BMF libraries have total over 3,000 compounds, which also include about 800 natural products. In a quantitative HTS platform, compounds were tested at seven different concentrations simultaneously, 0.3125, 0.625, 1.25, 2.5, 5, 10 and 20μM, except the NCC drugs, which were tested at a single concentration of 10μM. The initial hits of 337 compounds, about 10% of the total, were revealed to have a minimum of 30% inhibition at 10μM. To filter out cytotoxic agents and known luciferase inhibitors, we compared our screening results with those in the MDL Assay Explorer database, which contains dose response profiles of screenings for anti-proliferation and luciferase inhibition against the same libraries. Finally, 39 compounds, about 1% of the total, were identified as potential clean hits (Fig. 1). These potential hits (two from the NCC library, three from the LOPAC library and 34 from the combined MS, BMI and BMF libraries) are from many compound classes (Supplementary Fig. S5). The half-maximal inhibitory concentration (IC50) of the potential hits, except those from the NCC library, ranged from 1.18μM to 42.8μM (Supplementary Fig. S6 and S7). Among the top hits with high efficacy, there are known inhibitors of kinases upstream of c-Myc, such as Calphostin C, a protein kinase C (PKC) specific inhibitor (24, 25). We also see drugs that are not previously described to have anti-c-Myc activity, such as nitazoxanide (NTZ), an anti-parasitic drug; 4-chloromercuribenzoic acid (pCMB), a standard cysteine protease inhibitor; and fludarabine phosphate, a purine analog acting as an antimetabolite for chemotherapy (Supplementary Fig. S8). NTZ is especially interesting because it is a widely used off-label drug and well tolerated with very few side effects (26). In this study, we chose NTZ to further validate our sensor-based HTS system.
Figure 1.
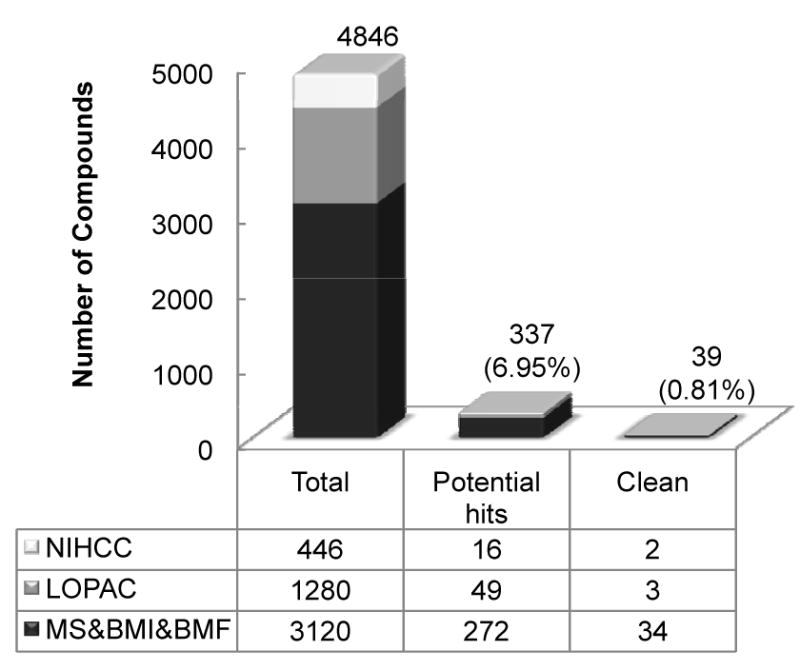
Summary of the HTS results. The contribution of each library to the total screened compounds, the potential hits and the clean hits is listed in the table. The total number of all libraries in each column is plotted above the table. The percentage of potential hits and clean hits is in parenthesis. The potential hits are screened compounds that have a minimum of 30% inhibitory effect at 10μM and the clean hits are those potential hits that are not cytotoxic or luciferase inhibitors.
Anti-c-Myc Efficacy of NTZ in Cell Culture
We first determined the IC50 of NTZ in vitro using the SK-ST sensor stable cells and the SK-FST firefly luciferase stable cells as a control for direct inhibitory effect on luciferase activity. NTZ induced dose dependent reduction of sensor signals in SK-ST cells after 16hrs treatment, while no significant changes occurred in SK-FST cells (Fig. 2A and 2B). The IC50 for the sensor signal inhibition of NTZ was about 100nM, which was about 2000- and 500-fold lower than that of c-Myc upstream kinase inhibitors, PD98059 and U0126 (2) respectively (Fig. 2C and 2D); and about 50-fold lower than that for a HMG-coA inhibitor, Atorvastatin (AT), which was recently found to be a potent c-Myc inhibitor (3, 21). Moreover, NTZ indeed induced dose dependent reduction of the endogenous phospho c-Myc and c-Myc proteins and the IC50 was consistent with that for the sensor signal (Fig. 3A). To examine the inhibitory spectrum of NTZ, we also tested its efficacy in other c-Myc associated tumor cell lines, lymphoma and osteosarcoma cells. NTZ induced dose dependent reduction of the endogenous phospho c-Myc and c-Myc proteins in both cell lines (Supplementary Fig. S9A and S9B), with an IC50 of 440nM for lymphoma cells and 12nM for osteosarcoma cells (Fig. 3B). These results revealed a potent efficacy of NTZ in c-Myc inhibition in different c-Myc associated cancer cells. Furthermore, inhibition of c-Myc at nanomolar range of NTZ resulted in slow cell growth rather than complete cell death. To determine the dose range of NTZ for cell toxicity, we performed a MTS assay in SKBR3 cells and determined that the half-maximal lethal dose (LD50) of NTZ was about 1μM (Supplementary Fig. S10), which was about 10-fold higher than its IC50. This wide range between LD50 and IC50 implies a safe therapeutic effect of NTZ.
Figure 2.
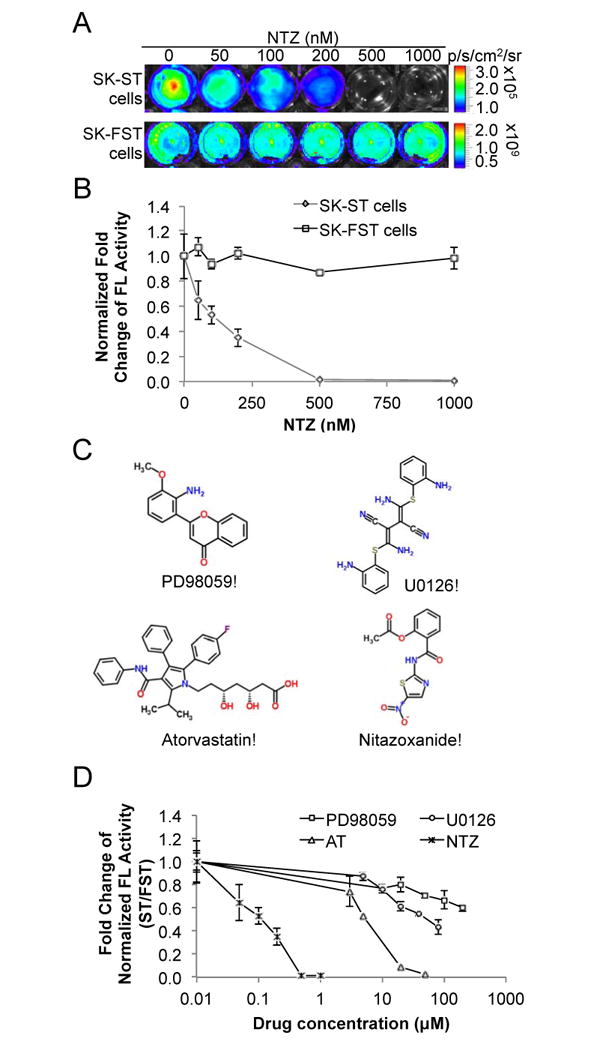
Efficacy of NTZ in cell culture. (A) SK-ST and SK-FST stable cells were treated with NTZ at indicated concentrations and subjected to BLI as described in Materials and Methods. (B) Plot of the fold change of FL activity after normalization to cell numbers at different concentrations of NTZ in SK-ST and SK-FST cells. (C) The chemical structure of NTZ and other three c-Myc inhibitors, PD98059, U0126 and Atorvastatin. (D) SK-ST and SK-FST cells were treated with indicated compounds at different concentrations. FL activity of the SK-ST cells was normalized to that of the SK-FST cells and the fold-change of the normalized FL activity was plotted against the compound concentration.
Figure 3.
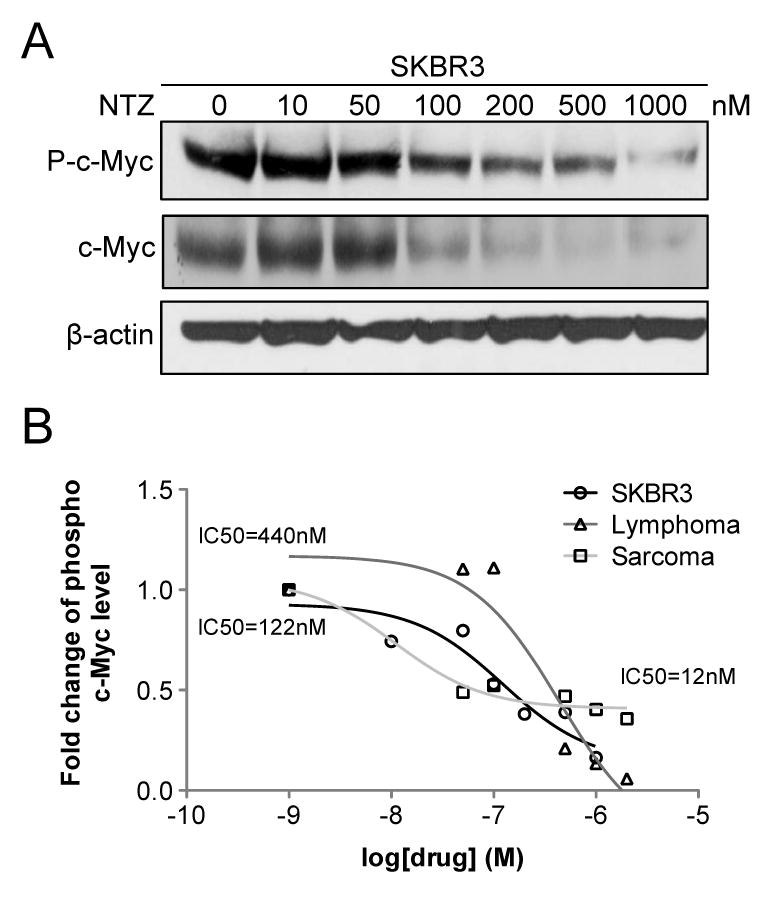
Inhibition of endogenous phosphorylation of c-Myc. (A) Western blot analysis of SKBR3 cells after NTZ treatment was performed using phospho c-Myc, c-Myc protein and β-actin antibodies. (B) NTZ induced dose dependent inhibition of phospho c-Myc level in all three tumor cell lines, SKBR3 (IC50=122nM), lymphoma (IC50=440nM) and sarcoma cells (IC50=12nM). The fold-change of phospho c-Myc level was plotted against the log scale of NTZ concentration.
Anti-tumor efficacy of NTZ in mouse tumor models
To determine whether NTZ could affect tumor growth as a single agent in vivo, we tested it in two tumor xenograft models. In one model, SK-ST cells were implanted subcutaneously into nude mice (N=12, Fig. 4A) to monitor the effect of NTZ on c-Myc activation, and in the other model, SK-FST cells were implanted (N=12, Fig. S11) to monitor cell numbers and the direct drug effect on FL activity. Since there has not been any in vivo study on effective dosage of NTZ in treating tumors, we chose a dose commonly used in animal models of protozoan infection that induces maximum growth inhibition without significant side effects (27, 28). Three days after implantation, mice were gavaged with NTZ (200mg/kg, N=6) in 100μl 0.5% CMC suspension or with the same volume of 0.5% CMC solution (N=6) twice a day for 27 days. The same regimen was repeated three times for each model. We found that the bioluminescent signal from SK-ST xenografts increased rapidly 10 days after implantation in the CMC group, but is greatly inhibited in the NTZ group (Fig. 4A and 4B). The signal of SK-FST xenografts slowly increased in both groups at the start, and about 18 days after implantation significant increase (P<0.05) of the signal was seen in the CMC group while sustained decrease of the signal was seen in the NTZ group (Supplementary Fig. S11). This signal change was consistent with the change in tumor size, where 18 days after implantation the average tumor size of the CMC group was significantly larger (P<0.001) than that of the NTZ group (Fig. 5). Twenty-seven days after implantation in a SK-ST xenograft model, we dissected out tumors from each group and observed drastic inhibition of tumor growth in the NTZ group by gross inspection and caliper measurements (P=0.00007) (Fig. 5A and 5B). H&E and Ki67 staining of the tumor samples indicated similar histology and cell proliferation pattern in the CMC and NTZ groups. However, the caspase-3 staining in the NTZ group gave average 23±8 positive cells in each sample (Fig. 6A), which is significantly greater than the average 5±2 positive cells in the CMC group (P<0.03). This indicates that there is a significant increase of apoptosis in the NTZ treated group. The endogenous phospho and c-Myc protein levels were also greatly reduced in the tumor samples from the NTZ group as compared with the CMC group (Fig. 6B). With this regimen, we didn’t observe the common side effects of NTZ in animal models of protozoan infection, such as diarrhea, vomiting and skin rash (7, 10). However, the mice had a 7-10% body weight loss in the NTZ group but not in the CMC group 12 days after implantation (Supplementary Fig. S12). This weight loss can be reversed upon removal of the NTZ treatment. Together, these results demonstrated an in vivo efficacy of NTZ in inhibiting c-Myc activity and tumor growth.
Figure 4.
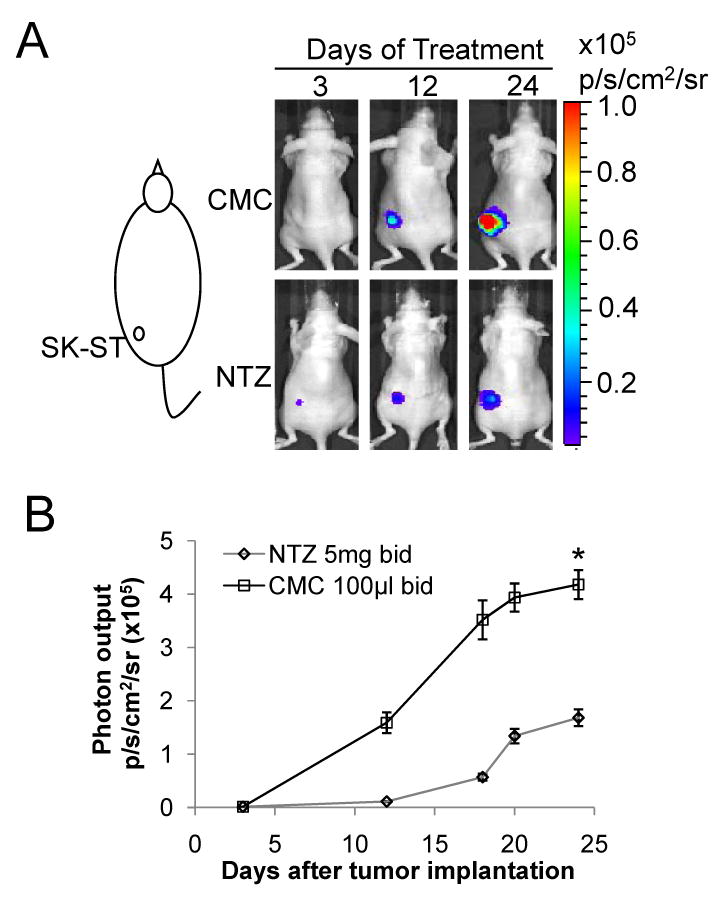
Efficacy of NTZ in a breast cancer xenograft mouse model (A) SK-ST stable cells were s.c. implanted as indicated and treated with NTZ or CMC as described in Materials and Methods. Representative images of FL imaging at indicated days of treatment are shown. (B) Photon output of SK-ST cells with NTZ or CMC treatment is plotted against days after implantation. * P<0.05.
Figure 5.
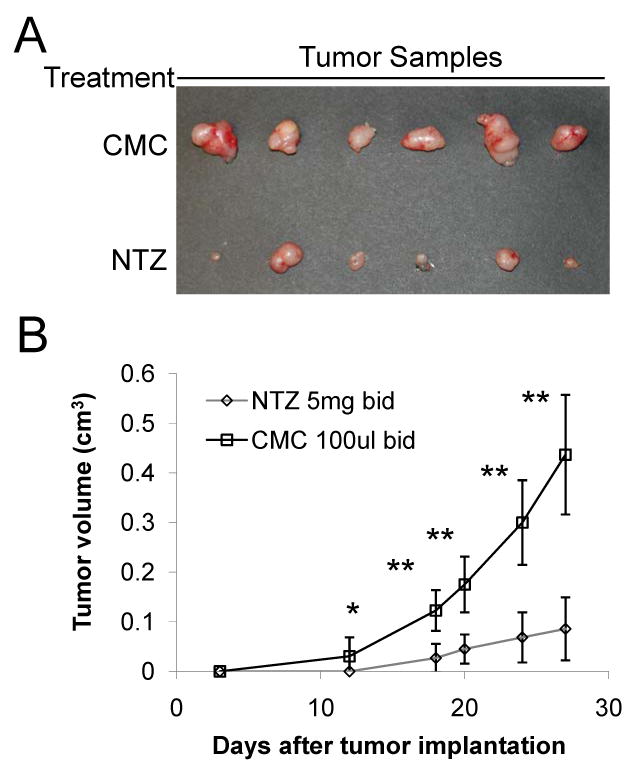
Inhibition of tumor growth by NTZ. (A) Representative photographs of dissected tumors from SK-ST xenograft mouse model with CMC or NTZ treatment 27 days after implantation. (B) Plot of caliper measured tumor sizes of NTZ or CMC treated groups in SK-ST xenograft mouse model at different days after implantation. * P<0.05, ** P<0.01.
Figure 6.
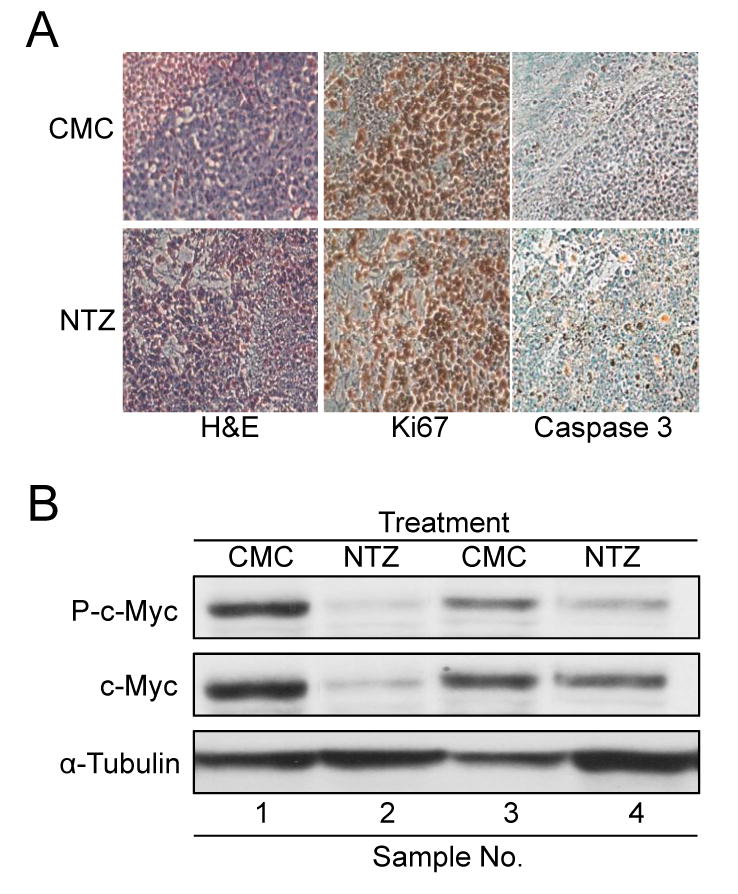
Immunohistochemistry and western blot analysis of NTZ treated tumor samples. (A) Representative images of H&E, Ki67 and caspase-3 IHC staining of tumor tissue samples from each group. (B) Western blot analysis of tumor tissue samples using phospho c-Myc, c-Myc and -tubulin antibodies.
DISCUSSION
Deregulation of c-Myc at the transcriptional level involves mutation, translocation and amplification of the myc gene, which makes targeting c-Myc at the gene expression level not an easy task. Regulation of c-Myc activity at the protein level is an attractive approach, but it is often indirect, since the protein lacks a clear ligand-binding domain for direct interaction (9). Thus an effective targeting of c-Myc protein requires good understanding of its signaling pathways and regulation networks. However, there are currently over 700 binary interactions of c-Myc according to the EBI IntAct, a comprehensive molecular interaction database, while new interactions keep adding to the list (10). Therefore, to identify an effective indirect target out of its complex network by examining the effect of each interaction is clearly labor intensive and often misses the network effect. Systematic profiling of drug inhibitory effect on c-Myc not only can help identify inhibitors of a specific interaction but also discover unexpected multiple or cross interactions, which in turn could reveal unknown function and molecular mechanisms involved in c-Myc signaling. Furthermore, profiling existing drugs for new use has become a more cost effective approach (29).
Efficient and large scale profiling of compound libraries is enabled by high throughput screening technology and a sensitive in vitro assay that can detect the target activity in a HTS format (30). We recently developed a c-Myc activation reporter system that is sensitive to compound inhibition induced changes in c-Myc activity and allows rapid and quantitative detection of those changes in both intact cells and living mice (16). To facilitate systematic compound screening against c-Myc, we further developed this reporter system into a robust cell-based assay and optimized it in a HTS platform (Supplementary Fig. S1-4). This new HTS system enables large-scale screening for compounds that regulate c-Myc activation in live cells. Furthermore, since the sensor-based assay measures changes at the level of the protein itself, it can capture upstream effects on c-Myc from both immediate and multi-level cross interactions. Therefore the targets of the resulted hits can be direct or indirect regulators of c-Myc activation. Although further low-throughput biological assays are needed to examine the c-Myc activation induced downstream effect, this system screens for upstream effectors and tends to identify more potent agents that potentially impact many downstream pathways of c-Myc rather than look for downstream effectors that only involve in a single or a few pathways.
We applied this new screening system on a quantitative HTS (qHTS) platform and screened against five different libraries of total about 5,000 compounds with known bioactivities. The qHTS platform allows compounds to be tested simultaneously in seven different concentrations. It not only facilitates rapid fitting and classification of the concentration-response curves for identification of compounds with a variety of potencies and efficacies, but also is more reliable than traditional single-concentration HTS due to the lower prevalence of false negative hits (30). Not surprisingly, among all the clean hits anti-neoplastic compounds are the most frequently occurred. Interestingly, the second most frequently occurred are anti-parasitic and anti-bacterial compounds (Supplementary Fig. S5). NTZ, one of the anti-parasitic drugs, was among the top potent hits (Supplementary Fig. S6).
NTZ is a thiazolide (Fig. 2C) that consists of a nitrothiazole moiety (left) and a salicylic acid moiety (right) linked via a peptide bond (middle). Studies in different disease models suggest that different sites on NTZ exert different functional activities. The nitro group in the nitrothiazole moiety is responsible for the activities against extracellular parasites and could be converted into a free radical that leads to cell death or interference in cell signaling pathways (31). The nitro group is also required for the inhibition of pyruvate ferredoxin oxidoreductase (PFOR) in anaerobic bacteria (32). The salicylic acid moiety, on the other hand, is responsible for the activities against intracellular parasites by interacting with thiazolide-binding proteins. More recent studies in other pathogens indicate that NTZ has a broad-spectrum of activity by interfering in various signaling pathways. Studies in HCV suggested that NTZ exhibits anti-viral activity by inducing PKR (protein kinase activated by double-stranded RNA) mediated phosphorylation of eIF2α that enhances host cell antiviral defenses (33). A study in Mycobacteria tuberculosis (Mtb) suggests that NTZ inhibits mTORC1 signaling pathway (34), which is known to regulate c-Myc activity. Another study in mouse macrophages showed inhibitory activity of NTZ against IL-6 production (35). Although there was no reported in vivo study on anti-neoplastic activity of NTZ, an in vitro study in human colon cancer cells suggested that NTZ inhibits tumor cell growth by interaction with glutathione-S-transferase P1 (GSTP1) (36). Interestingly, colon cancer has the highest myc over-expression among all human cancers (GenAtlas.org) and GSTP1 interferes the kinase activity of MAPK8 (37), which is an upstream regulator of c-Myc. Thus, the growth inhibition by NTZ in colon cancer cells could be a downstream effect of c-Myc inhibition. Our study, for the first time, revealed a potent efficacy of NTZ in c-Myc inhibition both in tumor cell lines and mouse tumor xenograft models.
To further explore the molecular mechanism of NTZ’s inhibitory effect on c-Myc, we first checked ChEMBL, a comprehensive bioactivity database for medicinal chemicals, and collected all experimentally identified NTZ targets. There are total 30 different human targets of NTZ, including MAPK1/ERK and TP53/P53 (Supplementary Fig. S13). ERK is the immediate upstream kinase of c-Myc, which phosphorylates and activates c-Myc, while the tumor suppressor protein P53 is often suppressed when c-Myc is activated. These in vitro data suggest potential pathways that NTZ acts on to inhibit c-Myc activation. Since both ERK and TP53 act upstream of c-Myc activation, these data also provide validation for our screening strategy for upstream inhibitors of c-Myc. Alternatively, we found another anti-parasitic family member, pyrvinium, has similar anti-neoplastic effect. It was shown that pyrvinium inhibits the cell growth of human colon cancer (38). Casien kinase 1 alpha (CK1α) was identified as the target of pyrvinium. Interestingly, one of CK1α’s targets, tau (39), is also a target of NTZ (Supplementary Fig. S13).
The wide spectrum of NTZ targets implies the complexity of its action. NTZ is supposed to bind its target to exert its function inside cells. Since c-Myc lacks a clear ligand-binding domain for direct interaction (40), NTZ most likely affects c-Myc activity indirectly, and possibly via multiple routes. Further experiments to elucidate the molecular mechanism include examining upstream regulatory factors of c-Myc, such as the MAPK signaling pathway (Supplementary Fig. S13); c-Myc interacting co-factors, such as Max; or those indirect regulators, such as HMG-coA or eIF2α. A more systematic approach toward mechanistic determination might include obtaining a gene expression profile of NTZ treatment using microarray analysis. Besides looking for alteration of gene expression of upstream regulators, this approach may also reveal expression profiles of downstream target genes and help pinpoint the effectors for NTZ induced apoptosis (Fig. 6A). More validation experiments for the anti-tumor efficacy of NTZ would include applying NTZ to various human cancer cell lines and tumor models, such as orthotopic or in-situ conditional transgenic models (41). All these would help determine the spectrum of NTZ efficacy against human cancers.
Further to its wide spectrum of activity, NTZ is extremely well tolerated. It was the first anti-parasitic compound developed for human use back in the mid-1990s (42) and is the only anti-protozoal drug approved for use in children. Indeed, there was a wide range between LD50 and EC50 of NTZ in SKBR3 breast cancer cells (Fig. 3 and Supplementary Fig. S10). The ratio between LD50 and IC50 is often referred as the therapeutic index of the drug (43) and the higher the ratio the safer the drug. Our study indicates that at the effective dose, NTZ inhibits c-Myc activation and induces apoptosis thus inhibits tumor growth in vivo mouse model, rather than bluntly kills cell. The safe pharmaceutical profile of NTZ for treatment of parasitic infections will likely help accelerate evaluation in any potential future clinical trials. Indeed, phase II clinical trials of NTZ anti-viral therapy were carried out before the understanding of its molecular mechanism of anti-viral activity (44). These trials provided valuable information about effective regimens that cannot be obtained in preclinical experiments. For example, the realization that a 4-week lead-in of NTZ monotherapy followed by combination therapy with a current anti-HCV agent provides sustained anti-viral efficacy (45). The combination approach in anti-viral therapy hints at the possible application of NTZ in anti-neoplastic therapy. In fact, most of the top hits from our screen were anti-neoplastic agents (Fig. S4), such as fludarabine phosphate (F-ara-A) (Supplementary Fig. S8) currently used for chronic lymphocytic leukemia (CLL). While it is interesting to figure out how a DNA analog has anti-c-Myc activity, it remains potentially of benefit to know whether combining F-ara-A and NTZ could improve clinical outcome of CLL.
Here we developed a new molecular imaging based quantitative high throughput screening approach. By utilizing it, we identified an anti-c-Myc effect of NTZ and validated its efficacy in both tumor cell lines and mouse xenograft models. Our study provides evidence for a potential new spectrum of clinical applications of NTZ. Moreover, the molecular imaging strategy coupled with systematic profiling of compound libraries promises to accelerate identification of new therapeutic agents or re-purposing of known drugs, as well as their in vivo validation in preclinical models of disease.
Supplementary Material
Acknowledgments
We would like to thank Dr. Carmel Chan for critical review and suggestions.
GRANT SUPPORT
This work is supported in part by NCI grants, R01 CA82214 (S.S. Gambhir) and ICMIC P50 CA114747 (S.S. Gambhir.). Dr. Fan-Minogue is supported by a NIH R25T CA118681 training grant. Dr. Massoud received partial salary support from the NIHR Cambridge Biomedical Research Center.
Footnotes
Conflicts of Interest: The authors disclose no potential conflicts of interest.
References
- 1.Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463(7283):899–905. doi: 10.1038/nature08822. Epub 2010/02/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eilers M, Eisenman RN. Myc's broad reach. Genes Dev. 2008;22(20):2755–66. doi: 10.1101/gad.1712408. Epub 2008/10/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jain M, Arvanitis C, Chu K, Dewey W, Leonhardt E, Trinh M, et al. Sustained loss of a neoplastic phenotype by brief inactivation of MYC. Science. 2002;297(5578):102–4. doi: 10.1126/science.1071489. Epub 2002/07/06. [DOI] [PubMed] [Google Scholar]
- 4.Soucek L, Whitfield J, Martins CP, Finch AJ, Murphy DJ, Sodir NM, et al. Modelling Myc inhibition as a cancer therapy. Nature. 2008;455(7213):679–83. doi: 10.1038/nature07260. Epub 2008/08/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hermeking H. The MYC oncogene as a cancer drug target. Curr Cancer Drug Targets. 2003;3(3):163–75. doi: 10.2174/1568009033481949. Epub 2003/05/29. [DOI] [PubMed] [Google Scholar]
- 6.Wang YH, Liu S, Zhang G, Zhou CQ, Zhu HX, Zhou XB, et al. Knockdown of c-Myc expression by RNAi inhibits MCF-7 breast tumor cells growth in vitro and in vivo. Breast Cancer Res. 2005;7(2):R220–8. doi: 10.1186/bcr975. Epub 2005/03/04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim HG, Miller DM. Inhibition of in vitro transcription by a triplex-forming oligonucleotide targeted to human c-myc P2 promoter. Biochemistry. 1995;34(25):8165–71. doi: 10.1021/bi00025a023. Epub 1995/06/27. [DOI] [PubMed] [Google Scholar]
- 8.Kimura S, Maekawa T, Hirakawa K, Murakami A, Abe T. Alterations of c-myc expression by antisense oligodeoxynucleotides enhance the induction of apoptosis in HL-60 cells. Cancer Res. 1995;55(6):1379–84. Epub 1995/03/15. [PubMed] [Google Scholar]
- 9.Vita M, Henriksson M. The Myc oncoprotein as a therapeutic target for human cancer. Semin Cancer Biol. 2006;16(4):318–30. doi: 10.1016/j.semcancer.2006.07.015. Epub 2006/08/29. [DOI] [PubMed] [Google Scholar]
- 10.Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, et al. BET Bromodomain Inhibition as a Therapeutic Strategy to Target c-Myc. Cell. 2011;146(6):904–17. doi: 10.1016/j.cell.2011.08.017. Epub 2011/09/06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011;478(7370):529–33. doi: 10.1038/nature10509. Epub 2011/10/04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berg T, Cohen SB, Desharnais J, Sonderegger C, Maslyar DJ, Goldberg J, et al. Small-molecule antagonists of Myc/Max dimerization inhibit Myc-induced transformation of chicken embryo fibroblasts. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(6):3830–5. doi: 10.1073/pnas.062036999. Epub 2002/03/138300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mo H, Henriksson M. Identification of small molecules that induce apoptosis in a Myc-dependent manner and inhibit Myc-driven transformation. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(16):6344–9. doi: 10.1073/pnas.0601418103. Epub 2006/04/163440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hydbring P, Bahram F, Su Y, Tronnersjo S, Hogstrand K, von der Lehr N, et al. Phosphorylation by Cdk2 is required for Myc to repress Ras-induced senescence in cotransformation. Proceedings of the National Academy of Sciences of the United States of America. 2009;107(1):58–63. doi: 10.1073/pnas.0900121106. Epub 2009/12/0580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fremin C, Meloche S. From basic research to clinical development of MEK1/2 inhibitors for cancer therapy. J Hematol Oncol. 2010;3:8. doi: 10.1186/1756-8722-3-8. Epub 2010/02/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fan-Minogue H, Cao Z, Paulmurugan R, Chan CT, Massoud TF, Felsher DW, et al. Noninvasive molecular imaging of c-Myc activation in living mice. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(36):15892–7. doi: 10.1073/pnas.1007443107. Epub 2010/08/1158920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cao Z, Fan-Minogue H, Bellovin DI, Yevtodiyenko A, Arzeno J, Yang Q, et al. MYC phosphorylation, activation, and tumorigenic potential in hepatocellular carcinoma are regulated by HMG-CoA reductase. Cancer Res. 2011;71(6):2286–97. doi: 10.1158/0008-5472.CAN-10-3367. Epub 2011/01/222860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Willmann JK, van Bruggen N, Dinkelborg LM, Gambhir SS. Molecular imaging in drug development. Nat Rev Drug Discov. 2008;7(7):591–607. doi: 10.1038/nrd2290. Epub 2008/07/05910. [DOI] [PubMed] [Google Scholar]
- 19.Tseng JR, Stuart D, Aardalen K, Kaplan A, Aziz N, Hughes NP, et al. Use of DNA microarray and small animal positron emission tomography in preclinical drug evaluation of RAF265, a novel B-Raf/VEGFR-2 inhibitor. Neoplasia. 2011;13(3):266–75. doi: 10.1593/neo.101466. Epub 2011/03/12660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chan CT, Paulmurugan R, Gheysens OS, Kim J, Chiosis G, Gambhir SS. Molecular imaging of the efficacy of heat shock protein 90 inhibitors in living subjects. Cancer Res. 2008;68(1):216–26. doi: 10.1158/0008-5472.CAN-07-2268. Epub 2008/01/02160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fukazawa T, Maeda Y, Matsuoka J, Yamatsuji T, Shigemitsu K, Morita I, et al. Inhibition of Myc effectively targets KRAS mutation-positive lung cancer expressing high levels of Myc. Anticancer Res. 2010;30(10):4193–200. Epub 2010/11/041930. [PubMed] [Google Scholar]
- 22.Zhang JH, Chung TD, Oldenburg KR. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen. 1999;4(2):67–73. doi: 10.1177/108705719900400206. Epub 2000/06/0670. [DOI] [PubMed] [Google Scholar]
- 23.Sigma-Aldrich. LOPAC1280™. A Versatile Library for Assay Validation and High Throughput Screening. Sigma Product Assets. 2004 [Google Scholar]
- 24.Kaneto H, Suzuma K, Sharma A, Bonner-Weir S, King GL, Weir GC. Involvement of protein kinase C beta 2 in c-myc induction by high glucose in pancreatic beta-cells. J Biol Chem. 2002;277(5):3680–5. doi: 10.1074/jbc.M109647200. Epub 2001/11/236800. [DOI] [PubMed] [Google Scholar]
- 25.Kobayashi E, Nakano H, Morimoto M, Tamaoki T. Calphostin C (UCN-1028C), a novel microbial compound, is a highly potent and specific inhibitor of protein kinase C. Biochem Biophys Res Commun. 1989;159(2):548–53. doi: 10.1016/0006-291x(89)90028-4. Epub 1989/03/15480. [DOI] [PubMed] [Google Scholar]
- 26.White CA., Jr Nitazoxanide: a new broad spectrum antiparasitic agent. Expert Rev Anti Infect Ther. 2004;2(1):43–9. doi: 10.1586/14787210.2.1.43. Epub 2004/10/1430. [DOI] [PubMed] [Google Scholar]
- 27.Zhao Z, Xue F, Zhang L, Zhang K, Fei C, Zheng W, et al. The pharmacokinetics of nitazoxanide active metabolite (tizoxanide) in goats and its protein binding ability in vitro. J Vet Pharmacol Ther. 2010;33(2):147–53. doi: 10.1111/j.1365-2885.2009.01119.x. Epub 2010/05/01470. [DOI] [PubMed] [Google Scholar]
- 28.Theodos CM, Griffiths JK, D'Onfro J, Fairfield A, Tzipori S. Efficacy of nitazoxanide against Cryptosporidium parvum in cell culture and in animal models. Antimicrob Agents Chemother. 1998;42(8):1959–65. doi: 10.1128/aac.42.8.1959. Epub 1998/08/019590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harrison C. Signatures for drug repositioning. Nat Rev Genet. 2011;12(10):668. doi: 10.1038/nrg3076. Epub 2011/09/17. [DOI] [PubMed] [Google Scholar]
- 30.Inglese J, Auld DS, Jadhav A, Johnson RL, Simeonov A, Yasgar A, et al. Quantitative high-throughput screening: a titration-based approach that efficiently identifies biological activities in large chemical libraries. Proc Natl Acad Sci U S A. 2006;103(31):11473–8. doi: 10.1073/pnas.0604348103. Epub 2006/07/2114730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hemphill A, Mueller J, Esposito M. Nitazoxanide, a broad-spectrum thiazolide anti-infective agent for the treatment of gastrointestinal infections. Expert Opin Pharmacother. 2006;7(7):953–64. doi: 10.1517/14656566.7.7.953. Epub 2006/04/29530. [DOI] [PubMed] [Google Scholar]
- 32.Sisson G, Goodwin A, Raudonikiene A, Hughes NJ, Mukhopadhyay AK, Berg DE, et al. Enzymes associated with reductive activation and action of nitazoxanide, nitrofurans, and metronidazole in Helicobacter pylori. Antimicrob Agents Chemother. 2002;46(7):2116–23. doi: 10.1128/AAC.46.7.2116-2123.2002. Epub 2002/06/121160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Elazar M, Liu M, McKenna SA, Liu P, Gehrig EA, Puglisi JD, et al. The anti-hepatitis C agent nitazoxanide induces phosphorylation of eukaryotic initiation factor 2alpha via protein kinase activated by double-stranded RNA activation. Gastroenterology. 2009;137(5):1827–35. doi: 10.1053/j.gastro.2009.07.056. Epub 2009/08/118270. [DOI] [PubMed] [Google Scholar]
- 34.Lam KK, Zheng X, Forestieri R, Balgi AD, Nodwell M, Vollett S, et al. Nitazoxanide Stimulates Autophagy and Inhibits mTORC1 Signaling and Intracellular Proliferation of Mycobacterium tuberculosis. PLoS Pathog. 2012;8(5):e1002691. doi: 10.1371/journal.ppat.1002691. Epub 2012/05/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hong SK, Kim HJ, Song CS, Choi IS, Lee JB, Park SY. Nitazoxanide suppresses IL-6 production in LPS-stimulated mouse macrophages and TG-injected mice. International immunopharmacology. 2012;13(1):23–7. doi: 10.1016/j.intimp.2012.03.002. Epub 2012/03/2230. [DOI] [PubMed] [Google Scholar]
- 36.Muller J, Sidler D, Nachbur U, Wastling J, Brunner T, Hemphill A. Thiazolides inhibit growth and induce glutathione-S-transferase Pi (GSTP1)-dependent cell death in human colon cancer cells. Int J Cancer. 2008;123(8):1797–806. doi: 10.1002/ijc.23755. Epub 2008/08/017970. [DOI] [PubMed] [Google Scholar]
- 37.Elsby R, Kitteringham NR, Goldring CE, Lovatt CA, Chamberlain M, Henderson CJ, et al. Increased constitutive c-Jun N-terminal kinase signaling in mice lacking glutathione S-transferase Pi. J Biol Chem. 2003;278(25):22243–9. doi: 10.1074/jbc.M301211200. Epub 2003/03/2222430. [DOI] [PubMed] [Google Scholar]
- 38.Thorne CA, Hanson AJ, Schneider J, Tahinci E, Orton D, Cselenyi CS, et al. Small-molecule inhibition of Wnt signaling through activation of casein kinase 1alpha. Nat Chem Biol. 2010;6(11):829–36. doi: 10.1038/nchembio.453. Epub 2010/10/08290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gao ZH, Seeling JM, Hill V, Yochum A, Virshup DM. Casein kinase I phosphorylates and destabilizes the beta-catenin degradation complex. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(3):1182–7. doi: 10.1073/pnas.032468199. Epub 2002/01/311820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Darnell JE., Jr Transcription factors as targets for cancer therapy. Nat Rev Cancer. 2002;2(10):740–9. doi: 10.1038/nrc906. Epub 2002/10/07400. [DOI] [PubMed] [Google Scholar]
- 41.Guttner Y, Windsor HM, Viiala CH, Dusci L, Marshall BJ. Nitazoxanide in treatment of Helicobacter pylori: a clinical and in vitro study. Antimicrob Agents Chemother. 2003;47(12):3780–3. doi: 10.1128/AAC.47.12.3780-3783.2003. Epub 2003/11/237800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fox LM, Saravolatz LD. Nitazoxanide: a new thiazolide antiparasitic agent. Clin Infect Dis. 2005;40(8):1173–80. doi: 10.1086/428839. Epub 2005/03/211730. [DOI] [PubMed] [Google Scholar]
- 43.Stanley TH. Anesthesia for the 21st century. Proc (Bayl Univ Med Cent) 2000;13(1):7–10. doi: 10.1080/08998280.2000.11927635. Epub 2006/01/070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rossignol JF, Kabil SM, El-Gohary Y, Elfert A, Keeffe EB. Clinical trial: randomized, double-blind, placebo-controlled study of nitazoxanide monotherapy for the treatment of patients with chronic hepatitis C genotype 4. Aliment Pharmacol Ther. 2008;28(5):574–80. doi: 10.1111/j.1365-2036.2008.03781.x. Epub 2008/07/15740. [DOI] [PubMed] [Google Scholar]
- 45.Rossignol JF, Elfert A, Keeffe EB. Treatment of chronic hepatitis C using a 4-week lead-in with nitazoxanide before peginterferon plus nitazoxanide. J Clin Gastroenterol. 2010;44(7):504–9. doi: 10.1097/MCG.0b013e3181bf9b15. Epub 2010/01/05040. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.