

Abstract
One of the main applications of solid-state NMR is to study the structure and dynamics of biopolymers, such as membrane proteins, under physiological conditions where the polypeptides undergo global motions as they do in biological membranes. The effects of NMR radiofrequency irradiations on nuclear spins are strongly influenced by these motions. For example, we previously showed that the MSHOT-Pi4 pulse sequence yields spectra with resonance line widths about half of those observed using the conventional pulse sequence when applied to membrane proteins undergoing rapid uniaxial rotational diffusion in phospholipid bilayers. In contrast, the line widths were not changed in microcrystalline samples where the molecules did not undergo global motions. Here, we demonstrate experimentally and describe analytically how some Hamiltonian terms are susceptible to sample motions, and it is their removal through the critical π/2 Z-rotational symmetry that confers the “motion adapted” property to the MSHOT-Pi4 pulse sequence. This leads to the design of separated local field pulse sequence “Motion-adapted SAMPI4” and is generalized to an approach for the design of decoupling sequences whose performance is superior in the presence of molecular motions. It works by cancelling the spin interaction by explicitly averaging the reduced Wigner matrix to zero, rather than utilizing the 2π nutation to average spin interactions. This approach is applicable to both stationary and magic angle spinning solid-state NMR experiments.
INTRODUCTION
Compared to other methods of protein structure determination, such as X-ray crystallography and solution NMR spectroscopy, solid-state NMR spectroscopy offers several advantages for applications to membrane proteins; in particular, solid-state NMR enables them to be studied in their native environment of liquid crystalline phospholipid bilayers under physiological conditions.1, 2, 3 This can be accomplished with protein-containing bilayer samples that are either magnetically or mechanically aligned relative to the magnetic field using Oriented Sample (OS) solid-state NMR or unoriented proteoliposomes samples using Rotationally Aligned (RA) solid-state NMR.4 In the samples for both types of solid-state NMR experiments, the proteins undergo rapid rotational diffusion about the lipid bilayer normal.5 The motional averaging of powder patterns from dipole-dipole and chemical shift interactions were observed in some of the earliest high-resolution solid-state NMR experiments on polycrystalline samples,6 in the 31P NMR spectra of phospholipids,7 and in the 13C′ NMR spectra of bacteriorhodopsin in bilayers.8
There are additional effects of motions on solid-state NMR spectra and radiofrequency (rf) pulses. Griffin and co-workers8, 9 observed considerable line broadening of resonances in Magic Angle Spinning (MAS) solid-state NMR spectra of bacteriorhodopsin undergoing rotational diffusion, and further exploited the motion effects in multiple pulse sequence. Similar effects have been observed in organic molecules, lipids, and membrane proteins.10, 11, 12, 13, 14, 15, 16, 17 Meanwhile, interferences between rotor rotation and pulse sequence have been utilized in many recoupling methods in MAS solid-state NMR.18, 19, 20
Although the line broadening effects of molecular motions in solid-state NMR spectra been well characterized, there have been relatively few studies focused on reduction of such line broadening under similar circumstances. For example, we have demonstrated a pulse sequence for static OS solid-state NMR21 that reduced 1H line widths by more than a factor of two compared to previous versions of the pulse sequence in samples where the proteins undergo fast rotational diffusion. The narrow 1H line widths enabled more accurate measurements of 1H chemical shift frequencies in solid-state NMR heteronuclear correlation (HETCOR) spectra and 1H–15N dipolar couplings in proton-detected local field (PDLF) spectra. Furthermore, the relatively narrow 1H resonance line widths enhanced the resolution in multidimensional spectra where the 1H chemical shift was one of the frequency dimensions. Interestingly, substantial line narrowing was only observed for membrane proteins undergoing rapid rotational diffusion, but not for a static crystal sample of N-acetylated leucine. The pulse sequence incorporated two modifications that combined to improve its performance. The first modification was to replace the continuous-wave (CW) heteronuclear decoupling with a single refocusing π pulse. While it had been shown by Waugh and Rothwell22 that continuous wave decoupling is susceptible to interference from molecular motion, by contrast a single π pulse is largely independent of any interference effects from protein motion, and the situation does not require further analysis. The second modification was the replacement of the frequency-switched Lee-Goldberg (FSLG) sequence by a Z-rotational magic sandwich sequence MSHOT-Pi4 for 1H–1H homonuclear decoupling.
In this article, the superior performance of MSHOT-Pi4 pulse sequence for membrane protein samples is explained and generalized, including (i) an analytical derivation that identifies the Hamiltonian terms susceptible to interference from sample motion and explains how they are eliminated in the MSHOT-Pi4 sequence; (ii) the critical role of the π/2 Z-rotational symmetry in MSHOT-Pi4 sequence and the experimentally verified prediction that MSHOT4 has better performance than either MSHOT2 sequence with π symmetry or MSHOT6 with π/3 symmetry; and (iii) the design and experimental spectra of a rotating-frame separated local field pulse sequence motion-adapted SAMPI4 developed from MSHOT-Pi4 with reduced interference from sample motions.
EXPERIMENTS AND METHODS
Sample preparation
The expression and purification of Pf1 coat protein has been described previously.23 Magnetically aligned bicelle samples of Pf1 coat protein were prepared with 6-O-PC (1,2-di-O-hexyl-sn-glycero-3-phosphocholine) and 14-O-PC (1,2-di-O-tetradecyl-sn-glycero-3-phosphocholine) as previously described.23, 24, 25
Solid-state NMR Spectroscopy
15N-detected solid-state NMR spectra were obtained on a 700 MHz Bruker Avance spectrometer equipped with a homebuilt 1H/15N double-resonance probe with a strip-shield to minimize heating of the lossy samples from high frequency, high power radiofrequency irradiations.26
For the HETCOR spectra of the membrane-bound form of Pf1 coat protein (Figure 3), all experiments were performed with 71.4 kHz 1H irradiation during t1 evolution and 50 kHz 1H and 15N irradiation elsewhere in the sequence. 3 cycles of SAMPI4 were used in the mixing period of the pulse sequence,21 which selectively transferred magnetization for resonances with 1H/15N dipolar couplings > 1 kHz. All three experiments had the same total t1 evolution time of 10.584 ms. Specifically, the spectra acquired with 2- and 4-phase cycled magic sandwich sequences (Figures 3a, 3b) had 64 complex t1 points with 168 μs dwell times; and the one with a 6-phase cycled magic sandwich (Figure 3c) had 43 complex t1 points with a 252 μs dwell time. The number of scans was increased from 40 to 60 in the 6-phase cycled magic sandwich spectrum to approximately match the total acquisition time in order to obtain comparative signal to noise levels. All experiments were processed identically without apodization applied in the indirect dimension where the 1H line width was measured. The 1H carrier frequency was 10.8 ppm with the water signal set at 4.7 ppm, and the 15N carrier frequency was 117 ppm by referencing to solid ammonium sulfate.
Figure 3.

Experimental spectra acquired on membrane-bound form of Pf1 coat protein in magnetically aligned bilayers with (a) MSHOT-4 (i.e., MSHOT-Pi4), (b) MSHOT-2, and (c) MSHOT-6 decoupling. All three sequences are magic sandwich sequences with the only difference being that MSHOT-Pi4 has 4-phase cycling, MSHOT-2 has 2-phase cycling, and MSHOT-6 has 6-phase cycling. From theoretical analysis, only the 4-phase cycling scheme possesses the critical π/2 Z-rotational symmetry and offers resistance to motion interference at both ωr = 2ω1 and ωr = ω1 frequencies. Agreeing with this prediction, experimental results show that MSHOT-Pi4 gives the narrowest line width.
The motion-adapted SAMPI4 spectra were obtained on the membrane-bound form of Pf1 coat protein (Figure 4a) using 49.0 kHz radiofrequency pulses on both 1H and 15N channels. Thirty-nine real t1 points were acquired in the indirect dimension giving a total evolution time of 4.65 ms for 1H–15N dipolar coupling. As the control experiment (Figure 4b), the original SAMPI4 pulse sequence27 was used with two dwells in each increment. 50 kHz radiofrequency pulse, 40 real t1 points, and the total t1 evolution time of 4.68 ms were used. The scaling factors for the two sequences are slightly different, as described in Sec. 4. The 1H carrier was at ∼9 ppm and 15N carrier at ∼120 ppm. The data processing and line width measurements were performed with NMRDraw28 and Sparky.29
Figure 4.

Experimental separated local field spectra acquired with (a) motion-adapted SAMPI4 sequence or (b) regular SAMPI4 sequence for membrane-bound form of Pf1 coat protein in magnetically aligned bicelle. One-dimensional spectral slices from the t1 dimension of the selected peaks are shown on the right side of the two-dimensional spectra. The two sequences give rise to similar line width and spectra.
Numerical simulation
The simulations were carried out with SIMPSON 2.0.30 The four-spin system used in the simulations included three 1H and one 15N nuclei. The three 1H nuclei had 10 kHz dipolar couplings between each pair, but only one of them had a 5 kHz dipolar coupling to the 15N atom. This same 1H atom had 1 kHz chemical shift offset with respect to the 1H carrier, while the other two were on resonance. The 1H chemical shift was monitored in the indirect dimension and the magnetization was “transferred” for 15N detection through three SAMPI4 periods with 50 kHz irradiation, as in the HETCOR experiments. The 1H irradiation in the indirect dimension was set to 100 kHz for the B1 field of magic sandwich sequence and the Beff field of the Lee-Goldberg sequence. The sample rotation was set at various frequencies indicated on Figure 3, and the angle between the rotational axis and the magnetic field was chosen to be 80°. The time step over which the Hamiltonian was considered time independent was set at 0.5 μs.
THEORY
We start by formulating the Lee-Goldberg and magic sandwich homonuclear decoupling sequences without molecular motion with spherical tensor operators. Two different mechanisms of averaging homonuclear dipolar coupling are identified; the setting of the reduced Wigner matrices to zero, and the 2π pulse nutation averaging of a spin interaction. We then consider pulse sequences in the presence of molecular motion, where the ineffectiveness of the second mechanism is demonstrated. Finally, the discussion is extended to rotating-frame separated local field experiments where both homonuclear and heteronuclear dipolar couplings are present.
Magic sandwich and Lee-Goldberg decoupling for static sample
For the case of two dipolar-coupled like spins, such as two 1H nuclei, in the usual rotating frame in a high magnetic field (Zeeman interaction representation), the Hamiltonian of the system under rf irradiation can be written as
| (1) |
Using spherical tensor operator notations, the homonuclear dipolar coupling under high magnetic field takes the form31
| (2) |
where ωD and are the spatial and spin part of the dipolar coupling Hamiltonian, respectively. The conversions between spherical tensor operators and the Cartesian spin operators are
| (3a) |
| (3b) |
| (3c) |
| (3d) |
| (3e) |
In a basic magic sandwich period (Figure 1a),32 the central spin-lock period is flanked by a pair of ideal delta 90° Y and −Y pulses. The overall effect of the radiofrequency pulses can be described as a rotation with the Euler angles
| (4) |
where ω1 is the irradiation frequency during the spin-lock period. The rotation transforms the spin part of the dipolar coupling Hamiltonian through the transformation equation
| (5) |
where is the reduced Wigner matrix element. The time propagator can then be approximated using Average Hamiltonian Theory with stroboscopic observation from time t0 to t0 + τ,
| (6) |
where is the average Hamiltonian. With the central spin lock set to be τ = (2π/ω1), and therefore the radiofrequency pulses do not cause a net rotation of the spins. The first order term of under the Magnus expansion can be separated into five components according to the “m” values in the spherical tensors
| (7) |
| (8a) |
| (8b) |
| (8c) |
In the rigid lattice limit or the fast motion limit, where the frequency of the motion is much faster than 1/τ, the spatial part of the dipolar coupling Hamiltonian, ωD, is time independent. In Sec. 3B, the case when the frequency of motion is near the time scale of 1/τ will be discussed. Here, we treat ωD as a constant, and evaluate the five components of the first order average Hamiltonian.
Figure 1.
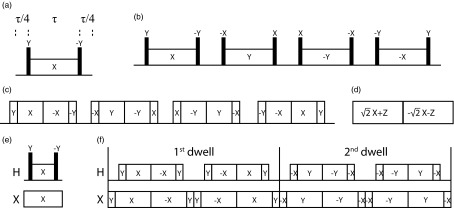
Pulse sequence elements discussed in the article. (a) Basic magic sandwich sequence element. (b) Basic element of MSHOT-4. (c) Experimental implementation of MSHOT-4 (MSHOT-Pi4 sequence). (d) Frequency-switched Lee-Goldberg sequence element. (e) Separated local field sequence with basic magic sandwich elements. (f) Motion-adapted SAMPI4 sequence, where the first and second dwells are shown, and the next two dwells have all pulse phases increase by 180° and so forth.
The m = ±1 and m = ±2 components both vanish to zero, but by two different mechanisms. The m = ±1 components are zero due to the corresponding reduced Wigner matrix . The elimination of m = ±2 components, however, is not because of the reduced Wigner matrix, which is ; instead, it is due to the term exp {−i ± 2ω1t} integrated to zero when τ = (2π/ω1). Notably, it is the difference between these two mechanisms of averaging that will influence the pulse sequence performance when molecular motion is present, as discussed in Sec. 3B. Only the time-invariant m = 0 component survives, and since , it gives . The magic sandwich sequence removes the homonuclear dipolar coupling by setting the total radiofrequency-free time to be . In practice, the magic sandwich is often designed with reflection symmetry33 and is capable of cancelling higher order Hamiltonians. Here, we limit our discussion to the first order average Hamiltonian.
Homonuclear decoupling under the Lee-Goldberg condition (Figure 1d)34, 35 can be evaluated using the same sets of formulae. The spin-lock pulse at the magic angle θm can be described as a rotation with the Euler angles
| (9) |
where ωeff is the frequency of the effective field resulting from both B1 field and offset. The resulting first order average Hamiltonian of homonuclear dipolar coupling is
| (10) |
| (11a) |
| (11b) |
| (11c) |
Out of the five reduced Wigner matrices, only . Consequently, only the m = 0 component is averaged to zero through the first mechanism, and the other four components m = ±1 and m = ±2 are averaged through the second mechanism.
In summary, two different mechanisms of eliminating homonuclear dipolar coupling are distinguished, one by explicitly setting the reduced Wigner matrixes to zero, and the other by performing a 2π nutation on the spin interaction. The magic sandwich and Lee-Goldberg decoupling sequences eliminate the homonuclear dipolar coupling by both mechanisms for different “m” components. As shown below, the second mechanism becomes ineffective in the presence of molecular motion, and this distinguishes the pulse sequence performance when applied to membrane proteins undergoing fast rotational diffusion about the bilayer normal.
Pulse sequence performance under molecular motion and the MSHOT-Pi4 sequence
In a molecule undergoing rotational motion at a frequency ωr, the spatial part of the dipolar coupling Hamiltonian ωD(t) becomes time dependent. Since ωD(t) is a second-rank tensor operator, it can be written as a linear combination of the dipolar couplings tensors (A2p) in the molecular frame
| (12) |
Consequently, the time-dependent component exp {−ipωrt} interferes with the pulse nutation in the calculation of an average Hamiltonian (Eqs. 8a, 8b, 8c, 11a, 11b, 11c). In the fast motion regime (ωr ≫ ω1), the interference is negligible. However, when ωr ≈ ω1 and/or ωr ≈ 2ω1, the second mechanism of averaging homonuclear dipolar coupling becomes ineffective, since the integration of is non-zero when mω1 + pωr ≈ 0. As a result, the m = ±2 terms in magic sandwich decoupling (Eq. 8c) and both m = ±1 and m = ±2 terms in Lee-Goldberg decoupling (Eqs. 11b, 11c) become non-zero over time τ, leaving residual homonuclear dipolar coupling in the first-order average Hamiltonian. Table 1 summarizes these possible non-zero terms. These two conditions are analogous to the single-quantum rotary resonance19 and the double-quantum HORROR condition,36 with the difference being that (i) the angle β is not at magic angle, and (ii) ωr represents a random molecular motion rather than a specifically set rotor speed.
Table 1.
The list of undesirable first-order average Hamiltonian terms generated from the interference between sample rotational motion and radiofrequency (rf) pulse sequence.
| ωr = 2ω11 | ||||
|---|---|---|---|---|
| Lee-Goldberg decoupling | {m = ±2, p = ±1}2 3, 1n3 | {m = ±1, p = ±1}3, | {m = ±1, p = ±2} | |
| {m = ±2, p = ±2} | ||||
| Magic sandwich decoupling | {m = ±2, p = ±1}3 | {m = ±2, p = ±2} | None | |
| MSHOT-Pi4 | None | None | None | |
| SAMPi4 | Homodipole | {m = ±2, p = ±1}3 | {m = ±2, p = ±2} | None |
| Heterodipole | {m = ±2, p = ±1}3 | {m = ±2, p = ±2} | None | |
| Motion-adapted SAMPi4 | Homodipole | None | None | None |
| Heterodipole | None | None | None |
ωr is the simplified rotational diffusion frequency of the membrane protein sample, and ω1 is the frequency of the rf pulses.
m is the spin-part of the Hamiltonian and is related to the rf pulse frequency. p is the space-part of the Hamiltonian and is related to the rotational frequency.
These terms would disappear for ideal perpendicularly aligned bicelles, i.e., the rotational axis is perpendicular to the magnetic field and there is no “wobbling” motion of the bicelle.
Figure 1b shows one of the magic sandwich variants with Z-rotational symmetry (MSHOT) sequences, MSHOT-4, which has 4 steps of Z-rotation and consequently a phase difference of π/2 among them. The MSHOT-Pi4 sequence21 is the practical implementation of MSHOT-4. The MSHOT pulse sequence, which stands for magic sandwich with high order truncation, was initially introduced as a way to eliminate higher order average Hamiltonians.37 Due to the special property of tensor operators, Z-rotation of the pulse phase was shown to modify the average Hamiltonian by adding only a phase factor e−imϕ to the Hamiltonian. For example, the second block in MSHOT-4 has a phase shift of ϕ = +π/2 along the Z axis, and it results in e−imϕ = −1 for m = ±2 components. The m = ±2 components in the second block thus become the inverse of the m = ±2 components in the first block
| (13) |
If ωD(t) takes the form shown in Eq. 12 and is assumed to have the same constant ωr frequency as the first block, then these two blocks cancel, averaging the m = ±2 components to zero. Thus, all first-order homonuclear dipolar coupling terms are fully averaged in a full MSHOT cycle. Notably, in an experimental situation ωD(t) is more complicated than that expressed in Eq. 12, since the molecular motion is random rather than at a constant frequency, ωr. Therefore, m = ±2 components cannot be fully averaged to zero, and the interference between rf pulses and molecular motions can never be fully eliminated. A more rigorous quantum mechanical treatment of random molecular motion may involve the use of stochastic Liouville equation,38, 39, 40 however, in this article we use the simpler average Hamiltonian theory aiming to differentiate pulse sequences’ susceptibility to molecular motion.
Notably, those MSHOT sequences that do not possess π/2 Z-rotations would not be able to cancel the m = ±2 components, and therefore would still be subject to some interference by molecular motion. Examples include MSHOT-2 and MSHOT-6, and experimental spectra from these sequences are included in Sec. 4. The results in MSHOT-6 are particularly interesting; from the original derivation of Z-rotational sequence, it is capable of cancelling residual homonuclear dipolar coupling to the same order as MSHOT-437 and should perform equally well. However, we predict that MSHOT-4 is superior to MSHOT-6 in the presence of molecular motions.
In the case of Lee-Goldberg decoupling, both m = ±1 and m = ±2 components become non-zero in the presence of molecular motion. The situation is not alleviated with the reflection symmetry that is usually imposed in FSLG41 (Fig. 1d) or phase-modulated Lee-Goldberg (PMLG)42 methods. The two symmetrical parts of the sequence correspond to the rotation of R(ωefft, θm, 0) and R(−ωefft, θm, 0), and they lead to the same reduced Wigner matrices that cannot cancel each other. As a consequence, both FSLG and PMLG remain susceptible to interferences from molecular motion.
Rotating-frame separated local field experiment under molecular motion
Polarization Inversion Spin Exchange at the Magic Angle (PISEMA)43 is distinguished from the original laboratory-frame separated local field experiments, because the magnetization is locked in the rotating frame by the radio-frequency irradiation, and the heteronuclear dipolar coupling is encoded in the flip-flop Hamiltonian I+S− + I−S+ rather than the laboratory-frame term IzSz. The rotating-frame separated local field experiments are generally found to give higher resolution, because of T1ρ being longer than T2 and the dipolar truncation effect;44 as a result, they are widely used and many variants have been designed since the original version of PISEMA was introduced, for example, SAMPI427 and HIMSELF.45
The basic magic sandwich element in SAMMY46 or SAMPI427 with two unlike spins A and B is shown in Figure 1e. The heteronuclear dipolar coupling HIS(t) is with and being the first rank spherical tensor operator of spins A and B, respectively. During the central spin-lock period, HIS(t) evolves at the exact Hartmann-Hahn match condition and results in the following first order average Hamiltonian:
| (14) |
where reduced Wigner matrix elements are and . Different from the single spin situation, the sum of “m” and “n” values is used here to categorize the average Hamiltonian
| (15a) |
| (15b) |
| (15c) |
| (15d) |
With τ = 2π/ω1 the double-quantum m + n = ±2 terms vanish through the first mechanism, i.e., exp {−i ± 2ω1t} integrates to zero. Only the zero-quantum m + n = 0 component survives and gives rise to the familiar flip-flop Hamiltonian . In the presence of molecular motion, however, m + n = ±2 would not vanish since exp {−ipωrt} interferes with the averaging of the exp {−i ± 2ω1t} term.
Similarly, a motion-adapted version of SAMPI4 sequence can be designed by rotating the pulse sequence phase of both nuclei by π/2 across the four quadrants. Figure 1f shows a practical example of this motion-adapted SAMPI4 pulse sequence. It can be readily shown that e−imϕe−inϕ = −1 for all the m + n = ±2 terms after ϕ = π/2 rotation, while the m + n = 0 term remains the same. Since the nucleus I (1H) now experiences the exact same sequence as MSHOT-PI4, the cancellation of 1H–1H homonuclear dipolar coupling should be also motion adapted. However, in our initial testing, the performance of the sequence is similar to the original SAMPI4 sequence.
Rotational diffusion of membrane proteins in aligned bicelle
The principle of motion interference is highly relevant to acquiring high-resolution NMR spectra of membrane proteins and other biopolymers, many of which undergo global reorientation about a single axis. First, the rotational diffusion of the protein about the bilayer normal has been measured to occur at ∼106 Hz by NMR and other techniques,8, 47, 48, 49 which is close to the relevant frequencies generated by the radiofrequency irradiations in the pulse sequences (40 kHz–200 kHz). Second, any types of motions that produce a spatial rotational factor exp {−imωrt} have the potential to interfere with the pulse sequence. Therefore, many other types of motion, especially local segmental motions, can be translated into rotational motion of membrane proteins and analyzed with the same method.
In the specific case of a membrane protein, the rotational diffusion always occurs about the axis defined by the bilayer normal. For magnetically aligned bicelles, the rotational axis would exactly align either parallel or perpendicular to the direction of the magnetic field. This results in β = 0 for the parallel bicelle and β = π/2 for the perpendicular bicelle under ideal situations. In Eq. 12, , where and . Consequently, all the time-dependent parts of ωD(t) (p = ±1 and p = ±2 components) vanish for a parallel bicelle, and p = ±1 but not p = ±2 components vanish for a perpendicular bicelle. Therefore, motion in an ideal parallel bicelle does not interfere with the pulse sequence performance; and in an ideal perpendicular bicelle, motion only interferes through p = ±2 components. In practical situations, however, “wobbling” motions of the bicelle often exist and could cause the fluctuation of the rotational diffusion axis. Consequently, the deviation of β angle away from 0 and π/2 re-introduces the time-dependent components as well as the motion interference to pulse sequence performance. For magnetically aligned bicelles, the existence of the “wobbling” motion can be quantified by the order parameter, which is usually less than unity (∼0.85).50, 51 By contrast, in mechanically aligned bilayers, the order parameter is usually equal to 1.0. It could suggest that the “wobbling” of the rotational diffusion axis is minimal in mechanically aligned bilayers, which therefore is potentially less susceptible to motion interference.
RESULTS AND DISCUSSION
Numerical simulation of 1H line width at various sample rotation rates
Three pulse sequences are compared, including (i) the conventional HETCOR sequence FSLG-CW,27, 52 which uses FSLG for 1H–1H homonuclear decoupling and CW irradiation for 1H–15N heteronuclear decoupling, and (ii) the recently described MSHOT-Pi4/Pi.21 The MSHOT-Pi4 sequence (Figure 1c) was the practical implementation of the basic MSHOT-4 sequence with the added inverse spin-lock components for reducing the effect of radiofrequency field inhomogeneity and the finite pulse compensation scheme adapted from the SAMPI4 sequence. The MSHOT-Pi4 sequence on the 1H channel was combined with the single π pulse on the 15N channel, and thus the total sequence was named MSHOT-Pi4/Pi. In addition, the third sequence, a two-phase Z-rotational MSHOT (MSHOT-2), is simulated as well; the only difference between it and the MSHOT-Pi4 sequence is that pulse phases are only X and −X, rather than being cycle through all four quadrants. The two phase cycled magic sandwich sequence was first described in the TREV-8 sequence33 and the current version MSHOT-2 sequence uses the same finite pulse compensation scheme as SAMPI427 and MSHOT-Pi4.21
Numerical simulations of the effects of motion on the radiofrequency irradiations were performed with the SIMPSON package30 by setting the sample to rotate at various constant speeds (Figure 2). Two-dimensional 1H–15N HETCOR spectra were simulated for three dipolar coupled 1H and one 15N spins, and the indirect dimension was used to analyze the performance of the sequences, which is similar to the strategy previously used to develop the MSHOT-Pi4/Pi pulse sequence.21
Figure 2.

Numerical simulation of 1H chemical shift line width at various sample rotation rates with SIMPSON software. (a) FSLG and continuous wave (CW) for homonuclear and heteronuclear decoupling. (b) MSHOT-2 (magic sandwich with two phase cycling, TREV-8) and a single π refocusing pulse for homonuclear and heteronuclear decoupling. (c) MSHOT-4 (magic sandwich with four phase cycling, MSHOT-Pi4) and a single π refocusing for homonuclear and heteronuclear decoupling. Effective field of all three homonuclear decoupling sequences are set at 100 kHz, and therefore the m = ±1 and m = ±2 components of the average Hamiltonian falls at 100 kHz and 200 kHz. Line broadening occurs when the rotational frequency of the nuclei coincides with these two frequencies. From theoretical analysis, interference with FSLG decoupling occur at both frequencies, TREV-8 has reduced interference at 100 kHz frequency, and MSHOT-Pi4 is resistant to interference at both frequencies.
The purpose of comparing the three sequences is that, from the theoretical analysis, the motion interference with FSLG decoupling occurs for both m = ±1 and m = ±2 components, MSHOT-2 is interfered only by the m = ±1 component, and MSHOT-4 is resistant to both sources of interference. Therefore, MSHOT-2 should have less interference at 100 kHz and MSHOT-4 should be free from interference at both frequencies. From the result, the two interference points can be clearly visible, and for MSHOT-Pi4, the two interference points are largely removed as predicted.
Here, we simulate the rotational motion analogous to the case of off-magic-angle spinning of the sample. The angle between the magnetic field and the axis of rotation is arbitrarily chosen to be 80° in order to mimic the case of perpendicular bicelle with wobbling motion, and the sample motion is simulated as a constant rotation. While the rotational diffusion of the protein sample can be simulated more rigorously as a random-walk motion, the common numerical simulation software for solid-state NMR, for example, SIMPSON30 and Spinevolution,53 are not equipped with this option. In our simulation, the interference of m = ±1 and m = ±2 components occurs only in a very narrow window at 100 kHz and 200 kHz. With more randomized motion, the spectral interference is likely to occur at a broader range of frequencies and with a less pronounced decrease of intensity. In addition, the ability of these pulse sequences to remove interference from motion would be compromised by the randomness of dipolar fluctuations.
Experimental comparison of MSHOT-2, MSHOT-4, and MSHOT-6
One prediction described in Sec. 3 is the critical role of Z-rotation symmetry at π/2 phase for removing the motion interference, and Z-rotation at other phases would not suffice. Here, MSHOT-2, MSHOT-4, and MSHOT-6 sequences are evaluated experimentally for homonuclear decoupling with membrane-bound form of Pf1 coat protein in magnetically aligned bicelles (Fig. 3). Two-dimensional HETCOR spectra were acquired back-to-back on the same sample, and from the measured line width, MSHOT-4 shows better resolution than both MSHOT-2 and MSHOT-6. This provides an important validation for the analytically derived principle of motion interference.
Experimental spectra of motion-resistant separated local field experiments
The resolution and resonance line widths in the heteronuclear dipolar coupling dimensions are compared for spectra acquired with motion-adapted SAMPI4 and the original SAMPI4 sequence on the membrane-bound form of Pf1 coat protein (Fig. 4). However, only slight improvement is observed between the two pulse sequences.
The first plausible reason for the similar performance between motion-adapted SAMPI4 and the original SAMPI4 is that separated local field spectra are usually acquired with lower levels of radiofrequency power due to the limitations of the low-γ15N channel of the probe. With our 5 mm static low-E probes, the B1 field for the separated local field spectra is usually around 50 kHz, while in the HETCOR experiments the 1H B1 field is typically ∼80 kHz. Since the rotational diffusion rate is around 105–106 Hz, the separated local field experiments may fortuitously avoid most of the motion interference.
Another reason for the similar performance between the two sequences is that the motion-adapted SAMPI4 sequence has 90° or 180° pulses on the 15N channel in the window where 1H are not irradiated. Compared to the simple inversion of 15N spin-lock in the original SAMPI4 sequence, these 90° pulses are likely to produce undesired terms in the average Hamiltonian. In fact, the inversion of 15N spin-lock in the middle of the original SAMPI4 sequence is beneficial in cancelling higher order or cross terms.27 We have tested replacing these simple 90° and 180° by composite pulses. For example, a composite pulse of 90270901809090 can produce a 90° rotation on Z axis54 and a pulse of 90−45904590−45 can produce 180° rotation on X axis.55 Both can fit into the short window of 3π/2 duration between the 1H spin lock periods. However, as these sequences lead to a variety of terms in the Hamiltonian, none of them have shown a significantly superior performance to the current version of motion-adapted SAMPI4 or the original SAMPI4.
For a separated local field experiment with ideal magic sandwich sequences (assuming the π/2 pulses are infinitely short), the heteronuclear dipolar coupling evolves during the dual-channel spin-lock period, which constitutes 2/3 of the total dwell. Therefore, the scaling factor of the pulse sequence is 0.67. The scaling factor for SAMPI4 was derived analytically; because the heteronuclear dipolar coupling evolves more than 2/3 of the dwell time (1.0927 times),27 and consequently, the scaling factor is 0.73. For the motion-adapted SAMPI4, the scaling factor was measured experimentally with respect to the spectra acquired by the original SAMPI4 sequence, and it was found that the dipolar coupling evolves less than 2/3 of the dwell time (0.92 times), resulting in a measured scaling factor of 0.61.
CONCLUSION
An analytically derived expression accounts for the previous observations of superior line narrowing of the MSHOT-Pi4 sequence for membrane protein samples. The derivation leads to a generalized principle for the design of the motion-adapted pulse sequences, which is to cancel the spin interaction explicitly by setting the reduced Wigner matrix to zero; and it also unveils the role of the critical π/2 phase in the Z-rotational symmetry of MSHOT-Pi4 sequence. This principle is further extended to the design of separated local field pulse sequence and the resulting motion-adapted SAMPI4 is tested experimentally.
References
- Park S. H., Das B. B., Casagrande F., Tian Y., Nothnagel H. J., Chu M., Kiefer H., Maier K., De Angelis A. A., Marassi F. M., and Opella S. J., Nature 491, 779 (2012). 10.1038/nature11580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opella S. J., Annu. Rev. Anal. Chem. 6, 305 (2013). 10.1146/annurev-anchem-062012-092631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H.-X. and Cross T. A., Annu. Rev. Biophys. 42, 361 (2013). 10.1146/annurev-biophys-083012-130326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das B. B., Nothnagel H. J., Lu G. J., Son W. S., Tian Y., Marassi F. M., and Opella S. J., J. Am. Chem. Soc. 134, 2047 (2012). 10.1021/ja209464f [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cone R. A., Nature New Biol. 236, 39 (1972). 10.1038/newbio236039a0 [DOI] [PubMed] [Google Scholar]
- Mehring M., Griffin R. G., and Waugh J. S., J. Chem. Phys. 55, 746 (1971). 10.1063/1.1676141 [DOI] [Google Scholar]
- McLaughlin A. C., Cullis P. R., Hemminga M. A., Hoult D. I., Radda G. K., Ritchie G. A., Seeley P. J., and Richards R. E., FEBS Lett. 57, 213 (1975). 10.1016/0014-5793(75)80719-8 [DOI] [PubMed] [Google Scholar]
- Lewis B. A., Harbison G. S., Herzfeld J., and Griffin R. G., Biochemistry 24, 4671 (1985). 10.1021/bi00338a029 [DOI] [PubMed] [Google Scholar]
- Long J. R., Sun B. Q., Bowen A., and Griffin R. G., J. Am. Chem. Soc. 116, 11950 (1994). 10.1021/ja00105a039 [DOI] [Google Scholar]
- Suwelack D., Rothwell W. P., and Waugh J. S., J. Chem. Phys. 73, 2559 (1980). 10.1063/1.440491 [DOI] [Google Scholar]
- Spiess H. W., Colloid Polym. Sci. 261, 193 (1983). 10.1007/BF01469664 [DOI] [Google Scholar]
- Schmidt A., Smith S. O., Raleigh D. P., Roberts J. E., Griffin R. G., and Vega S., J. Chem. Phys. 85, 4248 (1986). 10.1063/1.451796 [DOI] [Google Scholar]
- Palmer A. G., Williams J., and McDermott A., J. Phys. Chem. 100, 13293 (1996). 10.1021/jp9606117 [DOI] [Google Scholar]
- McMillan D. E., Hazendonk P., and Hodgkinson P., J. Magn. Reson. 161, 234 (2003). 10.1016/S1090-7807(03)00015-6 [DOI] [PubMed] [Google Scholar]
- Bain A. D., Prog. Nucl. Magn. Reson. Spectrosc. 43, 63 (2003). 10.1016/j.pnmrs.2003.08.001 [DOI] [Google Scholar]
- Reichert D., in Annual Reports on NMR Spectroscopy, edited by Webb G. A. (Academic Press, 2005), p. 159. [Google Scholar]
- Fares C., Qian J., and Davis J. H., J. Chem. Phys. 122, 194908 (2005). 10.1063/1.1899645 [DOI] [Google Scholar]
- Raleigh D. P., Levitt M. H., and Griffin R. G., Chem. Phys. Lett. 146, 71 (1988). 10.1016/0009-2614(88)85051-6 [DOI] [Google Scholar]
- Oas T. G., Griffin R. G., and Levitt M. H., J. Chem. Phys. 89, 692 (1988). 10.1063/1.455191 [DOI] [Google Scholar]
- Gullion T. and Schaefer J., J. Magn. Reson. 81, 196 (1989). 10.1016/0022-2364(89)90280-1 [DOI] [Google Scholar]
- Lu G. J., Park S. H., and Opella S. J., J. Magn. Reson. 220, 54 (2012). 10.1016/j.jmr.2012.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothwell W. P., and Waugh J. S., J. Chem. Phys. 74, 2721 (1981). 10.1063/1.441433 [DOI] [Google Scholar]
- Opella S. J., Zeri A. C., and Park S. H., Annu. Rev. Phys. Chem. 59, 635 (2008). 10.1146/annurev.physchem.58.032806.104640 [DOI] [PubMed] [Google Scholar]
- Park S. H., Marassi F. M., Black D., and Opella S. J., Biophys. J. 99, 1465 (2010). 10.1016/j.bpj.2010.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu G. J., Son W. S., and Opella S. J., J. Magn. Reson. 209, 195 (2011). 10.1016/j.jmr.2011.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C. H., Grant C. V., Cook G. A., Park S. H., and Opella S. J., J. Magn. Reson. 200, 74 (2009). 10.1016/j.jmr.2009.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevzorov A. A. and Opella S. J., J. Magn. Reson. 185, 59 (2007). 10.1016/j.jmr.2006.09.006 [DOI] [PubMed] [Google Scholar]
- Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., and Bax A., J. Biomol. NMR 6, 277 (1995). 10.1007/BF00197809 [DOI] [PubMed] [Google Scholar]
- Goddard T. D. and Kneller D. G., SPARKY 3, University of California, San Francisco; see http://www.cgl.ucsf.edu/home/sparky/.
- Bak M., Rasmussen J. T., and Nielsen N. C., J. Magn. Reson. 147, 296 (2000). 10.1006/jmre.2000.2179 [DOI] [PubMed] [Google Scholar]
- Mehring M. and Weberruss V. A., Object-Oriented Magnetic Resonance: Classes and Objects, Calculations and Computations (Academic Press, New York, 2001). [Google Scholar]
- Rhim W. K., Pines A., and Waugh J. S., Phys. Rev. B 3, 684 (1971). 10.1103/PhysRevB.3.684 [DOI] [Google Scholar]
- Takegoshi K. and McDowell C. A., Chem. Phys. Lett. 116, 100 (1985). 10.1016/0009-2614(85)80134-2 [DOI] [Google Scholar]
- Lee M. and Goldburg W. I., Phys. Rev. 140, A1261 (1965). 10.1103/PhysRev.140.A1261 [DOI] [Google Scholar]
- Goldburg W. I. and Lee M., Phys. Rev. Lett. 11, 255 (1963). 10.1103/PhysRevLett.11.255 [DOI] [Google Scholar]
- Nielsen N. C., Bildsoe H., Jakobsen H. J., and Levitt M. H., J. Chem. Phys. 101, 1805 (1994). 10.1063/1.467759 [DOI] [Google Scholar]
- Hohwy M. and Nielsen N., J. Chem. Phys. 106, 7571 (1997). 10.1063/1.473760 [DOI] [Google Scholar]
- Kubo R., J. Phys. Soc. Jpn. S26, 1 (1969). [Google Scholar]
- Levitt M. H. and Di Bari L., Phys. Rev. Lett. 69, 3124 (1992). 10.1103/PhysRevLett.69.3124 [DOI] [PubMed] [Google Scholar]
- Ghose R., Concepts Magn. Reson. 12, 152 (2000). [DOI] [Google Scholar]
- Bielecki A., Kolbert A. C., and Levitt M. H., Chem. Phys. Lett. 155, 341 (1989). 10.1016/0009-2614(89)87166-0 [DOI] [Google Scholar]
- Vinogradov E., Madhu P. K., and Vega S., Chem. Phys. Lett. 314, 443 (1999). 10.1016/S0009-2614(99)01174-4 [DOI] [Google Scholar]
- Wu C. H., Ramamoorthy A., and Opella S. J., J. Magn. Reson., Ser. A 109, 270 (1994). 10.1006/jmra.1994.1169 [DOI] [Google Scholar]
- Gan Z., J. Magn. Reson. 143, 136 (2000). 10.1006/jmre.1999.1971 [DOI] [PubMed] [Google Scholar]
- Dvinskikh S. V., Yamamoto K., and Ramamoorthy A., J. Chem. Phys. 125, 034507 (2006). 10.1063/1.2212939 [DOI] [PubMed] [Google Scholar]
- Nevzorov A. A. and Opella S. J., J. Magn. Reson. 164, 182 (2003). 10.1016/S1090-7807(03)00240-4 [DOI] [PubMed] [Google Scholar]
- Park S. H., Mrse A. A., Nevzorov A. A., De Angelis A. A., and Opella S. J., J. Magn. Reson. 178, 162 (2006). 10.1016/j.jmr.2005.08.008 [DOI] [PubMed] [Google Scholar]
- Guigas G. and Weiss M., Biophys. J. 91, 2393 (2006). 10.1529/biophysj.106.087031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fooksman D. R., Edidin M., and Barisas B. G., Biophys. Chem. 130, 10 (2007). 10.1016/j.bpc.2007.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prosser R. S., Hwang J. S., and Vold R. R., Biophys. J. 74, 2405 (1998). 10.1016/S0006-3495(98)77949-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders C. R., Hare B. J., Howard K. P., and Prestegard J. H., Prog. Nucl. Magn. Reson. Spectrosc. 26, 421 (1994). 10.1016/0079-6565(94)80012-X [DOI] [Google Scholar]
- Ramamoorthy A., Wu C. H., and Opella S. J., J. Magn. Reson., Ser. B 107, 88 (1995). 10.1006/jmrb.1995.1063 [DOI] [PubMed] [Google Scholar]
- Veshtort M. and Griffin R. G., J. Magn. Reson. 178, 248 (2006). 10.1016/j.jmr.2005.07.018 [DOI] [PubMed] [Google Scholar]
- Freeman R., Frenkiel T. A., and Levitt M. H., J. Magn. Reson. 44, 409 (1981). 10.1016/0022-2364(81)90181-5 [DOI] [Google Scholar]
- Madhu P. K., Zhao X., and Levitt M. H., Chem. Phys. Lett. 346, 142 (2001). 10.1016/S0009-2614(01)00876-4 [DOI] [Google Scholar]