

Abstract
Rationale
Endothelial microRNA-126 (miR-126) modulates vascular development and angiogenesis. However, its role in the regulation of smooth muscle cell (SMC) function is unknown.
Objective
To elucidate the role of miR-126 secreted by endothelial cells (ECs) in regulating SMC turnover in vitro and in vivo, as well as the effects of shear stress (SS) on the regulation.
Methods and Results
Co-culture of SMCs with ECs or treatment of SMCs with conditioned media from static EC monoculture (EC-CM) increased SMC miR-126 level and SMC turnover; these effects were abolished by inhibition of endothelial miR-126 and by the application of laminar SS (LSS) to ECs. SMC miR-126 did not increase when treated with EC-CM from ECs subjected to inhibition of miR biogenesis, nor with CM from sheared ECs. Depletion of extracellular/secreted vesicles in EC-CM did not affect the increase of SMC miR-126 by EC-CM. Biotinylated miR-126 or FLAG-tagged Ago2 transfected into ECs were detected in the co-cultured or EC-CM treated SMCs, indicating a direct EC-to-SMC transmission of miR-126 and Ago2. Endothelial miR-126 represses FOXO3, BCL2, and IRS1 mRNAs in the co-cultured SMCs, suggesting the functional roles of the transmitted miR-126. Systemic depletion of miR-126 in mice inhibited neointimal lesion formation of carotid arteries induced by cessation of blood flow. Administration of EC-CM or miR-126 mitigated the inhibitory effect.
Conclusions
Endothelial miR-126 acts as a key intercellular mediator to increase SMC turnover, and its release is reduced by atheroprotective LSS.
Keywords: Atherosclerosis, endothelial cell, extracellular miR-126, shear stress, smooth muscle cell
Introduction
Vascular smooth muscle cells (SMCs) are highly proliferative and migratory during the early embryonic stages of vasculogenesis, but become differentiated and quiescent in adult blood vessels 1. They can be activated and regain their highly proliferative characteristics under certain pathophysiological conditions, such as vascular injury and atherogenesis, thus contributing to the thickening and stiffening of the arterial wall 2. During atherogenesis, activated SMCs migrate into the intima, where they come into close contact with endothelial cells (ECs) lining the vessel wall 3. In the atherogenic vessel, SMC apoptosis occurs and is followed by proliferation of the remaining cells, thus inducing destabilization and vulnerability of the lesion plaques 4. The present study was designed to study the effects of EC-SMC interaction on SMC proliferation/apoptosis and neointimal formation and the underlying mechanisms, as well as the actions of different patterns of shear stress (SS) on these effects.
MicroRNAs (miRs) are noncoding single-stranded RNA molecules of ∼22 nucleotides that regulate mRNAs by binding to target sequences of mRNAs to cause their degradation or translation repression 5. In addition to their primary intracellular locations, miRs have recently been found extracellularly in plasma and other body fluids of humans and mice 6-9. Changes in extracellular circulating miR level and activity have been linked to pathologies of cardiovascular disorders, including acute myocardial infarction, heart failure, and coronary artery disease 10-12, suggesting the possibility that extracellular miRs may have diagnostic or prognostic values in cardiovascular diseases. Recent studies reveal that miRs could be delivered to recipient cells with functional targeting capabilities, leading to altered gene expression in the recipient cells 7, 13, 14. These miRs may be involved in intercellular communication and intracellular signal transduction.
MiR-126 is highly expressed in ECs to modulate vascular development and angiogenesis 15-17. MiR-126 expression in rat carotid artery is significant under normal condition 18 and is markedly increased after angioplasty 18, suggesting a potential modulatory role of miR-126 in vascular pathogenesis. The functional relevance of miR-126 in vascular SMCs has not yet been reported. Our present study shows that EC-secreted miR-126 regulated SMC gene expression and cellular functions via a paracrine effect, and that laminar SS (LSS) applied to ECs attenuated this effect; Argonaute2 (Ago2), the effecter component of the miRNA-induced silencing complex (miRISCs), associated with the EC-secreted miR-126 and facilitated its transmission from ECs to SMCs; knockout of miR-126 (KO) in mouse inhibited neointimal hyperplasia in the carotid arteries induced by cessation of blood flow. These results demonstrate that miR-126 plays an important role in EC-SMC interaction to modulate SMC function and vascular homeostasis.
Methods
The detailed methods are described in Supplementary Methods.
EC-SMC co-culture, and parallel-plate co-culture flow system
EC-SMC co-culture was established by plating human umbilical vein ECs (HUVECs) and human umbilical artery SMCs (HUASMCs) on the lower and upper sides, respectively, of a 10-μm-thick porous membrane with 0.4-μm pores in a transwell cell culture insert (Becton Dickinson). ECs and SMCs were maintained in their respective media supplemented with 2% FBS. The insert was incorporated into a parallel-plate flow chamber that contains a polycarbonate insert holder and is connected to a perfusion loop system. ECs were exposed to either LSS with a high level of mean SS at 12 dynes/cm2 or oscillatory SS (OSS) with a low level of mean SS at 0.5 dynes/cm2 and a superimposed sinusoidal oscillation with a frequency of 1 Hz and a peak-to-peak amplitude of ±4 dynes/cm2.
Animal model
Animal surgical procedures were performed in accordance with National Institutes of Health guidelines and were approved by the UC San Diego IACUC. Mice were subjected to carotid artery ligation as previously described19. Some mice were injected intravenously (iv) twice per week for 4 weeks with 200 μl of EC conditioned media (EC-CM) from 3-day monocultured static HUVECs or with control media. Some mice received a local administration of miR-126 mimics (PRE126) or negative control mimics (PREC) (2 μg) mixed with pluronic gel [25% (w/v), Pluronic F127, Sigma]. After 4 weeks of ligation, the ligated and unligated control carotid arteries were harvested and subjected to histology and immunostaining analyses of the vessels.
Statistical analysis
Data are expressed as mean ± SEM from three independent experiments. Statistical analysis was performed by Student's t-test for two groups of data and by one-way ANOVA for multiple comparisons. Statistical significance among multiple groups was determined by post hoc analysis (Tukey HSD test). P<0.05 was considered statistically significant.
Results
The increase of miR-126 level in the SMCs co-cultured with ECs is suppressed by LSS on ECs
By utilizing a parallel-plate co-culture flow system (Figure 1A), we determined the miR-126 levels in SMCs in response to co-culture with ECs and flow shear application to ECs. Under static condition, the co-culture of SMCs with ECs significantly increased the SMC mature form of miR-126, but not the primary transcript of miR-126 (pri-miR-126) (Figure 1B), indicating that the effect of EC co-culture on SMC miR-126 occurs at post-transcriptional level. Atheroprotective LSS (12 dynes/cm2) applied to ECs suppressed the level of miR-126 in the co-cultured SMCs, and this suppression lasted for at least 24 hr (Figure 1B). MiR-126 level in the EC conditioned media (CM) was markedly attenuated by exposure of ECs to LSS for 24 hr (Figure 1C), suggesting that the effects of LSS on miR-126 in SMCs are mediated at least partially through alterations in miR-126 secretion from ECs. Treatment of SMCs with CM from static ECs caused an enrichment of SMC miR-126; this enrichment was absent with CM obtained from ECs exposed to LSS for 24 hr (Figure 1D). The miR-126 level in ECs did not show significant difference between static mono-cultured and co-cultured ECs (Figure 1E), nor between static and LSS-treated mono-cultured ECs (Figure 1F). These results indicate that co-culture of SMCs with ECs does not induce miR-126 expression in ECs and that the inhibitory effect of LSS on miR-126 enrichment in the co-cultured SMCs does not involve a down-regulation of miR-126 in ECs. Together with the finding that LSS did not change the primary or mature miR-126 in the co-cultured ECs (Figure 1G), our results indicate that SS and SMC co-culture have no synergistic effects on the transcriptional regulation of miR-126 in ECs. These results demonstrate that the induction of SMC miR-126 by co-culture with ECs and the suppression of this induction by LSS on ECs are mediated by intercellular humoral mechanisms.
Figure 1. Atheroprotective SS to ECs suppresses miR-126 in the co-cultured SMCs.

(A), Schematic diagrams of EC/SMC co-culture and flow system. (B), HUVECs and HUASMCs were seeded on the lower and upper sides of a membrane, respectively (EC/SMC). Controls had SMCs but no ECs on the other side (Ø/SMC). The temporal expression levels of primary (pri-miR-126) and mature (miR-126) forms of miR-126 were determined by quantitative RT-PCR. * P < 0.05 vs. Ø/SMC 0 hr Static. (C), The levels of miR-126 in the 24-hr static or sheared media. * P < 0.05 vs. static control. (D), The levels of miR-126 in SMCs incubated with medium 199 supplemented with 2% FBS (CL Media), or conditioned medium from static or sheared ECs (24-hr shearing) (EC-CM). * P < 0.05 vs. CL Media. (E), miR-126 expression in ECs mono-culture or SMC-co-culture for 24 hr. (F), miR-126 expressisons in ECs exposed to LSS for 24 hr. (G), The levels of pri-miR-126 and miR-126 in ECs co-culture with SMCs under 24 hr-static or -LSS conditions.
In contrast to LSS, atheroprone oscillatory SS (OSS) (0.5±4 dynes/cm2) augmented the accumulation of mature miR-126 and also the pri-miR-126 level in SMCs (Online Figure IA), without a significant change of miR-126 level in EC-CM (Online Figure IB). Although OSS caused an increase in the SMC miR-126, it did not change the primary or mature miR-126 in the co-cultured ECs (Online Figure IC). These results suggest that OSS engenders a different regulatory machinery to modulate miR-126 secretion from ECs.
We also tested the flow-regulation of miR-126 enrichment in vivo. RNAs isolated from the descending thoracic aorta (TA) and the aortic arch (AA) of mice were subjected to determination of miR-126 levels in the vessel walls. Endothelia were denuded by perfusion with PBS buffer followed by air massage before excision of the vessels. A 3-fold higher expression of miR-126 was observed in the athero-susceptible AA (Online Figure IIA), which experiences disturbed and low SS, in comparison with the TA, which is exposed to laminar flow. The expression of the endothelial marker PECAM1 in AA was lower than that in TA (Online Figure IIB), indicating that the increased miR-126 level in AA is in the media layer and not due to contamination from the undenuded endothelia. These results validate the differential modulations of SMC miR-126 levels by different flow patterns in vivo.
ECs and SS regulate expressions of FOXO3, BCL2, and IRS1, the direct targets of miR-126, in SMCs
We tested whether the modulations of SMC miR-126 lead to consequential changes of the miR-126-targeted gene expressions in SMCs. FOXO3, BCL2, and IRS1 are miR-126 targets predicted by bioinformatics. MiRISC pull-down assay in SMCs validated the association of FOXO3, BCL2, and IRS1 mRNAs with Ago2-containing miRISCs in SMCs (Figure 2A). The association of these miR-126 targets with miRISCs was reduced by transfection of SMCs with miR-126 inhibitor (AM126) and enhanced by miR-126 mimics (PRE126) (Figure 2A). The role of miR-126 in the repression of FOXO3, BCL2, and IRS1 mRNAs in SMCs was also demonstrated by the up- and down-regulations of the transcripts of these genes in SMCs by AM126 and PRE126, respectively (Online Figure III). These effects of miR-126 were further validated by the responses of FOXO3, BCL2, and IRS1 3′ untranslated region (3′UTR) reporters to the manipulation of miR-126 in SMCs. At 24 hr post-transfection of SMCs with PRE126, there was a significant repression of the luciferase reporter activities of FOXO3, BCL2, and IRS1 wild-type 3′UTR constructs, as compared with the control vector without 3′UTR, but this repression was not seen with the mutated 3′UTR (miR-126 seeding sites substituted with scrambled sequences) (Figure 2B). In complementary experiments, there was a significant de-repression on the wild-type (but not the mutated) 3′UTR reporter activities of those three genes when the SMCs were transfected with AM126, as compared with AMC (Figure 2B). As expected, incubation of SMCs with EC-CM collected from mono-cultured ECs significantly inhibited the reporter activities of the wild-type 3′UTRs, but not the mutated 3′UTRs (Figure 2C). Repression and de-repression of FOXO3, BCL2, and IRS1 were observed when the SMCs were co-cultured with static and sheared ECs, respectively, in comparison to the mono-cultured SMCs (Figure 2D). These results indicate that ECs and SS regulate expressions of FOXO3, BCL2, and IRS1, the direct targets of miR-126, in SMCs.
Figure 2. ECs and SS regulate expressions of miR-126 targets in SMCs.

(A), The mRNA levels of FOXO3, BCL2, and IRS in Ago2-immunocomplexes in SMCs 48 hr post-transfection with PRE126 or AM126. * P < 0.05 vs. cells transfected with negative controls (PREC or AMC). (B), The relative luciferase levels in SMCs co-transfected with PRE126 (Left), AM126 (Right), or the negative control and luciferase reporter plasmids: empty vector only, wild type 3′-UTR of FOXO3, BCL2, or IRS1, and mutants of 3′-UTR of FOXO3, BCL2, or IRS1. (C), The relative luciferase levels in SMCs transfected with the indicated luciferase reporter plasmids and then incubated with control media or EC-CM. * P < 0.05 vs. cells transfected with empty vector. #P < 0.05 vs. cells transfected with plasmids harboring the wild type 3′-UTRs. (D), The protein levels of target genes in SMCs mono-cultured (Ø/SMC) or co-cultured with ECs (EC/SMC) kept under static condition or exposed to LSS. Images are representative of triplicate experiments with similar results.
Transmission of miR-126 from ECs to SMCs in a paracrine manner
We hypothesized that the miR-126 enrichment in SMCs following EC-co-culture is induced by a humoral transmission of miR-126 from ECs to SMCs. To test this hypothesis, a synthetic miR-126 (a 22-oligomer labeled with psoralen-biotin conjugate) was transfected into ECs, and the culture media were refreshed 8 hr post-transfection to remove the free oligonucleotides. The EC-CM was collected 24 hours after the refreshment. Transfection of ECs with this biotinylated miR-126 (B-miR-126) led to a marked enrichment of B-miR-126 in ECs and in the EC-CM in comparison to the mock transfection (Figure 3A). Incubation of SMCs with the EC-CM derived from ECs transfected with the B-miR-126 led to the presence of B-miR-126 in SMCs (Figure 3B). The fluorescent images identified the cytoplasmic presence of B-miR-126 in both ECs and SMCs (Figure 3B), suggesting a direct EC-to-SMC transmission of miR-126. Northern blot confirmed the presence of B-miR-126 in both ECs and co-cultured SMCs (Figure 3C). In SMCs co-cultured with ECs transfected with B-miR-126 or mock control, the B-miR-126 showed a 2.19 ± 0.18 fold enrichment as compared with the mock co-cultured SMCs (Figure 3D).
Figure 3. ECs transfer exogenous miR-126 to SMCs.

SMCs were incubated with CM derived from ECs transfected with biotinylated synthetic miR-126 (B-miR-126) or no-miRs mock control (Mock) for 3 hr. (A),The levels of B-miR-126 in ECs or the EC-CM were determined by pull down experiments with streptavidin-agarose beads followed by quantitative RT-PCR, (B) and B-miR-126 in ECs and SMCs were detected with QDot-605-streptavidin conjugates. * P < 0.05 vs. mock control. (C and D), SMCs were co-cultured with ECs transfected with B-miR-126 or mock control for 24 hr; the levels of B-miR-126 in SMCs were determined by Northern blot (C) or pull down experiments with streptavidin-agarose beads (D). * P < 0.05 vs. mock control.
EC-secreted vesicle-free miR-126 regulates SMC gene expression
To determine the possible EC-secreted constituents responsible for the increase of miR-126 in the recipient SMCs and to characterize the form of the transmitted miR-126, we exposed the EC-CM to deoxyribonuclease (DNase), ribonclease (RNase), or proteinase K (PK) digestions. The ability of EC-CM to increase miR-126 abundance in SMCs was abolished by RNase and reduced by PK, but unaffected by DNase (Figure 4A). These results suggest that RNAs and RNA-protein complexes are transmitted into SMCs, but not DNA components or the vesicle-protected RNA. To study the involvement of miR biogenesis, we knocked down Drosha, Dicer, and Ago2, which are the three key proteins required for the processing and maturation of miRs, in ECs through siRNA-mediated gene silencing. The impairment of miR-126 synthesis in ECs by si-Drosha/Dicer/Ago2(EIF2C2) blocked the miR-126 enrichment in SMCs in response to EC-CM (Figure 4B). We also found that the manipulation of miR-126 in ECs by using AM126 or PRE126 significantly decreased or increased, respectively, the miR-126 enrichment in SMCs treated with EC-CM (Figure 4C). To further verify the transmission of miR-126 from ECs to SMCs, we isolated the lung ECs from either miR-126+/+ (WT) mice or miR-126-/- (KO) mice and co-cultured those ECs with human SMCs. We found that co-culture with the WT ECs increased the miR-126 levels in SMCs, but co-culture with the KO ECs did not (Figure 4D).
Figure 4. Functional miR-126 transmission from ECs to SMCs is independent of vesicles.

(A), The levels of miR-126 in SMCs incubated with CL media or the EC-CM treated with DNase I, RNase A, or PK for 3 hr. * P < 0.05 vs. CL Media. (B), The levels of miR-126 in ECs transfected with control siRNA (siCL) or siRNA against Drosha (siDROSHA), Dicer (siDICER1), or Ago2 (siEIF2C2), or in SMCs incubated with CL media or EC-CM derived from those transfected ECs. (C), The levels of miR-126 in SMCs incubated EC-CM derived from ECs transfected with AM126, PRE126 or the negative controls. * P < 0.05 vs. siCL or CL media. #P < 0.05 vs. the AMC or PREC EC-CM. (D), The levels of miR-126 in SMCs co-cultured with lung ECs isolated from wild-type (WT) or miR-126 knockout mice (KO). * P < 0.05 vs. Ø/SMC. EC-CM was ultracentrifuged to fractionate the components in spin-down-pellets (Pellet, which was reconstituted with M199), or the remaining supernatant (Super). The levels of miR-126 in (E), the fractionated EC-CM or in (F), SMCs incubated with total or fractionated EC-ECM for 3 hr. (G), the transcript levels, or (H), protein levels of indicated genes in SMCs incubated with the fractioned EC-CM. Images are representative of triplicates with similar results. * P < 0.05 vs. CL media.
We separated the extracellular vesicles from the HUVEC-derived CM by ultracentrifugation and assayed the miR-126 levels in the vesicle-containing pellets and the vesicle-poor supernatant. Approximately 60% of the extracellular miR-126 was found to be present in the supernatant, and about 40% was in the pellet (Figure 4E), demonstrating that the EC-derived extracellular miR-126 is not restricted to the vesicles. Treatment of SMCs with the supernatant led to an increase of SMC miR-126 level comparable to that resulting from the unseparated EC-CM, but such an increase of miR-126 was not observed in SMC treated with the reconstructed media containing the precipitated pellet (Figure 4F). These observations are consistent with the result of Fig. 4a in suggesting that the EC-derived vesicle-free miR-126 is the main contributor to the miR-126 enrichment in SMCs.
To further examine the roles of miR-126 in the vesicle vs. vesicle-free components of EC-CM in the EC-induced repression of SMC genes, the SMCs were treated with total EC-CM, supernatant, or pellet. The total EC-CM and the vesicle-free supernatant reduced the transcript and protein levels of FOXO3, BCL2, and IRS1 in SMCs (Figures 4G and H). In contrast, the EC-derived vesicle-containing pellet did not significantly alter the expressions of these miR-126 targets (Figures 4G and H). These results show that ECs transmit functional vesicle-free miR-126 to the recipient SMCs to regulate the miR-126 targeted genes.
A recent study demonstrated that suppression of the ceramide signaling pathway through chemical inhibition of nSMase2 with GW4869 resulted in the attenuation of cellular miR export by exosomes and a significant reduction of specific extracellular miRs 20. In this study, we found that treatment of ECs with 20 μmol/L GW4869 for 2 hr did not affect the increase of miR-126 in the co-cultured SMCs (Online Figure IVA), indicating that the ceramide-triggered secretion of exosomes is not required for the transmission of endogenous miR-126 from ECs to SMCs. It has also been reported that apoptotic body can mediate the transmission of miR-126 from serum-starved apoptotic ECs to the nearby ECs 13. We tested the involvement of apoptotic body in mediating miR-126 delivery from ECs to SMCs by pretreatment of ECs with apoptosis inhibitor (AI) at a concentration of 50 or 100 μg/ml one hr before co-culture. AI treatment changed neither the miR-126 level in EC-CM nor that in the co-cultured SMCs (Online Figure IVB), indicating that miR-126 transmission from ECs to SMCs is not mediated by apoptotic body.
Ago2 carries miR-126 and facilitates its intercellular transmission
Since protease-sensitive components are involved in the transmission of miR-126 from ECs to SMCs (Figure 4A), we next identified the RNA-binding proteins that may stabilize the miRs secreted by ECs and thereby facilitate its uptake by SMCs. Ago2 has been reported to associate with miRs in the circulating blood independent of vesicles 21. We then tested the possibility that Ago2 and miR-126 can form an extracellular complex with a vesicle-excluded mechanism.
Co-immunoprecipitation experiments revealed that Ago2, but not Ago1, exhibited a strong association with miR-126 in the EC-CM (Figure 5A), suggesting that extracellular miR-126 is bound to Ago2. Moreover, when the bovine aortic ECs (BAECs) expressing eGFP-Ago2 fusion proteins were co-cultured with SMCs for 6 hr, the eGFP fluorescence can be detected in both ECs and SMCs (Figure 5B), indicating the transmission of Ago2 from ECs to the recipient SMCs. To confirm the Ago2-coordinated delivery of miR-126 from ECs to SMCs, BAECs co-transfected with synthetic miR-126 and FLAG-Ago2, or the negative controls FLAG-histone deacetylase 4 (HDAC4) (FLAG-CL1) and FLAG-HDAC5 (FLAG-CL2) that do not contain miR-126 seeding sequences, were co-cultured with SMCs and subjected to immunoprecipitation. As expected, anti-FLAG antibody was effective in capturing cellular miR-126 in ECs transfected with the FLAG-Ago2 construct, but not the FLAG-CL1 or -CL2 constructs (Online Figure VA). We detected a significantly larger amount of miR-126 associated with FLAG-Ago2 in SMCs as compared to FLAG-CL1 or -CL2, which exhibited a basal miR-126 enrichment that is probably attributable to the overexpression of synthetic miR-126 in the co-cultured ECs (Online Figure VB). The immunoprecipitation specificity was confirmed using anti-FLAG antibody and IgG control (Online Figure VC).
Figure 5. Ago2 associates with miR-126 and facilitates its transmission.

(A), The levels of miR-126 in EC-CM immunocomplexes of Ago2 or Ago1, or control IgG. * P < 0.05 vs. IgG control. (B), SMCs were co-cultured with BAECs transfected with a plasmid encoding the eGPF-Ago2. The eGFP-Ago2 proteins were visualized in live ECs or SMCs by microscopy. (C), The protection of miR-126 with pre-incubation of BSA or recombinant human Ago2 protein (rAgo2) for 30 min followed by RNase A digestion for 15 min. * P < 0.05 vs. miR-126 only. (D), The enrichment of miR-126 in SMCs treated with miR-126 pre-incubated with BSA or rAgo2 for 3 hr. * P < 0.05 vs. miRs only. (E), The levels of Ago2 in ECs (left) or EC-CM (right) under static or LSS conditions for 24 hr. (F), The levels of miR-126 in Ago2-immunocomplex in EC-CM under static of LSS conditions. Images are representative of triplicates with similar results. Semi-quantification results are shown in the lower panels. * P < 0.05 vs. Static.
To determine whether Ago2 affects miR-126 enrichment in SMCs via miR-126 transmission from ECs to SMCs in addition to its inhibition of miR-126 synthesis in ECs (Figure 4B), we co-transfected ECs with synthetic miR-126 and Ago2-specific siRNA (siEIF2C2) to circumvent the miR-126 biosynthesis. SMCs were treated with EC-CM collected from ECs transfected with miR-126/siEIF2C2 or negative control siRNA, control without miR-126, or mock transfection. Depletion of Ago2 in ECs attenuated the increase of miR-126 in SMCs in response to EC-CM with high miR-126 content (Online Figure VIA), suggesting that Ago2 affected miR-126 transmission in addition to its synthesis. Complementary experiments revealed that knockdown of Ago2 with siEIF2C2 also impaired the delivery of cel-miR-39 from ECs to the SMCs (Online Figure VIB), indicating the requirement of Ago2 in mediating the extracellular transfer of another miR from EC to SMC.
To address whether Ago2 contributes to the stability of miR-126 in the extracellular environment, we tested its ability in protecting miR-126 from RNase digestion. Synthetic miR-126 was incubated with the recombinant human Ago2 (rAgo2), BSA control, or left alone, and then subjected to RNase digestion. A marked increase of RNase resistance was seen when miR-126 was incubated with rAgo2, compared with the miR-126/BSA mixture or miR-126 only (Figure 5C). To investigate whether Ago2 facilitates miR uptake by the recipient cells, SMCs were treated with synthetic miR-126 or cel-miR-39 that had been pre-incubated with rAgo2 or BSA. The resulted high levels of miR-126 and cel-miR-39 in SMCs indicate that Ago2 positively contributed to miR-126 and cel-miR-39 uptakes by SMCs (Figure. 5D and Online Figure VIC). BSA also moderately increased the entry of cel-miR-39 into SMCs (Online Figure VIC), suggesting a non-specific effect of RNA-protein interaction in mediating the transmission of miRs between cells. Complementary experiments indicated that endogenous miR-126, -21, -221, and -155 were associated with Ago2 in EC-CM, and that these associations were not decreased by the treatment of ECs with nSMase2 inhibitor GW4869 (Online Figure VII), suggesting that the Ago2 facilitated miRs transmission may not be miR-126 specific.
SS regulates expression and secretion of Ago2 in ECs
As LSS regulates miR-126 secretion in ECs and hence the EC-to-SMC transmission of miR-126 (Figures. 1B and C), we reasoned that LSS may also regulate Ago2 secretion in ECs. LSS slightly decreased the expression level of Ago2 in ECs, but markedly reduced the abundance of Ago2 and the Ago2-bound-miR-126 level in the EC-CM (Figures 5E and F). Thus, the results suggest that the LSS-induced reduction of miR-126 release from ECs and the subsequent decrease of its uptake by SMCs are likely due to the decreases in EC expression and secretion of Ago2. Although OSS also decreased Ago2 expression and secretion and the Ago2-bound-miR-126 in the EC-CM (Online Figures. VIIIA - C), it did not decrease miR-126 level in CM (Online Figure IB); it is possible that OSS also regulates other pathways to modulate miR-126 transmission.
EC co-culture and LSS modulate SMC turnover via miR-126
Our data indicate that the EC-secreted miR-126 inhibits its targets FOXO3, BCL2, and IRS1 in the recipient SMCs. Since FOXO3, BCL2, and IRS1 are involved in cell proliferation, cell cycle progression, and apoptosis 22-24, we sought to identify the link between miR-126 transmission and the cellular functions modulated by EC-secreted miR-126 in SMCs. SMCs co-cultured with ECs had increased cell percentage with strong nuclear expression of proliferating cell nuclear antigen (PCNA), as compared with mono-cultured control SMCs (Figure 6A). Inhibition of miR-126 in ECs or exposure of ECs to LSS significantly reduced nuclear PCNA expression in SMCs (Figure 6A). The miR-126-mediated alteration of PCNA expression was confirmed by Western blot (Figure 6B). Furthermore, EC co-culture up-regulated cyclinA and down-regulated the cyclin-dependent kinase inhibitor p21 in SMCs (Figure 6B). These EC-induced changes in gene expression of SMC were reversed by LSS or inhibition of miR-126 in the ECs (Figure 6B) or in the SMCs (Online Figure IXB). Cell cycle analysis indicates that application of LSS to ECs not only abolished the increased percentage of SMCs in S phase in the co-culture, but actually decreased it to a level lower than that of the mono-culture control. Inhibition of miR-126 in ECs also reversed the cell cycle progression resulting from EC co-culture (Figure 6C). We also found that the percentage of apoptotic SMCs was increased by co-culture with ECs and that this increase was attenuated by LSS or inhibition of miR-126 in ECs or SMCs (Figure 6D and Online Figure IXD). Taken together, these results suggest that the EC-derived miR-126 increases cell proliferation, cell cycle progression, and apoptosis of co-cultured SMCs. Under LSS, these impaired SMC functions are corrected by the inhibition of humoral transmission of miR-126 from ECs to SMCs.
Fig. 6. SS-imposed ECs modulates SMCs turnover through miR-126.
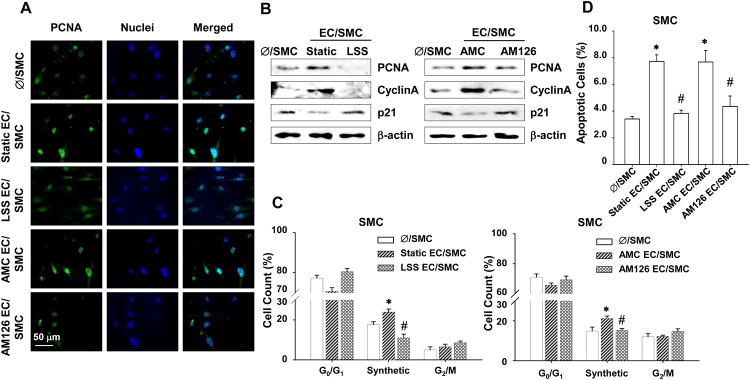
SMCs were mono-cultured or co-cultured with: (1) ECs kept under static condition or exposed to LSS for 24 hr or (2) ECs transfected with AMC or AM126. (A), PCNA in the SMCs was detected by immunostaining. (B), Protein levels of PCNA, cyclinA, and p21 in SMCs were assayed by Western blot. (C and D), SMC proliferation and apoptosis analyzed for propidium iodide stained DNA content and Annexin V stained membrane flip, respectively. * P < 0.05 vs. Ø/SMC. #P < 0.05 vs. Static EC/SMC or AMC EC/SMC.
To characterize the mechanisms by which miR-126 promotes SMC turnover, the three direct targets of miR-126, FOXO3, BCL2, and IRS1 25, were overexpressed in SMCs, which were then co-cultured with ECs. Overexpression of any of these three genes attenuated the EC-co-culture induced expression of PCNA, but not CyclinA, and reversed the effects of co-culture on expression of p21 (Online Figure XA). Overexpression of FOXO3 abolished the increased percentage of SMCs in S phase in the co-culture (Online Figure XB), whereas overexpression of BCL2 inhibited the increased percentage of SMCs undergoing apoptosis induced by co-culture (Online Figure XC). These results suggest that the miR-126-induced SMC proliferation, cell cycle progression, and apoptosis are at least partially attributable to its repression of FOXO3, BCL2, and IRS1.
MiR-126 promotes neointimal formation in mouse carotid artery after ligation
We employed the miR-126 knockout (KO) mice with a ligation-induced neointimal growth model 19 to further determine the effect of miR-126 on the formation of intimal lesion. The left common carotid artery of miR-126 wildtype (WT) or KO mice was ligated near the bifurcation to stop the blood flow, and the right carotid artery was sham operated as a control. Both sides of carotid arteries were isolated 4-weeks post-surgery for the measurements of miR-126 and other molecules and the analysis of histology. In the WT mice, ligation of carotid arteries increased the miR-126 level in the vessels (Figure 7A), promoted the abundance of miR-126 in the intima-media layer (Fig. 7b), and induced neointimal thickening. The presence of endothelium on the luminal surfaces of ligated and unligated carotid arteries was verified by immunostaining for PECAM1 (Online Figure XI). In the KO mice, the ligated arteries displayed a marked reduction in neointimal growth, as indicated by elastic staining (Figure 7C), and the proliferation of SMCs, as indicated by SM-actin (SMA) and PCNA immunostaining, in comparison with those of WT mice (Figures. 7D and E).
Fig. 7. Depletion of miR-126 in mice inhibits SMC proliferation and neointimal formation in arteries after cessation of blood flow.

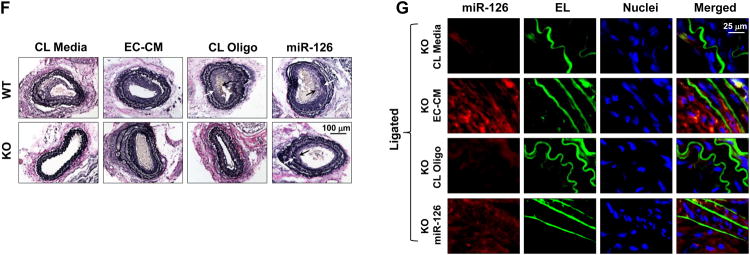
(A), The miR-126 expression levels in mouse carotid arteries with or without ligation. Vascular ECs were denuded prior to RNA isolation. * P < 0.05 vs Unligated control. (B), In situ hybridization of miR-126 in ligated or unligated carotid arteries from miR-126 wildtype (WT) mice. EL, elastic lamina, indicated by Yellow arrows. (C), Representative images of modified Verhoeff Van Gieson stained ligated or unligated carotid arteries from WT or miR-126 knockout (KO) mice. Arrows indicate the thickened neointima. (D and E), Representative images of immunofluorescent-stained proliferating (PCNA) SMC (SM-Actin) in ligated or unligated carotid arteries from WT and KO mice. (F), Representative images of modified Verhoeff Van Gieson stained ligated carotid arteries from WT or KO mice treated with control (CL) media or EC-CM; or with local delivery of CL oligo or miR-126 mimics (PRE126). Arrows indicate the thickened neointima. (G), In situ hybridization of miR-126 in ligated carotid arteries from KO mice with treatment of EC-CM, miR-126, or the respective controls.
We reintroduced miR-126 into KO mice by repetitive intravenous injection (twice a week) of CM from 3-day cultured ECs or local delivery of pluronic gel-loaded PRE126 to the ligated arteries beginning immediately after ligation. Carotid arteries of the KO mice that received long-term treatment with EC-CM exhibited an increase in neointimal growth in response to ligation compared to those treated with control media (Figure 7F). Local delivery of miR-126 in KO mice also enhanced neointimal formation (Figure 7F). Those outcomes coincide with the abundance of miR-126 in the arteries, as indicated by in situ hybridization detection in carotid arteries from KO mice with or without the administration of miR-126 (Figure 7G). The results from these experiments establish the atheroprone role of miR-126 in SMC function.
Discussion
Endothelial miR-126 has been shown to play a role in governing vascular integrity and angiogenesis 15, 16. While miR-126 serves as a beneficial regulator for EC homeostasis, the functions of miR-126 in the neighboring SMCs remain unclear. In the present study, we have demonstrated that miR-126 serves as a paracrine mediator secreted by the EC to act on the co-cultured SMC to modulate its gene expression and function toward atherogenic phenotype. The paracrine humoral transmission from EC to SMC is proven by using the exogenous B-miR-126 (Figures. 3A and B),which provides direct evidence that the EC is the origin of miR-126 transmission and that cell-cell communication between ECs and SMCs can be mediated by the humoral transmission of a miR. We also showed that the increase in SMC miR-126 upon co-culture with EC or in response to EC-CM was changed in the expected directions by manipulating the amount or biogenesis of miR-126 in ECs (Figures. 4B - D), thus further confirming that the EC is the origin of the miR-126 enrichment in co-cultured SMCs. The inhibition of FOXO3, BCL2, and IRS1 mRNAs in SMCs by miR-126 and co-culture with ECs (Figure 2) indicates that the transmission of miR-126 from ECs to SMCs mediates the EC-regulation of SMC gene expression.
Several recent studies found that vesicle-mediated transfer of miRs can transduce signals between cells 9, 13, 14, 20. Dimmeler and colleagues showed that ECs transfer the vesicle-packaged miR-143/145 to SMCs to enhance the repression of de-differentiation-associated genes in SMCs 14. Weber and colleagues reported that EC-derived apoptotic bodies convey paracrine alarm signals to the nearby recipient ECs 13. Our data indicate that RNAs and RNA-protein complexes are transmitted into SMCs, but not DNA components or the vesicle-protected RNA (Figure 4A). Treatments of SMCs with the fractions of the ultra-centrifuged EC-CM showed that the vesicle-free supernatant caused an increase in SMC miR-126 (Figure 4F), indicating that the EC-derived vesicle-free miR-126 is the main contributor to the miR-126 enrichment in SMCs. Besides miR-126, miR-21, -221, and -155 were also detected to be associated with Ago2 in EC-CM (Online Figure VII), supporting the hypothesis that the Ago2-facilitied miRs transmission is a general mechanism regulating cell-cell communications. The extracellular Ago2-associated miR-143 or -145 was undetectable (Online Figure VII), consistent with previous report indicating that EC-secreted miR-143 and -145 were packaged into vesicles 14. The current study provides an alternative mechanism that is distinct from those in previously reports in which the EC-specific miRs are exported and delivered through exosomal or apoptotic pathways.
Turchinovich et al. 6 and Arroyo et al. 21 reported that most miRs in human peripheral blood are ultrafiltratable together with Ago2 6, 21 and that miR-16, -21, -24 6 or miR-16, 92a, let-7a 21 are associated with Ago2 in the culture media of MCF7 cells or in the human plasma. Other proteins that bind miRs and deliver miRs to recipient cells independent of vesicles include NPM1 (nucleophosmin) 8 and HDL 7. Our results showed that Ago2 exhibited a strong association with miR-126 in the CM (Figure 5A) and the recipient SMCs (Figure 5B), and also carried miR-21, -221, -155, (Online Figure VII) and probably other miRs in the EC-CM. The amount of miR-126 associated with Ago2 was not decreased when the exosomes- or apoptotic body-related miR export pathways were chemically inhibited in ECs (Online Figure IV). Moreover, our results show that Ago2 positively contributed to the stability of miRs and their uptake by SMCs (Figure 5C and D, Online Figure VIC). The enhancement of cel-miR-39 uptake by Ago2 indicates that this effect of Ago2 is not limited to miR-126. Our data also demonstrate that LSS inhibits the expression and secretion of Ago2 in ECs (Figures 5E and F). These findings may provide a mechanistic explanation of how LSS regulates the miR-126 release as well as its transmission from ECs to SMCs. Our study along with the previous findings 6-8, 24 support a notion that both vesicle-dependent and -independent pathways may be involved in the humoral transmission of miRs.
In our EC/SMC co-culture flow system, ECs and SMCs were separated by a porous membrane with a 0.4- μm pore size that does not allow physical contact between ECs and SMCs, but allows communications via humoral transmission through the short distance between these cells. This system has been used previously to investigate the interaction of endothelium and SMCs under physiological and pathophysiological conditions 3, 26. Co-culture of static ECs with SMCs is known to induce SMC activation 3, 27-29, but the underlying molecular mechanisms and signaling pathways were unclear. Our current findings indicate that miR-126 controls SMC gene expression in a paracrine manner to mediate the EC-promotion of SMC turnover in terms of cell proliferation, enhancement of synthetic phase of the cell cycle, and apoptosis (Figure 6). This EC-promotion of SMC turnover was abolished or attenuated by the application of LSS to the EC or inhibition of miR-126 in either ECs (Fig. 6) or SMCs (Online Figure IX). However, overexpression of miR-126 by PRE126 alone in mono-cultured SMCs did not activate the cells (data not shown). Such non-symmetrical phenotypic effects have been reported in other miR studies that although miR depletion caused phenotypical changes, miR overexpression showed little effects. For example, miR-133a KO mice had increased proliferation and apoptosis of cardiomyocytes compared with WT, but cardiac overexpression of miR-133a did not affect apoptosis 30; Inhibition of miR-222 in cultured SMCs increased expressions of p21 and p57, but overexpression of miR-222 had no impact on p57 mRNA level 31. The mechanisms underlying the function of certain miRs seem to be context-dependent. In our case the functional outcomes caused by the alteration in miR-126 levels in SMCs resulting from EC co-culture vs. miR-126 overexpression might reflect the diversity of regulatory pathways triggered by different physiological/pathophysiological microenvironments. The actions of LSS in suppressing EC miR-126 secretion and hence the consequential changes of SMC functions provide further explanation of the miR-mediated atheroprotective effects of LSS.
Vascular SMC proliferation and apoptosis are the critical cellular events in vascular neointimal lesion formation in relation to restenosis and atherosclerosis 4, 32. The lesion originates and develops proferentially at sites of curvature, branching, and bifurcation in arteries where complex hemodynamic conditions are prominent 33. In this study, a 3-fold higher expression of miR-126 was observed in the athero-susceptible AA (Online Figure II), which experiences disturbed and low SS, in comparison with TA, where the SS is laminar and non-disturbed. Cessation of blood flow in carotid arteries also up-regulates the level of miR-126 in the vessel wall (Figures 7A and B). Furthermore, this study provides evidence that neointimal lesion formation after disruption of LSS in mice carotid arteries can be suppressed by systemic depletion of miR-126 (Figures 7C - F). These findings help to identify the contribution of miR-126 in neointimal hyperplasia and to establish the atheroprone role of miR-126 in SMCs despite its beneficial effects on endothelial functions.
Supplementary Material
Novelty and Significance.
What Is Known?
In endothelial cells microRNA (miR)-126 participates in maintaining vascular integrity, enhancing angiogenesis and inhibiting inflammation, but its action on smooth muscle cells (SMCs) is unknown.
The laminar shear stress seen in the atheroprotected regions of the vasculature is anti-atherogenetic.
Application of laminar shear stress to endothelial cells co-cultured with SMCs decreases SMC proliferation and promotes a change in SMC phenotype from synthetic to quiescent state.
What New Information Does This Article Contribute?
MiR-126 can be transmitted from endothelial cells to co-cultured SMCs via a paracrine mechanism that involves the intercellular communicating molecule Argonaute 2, to promote SMC proliferation.
Laminar shear stress applied to endothelial cells inhibits transmission of miR-126 from endothelial cells to co-cultured SMCs, thus suppressing SMC proliferation.
Curtailment of blood flow (decrease in lamina shear stress) induces proliferative pro-atherosclerotic changes in SMCs in wild-type mice, and this action is abrogated in miR-126-null mice.
Endothelial miR-126 has been shown to play a role in maintaining vascular integrity and facilitating angiogenesis, thus serving as an important regulator of EC homeostasis. Nevertheless, the functions of miR-126 in the neighboring SMCs are not known. We demonstrate that miR-126 can be secreted by endothelial cells and then taken up by the co-cultured SMCs to promote SMC proliferation, cell cycle progression, and apoptosis. Exposure of endothelial cells to laminar sheer stress inhibits its miR-126 secretion to abrogate the atherogenic actions of miR-126 on the co-cultured SMCs. This anti-atherogenic effect of shear stress mediated by inhibition of miR-126 secretion in vitro was confirmed in vivo in a carotid artery ligation model in which neointimal lesion formation seen in wildtype mice was abrogated in miR-126-null mice. These results indicate that the athero-protective hemodynamic force acting on endothelial cells exerts its anti-atherogenic effect on SMCs by inhibiting the paracrine secretion of miR-126 by endothelial cells.
Acknowledgments
We thank Dr. Shusheng Wang from Tulane University and Dr. Eric N. Olson from University of Texas Southwestern Medical Center for kindly providing us the miR-126 KO mice. We thank Dr. Michael J. Quon from University of Maryland for kindly providing us the human IRS1 construct.
Sources of funding: This work was supported in part by National Institutes of Health Research Grants HL106579 (to S. C., J. S. and Shankar Subramaniam), HL89940 (to J. S.), HL108735 (to S. C., J. S., and Shankar Subramaniam) and National Science Council Grant NSC-99-2911-I-009-101 (to S.C. and J.-J.C.).
Non-standard Abbreviations and Acronyms
- 3′UT
3′, untranslated region
- Ago2
Argonaute2
- AM126
anti-microRNA-126 inhibitor
- AMC
negative control inhibitor
- BAEC
bovine aortic endothelial cell
- DNase
deoxyribonuclease
- EC
endothelial cell
- EC-CM
endothelial cell-conditioned media
- HUASMC
human umbilical artery smooth muscle cell
- HUVEC
human umbilical vein endothelial cell
- KO
miR-126 knockout
- LSS
laminar shear stress
- microRNA
miR
- miRISCs
miRNA-induced silencing complex
- OSS
oscillatory shear stress
- PCNA
proliferating cell nuclear antigen
- PK
proteinase K
- PRE126
microRNA-126 mimics
- PREC
negative control mimics
- pri-miR-126
primary transcript of microRNA-126
- rAgo2
Argonaute2 human recombinant protein
- RNase
ribonuclease
- SMC
smooth muscle cell
- WT
miR-126 wild-type
Footnotes
Disclosures: None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 2.Doran AC, Meller N, McNamara CA. Role of smooth muscle cells in the initiation and early progression of atherosclerosis. Arterioscler Thromb Vasc Biol. 2008;28:812–819. doi: 10.1161/ATVBAHA.107.159327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tsai MC, Chen L, Zhou J, Tang Z, Hsu TF, Wang Y, Shih YT, Peng HH, Wang N, Guan Y, Chien S, Chiu JJ. Shear stress induces synthetic-to-contractile phenotypic modulation in smooth muscle cells via peroxisome proliferator-activated receptor alpha/delta activations by prostacyclin released by sheared endothelial cells. Circ Res. 2009;105:471–480. doi: 10.1161/CIRCRESAHA.109.193656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clarke MC, Figg N, Maguire JJ, Davenport AP, Goddard M, Littlewood TD, Bennett MR. Apoptosis of vascular smooth muscle cells induces features of plaque vulnerability in atherosclerosis. Nat Med. 2006;12:1075–1080. doi: 10.1038/nm1459. [DOI] [PubMed] [Google Scholar]
- 5.Bartel DP. Micrornas: Genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 6.Turchinovich A, Weiz L, Langheinz A, Burwinkel B. Characterization of extracellular circulating microrna. Nucleic Acids Res. 2011;39:7223–7233. doi: 10.1093/nar/gkr254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vickers KC, Palmisano BT, Shoucri BM, Shamburek RD, Remaley AT. Micrornas are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat Cell Biol. 2011;13:423–433. doi: 10.1038/ncb2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang K, Zhang S, Weber J, Baxter D, Galas DJ. Export of micrornas and microrna-protective protein by mammalian cells. Nucleic Acids Res. 2010;38:7248–7259. doi: 10.1093/nar/gkq601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iguchi H, Kosaka N, Ochiya T. Secretory micrornas as a versatile communication tool. Commun Integr Biol. 2010;3:478–481. doi: 10.4161/cib.3.5.12693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fichtlscherer S, De Rosa S, Fox H, Schwietz T, Fischer A, Liebetrau C, Weber M, Hamm CW, Roxe T, Muller-Ardogan M, Bonauer A, Zeiher AM, Dimmeler S. Circulating micrornas in patients with coronary artery disease. Circ Res. 2010;107:677–684. doi: 10.1161/CIRCRESAHA.109.215566. [DOI] [PubMed] [Google Scholar]
- 11.Fichtlscherer S, Zeiher AM, Dimmeler S. Circulating micrornas: Biomarkers or mediators of cardiovascular diseases? Arterioscler Thromb Vasc Biol. 2011;31:2383–2390. doi: 10.1161/ATVBAHA.111.226696. [DOI] [PubMed] [Google Scholar]
- 12.Dimmeler S, Zeiher AM. Circulating micrornas: Novel biomarkers for cardiovascular diseases? Eur Heart J. 2010;31:2705–2707. doi: 10.1093/eurheartj/ehq221. [DOI] [PubMed] [Google Scholar]
- 13.Zernecke A, Bidzhekov K, Noels H, Shagdarsuren E, Gan L, Denecke B, Hristov M, Koppel T, Jahantigh MN, Lutgens E, Wang S, Olson EN, Schober A, Weber C. Delivery of microrna-126 by apoptotic bodies induces cxcl12-dependent vascular protection. Sci Signal. 2009;2:ra81. doi: 10.1126/scisignal.2000610. [DOI] [PubMed] [Google Scholar]
- 14.Hergenreider E, Heydt S, Treguer K, Boettger T, Horrevoets AJ, Zeiher AM, Scheffer MP, Frangakis AS, Yin X, Mayr M, Braun T, Urbich C, Boon RA, Dimmeler S. Atheroprotective communication between endothelial cells and smooth muscle cells through mirnas. Nat Cell Biol. 2012;14:249–256. doi: 10.1038/ncb2441. [DOI] [PubMed] [Google Scholar]
- 15.Wang S, Aurora AB, Johnson BA, Qi X, McAnally J, Hill JA, Richardson JA, Bassel-Duby R, Olson EN. The endothelial-specific microrna mir-126 governs vascular integrity and angiogenesis. Dev Cell. 2008;15:261–271. doi: 10.1016/j.devcel.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fish JE, Santoro MM, Morton SU, Yu S, Yeh RF, Wythe JD, Ivey KN, Bruneau BG, Stainier DY, Srivastava D. Mir-126 regulates angiogenic signaling and vascular integrity. Dev Cell. 2008;15:272–284. doi: 10.1016/j.devcel.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuhnert F, Mancuso MR, Hampton J, Stankunas K, Asano T, Chen CZ, Kuo CJ. Attribution of vascular phenotypes of the murine egfl7 locus to the microrna mir-126. Development. 2008;135:3989–3993. doi: 10.1242/dev.029736. [DOI] [PubMed] [Google Scholar]
- 18.Ji R, Cheng Y, Yue J, Yang J, Liu X, Chen H, Dean DB, Zhang C. Microrna expression signature and antisense-mediated depletion reveal an essential role of microrna in vascular neointimal lesion formation. Circ Res. 2007;100:1579–1588. doi: 10.1161/CIRCRESAHA.106.141986. [DOI] [PubMed] [Google Scholar]
- 19.Kumar A, Lindner V. Remodeling with neointima formation in the mouse carotid artery after cessation of blood flow. Arterioscler Thromb Vasc Biol. 1997;17:2238–2244. doi: 10.1161/01.atv.17.10.2238. [DOI] [PubMed] [Google Scholar]
- 20.Kosaka N, Iguchi H, Yoshioka Y, Takeshita F, Matsuki Y, Ochiya T. Secretory mechanisms and intercellular transfer of micrornas in living cells. J Biol Chem. 2010;285:17442–17452. doi: 10.1074/jbc.M110.107821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arroyo JD, Chevillet JR, Kroh EM, Ruf IK, Pritchard CC, Gibson DF, Mitchell PS, Bennett CF, Pogosova-Agadjanyan EL, Stirewalt DL, Tait JF, Tewari M. Argonaute2 complexes carry a population of circulating micrornas independent of vesicles in human plasma. Proc Natl Acad Sci U S A. 2011;108:5003–5008. doi: 10.1073/pnas.1019055108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee HY, Chung JW, Youn SW, Kim JY, Park KW, Koo BK, Oh BH, Park YB, Chaqour B, Walsh K, Kim HS. Forkhead transcription factor foxo3a is a negative regulator of angiogenic immediate early gene cyr61, leading to inhibition of vascular smooth muscle cell proliferation and neointimal hyperplasia. Circ Res. 2007;100:372–380. doi: 10.1161/01.RES.0000257945.97958.77. [DOI] [PubMed] [Google Scholar]
- 23.Bennett MR, Evan GI, Schwartz SM. Apoptosis of human vascular smooth muscle cells derived from normal vessels and coronary atherosclerotic plaques. J Clin Invest. 1995;95:2266–2274. doi: 10.1172/JCI117917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Radhakrishnan Y, Busby WH, Jr, Shen X, Maile LA, Clemmons DR. Insulin-like growth factor-i-stimulated insulin receptor substrate-1 negatively regulates src homology 2 domain-containing protein-tyrosine phosphatase substrate-1 function in vascular smooth muscle cells. J Biol Chem. 2010;285:15682–15695. doi: 10.1074/jbc.M109.092270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim JA, Yeh DC, Ver M, Li Y, Carranza A, Conrads TP, Veenstra TD, Harrington MA, Quon MJ. Phosphorylation of ser24 in the pleckstrin homology domain of insulin receptor substrate-1 by mouse pelle-like kinase/interleukin-1 receptor-associated kinase: Cross-talk between inflammatory signaling and insulin signaling that may contribute to insulin resistance. J Biol Chem. 2005;280:23173–23183. doi: 10.1074/jbc.M501439200. [DOI] [PubMed] [Google Scholar]
- 26.Chiu JJ, Chen LJ, Lee PL, Lee CI, Lo LW, Usami S, Chien S. Shear stress inhibits adhesion molecule expression in vascular endothelial cells induced by coculture with smooth muscle cells. Blood. 2003;101:2667–2674. doi: 10.1182/blood-2002-08-2560. [DOI] [PubMed] [Google Scholar]
- 27.Nackman GB, Fillinger MF, Shafritz R, Wei T, Graham AM. Flow modulates endothelial regulation of smooth muscle cell proliferation: A new model. Surgery. 1998;124:353–360. ; discussion 360-351. [PubMed] [Google Scholar]
- 28.Imanishi T, Hano T, Nishio I, Han DK, Schwartz SM, Karsan A. Apoptosis of vascular smooth muscle cells is induced by fas ligand derived from endothelial cells. Jpn Circ J. 2001;65:556–560. doi: 10.1253/jcj.65.556. [DOI] [PubMed] [Google Scholar]
- 29.Vouyouka AG, Lin L, Basson MD. Pressure and endothelial coculture upregulate myocytic fas-fasl pathway and induce apoptosis by way of direct and paracrine mechanisms. Am J Surg. 2005;190:780–786. doi: 10.1016/j.amjsurg.2005.07.020. [DOI] [PubMed] [Google Scholar]
- 30.Liu N, Bezprozvannaya S, Williams AH, Qi X, Richardson JA, Bassel-Duby R, Olson EN. Microrna-133a regulates cardiomyocyte proliferation and suppresses smooth muscle gene expression in the heart. Genes Dev. 2008;22:3242–3254. doi: 10.1101/gad.1738708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu X, Cheng Y, Zhang S, Lin Y, Yang J, Zhang C. A necessary role of mir-221 and mir-222 in vascular smooth muscle cell proliferation and neointimal hyperplasia. Circ Res. 2009;104:476–487. doi: 10.1161/CIRCRESAHA.108.185363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ferns GA, Avades TY. The mechanisms of coronary restenosis: Insights from experimental models. Int J Exp Pathol. 2000;81:63–88. doi: 10.1046/j.1365-2613.2000.00143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ku DN, Giddens DP, Zarins CK, Glagov S. Pulsatile flow and atherosclerosis in the human carotid bifurcation. Positive correlation between plaque location and low oscillating shear stress. Arteriosclerosis. 1985;5:293–302. doi: 10.1161/01.atv.5.3.293. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.