

Abstract
Ebola virus and Sudan virus are members of the family Filoviridae of nonsegmented negative-strand RNA viruses (‘filoviruses’) that cause severe hemorrhagic fever with fatality rates as high as 90%. Infection by filoviruses requires membrane fusion between the host and the virus; this process is facilitated by the two subunits of the envelope glycoprotein, GP1 (the surface subunit) and GP2 (the transmembrane subunit). The membrane proximal external region (MPER) is a Trp-rich segment that immediately precedes the transmembrane domain of GP2. In the analogous glycoprotein for HIV-1 (gp41), the MPER is critical for membrane fusion and is the target of several neutralizing antibodies. However, the role of the MPER in filovirus GP2 and its importance in membrane fusion has not been established. Here, we characterize the conformational properties of peptides representing the GP MPER segments of Ebola virus and Sudan virus in the presence of micelle-forming surfactants and lipids, at pH 7 and pH 4.6. Circular dichroism (CD) spectroscopy and tryptophan fluorescence indicate that the GP2 MPER peptides bind to micelles of sodium dodecyl sulfate (SDS) and dodecylphosphocholine (DPC). Nuclear magnetic resonance (NMR) spectroscopy of the Sudan virus MPER peptide revealed that the residues 644–651 interact directly with DPC, and that this interaction enhances helical conformation of the peptide. The Sudan virus MPER peptide was found to moderately inhibit cell entry by a GP-pseudotyped vesicular stomatitis virus, but did not induce leakage of a fluorescent molecule from large unilammellar vesicle comprised of 1-palmitoyl-2-oleoylphostatidyl choline (POPC) or cause hemolysis. Taken together, this analysis suggests the filovirus GP MPER binds and inserts shallowly into lipid membranes.
Members of the family Filoviridae of nonsegmented negative-strand RNA viruses are taxonomically classified into two genera – Ebolavirus and Marburgvirus (‘filoviruses’) (1, 2). Two prototypic ebolaviruses, Ebola virus (EBOV) and Sudan virus (SUDV) were first identified in 1976 when outbreaks occurred in Zaire (known today as the Democratic Republic of Congo) and Sudan. Filovirus infection causes severe hemorrhagic fever; EBOV and SUDV infections have been associated with high case fatality rates (50–90% in some outbreaks) (3–5).
Filoviruses enter cells using “class I” viral fusion proteins, defined by the formation of an α-helical six-helix bundle by the glycoprotein ectodomain (6 – 8). GP is the envelope glycoprotein and consists of GP1 and GP2, the receptor-binding and fusion subunits, respectively (9, 10). GP1 and GP2 are generated from the GP precursor by furin cleavage and remain associated on the prefusion spike via disulfide bonds (10).
EBOV entry, which has been well-characterized, is initiated by binding of GP1 to adherence factors and uptake of the viral particle into the endosome (11). Once in the endosome, host cysteine proteases cathepsins L and B proteolytically cleave GP1 removing all but a ~17kD fragment (12). This cleavage event is thought to expose a receptor-binding domain (RBD) on GP1 that engages a putative host receptor, the endosomal cholesterol transporter Niemann-Pick C1 (NPC-1), to trigger membrane fusion (13–16). Next, the fusion loop (FL) of GP2 inserts into the endosomal membrane, leading to an extended intermediate conformation in which GP2 spans both viral and host endosomal membranes (6, 7). The heptad repeat regions of GP2 then fold into the highly stable six-helix bundle that brings the viral and host membranes into proximity (17–19). The energy released from this folding event is thought to decrease the kinetic barrier for membrane fusion, similar to other class I glycoproteins such as those from HIV-1 and influenza (6, 7). Presumably, this folding event yields a ‘hemifusion’ intermediate in which the outer leaflets of the two bilayers are fused but the inner bilayer is not. Subsequent events result in formation of a fusion pore through which the viral contents are delivered into the cellular cytosol.
The transition from the hemifusion intermediate to formation of the fusion pore is thought to be the rate-limiting step in some systems (20). Studies on the HIV-1 gp41 fusion peptide (analogous to the EBOV GP2 FL) and the membrane-proximal external region (MPER), a Trp-rich segment that lies between the CHR and the transmembrane (TM) domain, indicate that these segments have membrane-binding activity (21 – 26). It has been proposed that the gp41 MPER induces lipid mixing by forming a kinked a-helix that embeds along the membrane surface as a result of its high tryptophan content (27, 28). These results suggest that the fusion peptide and MPER are involved in promoting late fusion events for HIV-1 gp41, possibly by facilitating the hemifusion to fusion pore transition (26, 29). Furthermore, the native MPER sequence of gp41 is required for infection and is the target of broadly neutralizing antibodies (21, 27, 28).
In the EBOV GP2, NMR structures of a peptide representing the FL at neutral and low pH revealed that it adopts an extended loop structure at pH 7 and bends 90° at pH 5.5 to adopt a membrane lytic active form (30). However, the GP2 MPER has not been characterized in detail. The MPER was not included in any of three reported GP2 core domain structures in the post-fusion conformation, so there is limited structural information available for this region (17 – 19). Sáez-Cirión et al. conducted studies on a peptide representing the ‘pre-TM’ region that contains the MPER, and concluded this peptide is largely α-helical and membrane destabilizing (31). However, these structural studies were largely low-resolution (infrared-spectroscopy), and the behavior under low pH conditions was not explored (32).
Here we describe detailed characterization of peptides representing the GP2 MPER. Peptides of the EBOV and SUDV MPER regions were synthesized and characterized using circular dichroism (CD), tryptophan fluorescence, and nuclear magnetic resonance (NMR) in the presence of sodium dodecyl sulfate (SDS) and dodecylphosphocholine (DPC). These peptides were functionally characterized by performing viral entry inhibition assays with VSV-pseudotyped virus, vesicle leakage assays using large unilamellar vesicles (LUVs), and hemolysis.
Materials and Methods
Materials
N,N-dimethylformamide (DMF), dichloromethane (DCM), hydroxybenzotriazole (HOBt), 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) were obtained from AGTC Bioproducts, Wilmington, MA. N,N-diisopropylethylamine (DIEA), acetic anhydride, trifluoroacetic acid (TFA), thioanisole, 1,2-ethanedithiol, trifluoroethanol (TFE), and 36.5–38% hydrochloric acid and sodium dodecyl sulfate (SDS) were obtained from Sigma-Aldrich, St. Louis, MO. Fmoc-protected amino acids were obtained from Applied Biosystems/Life Technologies Corporation, Carlsbad, CA or AGTC Bioproducts. Rink Amide AM Resin (200–400 mesh) was obtained from NovaBiochem, Hohenbrunn, Germany. Acetonitrile was obtained from Fisher Scientific, Pittsburgh, PA. Dodecylphosphocholine (DPC), 1-palmitoyl-2-oleoylphostatidyl choline (POPC), and Liposofast Mini Extrusion system were obtained from Avanti Polar Lipids, Inc., Alabaster, AL. GP-pseudotyped virus was obtained from Dr. Kartik Chandran. HyClone (Logan, UT) Dulbecco’s Modified Eagles Medium + 4mM Glutamine + 4.5 g/L Glucose was used to perform the peptide inhibition assays. For the calcein leakage assays, phosphate-buffered saline (PBS) was obtained from Thermo Scientific, Amarillo, TX, N-[Tris(hydroxymethyl)methyl]-2-aminoethanesulfonic acid (TES buffer), sodium citrate, calcein, and Triton X-100 were obtained from Sigma-Aldrich. Fresh human red blood cells (hRBCs) were obtained from Research Blood Components, LLC (Brighton, MA).
Peptide Synthesis and Purification
Peptides were synthesized on an Applied Biosystems 433A synthesizer using Nα-Fmoc protecting goup strategy. Rink amide resin (0.1 mmol) was loaded into the reaction vessel and each Fmoc-protected amino acid was loaded into reaction cartridges at 3-fold molar excess. The N-terminus of the peptides were acetylated using 50% acetic anhydride/DCM. Simultaneous side chain deprotection and cleavage was carried out by treatment with 95% TFA, 2.5% thioanisole, and 2.5% 1,2-ethanedithiol for 3 h. After the resin was filtered, the crude peptide was precipitated, washed twice with cold diethyl ether, and pelleted. The pellet was suspended in a water/acetonitrile mixture and then lyophilized. Crude lyophilized peptide was redissolved in water/acetonitrile and purified by reverse-phase HPLC on a Vydac C18 column (10 μm, 25 × 21.2 mm) with water/acetonitrile mobile phases containing 0.1% TFA. Purified peptide was lyophilized and redissolved in 10mM phosphate buffer, pH 7.1. For NMR, viral entry inhibition, and vesicle leakage assays, the trifluoroacetate counterions were exchanged for chloride ions by dissolving lyophilized peptide in 50 mM hydrochloric acid and lyophilizing. This process was repeated twice. Peptide purity was determined using analytical reverse-phase HPLC and estimated to be > 90% in for both peptides. Peptide mass was confirmed using MALDI-TOF. Concentrations were determined by absorbance at 280nm using an extinction coefficient of 22,000 M−1 cm−1.
Circular Dichroism Spectroscopy
All spectra were acquired on a Jasco J-815 spectrometer with a 1 mm quartz cuvette. Peptides were dissolved to a concentration of 44.5 μM in the appropriate buffer (10 mM phosphate buffer, pH 7.1 or 10 mM sodium acetate, pH 4.6). Wavelength scans were obtained in duplicate at 0.5 nm step size and a speed of 2 nm/sec from 190 – 260nm. Background scans without peptide were also obtained and subtracted from the wavelength scans prior to converting mdeg units to mean residue ellipticity (θ). For the TFE experiments, a 60% TFE solution was made in the appropriate buffer. SDS titrations were performed by first obtaining a wavelength scan with peptide in the appropriate buffer without detergent; SDS was then added from a 100 mM stock solution in 0.5 mM increments from 1 mM to 10 mM and the spectra recorded. Background scans were taken independently for each concentration of SDS. DPC titrations were performed in a similar manner from a 200 mM stock solution in 0.1 mM increments from 0.1 mM to 2 mM. Relative CD signals in Figure 3 were calculated by obtaining θ values at 200 nm and 222 nm for each concentration of SDS/DPC using the following equation: Relative CD Signal = 1−(θ− θminimum)/ θmaximum (values for θmaximum and θminimum are included in the Supporting Information).
Figure 3.
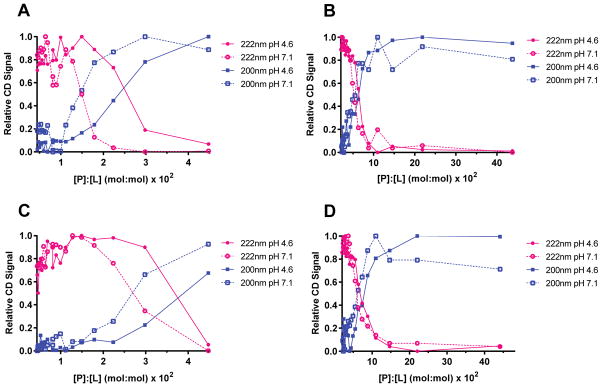
CD signal at 200nm and 222nm as a function of [P]:[L]. Relative CD signals were calculated by values at 200 nm and 222 nm for each concentration of SDS/DPC using the following equation: Relative CD Signal = 1−(θ−θminimum)/θmaximum. (A) S-MPER with SDS. (B) S-MPER with DPC. (C) Z-MPER with SDS. (D) Z-MPER with DPC.
Tryptophan Fluorescence
Fluorescence spectra were recorded on a Horiba Jobin Yvon fluorometer. An excitation wavelength of 295 nm was used and the emission spectra were recorded from 300 – 450 nm with slit widths of 5 nm. For all measurements, background readings were measured for buffer alone and with appropriate concentration of detergent. For full spectra, 1.5 μM S-MPER was added to either 10mM phosphate buffer, pH 7.1 or 10mM sodium acetate, pH 4.6 with or without 20mM of SDS or DPC. DPC titrations were carried out with 1 μM of peptide in the appropriate buffer. DPC was added in 0.2 mM increments from 0.2 to 3.0 mM. SDS was added in 0.2 mM increments from 0.2 to 4.0 mM. Background was subtracted from each data set before plotting.
Nuclear Magnetic Resonance (NMR) Spectroscopy
NMR experiments on ~2mM S-MPER peptide in 5mM NaOAc pH4.6, H2O:D2O (90:10) were acquired at 298K on a Bruker Avance II operating at a nominal frequency of 800MHz equipped with a triple resonance (1H, 13C, 15N) cryogenic probe including Z-axis pulse field gradients. Sequence specific assignments were obtained from 2D 1H,1H TOCSY (15 and 80ms mixing time), 2D 1H, 1H NOESY (400ms mixing time), 2D 1H,13Caliphatic HSQC, 2D 1H,13Caromatic HSQC and 2D 1H,15N HSQC spectra. For S-MPER peptide in 200mM 2H-DPC, 5mM NaOAc pH4.6, NMR experiments were acquired at 310K on a Bruker Avance II operating at a nominal frequency of 900MHz equipped a triple resonance cryogenic probe. Sequence specific assignments were obtained from 2D 1H,1H TOCSY (15 and 80ms mixing time), 2D 1H,1H NOESY (100 and 400ms mixing time), 2D 1H,13C aromatic HSQC and 2D 1H, 15N HSQC spectra in 90% H2O and a 2D 1H, 13Caliphatic HSQC spectrum in 100% D2O. The 2D 1H,1H TOCSY/NOESY were acquired with Excitation Sculpting water suppression (33). For the 5-doxyl stearate experiments a stock solution of the paramagnetic agent was dissolved in CHCl3:Methanol (2:1) and the correct amount for 6 or 9 mM was transferred to an Eppendorf tube, dried under argon gas and the peptide, previously dissolved in 200mM DPC micelles (100% D2O) was added. 2D 1H,13C aliphatic and 1H,13C aromatic HSQC spectra were acquired on a Varian Inova 600MHz spectrometer equipped with a triple resonance cryoprobe at 310K. All of the 1H,13C/15N HSQC spectra were acquired as non-uniform sampled experiments (50% sampling) and processed using the Compressed Sensing algorithm in MDDNMR v2.1 (34). All spectra were processed using NMRpipe/NMRDraw (35) and the data analyzed using CCPN Analysis (36). Secondary chemical shifts for 13Cα and 13Cβ chemical shifts were calculated from the difference method between measured values and random-coil values of 13Cα and 13Cβ (37). Chemical shifts were referenced to DSS in the samples (38). The secondary structure propensity (SSP) score were calculated using the SSP program (39) using 1Hα, 13Cα and 13Cβ chemical shifts. Average chemical shift changes for the backbone amide 1H and 15N nuclei (Δδav) of the S-MPER peptide were calculated using Δδav = {[(ΔδH)2 + 0.2(ΔδN)2]/2}1/2, where ΔδN represents the change in amide nitrogen chemical shift, and H represents the change in the amide chemical shift (40)
Peptide Entry Inhibition Assays
Entry assays were performed essentially as described (41). Vero cells (7.5 × 104 cells) were plated in a 48-well plate in the presence of 2% (v/v) FBS and 1% (v/v) Pen-Strep the day prior to experiments, resulting in cell confluency the following day. Peptide was added to the appropriate wells. After 1h of pre-incubation with peptides, GP-pseudotyped virus was added to the wells and incubated an additional 1h. Infection was stopped with 20μM ammonium chloride to neutralize endosomal pH, and infection was scored the following day by manually counting eGFP-positive cells under a fluorescence microscope.
Leakage from Large Unilamellar Vesicles
POPC LUVs were generated by making a chloroform solution containing 152 mg POPC, drying under reduced pressure to form a thin film, and mixing with a 2 mL solution of 10 mM TES + 100 mM sodium citrate + 40 mM calcein. Following 20 freeze-thaw cycles, the liposome suspension was extruded through a 100 nm polycarbonate membrane (Whatman Nuclepore Track-Etch Membrane) 21 times as required, at room temperature. Gel filtration (ÄKTA FPLC with HiPrep™ 16/60 Sephacryl TM-S-500HR column) was used to remove any free calcein. Lipid concentration was determined by Stewart assay (42) and liposome size was confirmed by dynamic light scattering (DLS) on a DynaPro nanostar (Wyatt, Santa Barbara, CA). DLS showed hydrodynamic diameter of 100nm and polydispersity in the 15–30% range. The instrument setup used standard PBS parameters; the refractive index was 1.333 (589nm, 20°C), the viscosity was 1.019 cP, and the Cauchy coefficient was 3119nm2. Data were acquired in auto-attenuation mode, and processed with DYNAMIC V6TM software.
The calcein leakage assay was performed by incubating 100μM S-MPER (obtained by dilution from a 1.4 mM peptide stock solution) with 500μM LUVs for 30 min, at pH 7 and 4.6. Leakage was determined by measuring calcein fluorescence (I) at 520nm (λex = 485nm) on a fluorescence spectrometer (Horiba Jobin Yvon Fluorolog FL 3–22). At the end of the experiments, the LUVs were lysed completely with Triton X-100 (1% w/v). Percent leakage was calculated using the following equation: (I−I0)/(Itotal−I0) × 100, where I0 is the fluorescence intensity before addition of peptide, and Itotal is the fluorescence intensity after addition of Triton X-100. Leakage experiments using the fY-Magainin 2 peptide were performed in a similar manner, as described in ref. 41 Each experiment was repeated twice, independently. Data reflect the average of the two sets of experiments.
Hemolysis Assays
For the hemolytic assay, fresh hRBCs were centrifuged at 3500 rpm, and washed with PBS until the supernatant was clear. The hRBCs were resuspended and diluted to a final concentration of 1% v/v in PBS, pH 7.4 or 4.6, and used immediately. The PBS for this assay was composed of 0.14M NaCl, 2.7mM KCl, 10.1mM Na2HPO4, and 1.76mM KH2PO4, pH 7.4, in deionized water. To perform the assay, 5 μL of several dilutions of S-MPER in PBS, or PBS alone, was added to 500 μL hRBCs in PBS, gently shaken, and incubated at 37°C for 1 h. The incubated mixture was centrifuged at 3500 rpm for 10 min and the supernatant was diluted 1:1 (v/v) in water (50μL each) in a clear bottom sterile 96-well plate. Release of hemoglobin was monitored at 415nm using a plate-reader. To calculate the percentage of hemolysis, the following equation was used: (A415, peptide− A415, buffer)/(A415, complete hemolysis − A415, buffer), where complete hemolysis was determined by mixing hRBCs with 1% TritonX-100. The experiment was also performed with the positive control Gramicidin A, essentially as described (44). Each condition was performed in triplicate, and standard deviation error was calculated for each data point.
RESULTS
Design and Synthesis of EBOV MPER Peptides
The MPER of class I fusion proteins is defined as the tryptophan-rich region between the CHR and the TM of the transmembrane subunit. Crystallographic and limited proteolysis studies indicate the CHR α-helix ends at or around Asp632 for EBOV GP2, and in fact the sequence falls out of the canonical heptad repeat at this point (17–19). The TM is predicted by hydropathy analysis to begin at position ~658 for EBOV GP2 (31). As shown in Figure 1, the region from Asp632 to Trp651 (EBOV numbering) contains a high tryptophan content, particularly in the segment from Trp644 – Trp651 and therefore we focused on this segment. This region is well conserved among the five species within the genus Ebolavirus (Figure 1; note that for the type member of the species Reston Ebolavirus, this segment corresponds to residues Asp633 – Trp652). There are four conserved tryptophans at positions 644, 645, 648, and 651 (EBOV numbering). There is a proline residue near the N-terminus which may result in conformational constraints in this region (45). There are several other conserved residues with ranging physicochemical properties, all likely to contribute to the overall structure and function of this region. In particular, residues 643–651 of GP proteins derived from members of all Ebolavirus species but one (RESTV) are completely conserved; notably RESTV is not pathogenic in humans. Moreover, some positions are conserved with respect to charge, for example, residue 650 varies between arginine and lysine. The degree of conservation and high tryptophan content suggests that the Ebola virus MPER has membrane binding or perturbing activity, and is likely analogous to the HIV-1 MPER even though these two viruses are evolutionarily distinct.
Figure 1.

Amino acid alignment of GP2 MPER regions from type members of the five species of genus Ebolavirus (BDBV, Bundbugyo virus; TAFV, Thai Forest virus). Residues that are identical in at least four of the viruses are highlighted in gray (hydrophobic) or boxed (polar). For comparison, the MPER segments of FIV and HIV-1 gp41 are included. Z-MPER and S-MPER peptides correspond the sequences shown here for EBOV and SUDV, respectively. N- and C-termini of peptides were blocked with acetyl and amide groups, respectively.
The EBOV and SUDV MPER were chosen for this study because they are the most prevalent and pathogenic among the ebolaviruses. Peptides corresponding to these regions (Z-MPER for EBOV and S-MPER for SUDV) were produced using solid-phase peptide synthesis with standard Fmoc protecting group strategy. The N-termini were blocked with an acetyl group and the C-termini by an amide. Deprotection/cleavage and purification by RP-HPLC followed standard protocols. The identity of the purified peptides was verified by MALDI-TOF and purity was judged to be > 90% by analytical RP-HPLC (see Supporting Information).
Circular Dichroism (CD) Spectroscopy: Effects of Co-Solvents and Detergents
CD spectroscopy was used to assess the overall secondary structure of Z-MPER and S-MPER. The FL undergoes a conformational change at endosomal pH to a form that induces rapid lipid mixing (30). It is reasonable to predict that the MPER also forms a membrane-active conformation at endosomal pH that differs from its conformation at neutral pH. Therefore, CD spectra were recorded at pH 7.1 in 10mM phosphate buffer (PB) and pH 4.6 in 10mM sodium acetate (NaOAc) buffer to represent the conditions at the plasma membrane and endosomal membrane, respectively (Figure 2A). The CD signatures of both peptides had a pronounced minimum at 200nm and a slight shoulder at 220nm, but no other minima or maxima under both pH conditions. These CD spectra are indicative of a mixed or slightly helical conformation, although the curve shape may be obscured to some extent by far UV interference from the tryptophans of the peptides. However, the near UV spectrum did not appear to have any rotational properties (data not shown).
Figure 2.

CD spectra of Z-MPER and S-MPER in various conditions. (A) Peptides with or without 60% TFE. (B – E) Z-MPER titrations with SDS (B and C) or DPC (D and E) pH 4.6 and 7.1. Data were acquired in SDS concentrations up to 10mM in 0.5mM increments, and in DPC concentrations up to 2mM in 0.1mM increments. (F – I) Similar analysis of S-MPER.
To assess the structural properties of Z-MPER and S-MPER in the presence of a structure-promoting co-solvent, CD spectra were recorded at pH 7.1 and pH 4.6 in the presence of 60% (v/v) of trifluoroethanol (TFE) (Figure 2A). It is believed that TFE induces secondary structure in short peptides by promoting intramolecular hydrogen bonding (46). The CD spectra of Z-MPER and S-MPER in 60% TFE resulted in a slightly right-shifted minimum (to 201 nm) and the formation of a second minimum near 217 nm. Although these spectra do not reflect a specific secondary structure, the tendency toward a double minimum suggests that these peptides have helical propensities.
To determine whether Z-MPER and S-MPER interact with lipids, titrations with SDS and DPC were performed at pH 7.1 and pH 4.6. SDS forms negatively-charged micelles at a critical micelle concentration (CMC) of 8.3 mM and DPC forms zwitterionic micelles at a CMC of 1.1 mM (47, 48). Titrations of S-MPER with up to 10 mM SDS resulted in a lipid-dependent change in CD spectrum at both pH values, reflected by the formation of a double minimum (Figures 2B and 2C). Overall, there was a gradual decrease in amplitude of the 200 nm minimum with increasing SDS and simultaneously gradual appearance of a 222 nm minimum, eliminating the original 220 nm shoulder. An overlay of spectra recorded at various SDS concentrations revealed an isodichroic point at ~ 218 nm, suggesting the change in CD spectrum reflects a two-state conformational transition. Similar results were obtained upon titration of Z-MPER with SDS (Figure 2F and 2G).
Titration with up to 2 mM DPC resulted in a similar change to the CD spectra for both S-MPER and Z-MPER in both pH conditions (Figures 2D, 2E, 2H, and 2I). For both peptides, a minimum at 222 nm developed with increasing amounts of DPC and the minimum at ~200 nm decreased in amplitude; an isodichroic point was observed at ~215 nm. However, the ratios of the signal intensities for these two minima at high DPC concentrations contrasted with the signal ratios at high SDS; with 2 mM DPC, the 200 nm band was more intense than the 222 nm band whereas the opposite relative ratio of the 200 and 222 nm bands was observed in 10 mM SDS. In addition, increasing DPC concentration resulted in the appearance of a positive band at 233 nm; this positive band was not observed with increasing SDS concentrations.
When the normalized CD signals at 200 nm and 222 nm were plotted as a function of [Peptide]:[Lipid] ratio ([P]:[L]), the conformational transition, defined by the intersection of the 200 nm and 222 nm plots, occurs at [P]:[L] of 1.5 × 10−2 (3.3 mM SDS) at pH 7.1 and 2.5 × 10−2 at pH 4.6 (1.8 mM SDS) for S-MPER (44.5 μM peptide concentration, Figure 3A). Interestingly, these transitions occurred at SDS concentrations below the CMC in standard conditions (8.3 mM). Z-MPER was also affected by pH (experiments performed at identical peptide concentrations), undergoing apparent conformational change [P]:[L] of 2.6 × 10−2 (1.7 mM SDS) at pH 7.1 and 3.7 × 10−2 (1.2 mM SDS) at pH 4.6 (Figure 3B). Since SDS titration experiments for both S-MPER and Z-MPER were performed at identical peptide concentrations, it is likely that the observed differences in [P]:[L] at which the conformational transition occurs reflects innate differences in how the two peptides interact with SDS micelles. The sequences of S-MPER and Z-MPER differ in the predicted charge at neutral pH (−3 for S-MPER and −2 for Z-MPER) and in the overall pattern of negatively-charged residues (Asp) in the N-terminal half. Since SDS micelles contain anionic headgroups, it is reasonable to expect that lipid binding (and therefore conformational transition) would occur at lower [P]:[L] for Z-MPER since it has less overall charge. Furthermore, the [P]:[L] at which the conformation transition takes place appears to be pH-sensitive (occurring at lower [P]:[L] at pH 7.1 than at pH 4.6 for both peptides). This pH-dependence may reflect intrinsic conformational tendencies of the peptides in the context of anionic lipids. Alternatively, this behavior may result from differences in SDS micelle formation under different concentrations (the CMC for SDS is pH-dependent with lower CMC at lower pH). These two possibilities are not mutually exclusive, and therefore these effects may also be due to some combination of the two
Differences in behavior between the two peptides, and at the two pH conditions were less pronounced in titrations with DPC (Figure 3B, 3D). A conformational transition took place at [P]:[L] of 6.0 × 10−2 (0.7 mM DPC) at pH 4.6 and 5.0 × 10−2 (0.8 mM DPC) at pH 7.1 for S-MPER. For Z-MPER, these conformational changes took place at [P]:[L] of 7.4 × 10−2 (0.6 mM DPC) and 6.0 × 10−2 (0.7 mM DPC) for pH 4.6 and 7.1, respectively.
The CD signatures observed in the presence of high SDS and DPC do not match those of any of the canonical secondary structures, likely due to interference by tryptophan in a manner that is dependent on membrane context. Therefore, the spectra in Figure 2B – 2I do not provide direct secondary structural information. Nonetheless, it can be concluded that there is a conformational transition in these peptides upon addition of micelle-forming surfactants SDS and DPC. TFE appears to have some effect on the CD signature, in a manner that is distinct from effects observed in SDS or DPC.
Tryptophan Fluorescence Studies
The Trp-rich content of the GP2 MPER permits the use of Trp fluorescence to detect binding of S-MPER and Z-MPER to SDS and DPC micelles. The typical emission maximum wavelength (λem) of Trp-containing peptides is 354 nm in aqueous solution and shifts toward lower wavelengths in apolar environments (24–26, 49). Addition of 20 mM SDS or DPC to S-MPER at pH 7 resulted in a blue shift in λem from 354 nm to 347 nm (Figure 4A; emission spectra were obtained by exciting at 295nm.) A large increase in fluorescence intensity resulted upon addition of DPC, a phenomenon that is commonly observed when Trp-rich peptides interact with lipids containing a positive charge (50). A decrease in fluorescence intensity typically occurs when tryptophans interact with lipids containing a negatively-charged headgroup; this effect was observed upon addition of SDS to S-MPER. Similar results were obtained with Z-MPER (data not shown). Taken together, the changes in fluorescence intensity and the blue shift of λem are indicative of an interaction between S-MPER and the lipid headgroups of SDS and DPC.
Figure 4.

Effects of micelle-forming detergents on tryptophan fluorescence. (A) Emission spectra of S-MPER alone and in the presence of 20mM SDS or 20mM DPC. (B and C) Effects of SDS (B) or DPC (C) on emission at 354nm. (D) Change in emission maximum (λem) upon detergent binding for S-MPER.
Titration of S-MPER and Z-MPER with SDS and DPC were carried out at pH 7.1 and 4.6 to determine the [P]:[L] at which the binding interaction occurs. Titration of both peptides with SDS showed a decrease in fluorescence intensity at low [P]:[L] (<0.5 × 10−3), followed by an increase to maximum with a transition at [P]:[L] ~0.7 × 10−3. There were minor differences among the two peptides and pH conditions but the overall trends were similar. This contrasts with the behavior observed by CD, where there were differences in [P]:[L] at which transition occurred for the various peptides and pH conditions. Furthermore, the specific [P]:[L] at which binding was observed did not correlate with the transition observed by CD. These discrepancies could be a consequence of the fact that the fluorescence experiments are carried out at a much lower peptide concentration (1 μM vs. 44.5 μM for the CD studies). The initial decrease in fluorescence intensity is not correlated with the characteristic blue-shift in λem (Figure 4D shows a plot of λem vs. [P]:[L] for S-MPER), but the larger transition at [P]:[L] ~0.7 × 10−3 is correlated with the blue shift. Therefore, we conclude that the transition represents the binding event and effects at lower [P]:[L] may be due to other effects.
Titration of both peptides with DPC resulted in sigmoidal decrease in fluorescence intensity that was coupled with blue shift (Figure 4D). The transition for both peptides occurred at [P]:[L] of 0.6 × 10−3 (0.7 mM DPC) at pH 7.1 and 0.8 × 10−3 (0.9 mM DPC) at pH 4.6. The behavior of both S-MPER and Z-MPER were similar to one another and similar across both pH conditions, consistent with the observations by CD.
Together, results from these studies suggest that the GP2 MPER sequences have the potential to bind an insert into surfactant micelles, and that this binding event is correlated with a conformational transition in the MPER peptides. The behavior of the peptides as monitored by fluorescence and CD for binding to DPC were consistent, indicating that the binding and conformational transition is not dependent on pH and that peptides from the two species had similar activity. The titration of peptides with SDS yielded less straightforward results when monitored by fluorescence, and did not correlate precisely with effects observed by CD. These results may reflect the fact the SDS is anionic while DPC is zwitterionic and therefore closer to the native mammalian/viral membrane.
Nuclear magnetic resonance (NMR) analysis of S-MPER structure and detergent interactions
NMR spectroscopy was used to obtain more detailed, sequence-specific information on the secondary structure of the S-MPER peptide and its interaction with micelles. Essentially complete 1H, 13C, and 15N resonance assignments for both the peptide alone (Table S1 of Supporting Information), and for the peptide in the presence of 200 mM deuterated DPC (Table S2) were made using 2D TOCSY and NOESY spectra for sequential 1H assignments, with natural abundance 1H13C and 1H15N HSQC spectra being used to obtain the corresponding 13C and 15N chemical shifts (Figures S1–S3, respectively). In the absence of detergent, the peptide appeared essentially unstructured, with no NOEs indicative of secondary structure observed. Most backbone chemical shifts were near their random coil values, although they were slightly shifted towards helical values for Asn637 to Gly647 (Figure 5A), leading to a 10 to 40% helical propensity (Figure 5B) as defined by secondary structure propensity (SSP) chemical shift analysis (39). Upon addition of 200 mM DPC there were only small chemical shift changes for the N-terminal residues Asp632 through Pro636, a slight decrease from helical shifts in residues Trp644 to Asn643, and a significant increase towards helical secondary shifts for C-terminal residues Trp644 to Gln650 (Figure 5A), leading to loss of helical propensity for the residues 637 to 642, and an increased helical propensity of up to 60% from Trp644 onwards. Only a few readily identifiable backbone NOEs consistent with helical structure were observed; this is partly due to the limited chemical dispersion in the spectrum of this fairly repetitive sequence, but mainly a result of the peptide not forming a stable, fully structured conformation even in the presence of DPC.
Figure 5.

Nuclear magnetic resonance of S-MPER (A) Chemical shift deviations from random coil shifts for δCα-δCβ. (B) Secondary structure propensity (SSP) scores for S-MPER in H2O (black) and in 200mM DPC micelles (gray) calculated using 13Cα, 13Cβ and 1Hα chemical shifts. Positive values represent helical propensity. (C) Average amide chemical shift differences, {[(Δδ1H)2 + 0.2(Δδ15N)2]/2}1/2, for S-MPER in H2O vs. 200mM DPC micelles. The alternative chemical shift difference for the ambiguous assignment of the amide correlations of Q19/W20 is shown in gray. (D) Effect of a paramagnetic agent on S-MPER in 200mM DPC micelles in D2O for the correlation of the last protonated carbon of each amino acid in the aliphatic 1H,13C HSQC spectrum (except that the Cγ/Hγ and H/C3ε correlations were used for Pro and Trp). The ratio of integrated crosspeak intensities with and without 9mM 5-doxyl stearate is plotted along with the amino acid sequence. Residues where it was not possible to get reliable intensity ratios because of overlapping crosspeaks are marked with *. (E) α-Helical wheel representation of the C-terminal residues (12–20) of S-MPER color coded by % intensity loss on addition of 9 mM 5-doxylstearate; >50% brown, 35–50% green, <35% blue.
Significant detergent-induced chemical shift changes were observed for the backbone amide 1H15N resonances of the C-terminal segment of the S-MPER (Figure 5C), consistent with only those residues (Trp644–Trp651) interacting with the detergent micelle. Since these changes could result from interactions with detergent micelles, or from conformational changes, or a combination of the two, the lipophilic paramagnetic broadening agent 5-doxylstearate was used as a direct probe of membrane interactions. The doxyl nitroxide group at C5 of the C18 stearic acid is a moderately shallow depth probe that broadens and reduces the intensity of NMR cross peaks from groups on the surface and partially inserted into the detergent micelle in which it is incorporated. As shown in Figure 5D, the side chain resonances of the first 12 residues of S-MPER were unaffected by incorporation of 9 mM doxylstearate into the DPC micelle. Residues Trp644 to Trp651, however, showed significant reductions in intensity. These intensity changes varied with a periodicity of about three, with the four Trp (644, 645, 648 and 651) side chains showing the largest effects. Mapping these paramagnetic relaxation effects onto a helical wheel representation of the C-terminal half of the peptide (Figure 5E) shows that the data are most consistent with a generally helical conformation for this segment, with the face of the nascent helix that contains all four Trp residues forming the surface that interacts only shallowly with the detergent micelle.
Biological and Membrane Perturbing Assays
The MPER segments of HIV-1 and feline immunodeficiency virus (FIV) have antiviral activity, likely because of their membrane-lytic or membrane-binding activity (51, 52). We tested the potential of S-MPER to inhibit GP-mediated cell entry using a vesicular stomatitis virus pseudotyped to display SUDV GP on its surface in place of the native G protein (‘VSV-GP’). A broadly inhibiting Ebolavirus C-peptide (‘Tat-Ebo’) was used as a positive control (41). S-MPER was found to inhibit infection by ~45% at 300 μM and ~25% at 100 μM (Figure 6A) with no toxic effect to the cells at these concentrations. This inhibition was specific for the envelope glycoprotein of SUDV; the peptde had no activity against VSV containing its native envelope glycoprotein G (‘VSV-G’) (Figure 6B).
Figure 6.

Biological and membrane-perturbing assays with S-MPER. (A) Inhibition of VSV-GP entry by S-MPER. The potent entry inhibitor Tat-Ebo (ref. 41) was used as a positive control. (B) Control entry assay using VSV-G. (C) Leakage assays with 500 μM POPC LUVs. Experiments were performed twice independently with similar results, a representative data set is shown. (D) Hemolysis assays.
The ability of S-MPER to induce leakage of a self-quenching small molecule dye (Calcein) from LUVs comprised of POPC was examined. As shown in Figure 6C, S-MPER did not induce leakage at either pH 4.6 or 7.0 at up to 80 μM peptide concentrations (corresponding to a [P]:[L] of 0.16; the LUV concentration was 500 μM). A pH-sensitive variant of magainin 2 containing trifluorotyrosine (fY-Magainin 2) was used as a control; as previously reported, fY-Magainin 2 induced LUV leakage at pH 4.6 but not at pH 7.0 (43). However, Trp fluorescence experiments indicated that both S-MPER and fY-Magainin 2 bound LUVs to a similar degree at 10 μM peptide concentrations and pH 5.0 (Supporting Information). These results indicate that, while the S-MPER peptide is able to bind and insert into membranes, this effect does not induce leakage. These results are also consistent with the lack of cellular toxicity at high peptide concentrations (300 μM). To further explore potential membrane lytic effects in the context of a more complex membrane system, hemolysis assays were performed with S-MPER and the peptide Gramicidin A, which induces hemolysis at high concentrations, as a control (Figure 6D) (44). The S-MPER peptide did not have hemolytic properties at peptide concentrations up to 140 μM.
DISCUSSION
The studies performed on peptides representing the MPER of EBOV and SUDV revealed three overall conclusions: 1) As a peptide, the GP2 MPER binds to micelle-forming surfactants in a pH-independent manner with higher affinity for zwitterionic micelles; 2) A large conformational change to a more predominantly helical state occurs for the tryptophan-rich region of this peptide upon micelle-binding; 3) These peptides have modest viral entry inhibitory activity but do not induce leakage from LUVs.
The CD spectra of S-MPER and Z-MPER were difficult to interpret because they did not correspond to a signature for a specific secondary structure. CD signatures of unstructured peptides typically exhibit a minimum at 195 nm and, in some cases, a positive band at 212 nm. An α-helical secondary structure is characterized by a double minimum at 208 nm and 222 nm. The CD spectra of S-MPER and Z-MPER did not fit either of these signatures. Instead, the peptides in aqueous solution had a minimum at 200 nm and a shoulder at 220 nm and formed double minima at 200 nm and 222 nm in the presence of surfactant. Aromatic residues, particularly Trp, often result in far UV interference of CD spectra because they have fully allowed ππ* transitions, typically at ~220nm. Trp-rich peptides can therefore exhibit unusual CD signatures. Furthermore, the orientation of the tryptophan indole group affects the rotational direction and ellipticity of peptides (53). The high Trp content of Z-MPER and S-MPER is likely the cause of such unusual spectra.
Nonetheless, titrations of both MPER peptides with SDS and DPC revealed a conformational transition in the presence of micelle-forming surfactants, which was corroborated by the tryptophan fluorescence experiments that showed binding. Both surfactants induced a blue shift in λem from 354 nm to 346 nm indicating burial of the Trp residues in the surface of the micelles. The fluorescence intensities observed in Figure 3A typical for binding of SDS and DPC by Trp-rich peptides. Binding to DPC results in an increase in fluorescence intensity because the positive charge of the DPC headgroup destabilizes the charge transfer energy between the Trp indole group and its amide backbone which typically acts as the electron acceptor. The opposite is true for SDS, resulting in a decrease in fluorescence intensity (50).
The combination of CD and Trp fluorescence experiments led to two other conclusions. First, the interaction of Z-MPER and S-MPER with DPC micelles is higher affinity than with SDS. When analyzing the interactions with SDS, the shape of the fluorescence titration curves (Figure 4C) and the apparent pH-dependent conformational transition in the CD studies (Figure 3A, B) indicate that the peptides interact with SDS in a distinct manner from DPC. Hence, Z-MPER and S-MPER interact specifically with physiologically relevant zwitterionic membranes. The second conclusion drawn from these experiments was that Z-MPER and S-MPER appear to interact with DPC in a pH-independent manner. This finding contrasts with the reported behavior of the FL, which undergoes a pH-dependent conformational change to a membrane-active form that is competent to induce lipid mixing (30).
Although there are slight differences in the [P]:[L] at which conformational transitions occur in DPC, this is likely due to subtle changes in CMC. It has been previously shown that increasing salt concentration as well as lowering pH causes the CMC to decrease (47, 48, 54, 55); therefore, the slightly lower concentration of DPC required to induce a conformational change for both peptides at pH 4.6 is likely a result of the higher salt concentration and/or the low pH. However, it cannot be ruled out the MPER peptides exhibit pH-dependent conformational changes, similar to the FL. Interestingly, the SDS-induced conformational transitions were observed at surfactant concentrations much lower than the CMC. It is therefore possible that monomeric detergent induces conformational effects of S-MPER and Z-MPER, perhaps by preventing collapse of hydrophobic groups that would be preferred in pure aqueous solutions.
2D HSQC NMR studies revealed that residues 644–651 interact with DPC and adopt a more helical conformation upon DPC binding. Interestingly, these results are in agreement with the CD spectra observed in 60% TFE. When 60% TFE was added to S-MPER, the amplitude at 220 nm intensified but the 200 nm peak intensity did not change significantly, unlike spectra observed when SDS or DPC were added. Partial structuring in the presence of TFE is consistent with the conclusions based on NMR in DPC micelles that indicate only the C-terminal portion of S-MPER is prone to helix formation in lower polarity environments. Taken together, these data suggest that S-MPER has some helical propensity in water but contains significant helical content only in the presence of surfactants. These data support an earlier study on an EBOV-derived peptide, EBOC, that is composed of the ten C-terminal residues of Z- MPER plus the first six residues of the TM region (31). FTIR data from this work estimated the 18-residue EBOC to be 80% helical, suggesting that 14 residues form an α-helix. The first four residues of EBOC were shown by our NMR analysis of S-MPER to lack secondary structure, implying that it is the region we have identified as helical, plus the TM region, that forms a helix.
FIV, the feline immunodeficiency virus, is a lentivirus that has similar pathogenesis and entry mechanism to HIV-1. It has been shown that the FIV MPER has inhibitory activity against FIV. The rationale for this inhibitory activity is that the MPER peptide associates with the target cell membrane and interacts with the fusion peptide, blocking interaction with the MPER of the fusion subunit and hence blocking fusion (26, 49, 52, 55–58). This is a reasonable speculation because this has been demonstrated with the HIV-1 C-peptide inhibitor T20. T20 is a peptide representing one of the α-helical coiled-coils and functions by blocking formation of the trimer-of-hairpins (57). The HIV-1 MPER was tested for inhibitory activity as well, but was less effective (58). The inhibitory activity of S-MPER suggests that addition of this peptide is somehow interfering with the viral entry process, possibly in a manner similar to T20. It was found for the FIV MPER that a WX2WX2W motif is required for the membrane interaction responsible for its inhibitory activity (55). Interestingly, the Ebola virus MPER also contains this motif, WTGWRQW, which is strictly conserved among all species.
Although S-MPER had modest antiviral activity, it did not induce leakage from POPC LUVs. This behavior contrasts with EBOC, a peptide that corresponds to the ‘preTM’ region of GP2 and shown to have membrane lytic activity at [P]:[L] of ~10−2. EBOC is longer than the peptides examined here, encompassing six additional C-terminal residues. It is possible that inclusion of this additional segment mediates deep insertion into the lipid bilayer, resulting in membrane lytic activity for EBOC. However, the shorter S-MPER and Z-MPER peptides studied here insert more shallowly into membranes with only the Trp face of the nacent helix interacting with the surface of the DPC micelle in the 5-doxylstearate NMR experiments, and thus, while surfactant binding and conformational changes can be detected by CD, fluorescence, and NMR, the insertion into the lipid bilayer is not dramatic enough to result in membrane lytic activity.
Overall, the results presented here indicate that the MPER segments of EBOV and SUDV bind membrane surfaces and that this induces a conformational change in the Trp-rich segment. This behavior is similar to that of the HIV-1 gp41 MPER and, by analogy, suggests a role for the EBOV and SUDV MPER in membrane fusion. A definitive assignment of function for this region would require mutational studies in the context of a pseudovirus entry system. However, a potential role for the MPER is likely not dependent on pH, which contrasts to the pH-dependent membrane activity of the FL and recent studies by our group and others that suggest low pH induces membrane-active conformations in the isolated ectodomain (30, 59, 60).
Supplementary Material
Acknowledgments
FUNDING STATEMENT
This work was funded by the Albert Einstein College of Medicine, the National Institutes of Health (AI090249 to J. R. L., GM072085 to M. E. G., and AI088027 to K. C.), and the National Science Foundation (CHE1112188 to J. G.).
ABBREVIATIONS
- EBOV
Ebola virus
- SUDV
Sudan virus
- RBD
receptor-binding domain
- FL
fusion loop
- HIV-1
human immunodeficiency virus type-I
- MPER
membrane-proximal external region
- TM
transmembrane
- CD
circular dichroism
- NMR
nuclear magnetic resonance
- SDS
sodium dodecyl sulfate
- DPC
dodecylphosphocholine
- VSV
vesicular stomatitis virus
- LUV
large unilamellar vesicles
- POPC
1-palmitoyl-2-oleoylphosphatidyl choline
- RESTV
Reston virus
- TFE
trifluoroethanol
- CMC
critical micelle concentration
- [P]:[L]
[Peptide]:[Lipid] ratio
Footnotes
CONFLICT OF INTEREST STATEMENT
The authors declare no competing financial interest.
SUPPORTING INFORMATION AVAILABLE
Analytical HPLC traces of Z-MPER and S-MPER; θmin and θmin values used for normalized CD plots in Figure 3; peptide binding to LUVs; 1H, 13C, and 15N resonance assignments; 1H13C and 1H15N HSQC spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Kuhn JH, Bao Y, Bavari S, Becker S, Bradfute S, Brister JR, Bukreyev AA, Chandran K, Davey RA, Dolnik O, Dye JM, Enterlein S, Hensley LE, Honko AN, Jahrling PB, Johnson KM, Kobinger G, Leroy EM, Lever MS, Mühlberger E, Netesov SV, Olinger GG, Palacios G, Patterson JL, Paweska JT, Pitt L, Radoshitzky SR, Saphire EO, Smither SJ, Swanepoel R, Towner JS, van der Groen G, Volchkov VE, Wahl-Jensen V, Warren TK, Weidmann M, Nichol ST. Virus nomenclature below the species level: a standardized nomenclature for natural variants of viruses assigned to the family Filoviridae. Arch Virol. 2012;155:2083–2103. doi: 10.1007/s00705-012-1454-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sanchez A, Kiley MP, Holloway BP, Auperin DD. Sequence analysis of the Ebola virus genome: organization, genetic elements, and comparison with the genome of Marburg virus. Virus Res. 1993;29:215–240. doi: 10.1016/0168-1702(93)90063-s. [DOI] [PubMed] [Google Scholar]
- 3.Feldmann H, Geisbert TW. Ebola haemorrhagic fever. Lancet. 2011;377:849–862. doi: 10.1016/S0140-6736(10)60667-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sanchez A, Lukwiya M, Bausch D, Mahanty S, Sanchez AJ, Wagoner KD, Rollin PE. Analysis of human peripheral blood samples from fatal and nonfatal cases of Ebola (Sudan) hemorrhagic fever: cellular responses, virus load, and nitric oxide levels. J Virol. 2004;78:10370–10377. doi: 10.1128/JVI.78.19.10370-10377.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sullivan N, Yang ZY, Nabel GJ. Ebola virus pathogenesis: implications for vaccines and therapies. J Virol. 2003;77:9733–9737. doi: 10.1128/JVI.77.18.9733-9737.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harrison SC. Viral membrane fusion. Nat Struct Mol Biol. 2008;15:690–698. doi: 10.1038/nsmb.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.White JM, Delos SE, Brecher M, Schornberg K. Structures and mechanisms of viral membrane fusion proteins: multiple variations on a common theme. Crit Rev Biochem Mol Biol. 2008;43:189–219. doi: 10.1080/10409230802058320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miller EH, Chandran K. Filovirus entry into cells – new insights. Curr Opin Virol. 2012;2:206–214. doi: 10.1016/j.coviro.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee JE, Fusco ML, Hessell AJ, Oswald WB, Burton DR, Saphire EO. Structure of the Ebola virus glycoprotein bound to an antibody from a human survivor. Nature. 2008;454:177–182. doi: 10.1038/nature07082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee JE, Saphire EO. Ebolavirus glycoprotein structure and mechanism of entry. Future Virol. 2009;4:621–635. doi: 10.2217/fvl.09.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nanbo A, Imai M, Watanabe S, Noda T, Takahashi K, Neumann G, Halfmann P, Kawaoka Y. Ebolavirus is internalized into host cells via macropinocytosis in a viral glycoprotein-dependent manner. PLoS Path. 2010;6:e1001121. doi: 10.1371/journal.ppat.1001121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chandran K, Sullivan NJ, Felbor U, Whelan SP, Cunningham JM. Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science. 2005;308:1643–1645. doi: 10.1126/science.1110656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wong AC, Sandesara RG, Mulherkar N, Whelan SP, Chandran K. A forward genetic strategy reveals destabilizing mutations in the Ebolavirus glycoprotein that alter its protease dependence during cell entry. J Virol. 2010;84:163–175. doi: 10.1128/JVI.01832-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carette JE, Raaben M, Wong AC, Herbert AS, Obernosterer G, Mulherkar N, Kuehne AI, Kranzusch PJ, Griffin AM, Ruthel G, Dal Cin P, Dye JM, Whelan SP, Chandran K, Brummelkamp TR. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature. 2011;477:340–343. doi: 10.1038/nature10348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cote M, Misasi J, Ren T, Bruchez A, Lee K, Filone CM, Hensley L, Li Q, Ory D, Chandran K, Cunningham J. Small molecule inhibitors reveal Niemann-Pick C1 is essential for Ebola virus infection. Nature. 2011;477:344–348. doi: 10.1038/nature10380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miller EH, Obernosterer G, Raaben M, Herbert AS, Deffieu MS, Krishnan A, Ndungo E, Sandesara RG, Carette JE, Kuehne AI, Ruthel G, Pfeffer SR, Dye JM, Whelan SP, Brummelkamp TR, Chandran K. Ebola virus entry requires the host-programmed recognition of an intracellular receptor. EMBO J. 2012;31:1947–1960. doi: 10.1038/emboj.2012.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weissenhorn W, Carfi A, Lee KH, Skehel JJ, Wiley DC. Crystal structure of the Ebola virus membrane fusion subunit, GP2, from the envelope glycoprotein ectodomain. Mol Cell. 1998;2:605–616. doi: 10.1016/s1097-2765(00)80159-8. [DOI] [PubMed] [Google Scholar]
- 18.Malashkevich VN, Schneider BJ, McNally ML, Milhollen MA, Pang JX, Kim PS. Core structure of the envelope glycoprotein GP2 from Ebola virus at 1.9-A resolution. Proc Natl Acad Sci USA. 1999;96:2662–2667. doi: 10.1073/pnas.96.6.2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koellhoffer JF, Malashkevich VN, Harrison JS, Toro R, Bhosle RC, Chandran K, Almo SC, Lai JR. Crystal structure of the Marburg virus GP2 core domain in its postfusion conformation. Biochemistry. 2012;51:7665–7675. doi: 10.1021/bi300976m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Floyd DL, Ragains JR, Skehel JJ, Harrison SC, van Oijen AM. Single-particle kinetics of influenza virus membrane fusion. Proc Natl Acad Sci USA. 2008;105:15382–15387. doi: 10.1073/pnas.0807771105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Salzwedel K, West JT, Hunter E. A conserved tryptophan-rich motif in the membrane-proximal region of the human immunodeficiency virus type 1 gp41 ectodomain is important for Env-mediated fusion and virus infectivity. J Virol. 1999;73:2469–2480. doi: 10.1128/jvi.73.3.2469-2480.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lay CS, Ludlow LE, Stapleton D, Bellamy-McIntyre AK, Ramsland PA, Drummer HE, Poumbourios P. Role for the terminal clasp of HIV-1 gp41 glycoprotein in the initiation of membrane fusion. J Biol Chem. 2011;286:41331–41343. doi: 10.1074/jbc.M111.299826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu J, Deng Y, Dey AK, Moore JP, Lu M. Structure of the HIV-1 gp41 membrane-proximal ectodomain region in a putative prefusion conformation. Biochemistry (Mosc) 2009;48:2915–2923. doi: 10.1021/bi802303b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peisajovich SG, Epand RF, Pritsker 0M, Shai Y, Epand RM. The polar region consecutive to the HIV fusion peptide participates in membrane fusion. Biochemistry. 2000;39:1826–1833. doi: 10.1021/bi991887i. [DOI] [PubMed] [Google Scholar]
- 25.Epand RF, Sayer BG, Epand RM. The tryptophan-rich region of HIV gp41 and the promotion of cholesterol-rich domains. Biochemistry. 2005;44:5525–5531. doi: 10.1021/bi0500224. [DOI] [PubMed] [Google Scholar]
- 26.Reuven EM, Dadon Y, Viard M, Manukovsky N, Blumenthal R, Shai Y. HIV-1 gp41 transmembrane domain interacts with the fusion peptide: implication in lipid mixing and inhibition of virus-cell fusion. Biochemistry. 2012;51:2867–2878. doi: 10.1021/bi201721r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun ZY, Oh KJ, Kim M, Yu J, Brusic V, Song L, Qiao Z, Wang JH, Wagner G, Reinherz EL. HIV-1 broadly neutralizing antibody extracts its epitope from a kinked gp41 ectodomain region on the viral membrane. Immunity. 2008;28:52–63. doi: 10.1016/j.immuni.2007.11.018. [DOI] [PubMed] [Google Scholar]
- 28.Song L, Sun ZY, Coleman KE, Zwick MB, Gach JS, Wang JH, Reinherz EL, Wagner G, Kim M. Broadly neutralizing anti-HIV-1 antibodies disrupt a hinge-related function of gp41 at the membrane interface. Proc Natl Acad Sci U S A. 2009;106:9057–9062. doi: 10.1073/pnas.0901474106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Noah E, Biron Z, Naider F, Arshava B, Anglister J. The membrane proximal external region of the HIV-1 envelope glycoprotein gp41 contributes to the stabilization of the six-helix bundle formed with a matching N’ peptide. Biochemistry. 2008;47:13602–13611. doi: 10.1021/bi7023139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gregory SM, Harada E, Liang B, Delos SE, White JM, Tamm LK. Structure and function of the complete internal fusion loop from Ebolavirus glycoprotein 2. Proc Natl Acad Sci U S A. 2011;108:11211–11216. doi: 10.1073/pnas.1104760108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saez-Cirion A, Gomara MJ, Agirre A, Nieva JL. Pre-transmembrane sequence of Ebola glycoprotein. Interfacial hydrophobicity distribution and interaction with membranes. FEBS Lett. 2003;533:47–53. doi: 10.1016/s0014-5793(02)03747-x. [DOI] [PubMed] [Google Scholar]
- 32.Han Z, Licata JM, Paragas J, Harty RN. Permeabilization of the plasma membrane by Ebola virus GP2. Virus Genes. 2007;34:273–281. doi: 10.1007/s11262-006-0009-4. [DOI] [PubMed] [Google Scholar]
- 33.Hwang TL, Shaka AJ. Water suppression that works: excitation sculpting using arbitrary wave-forms and pulsed-field gradiatens. J Magn Reson A. 1995;112:275–279. [Google Scholar]
- 34.Kazimierczuk K, Orekhov VY. Accelerated NMR spectroscopy by using compressed sensing. Angew Chim Int Ed. 2011;50:5556–5559. doi: 10.1002/anie.201100370. [DOI] [PubMed] [Google Scholar]
- 35.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfiefer J, Bax A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 36.Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, Llinas M, Ulrich EL, Markley JL, Ionides J, Laue ED. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins. 2005;59:687–696. doi: 10.1002/prot.20449. [DOI] [PubMed] [Google Scholar]
- 37.Schwarzinger S, Kroon GJ, Foss TR, Wright PE, Dyson HJ. Random coil chemical shifts in acidic 8 M urea: implementation of random coil shift data in NMRView. J Biomol NMR. 2000;8:43–48. doi: 10.1023/a:1008386816521. [DOI] [PubMed] [Google Scholar]
- 38.Wishart DS, Bigam CG, Yao J, Abildgaard F, Dyson JH, Oldfield E, Markley JL, Sykes BD. 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J Biomol NMR. 1995;6:135–140. doi: 10.1007/BF00211777. [DOI] [PubMed] [Google Scholar]
- 39.Marsh JA, Singh VK, Jia Z, Forman-Kay JD. Sensitivity of secondary structure propensities to sequences differences between α- and γ-synuclein: implications for fibrillation. Protein Sci. 2006;15:2795–2804. doi: 10.1110/ps.062465306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Garrett DS, Seok YJ, Peterkofsky A, Clore GM, Gronenborn AM. Identification by NMR of the binding surface for the histidine-containing phosphocarrier protein HPr on the N-terminal domain of enzyme I of the Escherichia coli phosphotransferase system. Biochemistry. 1997;36:4393–4398. doi: 10.1021/bi970221q. [DOI] [PubMed] [Google Scholar]
- 41.Miller EH, Harrison JS, Radoshitzky SR, Higgins CD, Chi X, Dong L, Kuhn JH, Bavari S, Lai JR, Chandran K. Inhibition of Ebola virus entry by a C-peptide targeted to endosomes. J Biol Chem. 286:15854–15861. doi: 10.1074/jbc.M110.207084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stewart JC. Colorimetric determination of phospholipids with ammonium ferrothiocyanate. Anal Biochem. 1980;104:10–14. doi: 10.1016/0003-2697(80)90269-9. [DOI] [PubMed] [Google Scholar]
- 43.Wang F, Qin L, Wong P, Gao J. Facile synthesis of tetrafluorotyrosine and its application in pH triggered membrane lysis. Org Lett. 2011;13:236–239. doi: 10.1021/ol102610q. [DOI] [PubMed] [Google Scholar]
- 44.Wang F, Qin L, Pace CJ, Wong P, Malonis R, Gao J. Solubilized gramicidin A as potential systemic antibiotics. Chembiochem. 2012;13:51–55. doi: 10.1002/cbic.201100671. [DOI] [PubMed] [Google Scholar]
- 45.Kim MK, Kang YK. Positional preference of proline in α-helices. Protein Sci. 1999;8:1492–1499. doi: 10.1110/ps.8.7.1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Buck M. Trifluoroethanol and colleagues: cosolvents come of age. Recent studies with peptides and proteins. Q Rev Biophys. 1998;31:297–355. doi: 10.1017/s003358359800345x. [DOI] [PubMed] [Google Scholar]
- 47.Emerson MF, Holtzer A. On the ionic strength dependence of micelle number. II. J Phys Chem. 1967;71:1898–1907. doi: 10.1021/j100865a057. [DOI] [PubMed] [Google Scholar]
- 48.Palladino P, Rossi F, Ragone R. Effective critical micellar concentration of a zwitterionic detergent: a fluorimetric study on n-dodecyl phosphocholine. J Fluorescence. 2010;20:191–196. doi: 10.1007/s10895-009-0537-0. [DOI] [PubMed] [Google Scholar]
- 49.Esposito C, D’Errico G, Armenante MR, Giannecchini S, Bendinelli M, Rovero P, D’Ursi AM. Physicochemical characterization of a peptide deriving from the glycoprotein gp36 of the feline immunodeficiency virus and its lipoylated analogue in micellar systems. Biochim Biophys Acta. 2006;1758:1653–1661. doi: 10.1016/j.bbamem.2006.06.017. [DOI] [PubMed] [Google Scholar]
- 50.Callis PR, Liu T. Quantitative prediction of fluorescence quantum yields for tryptophan in proteins. J Phys Chem B. 2004;108:4248–4259. [Google Scholar]
- 51.Giannecchini S, Di Fenza A, D’Ursi AM, Matteucci D, Rovero P, Bendinelli M. Antiviral activity and conformational features of an octapeptide derived from the membrane-proximal ectodomain of the feline immunodeficiency virus transmembrane glycoprotein. J Virol. 2003;77:3724–3733. doi: 10.1128/JVI.77.6.3724-3733.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Apellaniz B, Ivankin A, Nir S, Gidalevitz D, Nieva JL. Membrane-proximal external HIV-1 gp41 motif adapted for destabilizing the highly rigid viral envelope. Biophys J. 2011;101:2426–2435. doi: 10.1016/j.bpj.2011.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Woody RW. Contributions of tryptophan side chains to the far-ultraviolet circular dichroism of proteins. Eur Biophys J. 1994;23:253–262. doi: 10.1007/BF00213575. [DOI] [PubMed] [Google Scholar]
- 54.Rahman A, Brown CW. Effect of pH on the critical micelle concentration of sodium dodecyl sulphate. Journal of Applied Polymer Science. 1983;28:1331–1334. [Google Scholar]
- 55.Corrin ML, Harkins WD. The effect of salts on the critical concentration for the formation of micelles in colloidal electrolytes. J Am Chem Soc. 1947;69:683–688. doi: 10.1021/ja01195a065. [DOI] [PubMed] [Google Scholar]
- 56.D’Errico G, Vitiello G, D’Ursi AM, Marsh D. Interaction of short modified peptides deriving from glycoprotein gp36 of feline immunodeficiency virus with phospholipid membranes. Eur Biophys J. 2009;38:873–882. doi: 10.1007/s00249-009-0454-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ingallinella P, Bianchi E, Ladwa NA, Wang YJ, Hrin R, Veneziano M, Bonelli F, Ketas TJ, Moore JP, Miller MD, Pessi A. Addition of a cholesterol group to an HIV-1 peptide fusion inhibitor dramatically increases its antiviral potency. Proc Natl Acad Sci U S A. 2009;106:5801–5806. doi: 10.1073/pnas.0901007106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ingale S, Gach JS, Zwick MB, Dawson PE. Synthesis and analysis of the membrane proximal external region epitopes of HIV-1. J Pept Sci. 2010;16:716–722. doi: 10.1002/psc.1325. [DOI] [PubMed] [Google Scholar]
- 59.Harrison JS, Higgins CD, Chandran K, Lai JR. Designed protein mimics of the Ebola virus glycoprotein GP2 α-helical bundle: stability and pH effects. Protein Sci. 2011;20:1587–1596. doi: 10.1002/pro.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brecher M, Schornberg KL, Delos SE, Fusco ML, Saphire EO, White JM. Cathepsin cleavage potentiates the Ebola virus glycoprotein to undergo a subsequent fusion-relevant conformational change. J Virol. 2012;86:364–372. doi: 10.1128/JVI.05708-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.