

Abstract
Objective:
To identify patients with GFPT1-related limb-girdle myasthenia and analyze phenotypic consequences of the mutations.
Methods:
We performed genetic analysis, histochemical, immunoblot, and ultrastructural studies and in vitro electrophysiologic analysis of neuromuscular transmission.
Results:
We identified 16 recessive mutations in GFPT1 in 11 patients, of which 12 are novel. Ten patients had slowly progressive limb-girdle weakness responsive to cholinergic agonists with onset between infancy and age 19 years. One patient (no. 6) harbored a nonsense mutation and a second mutation that disrupts the muscle-specific GFPT1 exon. This patient never moved in utero, was apneic and arthrogrypotic at birth, and was bedfast, tube-fed, and barely responded to therapy at age 6 years. Histochemical studies in 9 of 11 patients showed tubular aggregates in 6 and rimmed vacuoles in 3. Microelectrode studies of intercostal muscle endplates in 5 patients indicated reduced synaptic response to acetylcholine in 3 and severely reduced quantal release in patient 6. Endplate acetylcholine receptor content was moderately reduced in only one patient. The synaptic contacts were small and single or grape-like, and quantitative electron microscopy revealed hypoplastic endplate regions. Numerous muscle fibers of patient 6 contained myriad dilated and degenerate vesicular profiles, autophagic vacuoles, and bizarre apoptotic nuclei. Glycoprotein expression in muscle was absent in patient 6 and reduced in 5 others.
Conclusions:
GFPT1-myasthenia is more heterogeneous than previously reported. Different parameters of neuromuscular transmission are variably affected. When disruption of muscle-specific isoform determines the phenotype, this has devastating clinical, pathologic, and biochemical consequences.
Congenital myasthenic syndromes (CMS) are heterogeneous disorders in which the safety margin of neuromuscular transmission is compromised by one or more specific mechanisms. Most CMS are caused by defects in endplate (EP)-specific proteins.1 Recently, however, it became apparent that proteins distributed in many tissues, namely plectin,2 GFPT1,3 and DPAGT1,4 are also CMS targets. Both GFPT1 and DPAGT1 subserve glycosylation of nascent peptides.5 Mutations in either protein result in limb-girdle myasthenia with tubular aggregates in type 2 muscle fibers.
Glutamine-fructose-6-phosphate transaminase 1 (GFPT1) is the initial and rate-limiting enzyme in the biosynthesis of N-acetylglucosamine, an essential substrate for O- and N-glycosylation.6 A long muscle-specific isoform is encoded by GFPTL1. In 2011, a CMS due to mutations in GFPT1 was identified in 16 kinships,3 and phenotypic features of these patients were further documented in 2012.7 Another patient with GFPT1-myasthenia was reported in 2012.8 Analysis of parameters of neuromuscular transmission and detailed examination of the EP ultrastructure have not been available to date.
Herein, we report our findings in 11 patients with GFPT1-myasthenia. Using whole-exome and Sanger sequencing, we identified 12 novel mutations, performed histochemical studies in 9 patients, examined in vitro parameters of neuromuscular transmission in 5, and quantitatively analyzed 170 EP regions by electron microscopy in 6. We also found that when disruption of the muscle-specific GFPTL1 isoform determines the phenotype, it results in a severe autophagic myopathy, impairs the release and response to acetylcholine (ACh), abolishes glycoprotein expression in skeletal muscle, and has devastating clinical consequences.
METHODS
Standard protocol approval, registrations, and patient consents.
Eleven patients were investigated. All human studies were approved by the Institutional Review Board of the Mayo Clinic, and each patient gave informed consent to participate in the study.
Structural studies.
Intercostal muscle specimens were obtained from patients 1 to 5, anconeus muscle from patient 6, and brachial biceps from patient 8 and from control subjects without muscle disease undergoing thoracic surgery. Cryosections were used to colocalize the ACh receptor (AChR) and ACh esterase (AChE) as described.9 AChE was also visualized on teased, glutaraldehyde-fixed muscle fibers cytochemically.10 EPs were localized for electron microscopy11 and quantitatively analyzed12 by established methods. Peroxidase-labeled α-bgt was used for the ultrastructural localization of AChR.13 The number of AChRs per EP was measured with [125I]α-bgt.14
In vitro electrophysiologic studies.
Intracellular microelectrode studies were performed on intercostal muscle specimens of 5 patients. The amplitude of the miniature EP potential (MEPP), miniature EP current (MEPC), EP potential, and estimates of the quantal content of the EP potential (m) were measured as previously described.15,16 Single-channel patch-clamp recordings were performed on EPs of 3 patients as previously described.17,18
Mutation analysis.
Genomic DNA was isolated from blood and cDNA from muscle by standard methods. In 7 patients, mutations were identified by Sanger sequencing and in 4 by exome sequencing and confirmed by Sanger sequencing. Before identifying the mutations in GFPT1, we sequenced RAPSN, DOK7, and CHRNE in all patients, CHRNA1, CHRNB1, and CHNRD in patients 1–6 and 9–11, and MUSK in patient 6.
For exome sequencing, paired-end libraries were prepared following the manufacturer's protocol (Illumina, San Diego, CA and Agilent, Santa Clara, CA) using 3 µg of genomic DNA. Whole-exome capture was performed using the protocol for Agilent's SureSelect Human All Exon 51 or 71 MB v4 kit for patients 6 and 8 and SureSelect Human All Exon v2 kit for patients 3 and 5. The coverage was above 60× in all samples. We first looked at the genes that have been reported in association with CMS. All detected changes were confirmed by Sanger sequencing. Sanger sequencing was performed with PCR primers to sequence exons and flanking noncoding regions of GFPT1. Nucleotide numbering of cDNA was based on GeneBank accession number NM_002056.3 for the short isoform and NM_00124710.1 for the muscle-specific long isoform, with +1 corresponding to the A of the ATG translation initiation codon of the reference sequence. PCR-amplified fragments were purified with shrimp alkaline phosphatase and exonuclease I (USB) and sequenced with an ABI3730xI DNA sequencer (Applied Biosystems, Foster City, CA) using fluorescently labeled dideoxy terminators.
Immunoblot analysis.
Frozen muscle extracts of patients and controls were extracted and prepared for immunoblotting as previously described.19 After transfer to nitrocellulose or polyvinylidene fluoride membrane (Life Technologies, Carlsbad, CA), they were probed with rabbit polyclonal RL2 antibody that recognizes O-glycosylated proteins (dilution 1:500; Abcam, Cambridge, MA) and mouse monoclonal O-GlcNAc antibody CTD110.6 (dilution 1:200; Covance, Berkeley, CA) that recognizes O- and N-glycosylated proteins.20 The immunoblots were quantitatively analyzed using NIH Image 1.61 software and myosin heavy-chain of the transferred gel as a loading control. Immunoreactivity was detected using the alkaline phosphatase method as described.21
RESULTS
Clinical features.
Eleven unrelated patients, 4 women and 7 men, were investigated (see table e-1 on the Neurology® Web site at www.neurology.org). All had negative tests for anti-AChR antibodies. The age at onset varied from the time of birth to age 19 years (mean, 8.3 years), with 6 patients presenting before age 10 years. All except patient 6 were ambulatory with Medical Research Council grade 4 to 2 weakness of limb-girdle and 4 to 3 weakness of distal muscles with no involvement of the cranial muscles (figure 1C). The distal weakness typically involves the forearm extensor, intrinsic hand, and anterior leg muscles. The weakness is slowly progressive, although the rate of progression varies among patients. Each patient has a decremental EMG response. Nerve conduction studies were normal in all patients. Needle EMG studies revealed unstable small motor unit potentials typical of patients with myasthenia attributable to impulse blocking at EPs of variable numbers of fibers in motor units at a given instant that can mimic a myopathy.22 In addition, some muscles in patients 6 and 10 showed spontaneous electrical activity, and findings in patient 11 suggested a very chronic myopathy. The serum creatine kinase level, determined in 7 patients, was mildly elevated only in patient 7, who exercised regularly.
Figure 1. Patients with GFPT1-myasthenia and histochemical observations.

(A) Patient 6 during infancy and (B) at age 4 years. Note facial diplegia, open mouth, protruding tongue, lack of head control, and strabismus. (C) Patient 2 at age 12 years showing Gowers sign. (D) Large tubular aggregates in patient 4 and (E) small and infrequent tubular aggregates in patient 2 shown by NADH dehydrogenase reaction. (F) Acid phosphatase–positive autophagic vacuoles in patient 6. (G) Type 1 fiber preponderance in patient 6, ATPase at pH 4.3. (H–K) Face-on views of EP regions. (H–J) The EPs are composed of small grape-like regions. (I and J) Paired localization of AChE (green) and AChR (red) in frozen section. (H and K) Localized AChE on fixed, teased muscle fibers. Bars = 50 μm in panels D–G and 20 μm in H–K. AChE = acetylcholinesterase; AChR = acetylcholine receptor; ATPase = adenosine triphosphatase; EP = endplate; NADH = nicotinamide adenine dinucleotide.
Two patients presented at birth: patient 1 had repeated apneic spells during infancy and delayed motor milestones but his subsequent clinical course was benign, with weakness confined to the limb-girdle muscles. Patient 6 did not move in utero and was born after 37 weeks of gestation with apnea, poor cry and suck, severe hypotonia, multiple joint contractures, and cranial synostosis requiring surgery (figure 1A). At age 4 years, she had severe weakness of all except the ocular muscles, was bedfast, and required gastrostomy feeding and ventilatory support at night (figure 1B). At age 6 years, her clinical state was unchanged. The patient's mother had dermatomyositis and a high titer of anti-AChR antibodies but none recognizing the ACh-γ subunit (determined by Dr. Angela Vincent) and no myasthenic symptoms or EMG findings to suggest a neuromuscular transmission defect. The patient had repeatedly negative tests for AChR antibodies, including those in the neonatal period and later in life.
Five patients had similarly affected nuclear family members (figure e-1 and table e-1). Patients 10 and 11 were born to consanguineous parents. Patient 8 also had Usher syndrome type 1D associated with retinitis pigmentosa and hearing loss caused by heterozygous p.Gly2123Arg and p.Ile3210Thr mutations in the cadherin-related gene CDH23 (NM_022124.5) located on chromosome 10 at q22.1. According to Exome Variant Server (NHLBI Go Genome Sequencing Project [ESP], Seattle, WA http://evs.gs.washington.edu/EVS; December 2012), Gly2123Arg is not present, and p.Ile3210Thr is present in only 3 of 12,541 European and African American alleles. Patient 9 also had congenital aortic stenosis and Asperger syndrome and 2 sisters with learning disability. Patient 10 and 2 of his similarly affected uncles had high myopia.
Ten patients responded partially to pyridostigmine. Further improvement occurred in 5 patients with 3,4-diaminopyridine, in one with ephedrine, and in one with albuterol. Patient 6 remained severely disabled despite treatment with pyridostigmine and 3,4-diaminopyridine. Unlike patients with Dok7 myasthenia, none responded adversely to cholinergic therapy.
Of special historical interest, the clinical features of patient 4 and her brothers were reported in 1966 by Michael McQuillen under the rubric of familial limb-girdle myasthenia.23
Histochemical observations.
Conventional histochemical studies of limb or intercostal muscle specimens were available in patients 1 to 9. Patients 7 and 9 had a limb muscle biopsy before referral to our institution. Seven of 9 patients harbored tubular aggregates of the sarcoplasmic reticulum (SR) in type 2 fibers. These were abundant in 3 patients (figure 1D) but small and difficult to detect in 4 patients (figure 1E). In patient 6, the anconeus muscle showed abnormal variation of fiber size, endomysial fibrosis, sparse regenerating and necrotic fibers, and vacuolated fibers reacting strongly for acid phosphatase, indicating they were autophagic in origin (figure 1F). A small proportion of fibers in patients 1, 6, and 8 contained acid phosphatase–negative rimmed vacuoles of variable size distinct from tubular aggregates (figure e-2). Neurogenic features, consisting of fiber-type grouping and small fibers of either histochemical type, were present in patients 3, 5, and 8, but patient 3 was also diabetic. Muscle specimens of patients 2 and 6 displayed type 1 fiber preponderance (figure 1G).
AChE- or AChR-reacted EPs viewed face-on showed either a single or double round, comma-shaped or ring-like regions (figure 1K), or a cluster of small regions with a grape-like appearance (figure 1, H–J). No EPs had a normal pretzel shape. The grape-like nerve endings, however, are functionally different from those found on multiply innervated tonic muscle fibers of birds and reptiles that cannot propagate an action potential.24
In vitro electrophysiology and counts of AChRs per EP.
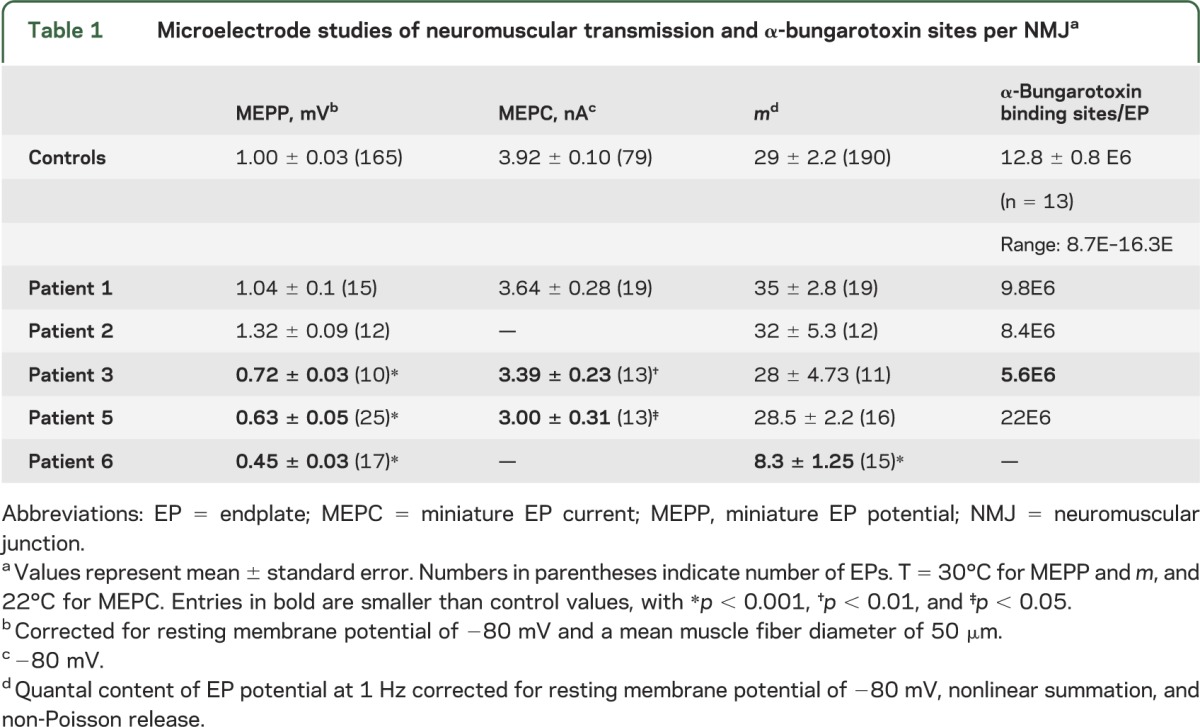
In vitro parameters of neuromuscular transmission were evaluated in patients 1, 2, 3, 5, and 6 (table 1). The synaptic response to ACh is reflected by the amplitude of the MEPP and MEPC. The MEPP amplitude was abnormally small in 3 patients, especially in patient 6; the MEPC amplitude, determined in patients 1, 3, and 5, was abnormally low in patients 3 and 5. Quantal release by nerve impulse was normal, except in patient 6 in whom it was 29% of normal. The number of α-bungarotoxin binding sites per EP fell below the normal range only in patient 3. Single-channel patch-clamp recordings in 3 patients revealed normal kinetic properties of the AChR ion channel.
Table 1.
Microelectrode studies of neuromuscular transmission and α-bungarotoxin sites per NMJa
EP ultrastructure.
We examined and quantitatively analyzed 170 EP regions of patients 1 to 6. On simple inspection, EPs of patient 5 showed no abnormality. In other patients, some EPs appeared normal but many had simplified postsynaptic regions with few, poorly developed, or no junctional folds (figure 2, A and B). The junctional sarcoplasm often harbored dilated vesicles. The density of AChR on the junctional folds, demonstrated by peroxidase-labeled α-bungarotoxin in patients 1, 2, and 4, was normal or mildly attenuated. Quantitative electron microscopy analysis of 170 EP regions revealed that the mean nerve terminal area, postsynaptic area, and postsynaptic membrane density were abnormally small in 5 patients, but in patient 5 only the postsynaptic membrane density was reduced (see table e-2).
Figure 2. Electron microscopy findings.

(A and B) Endplate regions in patients 1 and 2. Note nearly absent junctional folds and small nerve terminals. (C and D) Muscle fiber pathology in patient 6. (C) A large accumulation of dilated and degenerating vesicular profiles near a nucleus. (D) Multiple autophagic vacuoles harboring granular or globular debris and apoptotic nuclei, one of which is large and bizarre (asterisk). Bars = 1 μm in panels A, B, and D, and 2 μm in C.
Muscle fiber ultrastructure.
Muscle fiber ultrastructure was examined in patients 1 to 6. In patients 1 and 5, some fibers displayed pleomorphic myeloid structures (figure e-3). In patient 6, numerous fibers harbored dilated and degenerating vesicular profiles (figure 2C), multiple autophagic vacuoles, lipofuscin granules, other electron-dense bodies, and bizarre apoptotic nuclei (figure 2D).
Genetic analysis.
We identified 16 recessive mutations in GFPT1 in 11 patients, of which 12 are novel (see table 2, figure 3A, and figure e-1). Eight patients were compound heterozygous and patients 7, 10, and 11 were homozygous. Three mutations were frameshift (patients 1, 8, and 9) and 2 were nonsense (patients 5 and 6). In patient 6, the nonsense mutation was accompanied by a canonical splice-site mutation that disrupted the muscle-specific exon. In patient 3, the intronic mutation was 8 nucleotides before the start of exon 8. To further investigate the consequences of the splice-site mutations in patients 3 and 6, we isolated their cDNA. In patient 3, c.606-8A>G resulted in recognition of a new acceptor site before exon 8 and yielded 5 missense amino acids and a stop codon. In patient 6, c.686-2A>G eliminated the first 4 nucleotides of the muscle-specific exon and resulted in 56 missense amino acids and a stop codon. No DNA was available from deceased parents of patients 3, 4, and 8, but their phenotypic features strongly suggested that they harbored biallelic mutations, and 3 unaffected siblings of patient 8 carried 1 of the 2 mutations identified in the proband.
Table 2.
Identified mutations in 11 patients

Figure 3. GFPT1 mutations and immunoblots of muscle extracts.

(A) Scaled linear model of GFPT1 with 3 domains and observed mutations. Predicted peptides of the mutant transcripts of patient 6 are shown separately. Novel mutations are in bold type. Red box represents muscle-specific exon. Numbering is based on the short isoform of GFPT1. (B) Immunoblots demonstrating expression of the ∼70-kD glycosylated protein with RL2 (left panel) and O-GlcNAc (right panel) antibodies in control and 7 patient muscles. Expression levels are normalized for myosin level in the transferred gel and by comparison to controls. Glycosylated protein expression is absent in patient 6 and close to normal in patient 5. SIS = sugar isomerase domain.
Among the 8 missense mutations detected in patients 2, 4, 7, 8, 9, 10, and 11, 3 were previously reported.3 None of the novel mutations was observed in the NHLBI Exome Variant database. All mutated residues are evolutionary conserved across vertebrate species and predicted to be damaging by one or more software programs, including PolyPhen 2, SIFT, and Mutation Taster. The previously published c.*22C>A variant in the 3′ untranslated region observed in patients 1, 2, 3, and 5 is predicted to decrease expression of the affected allele.3
Immunoblot analysis.
Muscle extracts of patients 1–6 and 8 and of controls were immunoblotted with the RL2 antibody that recognizes epitopes of O-glycosylated proteins and O-GlcNAc antibody that recognizes epitopes of both O- and N-glycosylated proteins20 (figure 3B). The major band of glycosylated proteins in control and patient muscles appeared around 70 kD. With both antibodies, this band was absent in patient 6, reduced in patients 1–4 and 8, and normally expressed in patient 5.
DISCUSSION
Glycosylated proteins are widely distributed in many tissues and organs. Therefore, it is puzzling that mutations in GFPT1 selectively affect neuromuscular transmission and muscle fiber architecture. Patient 6 was born with cranial synostosis, patient 9 also had aortic stenosis and Asperger syndrome, and patient 10 and 2 of his similarly affected uncles had high myopia. However, we cannot ascertain that the associated disorders stemmed from defects in GFPT1.
In 10 patients, the weakness progressed gradually and involved limb-girdle and distal extremity muscles with a characteristic predilection for the anterior forearm, small hand, and anterior leg muscles. In contrast, patient 6 was permanently severely affected since birth.
Genotype-phenotype correlations are hindered in patients harboring biallelic mutations. In 4 patients, however, a single mutation determined the phenotype. Patients 7, 10, and 11 were respectively homozygous for Arg111Cys, Val668Ile, and Ile274Thr. Patient 7 presented at age 10 years and had moderate weakness at age 30; patient 10 presented in early childhood and patient 11 at age 15. Both had moderate to marked weakness in the third decade of life.
In patients 1, 2, 3, and 5, the c.*22C>A was accompanied by a frameshift, missense, splice-site, and nonsense mutation, respectively, and in patients 1, 3, and 5, the c.*22C>A was predicted to determine the phenotype. Paradoxically, patient 5 was less severely affected than patients 1 to 3: at age 49 years, he was still able to work, had well-preserved EP morphology (table e-2), and showed near-normal protein glycosylation (see figure 3B). The clinical diversity of patients harboring c.*22C>A as the phenotypic determinant is likely related to differences in their genetic background.
Patient 6 harbored a nonsense mutation and one that abrogated the function of the muscle-specific isoform. She had been clinically devastated since birth (figure 1, A and B), had an advanced autophagic myopathy (figure 2, C and D), had severely impaired neuromuscular transmission, and expressed no N- or O-glycosylated proteins in muscle (figure 3B). Thus, the muscle-specific exon of GFPT1 is essential for muscle fiber integrity and neuromuscular transmission.
Patient 8 is of special interest because she also had Usher syndrome type 1D associated with retinitis pigmentosa and hearing loss caused by mutations in the cadherin-related gene CDH23 on chromosome 10. Because GFPT1 is located on chromosome 2, the association with Usher syndrome must be attributable to chance. Interestingly, among previously reported patients with GFPT1 deficiency, one had retinitis pigmentosa and 2 siblings had juvenile macular degeneration. The genetic basis of their retinal disease was not determined.7
The infrequent acid phosphatase–negative rimmed vacuoles in patients 1, 6, and 8 (see figure e-2) likely arose from accumulation of degraded cytoplasmic material, as shown in figure e-3, that had dropped out from the frozen section during processing.
In 6 patients, we searched for structural and electrophysiologic correlates of the myasthenia (table 2 and table e-2). In all patients, the EPs were composed of small regions occurring singly or in grape-like clusters (figure 1, H–K), and in all except patient 5, many EPs were poorly developed with simplified postsynaptic membranes (figure 2, A and B). That many EPs were underdeveloped likely stems from hypoglycosylation and altered function of EP-specific glycoproteins, such as MuSK, agrin, and dystroglycans. Simplification of the postsynaptic membrane should decrease the input resistance of the EP and hence the MEPP amplitude.25 However, it was not reduced in patients 1 and 2, but was reduced in patient 5 whose junctional folds were well developed. In patient 5, the EP AChR content was 44% of normal, which is in accord with the 40% reduction of his MEPP. In patient 6, the 55% decrease of the MEPP amplitude and the marked decrease in quantal release by nerve impulse readily explain the defect in neuromuscular transmission.
To summarize, we found no correlation between EP morphology and physiology or between genotype and phenotype.
Tubular aggregates of the SR in type 2 fibers are a useful marker for both GFPT- and DPAGT1-myasthenia,4,7 but were absent in 2 patients and difficult to detect in 4. They also occur in the slow-channel myasthenic syndrome,26 in a late-onset myasthenic disorder with bulbar symptoms,27 in patients with painful muscle cramps on exercise,28–32 in some patients with exercise-induced hyperthermia and a positive caffeine contracture test,33 and in diverse other conditions. A recent study identifies mutations in STIM1 as the cause of a tubular aggregate myopathy.34 STIM1 is a glycosylated endoplasmic- and hence SR-associated Ca2+ sensor that augments Ca2+ uptake by the SR when the SR Ca2+ level is reduced.35 Functionally deficient STIM1 resulted in higher basal Ca2+ levels in patient cells than in controls. A possible explanation for proliferation of the SR tubules in GFPT1- and DPAGT1-myasthenia is that hypoglycosylation of STIM1 results in a Ca2+ shift from the SR to the cytosol. Moreover, impaired cytosolic Ca2+ homeostasis attributable to diverse causes may well be the common denominator for proliferation of the SR tubules in diverse myopathies.
Supplementary Material
GLOSSARY
- ACh
acetylcholine
- AChE
acetylcholinesterase
- AChR
acetylcholine receptor
- CMS
congenital myasthenic syndrome
- EP
endplate
- GFPT1
glutamine-fructose-6-phosphate transaminase 1
- MEPC
miniature endplate current
- MEPP
miniature endplate potential
- SR
sarcoplasmic reticulum
Footnotes
Supplemental data at www.neurology.org
AUTHOR CONTRIBUTIONS
D. Selcen contributed to study concept, data acquisition, analysis, and interpretation. X.-M. Shen contributed to data acquisition and analysis. M. Milone and J. Brengman contributed to data acquisition. K. Ohno contributed to data acquisition and analysis. F. Deymeer, R. Finkel, and J. Rowin contributed to data acquisition. A. Engel contributed to study concept, data acquisition, analysis, and interpretation.
STUDY FUNDING
Supported by NIH6277 and a research grant from the MDA to A.G.E., and MEXT and MHLW of Japan to K.O.
DISCLOSURE
D. Selcen, X.-M. Shen, M. Milone, and J. Brengman report no disclosures. K. Ohno received Grants-in-Aid from MEXT and MHLW of Japan. F. Deymeer, R. Finkel, and J. Rowin report no disclosures. A. Engel serves as an Associate Editor of Neurology® and is supported by research grants from NIH and the Muscular Dystrophy Association. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Engel AG. Current status of the congenital myasthenic syndromes. Neuromuscul Disord 2012;22:99–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Selcen D, Juel VC, Hobson-Webb LD, et al. Myasthenic syndrome caused by plectinopathy. Neurology 2011;76:327–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Senderek J, Muller JS, Dusl M, et al. Hexosamine biosynthetic pathway mutations cause neuromuscular transmission defect. Am J Hum Genet 2011;88:162–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Belaya K, Finlayson S, Slater C, et al. Mutations in DPAGT1 cause a limb-girdle congenital myasthenic syndrome with tubular aggregates. Am J Hum Genet 2012;91:1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Freeze HH, Eklund EA, Ng BG, Patterson MC. Neurology of inherited glycosylation disorders. Lancet Neurol 2012;11:453–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wellen KE, Thompson CB. A two-way street: reciprocal regulation of metabolism and signalling. Nat Rev Mol Cell Biol 2012;13:270–276 [DOI] [PubMed] [Google Scholar]
- 7.Guergueltcheva V, Muller JS, Dusl M, et al. Congenital myasthenic syndrome with tubular aggregates caused by GFPT1 mutations. J Neurol 2012;259:838–850 [DOI] [PubMed] [Google Scholar]
- 8.Huh SH, Kim HS, Jang HJ, Park YE, Kim DS. Limb-girdle myasthenia with tubular aggregates associated with GFPT1 mutations. Muscle Nerve 2012;46:600–604 [DOI] [PubMed] [Google Scholar]
- 9.Fambrough DM, Engel AG, Rosenberry TL. Acetylcholinesterase of human erythrocytes and neuromuscular junctions: homologies revealed by monoclonal antibodies. Proc Natl Acad Sci USA 1982;79:1078–1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gautron J. Ultrastructural cytochemistry of acetylcholinesterase [in French]. Microscopie 1974;21:259–264 [Google Scholar]
- 11.Engel AG. The muscle biopsy. In: Engel AG, Franzini-Armstrong C, editors. Myology, 3rd ed New York: McGraw-Hill; 2004:681–690 [Google Scholar]
- 12.Engel AG. Quantitative morphological studies of muscle. In: Engel AG, Franzini-Armstrong C, editors. Myology, 2nd ed New York: McGraw-Hill; 1994:1018–1045 [Google Scholar]
- 13.Engel AG, Lindstrom JM, Lambert EH, Lennon VA. Ultrastructural localization of the acetylcholine receptor in myasthenia gravis and in its experimental autoimmune model. Neurology 1977;27:307–315 [DOI] [PubMed] [Google Scholar]
- 14.Engel AG. The investigation of congenital myasthenic syndromes. Ann NY Acad Sci 1993;681:425–434 [DOI] [PubMed] [Google Scholar]
- 15.Engel AG, Nagel A, Walls TJ, Harper CM, Waisburg HA. Congenital myasthenic syndromes. I. Deficiency and short open-time of the acetylcholine receptor. Muscle Nerve 1993;16:1284–1292 [DOI] [PubMed] [Google Scholar]
- 16.Uchitel O, Engel AG, Walls TJ, Nagel A, Atassi ZM, Bril V. Congenital myasthenic syndromes. II. A syndrome attributed to abnormal interaction of acetylcholine with its receptor. Muscle Nerve 1993;16:1293–1301 [DOI] [PubMed] [Google Scholar]
- 17.Milone M, Hutchinson DO, Engel AG. Patch-clamp analysis of the properties of acetylcholine receptor channels at the normal human endplate. Muscle Nerve 1994;17:1364–1369 [DOI] [PubMed] [Google Scholar]
- 18.Shen XM, Brengman J, Sine SM, Engel AG. Myasthenic syndrome AChRα C-loop mutant disrupts initiation of channel gating. J Clin Invest 2012;122:2613–2621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Selcen D, Stilling G, Engel AG. The earliest pathologic alterations in dysferlinopathy. Neurology 2001;56:1472–1481 [DOI] [PubMed] [Google Scholar]
- 20.Isono T. O-GlcNAc-specific antibody CTD110.6 cross-reacts with N-GlcNAc-modified proteins induced under glucose deprivation. PLoS One 2011;6:e18959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ohno K, Tsujino A, Shen XM, et al. Choline acetyltransferase mutations cause myasthenic syndrome associated with episodic apnea in humans. Proc Natl Acad Sci USA 2001;98:2017–2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lindsley DB. Myographic and electromyographic studies of myasthenia gravis. Brain 1935;58:470–482 [Google Scholar]
- 23.McQuillen MP. Familial limb-girdle myasthenia. Brain 1966;89:121–132 [DOI] [PubMed] [Google Scholar]
- 24.Engel AG. The neuromuscular junction. In: Engel AG, Franzini-Armstrong C, editors. Myology, 3rd ed New York: McGraw-Hill; 2004:325–372 [Google Scholar]
- 25.Magleby KL. Neuromuscular transmission. In: Engel AG, Franzini-Armstrong C, editors. Myology, 3rd ed New York: McGraw-Hill; 2004:373–395 [Google Scholar]
- 26.Engel AG, Lambert EH, Mulder DM, et al. A newly recognized congenital myasthenic syndrome attributed to a prolonged open time of the acetylcholine-induced ion channel. Ann Neurol 1982;11:553–569 [DOI] [PubMed] [Google Scholar]
- 27.Morgan-Hughes JA, Lecky BR, Landon DN, Muray NM. Alterations in the number and affinity of junctional acetylcholine receptors in a myopathy with tubular aggregates: a newly recognized receptor defect. Brain 1981;104:279–295 [DOI] [PubMed] [Google Scholar]
- 28.Brumback RA, Staton RD, Susag ME. Exercise-induced pain, muscle stiffness, and tubular aggregates in skeletal muscle. J Neurol Neurosurg Psychiaty 1981;44:250–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morgan-Hughes JA, Mair WG, Lascelles PT. A disorder of skeletal muscle associated with tubular aggregates. Brain 1970;93:873–880 [DOI] [PubMed] [Google Scholar]
- 30.Orimo S, Araki M, Ishii H, Kurosawa T, Arai M, Hiyamuta E. A case of “myopathy with tubular aggregates” with increased muscle fibre sensitivity to caffeine. J Neurol 1987;234:424–426 [DOI] [PubMed] [Google Scholar]
- 31.Pierobon-Bormioli S, Armani M, Ringel S, et al. Familial neuromuscular disease with tubular aggregates. Muscle Nerve 1985;8:291–298 [DOI] [PubMed] [Google Scholar]
- 32.Shahrizaila N, Lim WS, Robson DK, Wills AJ. Tubular aggregate myopathy presenting with acute type II respiratory failure and severe orthopnea. Thorax 2006;61:89–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.De Paula M, Bartoli M, Courrier S, et al. Further heterogeneity in myopathy with tubular aggregates. Muscle Nerve 2012;46:984–985 [DOI] [PubMed] [Google Scholar]
- 34.De Paula M, Koch C, Attarian S, et al. Constitutive activation of STIM1 causes tubular aggregate myopathy. Am J Hum Genet 2013;92:271–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Soboloff J, Rothberg BS, Madesh M, Gill DL. STIM proteins: dynamic calcium signal transducers. Nat Rev Mol Cell Biol 2012;13:549–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.