

Abstract
MiR-122, a pivotal liver specific miRNA, has been implicated in several liver diseases including hepatocellular carcinoma (HCC) and hepatitis C and B viral infection. This study aimed to explore epigenetic regulation of miR-122 in human hepatocellular carcinoma (HCC) cells and to examine the effect of hepatitis C virus (HCV) and hepatitis B virus (HBV). We performed microRNA microarray analysis and identified miR-122 as the most up-regulated miRNA (6-fold) in human hepatocellular cancer cells treated with 5′aza-2′deoxycytidine (5-Aza-CdR, DNA methylation inhibitor) and 4-phenylbutyric acid (PBA, histone deacetylation inhibitor). Real-time PCR analysis verified significant upregulation of miR-122 by 5′aza and PBA in HCC cells, and to a lesser extent in primary hepatocytes. Peroxisome proliferator activated receptor-gamma (PPARγ) and retinoid X receptor alpha (RXRα) complex was found to be associated with the DR1 and DR2 consensus site in the miR-122 gene promoter which enhanced miR-122 gene transcription. 5-Aza-CdR and PBA treatment increased the association of PPARγ/RXRα, but decreased the association of its co-repressors (N-CoR and SMRT), with the miR-122 DR1 and DR2 motifs. The aforementioned DNA-protein complex also contains SUV39H1, a H3K9 histone methyl transferase, which downregulates miR-122 expression. Our findings establish a novel role of the PPARγ binding complex for epigenetic regulation of miR-122 in human HCC cells. Moreover, we show that hepatitis B virus X protein (HBX) binds PPARγ and inhibits the transcription of miR-122, whereas hepatitis C viral particles exhibited no significant effect; these findings provide mechanistic insight into reduction of miR-122 in patients with HBV but not with HCV infection.
Keywords: miR-122, PPARγ, HCC, epigenetic regulation, Hepatitis B virus X protein, HCV, liver, hepatocytes
INTRODUCTION
Hepatocellular carcinoma (HCC) is the most common primary malignant tumor of the liver and the third most common cause of cancer-related death worldwide(1). Major risk factors for HCC include infection of hepatitis C virus (HCV) and hepatitis B virus (HBV), alcoholic and nonalcoholic fatty liver diseases(2). The global burden of HCC is expected to increase in the next decades, with HCV infection responsible for the rising incidence of HCC in the United States and HBV infection as the leading cause of HCC globally. Most of the HCC risk factors are known to cause epigenetic changes such as DNA methylation and histone modification, although the precise gene targets and the underlying mechanisms remain incompletely defined.
MicroRNAs have emerged as important regulators of gene expression in both normal and disease states(3, 4). Recent evidence suggests deregulation of miRNAs in hepatocarcinogenesis and tumor progression(5, 6). In this context, it is noticeable that epigenetic modification is being recognized as a key mechanism for regulation of miRNA expression(7), although it remains largely unknown whether miRNAs in the liver are epigenetically regulated.
miR-122 is the most highly expressed miRNA in the liver(8) and is implicated in several important aspects of liver pathobiology, including hepatocarcinogenesis, HCV replication, lipid metabolism and iron homeostasis(9–13). miR-122 is known to bind 5′-UTR of the HCV genome and stimulate the translation of HCV RNA(13); accordingly, inhibition of miR-122 decreases HCV viral load in cultured cells and in chimpanzee model of HCV infection(14). The level of miR-122 is decreased in patients with HBV infection(15), although the mechanism for HBV-mediated reduction of miR-122 is not known. Deletion of miR-122 in mice has been shown to cause hepatosteatosis, liver inflammation and fibrosis, and ultimately hepatocellular carcinoma(16, 17). Several liver enriched transcription factors (LETEs) are known to regulate miR-122 expression(18, 19); however, little is known about epigenetic regulation of miR-122 expression in the liver. Additionally, while recent studies have documented the effect of miR-122 in the liver(20), it remains unknown how miR-122 expression is regulated in hepatocellular cancer cells and in liver diseases.
Peroxisome proliferator-activated receptor-γ (PPARγ) is a ligand-activated transcription factor that belongs to the nuclear hormone receptor superfamily(21). PPARγ forms a heterodimer with retinoid X receptor α (RXRα) and binds to the DNA response element consisting of a direct repeat of two hexanucleotides spaced by one or two nucleotide (DR1 or DR2 motif, respectively)(22, 23). In the absence of ligands, PPARγ/RXRα associate with corepressors such as the nuclear receptor corepressor protein (NCoR) and the silencing mediator of retinoid and thyroid hormone receptors (SMRT); this nuclear receptor co-repressor complex modify the chromatin environment through recruitment of histone deacetylase (HDAC) or histone methyltransferase (HMT), thereby downregulating transcriptional activity(24, 25). In the presence of PPARγ ligands, the corepressors become dissociated from PPARγ/RXRα, thus enabling gene transcription.
In the present study, we performed miRNA microarray analysis and our data showed that miR-122 is one of the most up-regulated miRNA in HCC cells treated with the epigenetic drugs (5-Aza-CdR and PBA). Given that the promoter region of miR-122 contains DR1 and DR2 motifs(19), we postulated that PPARγ/RXRα complex might be implicated in epigenetic regulation of miR-122 during hepatocarcinogenesis. Indeed, our experimental results demonstrate that PPARγ/RXRα associate with DR1 and DR2 motifs of the miR-122 promoter to regulate miR-122 expression in HCC cells and the effect is dependent on two PPARγ corepressors, N-CoR and SMRT, and a key HMT, SUV39H1. Moreover, our data show that hepatitis B virus X protein (HBX) binds PPARγ and inhibits the transcription of miR-122 gene, which provides mechanistic explanation for the intriguing differential regulation of miR-122 by hepatitis B and C viruses.
EXPERIMENTAL PROCEDURES
Cell culture and reagents
Cells were maintained at 37°C and 5% CO2. Human hepatocellular cancer cell lines (HepG2, Huh7 and Hep3B cells) were obtained from the American Type Culture Collection (Rockville, MD). HepG2 and Hep3B cells were cultured in minimum essential medium (MEM) and Huh7 cells in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS; Gibco) and antibiotic, respectively. Huh7.5 cells line was obtained from the laboratory of Charlie Rice (The Rockfeller University, New York) and were cultured in DMEM with 10% FBS and antibiotics. Primary human hepatocyte cultures were purchased from Lonza (Walkersville, MD) and cultured in collagen I coated plates (BD Bioscience, Bedford, MA) with hepatocyte basal medium supplemented with HCM SingleQuots growth factors (Lonza, Walkersville, MD). HepG2.2.15 cells were maintained in DMEM containing 10% FBS as previously described(26). The differentiated HepaRG cells(27) were purchased from Invitrogen (Carlsbad, CA) and maintained in William’s medium E with GlutaMax-1, supplemented with General Purpose Working Medium (Invitrogen). The immortalized untransformed human neonatal liver NeHepLxHT cells were purchased from American Type Culture Collection and cultured as described(28). The 5-Aza-2′-deoxycytidine (5-Aza-CdR), 4-phenylbutyric acid (PBA), Chaetocin and 9-cis retinoic acid were obtained from Sigma-Aldrich (St. Louis, MO). Phamacological PPARγ ligands (rosiglitazone, troglitazone, ciglitazone, 15-keto prostaglandin E2, 15-deoxy-12, 14-prostaglandin J2) were purchased from Cayman chemical (Ann Arbor, MI). Antibodies against di-methyl and tri-methyl histone H3K9, PPARγ (ChIP grade), SMRT and acetyl histone were obtained from Abcam (Cambridge, MA). Anti-N-CoR antibody was purchased from Millipore (Billerica, MA). Anti-C/EBPα, anti-Akt, anti-PTEN, anti-mTOR, anti-phospho-mTOR, anti-Smad3, anti-Smad 4 and anti-SAPK/JNK were obtained from Cell signaling (Beverly, CA). All other antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
Hepatitis C virus infection and detection
HCV virus infection was performed as described previously(29, 30). Huh7.5 cells were transfected with 20 μg of in vitro transcribed full-length HCV JFH1-GFP RNA by electroporation method. After 72 hours, the lysates and supernatant were collected by scraping and freeze-thaw cycle in dry-ice and infectious virus was clarified by centrifugation at 3,400 rpm for 5 minutes. Huh7.5 cells were plated on 6 well plates and the infectious culture medium containing HCV viral particles or control spent medium were added. 96 hours post-infection, the HCV infected GFP-positive cells were detected by fluorescence microscopy and quantitative real-time RT-PCR as previously described(29, 30) [the forward primer sequence is 5′-TCTTCACGCAGAAAGCGTCTA-3′; the reverse primer sequence is 5′-CGGTTCCGCAGACCACTATG-3′; the probe sequence is 5-′TGAGTGTCGTGCAGCCTCCAGGA-3′, labeled at the 5′ end with FAM (6-carboxyfluorescein) fluorophore reporter molecule and at the 3′ end with TAMRA (6-carboxytetramethylrhodamine) quencher molecule].
Hepatitis B virus infection and detection
The HepG2.2.15 cell line was used for production of Hepatitis B viral particles. HBV inoculum was prepared from freshly collected supernatants of HepG2.2.15 cells by ultracentrifugation in Beckman rotor at 40,000 rpm for 1 hr at 4°C. The pellet was resuspended in Williams E medium. HepaRG cells were incubated with concentrated infectious source diluted 2 fold in culture medium supplemented with 4% PEG 8000 for 20 hr at 37°C. At the end of the incubation, the cells were washed three times with the culture medium and maintained for 7 days (the medium was exchanged every 2 days). For detection of HBV DNA, the cells and supernatants were collected and DNA was extracted and purified with Qiagen DNeasy kit (Qiagen). The purified total DNA was used as template for quantitative RT-PCR. RT-PCR was performed using SYBR green PCR kit (Qiagen). Primers for amplification of the HBV DNA were 5′-ATCTTCTTGTTGGTTCTTCT-3′ (forward) and 5′-CTGAAAGCCAAACAGTGG-3′ (reverse). For detection of HBX, total RNA isolated using Trizol reagent (Invitrogen) was reversely transcribed with Superscript II RT reagent kit (Invitrogen, Carlsbad, CA), followed by quantitative RT-PCR. Primers for amplification of the HBX mRNA were 5′-TCTCAGCAATGTCAACGAC-3′ (forward) and 5-TTTATGCCTACAGCCTCCT-3′ (reverse), and for the glyceraldehyde phosphate dehydrogenase (GAPDH) mRNA were 5′-TTGCCATCAATGACCCCTTCA-3′ (forward) and 5′-GCCCCACTTGATTTTGGA-3′ (reverse).
RESULTS
Epigenetically regulated miRNAs in human hepatocellular carcinoma cells
To identify epigenetically regulated miRNAs in HCC, we performed miRNA microarray in human hepatocellular cancer cells (HepG2) treated with the DNA methylation inhibitor (5-aza-2′deoxycytidine, 5-Aza-CdR) and the histone deacetylase inhibitor (4-phenylbutyric acid, PBA). The microarray data were analyzed by using hierarchical clustering of the log2 value and displayed in a heatmap (Figure 1A). Out of 837 human miRNAs that were analyzed, 43 miRNAs were differentially expressed in 5-Aza-CdR and PBA treated cells compared to control vehicle treated cells (at the level of p<0.01). The up-regulated miRNAs include miR-122, miR-30e, miR-3922-5p, miR-125-5p, and miR-224; the down-regulated miRNAs include miR-654-3p, miR-4481, miR-133a, and miR-133b. Among these, miR-122 was identified as the most up-regulated miRNA (6.6 fold, Figure 1B).
Figure 1. The expression of miR-122 is epigenetically suppressed in HCC cells.

(A) MiRNA expression heat map depicting miRNAs differentially expressed (p<0.01) in HepG2 cells treated with control vehicle or with 5-Aza-CdR (3 μM) and PBA (3 mM) for 48 hours. (B) Summary of miRNA microarray data with at least 3 fold changes (compared to control) (p-value was calculated by ANOVA). (C) qRT-PCR analysis of mature miR-122 in HepG2 and Huh7 cells treated with control vehicle or with 5-Aza-CdR and PBA for 48 hours. The data were normalized to U6 RNA. (D) qRT-PCR analysis of pri-miR-122 in HepG2 and Huh7 cells treated with control vehicle or with 5-Aza-CdR and PBA for 48 hours. (E) qRT-PCR of miR-122 expression in HCC cell lines compared to human primary hepatocytes. (F) qRT-PCR of miR-122 expression in human primary hepatocytes treated with 5-Aza-CdR (3 μM) and PBA (3 mM) for 48 hours. The data represent mean + SD (***P<0.001, n = 3).
Given that miR-122 is the dominant hepatocyte-specific miRNA (accounting for approximately 70% of the liver’s total miRNAs)(8, 31) and its expression is decreased during hepatocarcinogenesis(16, 32), we elected to focus on epigenetic regulation of miR-122. As determined by qRT-PCR analysis, 5-Aza-CdR/PBA treatment increased the levels of mature miR-122 in two hepatocellular cancer cell lines, HepG2 and Huh7, by 11.6- and 4.2-folds, respectively (Figure 1C). 5-Aza-CdR and PBA treated HepG2 and Huh7 cells also showed significantly higher expression of pri-miR-122 level (6.3- and 5-fold increase, respectively) (Figure 1D), suggesting that epigenetic up-regulation of miR-122 occurs predominantly at the transcriptional level. We observed that primary human hepatocytes express much higher basal level of miR-122 compared to hepatocellular cancer cell lines (HepG2, Huh7 and Hep3B) (Figure 1E). However, in primary human hepatocytes, miR-122 expression was up-regulated to a lesser extent by 5-Aza-CdR/PBA (1.8-fold, Figure 1F). These results suggest that the expression of miR-122 in HCC cells is epigenetically suppressed to a greater extent than in primary hepatocytes.
5-Aza-CdR and PBA induce PPARγ/RXRα binding to DR1 and DR2 motifs of miR-122 gene promoter
The promoter region of miR-122 gene contains specific binding sites for the liver-enriched transcription factors (LETFs), such as hepatocyte nuclear factors-4α (HNF-4α) and CCAAT/enhancer-binding proteins (C/EBPs), which regulate miR-122 gene transcription(18, 19). To determine whether 5-Aza-CdR/PBA induce miR-122 expression through induction of these LETFs, we examined the protein levels of HNF-4α, C/EBPα and related signaling molecules. As shown in Supplemental Figure S1, the levels of these molecules were not significantly altered by 5-Aza-CdR and PBA treatment. We observed that the level of E-cadherin expression is increased in HepG2 cells treated with 5-Aza-CdR and PBA; this finding is consistent with the notion that E-cadherin expression is frequently suppressed by epigenetic mechanisms such as promoter hypermethylation in HCC(33).
Given that the promoter region of miR-122 contains DR1 and DR2 motifs which are recognized by several members of the nuclear hormone receptor superfamily including PPARγ(19), we performed DNA-pull down assay using biotinylated DR1 or DR2 oligonucleotides corresponding to the miR-122 promoter. As shown in Figure 2A, PPARγ and RXRα bound to miR-122 DR1 and DR2 motifs and the association was significantly enhanced by 5-Aza-CdR and PBA. In contrast, PPARα did not bind to either DR1 or DR2 consensus site. Transient transfection with a PPARγ expression vector further enhanced 5-Aza-CdR/PBA-induced PPARγ binding to miR-122 DR1 and DR2 motifs (Figure 2B). To further determine PPARγ association with miR-122 promoter DNA, we performed chromatin immunoprecipitation (ChIP) assay using three specific primer sets corresponding to the three DR1 and DR2 regions in miR-122 gene promoter. As shown in Figure 2C, 5-Aza-CdR/PBA treatment significantly enhanced PPARγ binding to the DR1 and DR2 regions of the miR-122 promoter. To analyze for miR-122 gene promoter transcription activity, we generated a luciferase reporter construct containing the DR1 and DR2 regions of the miR-122 promoter and our data showed that 5-Aza-CdR/PBA treatment significantly increased miR-122 gene promoter luciferase reporter activity (Figure 2D). Therefore, 5-Aza-CdR and PBA treatment induces PPARγ/RXRα complex association with the DR1 and DR2 element and this mechanism is implicated in 5-Aza-CdR/PBA-induced miR-122 expression.
Figure 2. PPARγ/RXRα complex in 5-Aza-CdR/PBA-induced miR-122 expression.

(A) 5-Aza-CdR/PBA induce the binding of endogenous PPARγ/RXRα to the DR1 and DR2 consensus site. (Upper panel) schematic representation of putative PPARγ/RXRα binding sites in human miR-122 gene promoter. (Mid panel) Equal amount of cell lysates from HepG2 cells were incubated with biotinylated double-stranded oligonucleotides corresponding to the DR1 and DR2 motifs in miR-122 promoter and with sterptavidin-agarose beads. The precipitated complexes were subjected to SDS-PAGE and Western blotting. (Lower panel) Western blot for PPARα in HepG2 cells with or without 5-Aza-CdR/PBA treatment. (B) 5-Aza-CdR/PBA induce PPARγ/RXRα binding to miR-122 DR1 and DR2 motifs in HepG2 cells with PPARγ overexpression. After transient transfection of PPARγ expression vector, the cells were treated with 5-Aza-CdR/PBA for 48 hours and the cell lysates were obtained for DNA pull down assay. (C) ChIP assay. The chromatin extracted from HepG2 cells treated with 5-Aza-CdR/PBA or control vehicle were subjected to immunoprecipitation with PPARγ antibody and the precipitates were subjected to qRT-PCR analysis using primers to amplify DR1 and DR2 regions as indicated in the schematic diagram (the arrowheads show the primer regions in the miR-122 promoter). Normal rabbit IgG was used as the negative control. (D) Effect of 5-Aza-CdR and PBA on miR-122 promoter luciferase activity in HepG2 and Huh7 cells. After transient transfection of miR-122-Luc promoter vectors, the cells were treated 5-Aza-CdR and PBA for 48 hours and the cell lysates were obtained for luciferase activity. (E) qRT-PCR for mature miR-122 in HepG2 cells treated with the PPARγ agonist (15-d-PGJ2, 15-keto-PGE2) or RXRα agonist (9-cis RA). The cells were transiently transfected with the PPARγ expression vector or control vector and the cells were incubated for 24 hours with DMSO or 10 μM agonist. qRT-PCR was performed to measure mature miR-122. (F) qRT-PCR for mature miR-122 in PPARγ overexpressed HepG2 cells treated with 10 μM of the PPARγ agonists (rosiglitazone, troglitazone and ciglitazone) or the vehicle control (DMSO). (G) qRT-PCR for mature miR-122 in NeHepLxHT cells transfected with the PPARγ siRNA or the PPARγ expression vector (Upper panel). Knockdown or overexpression of PPARγ in NeHepLxHT cells were confirmed by western blotting (Lower panel). The data represent mean + SD from (***P<0.001, n = 3).
As the activity of PPARγ/RXRα is influenced by specific ligands, we next examined the effect of PPARγ and RXRα ligands on miR-122 expression. For these experiments, HepG2 cells were treated with the PPARγ agonists, 15-deoxy-prostaglandin J2 (15d-PGJ2, 10 μM) or 15-keto-prostaglandin E2 (15-keto-PGE2, 10 μM), and the RXRα agonist, 9-cis-retinoic acid (9-cis RA, 10 μM). As shown in Figure 2E, the expression of miR-122 was increased by these three agonists and the effects were further augmented when PPARγ protein was overexpressed. Treatment with additional PPARγ agonists (rosiglitazone, troglitazone, ciglitazone) also increased the expression of miR-122 in PPARγ overexpressed HepG2 cells (Figure 2F). To evaluate the effects of PPARγ on miR-122 expression in non-malignant hepatocytes, NeHepLxHT cells (immortalized untransformed neonatal hepatocytes) were transfected with PPARγ siRNA or expression vector. As shown Figure 2G, knockdown of PPARγ decreased miR-122 expression, whereas overexpression of PPARγ increased it. These results demonstrate that miR-122 expression is positively regulated by PPARγ and RXRα in cells of hepatocyte origin.
5-Aza-CdR and PBA induce N-CoR and SMRT dissociation from PPARγ and DR1/DR2 complex
Given that N-CoR and SMRT are co-repressors of PPARγ(34), we performed DNA-pull down assay to determine their association with the miR-122 DR1 and DR2 motifs. Our data showed that 5-Aza-CdR and PBA treatment decreased the binding of N-CoR and SMRT to DR1 and DR2 oligonucleotides (Figure 3A). Accordingly, co-immunoprecipitation assay showed that 5-Aza-CdR and PBA treatment led to dissociation of N-CoR and SMRT from PPARγ (Figure 3B), although the protein levels of N-CoR and SMRT were not altered. These findings suggest that dissociation of N-CoR and SMRT from PPARγ and DR1/DR2 complex contribute to 5-Aza-CdR/PBA-induced miR-122 expression.
Figure 3. 5-Aza-CdR/PBA-induced miR-122 expression is associated with N-CoR/SMRT corepressor dissociation and inhibition of SUV39H1.

(A) Binding of endogenous N-CoR and SMRT to miR-122 DR1 and DR2 motifs in HepG2 cells treated with control vehicle or with 5-Aza-CdR/PBA. (B) Effect of 5-Aza-CdR and PBA on PPARγ association with N-CoR, SMRT and SUV39H1 in HepG2 cells. (Left panel) The cell lysates were immunoprecipitated with anti-PPARγ antibody followed by immunoblotting with indicated antibodies. (Right panel) Conventional western blotting using indicated antibodies. (C) Effect of 5-Aza-CdR and PBA on SUV39H1 expression in HepG2 and Huh7 cells. The cells were treated with 5-Aza-CdR/PBA for 48 hours and the cell lysates were obtained for Western blotting with anti-SUV39H1 antibody. (D) Binding of SUV39H1 to miR-122 DR1 and DR2 motifs. HepG2 cells were treated with 5-Aza-CdR and PBA for 48 hours and the cell lysates were incubated with biotinylated DR1 and DR2 oligonucleotides. The samples were subjected to western blotting using anti-SUV39H1. (E) The effect of SUV39H1 knockdown on miR-122 expression. HepG2 cells were transfected with two different siRNAs targeting SUV39H1 (100 nM) or mock siRNA as control. The efficiency of SUV39H1 knock-down was analyzed by western blotting 72 hours after transfection (left panel). qRT-PCR for mature miR-122 was performed (right panel). (F) The SUV39H1 inhibitor, chaetocin, increased miR-122 expression in HepG2 and Huh7 cells. The cells were treated with 200 nM chaetocin for 48 hours and qRT-PCR was performed to determine the level of miR-122. (G) Effect of 5-Aza-CdR/PBA on histone acetylation (chromatin immunoprecipitation assay). Chromatins extracted from HepG2 cells treated with 5-Aza-CdR/PBA or control vehicle were subjected to immunoprecipitation with anti-acetyl histone antibody. The precipitates were subjected to qRT-PCR analysis using primers to amplify DR1 and DR2 regions of the miR-122 gene promoter as indicated in the schematic diagram. The data represent mean + SD (***P<0.001, **P<0.01; n = 3).
The role of SUV39H1 and histone modification in miR-122 expression
Epigenetic regulation of gene expression is known to involve DNA methylation and histone modifications (acetylation and/or methylation). As miR-122 gene promoter contains no CpG island, we performed further experiments to determine whether histone modification might be involved in miR-122 regulation. As shown in Figure 3C, 5-Aza-CdR/PBA treatment decreased the level of SUV39H1, a H3K9 histone methyl transferase (HMT), in both HepG2 and Huh7 cells. Consistent with this, the association of SUV39H1 with miR-122 DR1 and DR2 motifs was also reduced after 5-Aza-CdR/PBA treatment (Figure 3D). Thus, SUV39H1 is a negative regulator for miR-122 gene expression; this assertion is consistent with the well-documented repression of gene transcription by SUV39H1 and its enzymatic products (H3K9 dimethyl and trimethyl)(35, 36). To further determine the role of SUV39H1 in miR-122 expression, we assessed miR-122 levels in cells transfected with SUV39H1 targeting siRNAs. As shown in Figure 3E, knockdown of SUV39H1 by two different siRNAs enhanced miR-122 expression by 5.3- and 4.3-folds, respectively. Similarly, inhibition of SUV39H1 by its pharmacological inhibitor, chaetocin, increased miR-122 expression in both HepG2 and Huh7 cells (Figure 3F). These findings are consistent with the observation that the levels of H3K9 dimethyl and trimethyl were decreased in human primary hepatocytes compared to hepatocellular cancer cells (Supplemental Figure S2). Furthermore, ChIP assay revealed that histone acetylation around DR1 and DR2 regions of the miR-122 promoter was increased in cells treated with 5-Aza-CdR and PBA (Figure 3G). Taken together, these results suggest the role of SUV39H1-mediated histone H3K9 methylation and histone acetylation in regulation of miR-122 expression.
Hepatitis C virus does not influence miR-122 expression
Hepatitis C and hepatitis B virus infection are major epigenetic factors associated with HCC. miR-122 has been shown to bind 5′-UTR of HCV RNA leading to HCV accumulation(13). To investigate whether HCV infection might influence miR-122 expression, we utilized Huh7.5 cells (which are optimal for HCV infection). In this system, more than 80% of the Huh7.5 cells become infected 96 hours after addition of the hepatitis C virus (clone JFH1-GFP), as visualized by GFP fluorescence; successful infection is also verified by qRT-PCR analysis for HCV RNA (Figure 4A). As shown in Figure 4B, the levels of miR-122 expression were not significantly different between HCV-infected and control cells. Likewise, HCV infection did not significantly alter miR-122 promoter luciferase reporter activity (Figure 4C). Thus, HCV infection does not significantly influence miR-122 expression.
Figure 4. Effect of HCV on miR-122 expression.
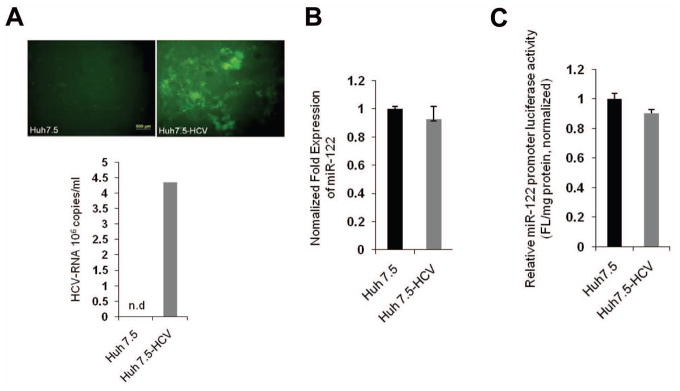
(A) Huh7.5 cells were infected with JFH1-GFP-HCV virus for 96 hours. (Upper panel) Fluorescence microscopy showing intracellular expression of GFP in cells infected with JFH1-GFP-HCV. (Lower panel) qRT-PCR analysis for HCV RNA in cells with or without JFH1-GFP-HCV infection. (B) qRT-PCR analysis for miR-122 in Huh7.5 cells with or without JFH1-GFP-HCV infection for 96 hours. The results were normalized to the level of U6 RNA. (C) miR-122 promoter luciferase reporter activity in Huh7.5 cells with or without JFH1-GFP-HCV infection for 96 hours.
Hepatitis B virus down-regulates miR-122 expression
The expression of miR-122 is known to be down-regulated in patient with HBV infection(15). To investigate the relationship between miR-122 expression and HBV, we utilized HepG2.2.15 cells, which are derived from HepG2 cells and characterized by having stable HBV expression and replication in culture systems(37). As shown in Figure 5A, successful HBV replication was confirmed by the presence of HBV DNA in HepG2.2.15 cells but not in HepG2 cells, whereas the expression of miR-122 was markedly down-regulated in HepG2.2.15 cells compared to HepG2 cells. Additionally, we performed HBV infection experiments by using supernatants of HepG2.2.15 cells in the presence of 4 % polyethylene glycol (PEG). As shown in Figure 5B and C, HBV infected HepaRG and primary human hepatocytes showed significantly decreased miR-122 compared to uninfected cells (optimal infection efficiency was confirmed by detecting HBV DNA and HBX mRNA in infected cells). These results suggest that HBV infection down-regulates the expression of miR-122.
Figure 5. Effect of HBV on miR-122 expression.

(A) HepG2.2.15 cells were cultured for 72 hours; DNA was extracted from HepG2.2.15 cells or supernatants (400 μl). HBV DNA was detected by qRT-PCR using HBV-specific primers (Left panel). HepG2 cells and their supernatants were used as negative controls. qRT-PCR analysis of mature miR-122 was performed in HepG2 and HepG2.2.15 cells (Right panel). (B) HepaRG cells were infected with HBV inoculums for 20 hours and incubated for 7 days. The medium was changed every 2 days. At the end of incubation, HBV DNAs were detected in the culture medium and HBV-infected cells (Left panel). Total RNAs extracted from uninfected or HBV-infected HepaRG cells were subjected to qRT-PCR analysis for miR-122 (Right panel). (C) Human primary hepatocytes were infected with HBV inoculums for 5 or 7 days. The HBX mRNA levels (Left panel) and miR-122 expression levels (Right panel) were determined by qRT-PCR. Levels of GAPDH mRNA and U6 were used as an internal control. The data represent mean + SD (***P<0.001, **P<0.01; n = 3).
The effect of hepatitis B virus X protein (HBX) on miR-122 expression
Given that hepatitis B virus X protein (HBX) plays a pivotal role in HBV-mediated hepatocarcinogenesis and that HBX is known to modulate transcription machinery via protein-protein interaction(38), we investigated the potential effect of HBX on miR-122 expression in our system. As shown in Figure 6A, transfection of HBX decreased miR-122 expression in HepG2 and Huh7 cells as well as in primary human hepatocytes. Accordingly, transfection of HBX also reduced miR-122 promoter luciferase reporter activity (Figure 6B). On the other hand, siRNA knockdown of HBX in HepG2.2.15 cells significantly increased the expression of miR-122 (Figure 6C). These results suggest that HBX protein is able to down-regulate miR-122 expression. Co-immunoprecipitation assay showed that HBX bound to PPARγ (Figure 6D), which is consistent with the previous report that HBX binds to the DNA binding domain of PPARγ and suppresses PPARγ-mediated transactivation(39). These observations suggest that HBX protein negatively regulates miR-122 expression through binding and inhibiting PPARγ. The role of PPARγ for suppression of miR-122 gene transcription is further corroborated by the observation that overexpression of PPARγ prevented HBX-induced reduction of miR-122 mature and pri-miRNA levels (Figure 6E and 6F). Taken together, these results provide mechanistic explanation for reduction of miR-122 in HBV-infected patients as recently reported by Wang and colleagues(15).
Figure 6. Effect of HBX on miR-122 expression.

(A) qRT-PCR analysis of miR-122 in HepG2 and Huh7 cells 48 hours after transfection with HBX expression vector or control vector (left and mid panel). Human primary hepatocytes were transfected with HBX for 72 hours and miR-122 expression levels were measured by qRT-PCR (right panel). (B) miR-122 promoter luciferase reporter activity in HepG2 and Huh7 cells 48 hours after transfection with HBX expression vector or control vector (***p<0.001). (C) Mock-siRNA and HBX-siRNA (100 nM) were transfected into HepG2.2.15 cells using lipofectamine. The levels of HBX protein (Left-upper panel) or mRNA (Left-lower panel) were detected by western blot and qRT-PCR (72 hours after transfection). The levels of miR-122 expression were measured by qRT-PCR in control and HBX-siRNA transfected HepG2.2.15 cells (Right panel). (D) HBX associates with PPARγ. HepG2 cells were transfected with the HBX expression vector or control vector and the cell lysates were immunoprecipitated with anti-PPARγ antibody followed by immunoblotting with anti-HBX antibody (upper panel). Conventional western blotting for HBX and β-actin in HBX transfected or vector control cells are shown at the lower panel. (E) qRT-PCR analysis for mature miR-122 in HepG2 cells transfected with HBX and/or PPARγ. The data represent mean + SD (***P<0.001, n = 3). (F) qRT-PCR analysis for pri-miR-122 in HepG2 cells transfected with HBX and/or PPARγ.
DISCUSSION
The present study discloses a novel epigenetic regulatory mechanism for miR-122 expression in HCC cells, which involves PPARγ/RXRα binding to DR1 and DR2 motifs of the miR-122 promoter. Our findings suggest that this process is influenced by the PPARγ co-repressors (N-CoR and SMRT) and by the histone methyl transferase (SUV39H1). We observe that PPARγ and RXRα bind to DR1 and DR2 motifs of the miR-122 promoter and their association is significantly increased in HCC cells treated with 5-Aza-CdR and PBA. The association is specific for PPARγ isoform, as PPARα did not bind to DR1 and DR2 motifs. Consistent with these findings, we observed that treatment with the PPARγ and RXRα agonists increased the expression of miR-122 in HCC cells. Additionally, overexpression and knockdown studies showed that PPARγ also regulated the expression of miR-122 in non malignant hepatocytes. These findings suggest that PPARγ and RXRα are positive regulators for miR-122 expression. On the other hand, we observed that 5-Aza-CdR and PBA treatment reduced the interaction of N-CoR/SMRT with PPARγ/RXRα and with DR1 and DR2 elements in the miR-122 promoter, suggesting that the PPARγ co-repressors, N-CoR and SMRT, are negative regulators for miR-122 expression. In addition, we found that 5-Aza-CdR and PBA treatment inhibited the expression of SUV39H1 (a H3K9 methyltransferase that catalyzes the formation of H3K9 dimethyl and trimethyl, leading to suppression of gene transcription) and decreased SUV39H1 binding to the DR1 and DR2 regions of the miR-122 promoter. The role of SUV39H1 for miR-122 suppression is further supported by the observation that knockdown or inhibition of SUV39H1 enhanced miR-122 expression in HCC cells. The latter finding is also corroborated by the observation that human primary hepatocytes contain lower levels of H3K9 dimethyl and trimethyl compared to HCC cells. Thus, SUV39H1 is another negative regulator for miR-122 expression in HCC cells. Collectively, our findings suggest that PPARγ and RXRα-mediated miR-122 expression is suppressed by N-CoR/SMRT/SUV39H1 in HCC cells (illustrated in Figure 7). It is plausible that reduction of SUV391 by 5-Aza-CdR and PBA may lead to dissociation of N-CoR/SMRT/SUV391 from the PPARγ/RXRα and DR1/DR2 binding complex, thus allowing transcription of the miR-122 gene. Furthermore, we observed that 5-Aza-CdR and PBA treatment also increased histone acetylation around miR-122 promoter regions. Therefore, epigenetic regulation of miR-122 in HCC cells is a complicated process which involves the PPARγ/RXRα/N-CoR/SMRT/SUV39H1/DR1/DR2 binding complex, histone acetylation, and histone H3K9 methylation.
Figure 7. Schematic illustration of mechanisms for epigenetic regulation of miR-122.

Epigenetic regulation of miR-122 in HCC cells and hepatocytes involves the PPARγ/RXRα/N-CoR/SMRT/SUV39H1/DR1/DR2 binding complex, histone acetylation, and histone H3K9 methylation. 5-Aza-CdR and PBA treatment inhibit SUV39H1 expression and cause dissociation of the corepressor complex (N-CoR/SMRT/SUV39H1) from PPARγ/RXRα/DR1/DR2, thus allowing PPARγ/RXRα-induced miR-122 gene transcription. Hepatitis B viral X protein inhibits PPARγ through direct binding and thus down-regulate miR-122 expression.
Previous studies have shown that miR-122 expression is associated with the level of essential liver-enriched transcription factors (LETFs) including HNF4α and C/EBPα(11, 18, 19). However, HNF4α and C/EBPα do not appear to be implicated in epigenetic silencing of miR-122 in HCC cells, as their levels were not significantly altered by 5-Aza-CdR and PBA treatment in our system. It is worth mentioning that C/EBPα is epigenetically silenced in several extrahepatic cancers including lung cancer and acute myelogenous leukemia(40, 41) and HNF4α is decreased due to epigenetic silencing of transcription factor 2 (TCF2) in ovarian cancer(42). Thus, susceptibility to epigenetic modifications may depend on specific tissue context and different tumor types.
Hepatitis C and B viruses are major epigenetic environmental factors that are closely associated with HCC development. In this study, we investigated the mechanism for differential regulation of miR-122 by HCV and HBV. It is well known that miR-122 is essential for HCV RNA accumulation by binding to the 5′-UTR of the viral genome. Previous reports have shown that miR-122 stimulates HCV viral protein translation and binds HCV RNA in association with Agonaute 2 (Ago2) protein thereby slowing decay of the viral RNA in infected cells. Silencing of miR-122 with a locked nucleic acid (LNA)-modified oligonucleotide leads to suppression of HCV viremia and improvement of HCV-induced liver pathology in chronic HCV infected chimpanzees model(14). However, whether HCV is able to regulate miR-122 expression was not known prior to the current study. In our study, we did not observe significant changes in miR-122 expression and promoter activity in Huh7.5 cells infected with HCV particles. Thus, HCV infection does not appear to significantly influence the level of miR-122, despite that miR-122 positively regulates HCV replication and stability. Our findings are also consistent with the previous report showing no correlation between HCV RNA and miR-122 levels in liver biopsies from patients with chronic hepatitis C(43).
In contrast to HCV infection, miR-122 was significantly down-regulated in patients with HBV infection(15). miR-122 is known to down-regulate cyclin G1 and interrupt cyclin G1 interaction with p53, thus abrogating p53-mediated inhibition of HBV replication(15). However, the mechanism for HBV-mediated down-regulation of miR-122 was not known prior to the current study. In this paper, we employed three HBV infection systems (stable HBV expression in HepG2.2.15 cells, transient HBV infection in HepaRG cells, and transient HBV infection in primary human hepatocytes); all of these systems showed that HBV infection down-regulated miR-122 expression. Our data indicate that HBX overexpression significantly decreases miR-122 gene promoter activity and down-regulates miR-122 expression, whereas siRNA knockdown of HBX enhances miR-122 expression. A previous study reports that HBX binds PPARγ and thus blocks nuclear localization and suppresses PPARγ-mediated transactivation(39). In our system, we also observed that HBX directly interacted with PPARγ, although we did not observe nuclear translocation of PPARγ by HBX, as PPARγ is localized in the nucleus and HBX is localized in both cytoplasm and nucleus in HepG2 cells (Supplemental Figure S3). Our findings provide novel evidence for HBX-mediated inhibition of miR-122 transcription by PPARγ.
In summary, this study provides the first evidence for epigenetic regulation of miR-122, a pivotal liver specific miRNA, by PPARγ/RXRα complex through interaction with the co-repressors and H3K9 histone methyltransferase (SUV39H1) in HCC cells. Our data also provide important mechanistic insight into differential regulation of miR-122 by HBV and HCV.
Supplementary Material
Acknowledgments
Supported by National Institutes of Health grants CA106280, CA102325, CA134568, and DK077776 (to T.W.).
The authors would like to thank Dr. Partha K Chandra and Dr. Sidhartha Hazari for technical support on HCV infection and detection.
Abbreviations
- 5-Aza-CdR
5′aza-2′deoxycytidine
- C/EBP
CCAAT/enhancer-binding protein
- ChIP
chromatin immunoprecipitation
- HBV
hepatitis B virus
- HBX
hepatitis B virus X protein
- HCC
hepatocellular carcinoma
- HCV
hepatitis C virus
- HDAC
histone deacetylase
- HMT
histone methyltransferase
- HNF-4α
hepatocyte nuclear factors-4α
- LETFs
liver-enriched transcription factors
- N-CoR
nuclear receptor corepressor protein
- PBA
4-phenylbutyric acid
- PPARγ
peroxisome proliferator activated receptor-gamma
- RXRα
retinoid X receptor alpha
- SMRT
silencing mediator of retinoid and thyroid hormone receptors
Footnotes
No conflicts of interest exist
References
- 1.El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132:2557–2576. doi: 10.1053/j.gastro.2007.04.061. [DOI] [PubMed] [Google Scholar]
- 2.El-Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011;365:1118–1127. doi: 10.1056/NEJMra1001683. [DOI] [PubMed] [Google Scholar]
- 3.Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 4.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 5.Borel F, Konstantinova P, Jansen PL. Diagnostic and therapeutic potential of miRNA signatures in patients with hepatocellular carcinoma. J Hepatol. 2012;56:1371–1383. doi: 10.1016/j.jhep.2011.11.026. [DOI] [PubMed] [Google Scholar]
- 6.Braconi C, Henry JC, Kogure T, Schmittgen T, Patel T. The role of microRNAs in human liver cancers. Semin Oncol. 2011;38:752–763. doi: 10.1053/j.seminoncol.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iorio MV, Piovan C, Croce CM. Interplay between microRNAs and the epigenetic machinery: an intricate network. Biochim Biophys Acta. 2010;1799:694–701. doi: 10.1016/j.bbagrm.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 8.Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, Tuschl T. Identification of tissue-specific microRNAs from mouse. Curr Biol. 2002;12:735–739. doi: 10.1016/s0960-9822(02)00809-6. [DOI] [PubMed] [Google Scholar]
- 9.Esau C, Davis S, Murray SF, Yu XX, Pandey SK, Pear M, Watts L, et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006;3:87–98. doi: 10.1016/j.cmet.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 10.Castoldi M, Vujic Spasic M, Altamura S, Elmen J, Lindow M, Kiss J, Stolte J, et al. The liver-specific microRNA miR-122 controls systemic iron homeostasis in mice. J Clin Invest. 2011;121:1386–1396. doi: 10.1172/JCI44883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coulouarn C, Factor VM, Andersen JB, Durkin ME, Thorgeirsson SS. Loss of miR-122 expression in liver cancer correlates with suppression of the hepatic phenotype and gain of metastatic properties. Oncogene. 2009;28:3526–3536. doi: 10.1038/onc.2009.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science. 2005;309:1577–1581. doi: 10.1126/science.1113329. [DOI] [PubMed] [Google Scholar]
- 13.Henke JI, Goergen D, Zheng J, Song Y, Schuttler CG, Fehr C, Junemann C, et al. microRNA-122 stimulates translation of hepatitis C virus RNA. EMBO J. 2008;27:3300–3310. doi: 10.1038/emboj.2008.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lanford RE, Hildebrandt-Eriksen ES, Petri A, Persson R, Lindow M, Munk ME, Kauppinen S, et al. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science. 2010;327:198–201. doi: 10.1126/science.1178178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang S, Qiu L, Yan X, Jin W, Wang Y, Chen L, Wu E, et al. Loss of microRNA 122 expression in patients with hepatitis B enhances hepatitis B virus replication through cyclin G(1) -modulated P53 activity. Hepatology. 2012;55:730–741. doi: 10.1002/hep.24809. [DOI] [PubMed] [Google Scholar]
- 16.Tsai WC, Hsu SD, Hsu CS, Lai TC, Chen SJ, Shen R, Huang Y, et al. MicroRNA-122 plays a critical role in liver homeostasis and hepatocarcinogenesis. J Clin Invest. 2012;122:2884–2897. doi: 10.1172/JCI63455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hsu SH, Wang B, Kota J, Yu J, Costinean S, Kutay H, Yu L, et al. Essential metabolic, anti-inflammatory, and anti-tumorigenic functions of miR-122 in liver. J Clin Invest. 2012;122:2871–2883. doi: 10.1172/JCI63539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zeng C, Wang R, Li D, Lin XJ, Wei QK, Yuan Y, Wang Q, et al. A novel GSK-3 beta-C/EBP alpha-miR-122-insulin-like growth factor 1 receptor regulatory circuitry in human hepatocellular carcinoma. Hepatology. 2010;52:1702–1712. doi: 10.1002/hep.23875. [DOI] [PubMed] [Google Scholar]
- 19.Li ZY, Xi Y, Zhu WN, Zeng C, Zhang ZQ, Guo ZC, Hao DL, et al. Positive regulation of hepatic miR-122 expression by HNF4alpha. J Hepatol. 2011;55:602–611. doi: 10.1016/j.jhep.2010.12.023. [DOI] [PubMed] [Google Scholar]
- 20.Jopling C. Liver-specific microRNA-122: Biogenesis and function. RNA Biol. 2012;9:137–142. doi: 10.4161/rna.18827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lemberger T, Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: a nuclear receptor signaling pathway in lipid physiology. Annu Rev Cell Dev Biol. 1996;12:335–363. doi: 10.1146/annurev.cellbio.12.1.335. [DOI] [PubMed] [Google Scholar]
- 22.Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr Rev. 1999;20:649–688. doi: 10.1210/edrv.20.5.0380. [DOI] [PubMed] [Google Scholar]
- 23.Okuno M, Arimoto E, Ikenobu Y, Nishihara T, Imagawa M. Dual DNA-binding specificity of peroxisome-proliferator-activated receptor gamma controlled by heterodimer formation with retinoid X receptor alpha. Biochem J. 2001;353:193–198. doi: 10.1042/0264-6021:3530193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu C, Markan K, Temple KA, Deplewski D, Brady MJ, Cohen RN. The nuclear receptor corepressors NCoR and SMRT decrease peroxisome proliferator-activated receptor gamma transcriptional activity and repress 3T3-L1 adipogenesis. J Biol Chem. 2005;280:13600–13605. doi: 10.1074/jbc.M409468200. [DOI] [PubMed] [Google Scholar]
- 25.Kato S, Yokoyama A, Fujiki R. Nuclear receptor coregulators merge transcriptional coregulation with epigenetic regulation. Trends Biochem Sci. 2011;36:272–281. doi: 10.1016/j.tibs.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 26.Tufan NL, Lian Z, Liu J, Pan J, Arbuthnot P, Kew M, Clayton MM, et al. Hepatitis Bx antigen stimulates expression of a novel cellular gene, URG4, that promotes hepatocellular growth and survival. Neoplasia. 2002;4:355–368. doi: 10.1038/sj.neo.7900241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gripon P, Rumin S, Urban S, Le Seyec J, Glaise D, Cannie I, Guyomard C, et al. Infection of a human hepatoma cell line by hepatitis B virus. Proc Natl Acad Sci U S A. 2002;99:15655–15660. doi: 10.1073/pnas.232137699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Watanabe T, Ishihara K, Hirosue A, Watanabe S, Hino S, Ojima H, Kanai Y, et al. Higher-order chromatin regulation and differential gene expression in the human tumor necrosis factor/lymphotoxin locus in hepatocellular carcinoma cells. Mol Cell Biol. 2012;32:1529–1541. doi: 10.1128/MCB.06478-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hazari S, Chandra PK, Poat B, Datta S, Garry RF, Foster TP, Kousoulas G, et al. Impaired antiviral activity of interferon alpha against hepatitis C virus 2a in Huh-7 cells with a defective Jak-Stat pathway. Virol J. 2010;7:36. doi: 10.1186/1743-422X-7-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu Q, Oei Y, Mendel DB, Garrett EN, Patawaran MB, Hollenbach PW, Aukerman SL, et al. Novel robust hepatitis C virus mouse efficacy model. Antimicrob Agents Chemother. 2006;50:3260–3268. doi: 10.1128/AAC.00413-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chang J, Nicolas E, Marks D, Sander C, Lerro A, Buendia MA, Xu C, et al. miR-122, a mammalian liver-specific microRNA, is processed from hcr mRNA and may downregulate the high affinity cationic amino acid transporter CAT-1. RNA Biol. 2004;1:106–113. doi: 10.4161/rna.1.2.1066. [DOI] [PubMed] [Google Scholar]
- 32.Tsai WC, Hsu PW, Lai TC, Chau GY, Lin CW, Chen CM, Lin CD, et al. MicroRNA-122, a tumor suppressor microRNA that regulates intrahepatic metastasis of hepatocellular carcinoma. Hepatology. 2009;49:1571–1582. doi: 10.1002/hep.22806. [DOI] [PubMed] [Google Scholar]
- 33.Matsumura T, Makino R, Mitamura K. Frequent down-regulation of E-cadherin by genetic and epigenetic changes in the malignant progression of hepatocellular carcinomas. Clin Cancer Res. 2001;7:594–599. [PubMed] [Google Scholar]
- 34.Cohen RN. Nuclear receptor corepressors and PPARgamma. Nucl Recept Signal. 2006;4:e003. doi: 10.1621/nrs.04003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lakshmikuttyamma A, Scott SA, DeCoteau JF, Geyer CR. Reexpression of epigenetically silenced AML tumor suppressor genes by SUV39H1 inhibition. Oncogene. 2010;29:576–588. doi: 10.1038/onc.2009.361. [DOI] [PubMed] [Google Scholar]
- 36.Rea S, Eisenhaber F, O’Carroll D, Strahl BD, Sun ZW, Schmid M, Opravil S, et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature. 2000;406:593–599. doi: 10.1038/35020506. [DOI] [PubMed] [Google Scholar]
- 37.Sells MA, Chen ML, Acs G. Production of hepatitis B virus particles in Hep G2 cells transfected with cloned hepatitis B virus DNA. Proc Natl Acad Sci U S A. 1987;84:1005–1009. doi: 10.1073/pnas.84.4.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yen TS. Hepadnaviral X Protein:Review of Recent Progress. J Biomed Sci. 1996;3:20–30. doi: 10.1007/BF02253575. [DOI] [PubMed] [Google Scholar]
- 39.Choi YH, Kim HI, Seong JK, Yu DY, Cho H, Lee MO, Lee JM, et al. Hepatitis B virus X protein modulates peroxisome proliferator-activated receptor gamma through protein-protein interaction. FEBS Lett. 2004;557:73–80. doi: 10.1016/s0014-5793(03)01449-2. [DOI] [PubMed] [Google Scholar]
- 40.Tada Y, Brena RM, Hackanson B, Morrison C, Otterson GA, Plass C. Epigenetic modulation of tumor suppressor CCAAT/enhancer binding protein alpha activity in lung cancer. J Natl Cancer Inst. 2006;98:396–406. doi: 10.1093/jnci/djj093. [DOI] [PubMed] [Google Scholar]
- 41.Jost E, do ON, Wilop S, Herman JG, Osieka R, Galm O. Aberrant DNA methylation of the transcription factor C/EBPalpha in acute myelogenous leukemia. Leuk Res. 2009;33:443–449. doi: 10.1016/j.leukres.2008.07.027. [DOI] [PubMed] [Google Scholar]
- 42.Terasawa K, Toyota M, Sagae S, Ogi K, Suzuki H, Sonoda T, Akino K, et al. Epigenetic inactivation of TCF2 in ovarian cancer and various cancer cell lines. Br J Cancer. 2006;94:914–921. doi: 10.1038/sj.bjc.6602984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sarasin-Filipowicz M, Krol J, Markiewicz I, Heim MH, Filipowicz W. Decreased levels of microRNA miR-122 in individuals with hepatitis C responding poorly to interferon therapy. Nat Med. 2009;15:31–33. doi: 10.1038/nm.1902. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.