

Abstract
Objective
The present study was performed to investigate in a model of malignant hypertension if the antihypertensive actions of soluble epoxide hydrolase (sEH) inhibition are nitric oxide (NO)-dependent.
Methods
ANG II-dependent malignant hypertension was induced through dietary administration for 3 days of the natural xenobiotic indole-3-carbinol (I3C) in Cyp1a1-Ren-2 transgenic rats. Blood pressure (BP) was monitored by radiotelemetry and treatment with the sEH inhibitor [cis-4-[4-(3-adamantan-1-yl-ureido)-cyclohexyl-oxy]-benzoic acid (c-AUCB)] was started 48h before administration of the diet containing I3C. In separate groups of rats, combined administration of the sEH inhibitor and the nonspecific NO synthase inhibitor [Nω-nitro-l-arginine methyl ester (L-NAME)] on the course of BP in I3C-induced and noninduced rats were evaluated. In addition, combined blockade of renin–angiotensin system (RAS) was superimposed on L-NAME administration in separate groups of rats. After 3 days of experimental protocols, the rats were prepared for renal functional studies and renal concentrations of epoxyeicosatrienoic acids (EETs) and their inactive metabolites dihydroxyeicosatrienoic acids (DHETEs) were measured.
Results
Treatment with c-AUCB increased the renal EETs/DHETEs ratio, attenuated the increases in BP, and prevented the decreases in renal function and the development of renal damage in I3C-induced Cyp1a1-Ren-2 rats. The BP lowering and renoprotective actions of the treatment with the sEH inhibitor c-AUCB were completely abolished by concomitant administration of L-NAME and not fully rescued by double RAS blockade without altering the increased EETs/DHETEs ratio.
Conclusion
Our current findings indicate that the antihypertensive actions of sEH inhibition in this ANG II-dependent malignant form of hypertension are dependent on the interactions of endogenous bioavailability of EETs and NO.
Keywords: cytochrome P-450 metabolites, epoxyeicosatrienoic acids, malignant hypertension, nitric oxide, nitric oxide synthase, renin–angiotensin system, soluble epoxide hydrolase
Introduction
Epoxyeicosatrienoic acids (EETs) which are synthesized from arachidonic acid by cytochrome P450 (CYP) epoxygenase enzymes are increasingly recognized as important regulators of cardiovascular and renal function [1–3]. It has been shown that the approach to increase EETs' tissue levels by preventing their degradation to the biologically inactive dihydroxyeicosatrienoic acids (DHETEs) by soluble epoxide hydrolase (sEH) inhibition displays antihypertensive actions [3–7]. We have recently shown that substantial increase in the level of endogenous EETs in the presence of sEH inhibition markedly attenuate the development of angiotensin II (ANG II)-dependent form of malignant hypertension particularly by the improvement of renal hemodynamics and increased sodium excretion [8,9]. These findings are in accordance with results from previous studies proposing that the EETs-mediated antihypertensive actions are mainly due to their direct vasodilatory effects and their direct influence on renal tubular transport of sodium [1–3,5,6,10].
However, a recent study seriously questioned the generally accepted notion of a direct vasodilatory action of EETs and consequently questioned the generally accepted hypothesis regarding the underlying mechanism(s) responsible for the blood pressure (BP) lowering actions of sEH inhibition [1–3,10]. Hercule et al.[11] demonstrated that CYP-derived eicosanoids produce vasodilatory effects largely through their ability to activate endothelial nitric oxide (NO) synthase (eNOS) and NO release in the vasculature. On the basis of this study, the hypothesis has been proposed that the BP-lowering effects of sEH inhibition are produced through EETs-mediated increased NO bioavailability [11].
Accordingly, to test the hypothesis that the antihypertensive and renoprotective actions of sEH inhibition are NO-dependent, we evaluated the effects of chronic treatment with the sEH inhibitor cis-4-[4-(3-adamantan-1-yl-ureido)cyclohexyloxy]benzoic acid (c-AUCB) on the development of hypertension, renal function and hypertension-induced end-organ damage in a model of ANG II-dependent malignant hypertension under conditions of an isolated sEH inhibition and under conditions of simultaneous administration of c-AUCB and the nonspecific NO synthase (NOS) inhibitor Nω-nitro-l-arginine methyl ester (L-NAME).
Furthermore, to gain more detailed insight into the role of intrarenal interaction of CYP-derived metabolites with the renin–angiotensin system (RAS) and NO in the pathophysiology of ANG II-dependent malignant form of hypertension, we determined the intrarenal concentrations of ANG II, EETs and DHETEs and urinary excretion of nitrate/nitrite (NOX), 8-isoprostane (UISO), hydrogen peroxide (H2O2) and thiobarbituric acid-reactive substances (TBARS) in normotensive and hypertensive rats.
Finally, to further elucidate the role of an interaction of the RAS and NO in the development of hypertension, renal functional impairment and hypertension-associated end-organ damage in the model of ANG II-dependent malignant hypertension simultaneous blockade of NOS activity and of the RAS has been evaluated.
Methods
The studies were performed in accordance with the guidelines and practices established by the Institute for Clinical and Experimental Medicine Animal Care and Use Committee.
Animals and diets
As a model of malignant hypertension a recently generated inbred transgenic rat with inducible hypertension [strain name: TGR(Cyp1a1Ren-2)] was used. The Cyp1a1-Ren-2 transgenic rat was generated by inserting the mouse Ren-2 renin gene, fused to the cytochrome P-450 (Cyp1a1) promoter, into the genome of the Fischer 344 rat. The Cyp1a1 promoter is not constitutively expressed in the liver; however, after exposure to various natural xenobiotics, such as indole-3-carbinol (I3C) the expression of the Cyp1a1 promoter is rapidly enhanced with a marked increase of the expression of the Ren-2 renin gene in the liver, with a subsequent increase in ANG II levels [12]. We and others [13–15] have demonstrated that renin gene expression and subsequently the degree of hypertension can be precisely regulated in a dose-dependent and time-dependent way. It has been shown that chronic dietary administration of I3C at a dose of 0.3% induces malignant hypertension with marked activation of the endogenous RAS. In addition, this is characterized by a rapid increase in BP, pressure diuresis and natriuresis, severe renal vasoconstriction and ischemia. Therefore, the Cyp1a1-Ren-2 transgenic rat can be considered as a model of ANG II-dependent malignant hypertension [13–17].
All animals used in the present study were bred at the Center for Experimental Medicine of the Institute of Clinical and Experimental Medicine from stock animals supplied from the Center for Cardiovascular Science, University of Edinburgh, UK (we acknowledge the generous gift of Professor Mullins). All diets used in the present study were produced by SEMED (Prague, Czech Republic). The rats were fed either a rat chow without I3C (noninduced groups) or a rat chow containing 0.3% I3C (I3C-induced groups). It has been shown that I3C is a dietary supplement, which in transgene-negative rats, does not exhibit any harmful biological effects but causes a very strong induction of Cyp1a1 in TGR (Cyp1a1 Ren-2) through the activation of aryl hydrocarbon receptor, which is a basic helix-loop-helix-transcription factor that binds to the Cyp1a1 promoter [12].
Chemicals
The sEH inhibitor c-AUCB was prepared freshly and given in drinking water at a dose of 26 mg/l as described previously [8,9]. Briefly, the crystalline c-AUCB (26 mg) was dissolved in ethanol (7 ml) and cyclodextrin (150 mg) and after a 5-min sonication period, this solution was added to 1 l of tap water. Hydrogencarbonate (3ml/l) was added to ensure that the water did not become acidic as low pH could cause the compound to precipitate. This dose of c-AUCB was used in our recent studies and we found that it exhibited significant antihypertensive actions and substantially increased tissue concentrations of EETs [8,9]. We selected c-AUCB for the work to be consistent with our earlier studies [6–9]. L-NAME was given in drinking water at a dose of 600mg/l, which is the dose that maximally blocked NOS activity as has been demonstrated previously [18,19].
A combination of angiotensin-converting enzyme inhibitor (ACEI) trandolapril (Gopten, Abbot, Prague, Czech Republic) at a dose of 6mg/l drinking water and ANG II receptor subtype 1 (AT1) antagonist losartan (Lozap; Zentiva, Prague, Czech Republic) at a dose of 100 mg/l drinking water was used to block the activity of the RAS. Our recent study has demonstrated that these high doses of ACEI and AT1 completely block the development of ANG II-dependent form of hypertension and provide greater cardioprotection and renoprotection than the doses used routinely for the treatment of hypertension [20].
Experimental design
Studies in TGR (Cyp1a1-Ren-2) with chronic sEH inhibition, simultaneous sEH and NOS inhibition, and simultaneous RAS and NOS blockade.
Series 1: effects on blood pressure, albuminuria, urinary excretion of nitrate/nitrite, 8-isoprostane, hydrogen peroxide and thiobarbituric acid-reactive substances, renal tissue concentrations of epoxyeicosatrienoic acids, dihydroxyeicosatrienoic acids and angiotensin II as well as on renal morphology
In accordance with the recommendations for BP measurements in experimental animals, we employed a radiotelemetry system for direct BP measurements [21]. Male Cyp1a1-Ren-2 transgenic rats, initial weight 330 ± 6 g, were implanted with radiotransmitters TA11PA-C40 (Data Sciences, St. Paul, Minnesota, USA) to continuously monitor BP by a telemetric device throughout the experimental period as described in detail in our previous studies [8,9]. After 10 days of recovery from surgery basal BP was recorded for 3 days, then the treatment either with the sEH inhibitor c-AUCB or the combination of ACEI and AT1 receptor antagonist (Gopten + Lozap) was initiated. After 48 h feeding, the diet with or without I3C was started and in appropriate groups of animals administration of L-NAME in the drinking water began and this protocol continued for 3 days. This protocol was chosen, as we and others [8,9,12–15,17] demonstrated that when 0.3% I3C is employed, after 3 days I3C-induced groups exhibited a maximal degree of activation of the renin gene and the level of hypertension reached its upper limit. Urinary collections were performed in metabolic cages at the end of the experiment to assess daily albuminuria, NOX, UISO, H2O2 and TBARS (which are used for the assay of malondialdehyde, a product of lipid peroxidation, another marker for endogenous superoxide activity) excretion as described previously [7–9,22–24]. At the end, rats were sacrificed by decapitation and plasma and tissue ANG II levels were measured by radioimmunoassay as described previously [8,9]. The level of the arachidonic acid metabolites EETs and DHETEs were measured in the kidney cortex. The samples were extracted, separated by reverse-phase, highperformance liquid chromatography, and analyzed by negative-mode electrospray ionization and tandem mass spectroscopy as described previously [7–9].
To assess the renal glomerular damage, the right kidney was quickly removed, fixed in 4% formaldehyde, and embedded in paraffin. The sections stained with hematoxylin (eosin and PAS (periodic acid – Schiff reaction) were examined and evaluated in a blind-test fashion. One hundred glomeruli in each kidney were examined on a semi-quantitative scale as described previously [9,20,25]: grade 0, normal finding; grade 1–4, glomerular sclerotic area in range 25–100%. The glomerulosclerosis index (GSI) was calculated using the following formula:
where, nx is the number of glomeruli in each grade of glomerulosclerosis.
Tubulointerstitial injury was evaluated as defined by Nakano et al.[26] At least 30 random and nonoverlaping cortical fields were evaluated using 20 × objective. Tubulointerstitial injury was defined as tubular dilatation, atrophy, cast formation and sloughing of tubular epithelial cells or thickening of tubular basement membrane and scaring as follows grade 0, no injury; grade 1, tubulointerstitial injury in less than 25%; grade 2, (25–50%); grade 3, (>50%). This approach is now routinely used in our laboratory and allows comparison of the present results with those of our previous and also future studies evaluating the pathophysiology of hypertension-associated end-organ damage [7,9,13,20,25,27].
The following experimental groups were examined in TGR (Cyp1a1-Ren-2):
I3C-induced + untreated (n = 7)
I3C-induced + c-AUCB (n = 7)
I3C-induced + L-NAME (n = 7)
I3C-induced + L-NAME + c-AUCB (n = 7)
I3C-induced + L-NAME + RAS blockade (n = 7)
Noninduced + untreated (n = 6)
Noninduced + c-AUCB (n = 6)
Noninduced + L-NAME (n = 7)
Noninduced + L-NAME + c-AUCB (n = 7)
Noninduced + L-NAME + RAS blockade (n = 6)
Series 2: effects on renal hemodynamics and excretory function
The following experimental groups, exposed to the same protocol as TGR (Cyp1a1-Ren-2) in series 1, were examined:
I3C-induced + untreated (n = 8)
I3C-induced + c-AUCB (n = 9)
I3C-induced + L-NAME (n = 9)
I3C-induced + L-NAME + c-AUCB (n = 9)
I3C-induced + L-NAME + RAS blockade (n = 9)
Noninduced + untreated (n = 8)
Noninduced + c-AUCB (n = 8)
Noninduced + L-NAME (n = 9)
Noninduced + L-NAME + c-AUCB (n = v9)
Noninduced + L-NAME + RAS blockade (n = 8)
At the end of the experimental protocol, rats were anesthetized and acute clearance experiments were performed as described in detail in our previous studies to determine renal hemodynamics and excretory parameters [8,9].
Statistical analysis
All values are expressed as means ± SEM. Graph-Pad Prism software (Graph Pad Software, San Diego, California, USA) was used and statistical analysis was performed using Student's t-test, Wilcoxon's signed-rank test for unpaired data, or one-way analysis of variance (ANOVA) when appropriate. ANOVA for repeated measurements, followed by Student– Newman–Keuls test was performed for the analysis within groups (e.g. before and after either I3C or L-NAME administration). Values exceeding the 95% probability limits (P < 0.05) were considered statistically significant.
Results
Chronic inhibition of sEH, simultaneous inhibition of sEH and NOS, and simultaneous inhibition of the RAS and NOS
Series 1: effects on blood pressure, albuminuria, urinary nitrate/nitrite, 8-isoprostane, hydrogen peroxide and thiobarbituric acid-reactive substances, renal glomerular and tubulointerstitial damage as well as tissue concentrations of epoxyeicosatrienoic acids, dihydroxyeicosatrienoic acids and angiotensin II
As shown in Fig. 1a and b, I3C induction of the renin gene resulted in severe hypertension, the mean arterial pressure (MAP) increased from basal 108 ± 3 to final 163 ± 4 mmHg. This was associated with a marked loss of body weight from 334 ± 5 to 293 ± 5g (P < 0.05). Administration of L-NAME in I3C-induced rats temporarily augmented BP increases as compared with untreated I3C-induced rats (163 ± 4vs. 128 ± 3 mmHg, P < 0.05, on day 1 after I3C administration), but then MAP was similar as observed in untreated I3C-induced animals. Treatment with c-AUCB attenuated the development of hypertension and the loss of body weight in I3C-induced rats as compared with untreated I3C-induced rats. In contrast, combined treatment with c-AUCB and L-NAME in I3C-induced rats did not exhibit any reduction in BP or prevention of loss of body weight when compared with untreated I3C-induced rats. The combined RAS blockade and L-NAME treatment abolished the increases in MAP and decreases in body weight in I3C-induced rats.
FIGURE 1.
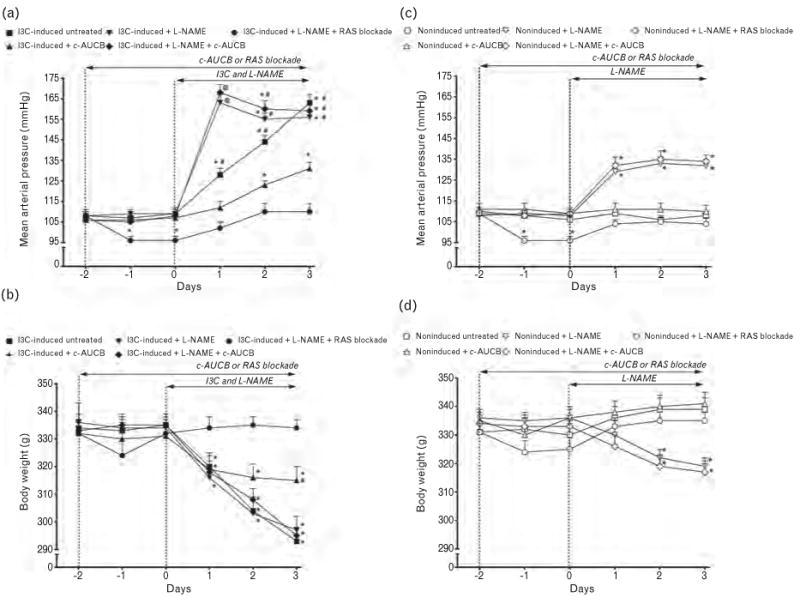
Course of mean arterial pressure (measured by radiotelemetry) (a and c) and body weight (b and d) in indole-3-carbinol (I3C)-induced and noninduced Cyp1a1-Ren-2 transgenic rats and the effects of isolated cis-4-[4-(3-adamantan-1-yl-ureido)cyclohexyloxy]benzoic acid (c-AUCB) or Nω-nitro-l-arginine methyl ester (L-NAME) administration and either combined treatment with c-AUCB and L-NAME or combined RAS blockade and L-NAME treatment in these rats. *P < 0.05 vs. basal values. #P < 0.05 vs. c-AUCB unmarked values at the same time point. @P < 0.05 vs. all values at the same time point.
As shown in Fig. 1c, MAP in noninduced rats remained within the normotensive range throughout the whole experimental period and was not affected by c-AUCB treatment. Similarly, body weight in the two experimental groups exhibited a slight, but not significant increase during the experimental period (Fig. 1d). Administration of L-NAME in noninduced rats resulted in rapid and marked elevations in MAP from 108 ± 2 to 132 ± 3 mmHg (P < 0.05) and was associated with a significant loss of body weight from 336 ± 3 to 319 ± 3g (P < 0.05). The combined treatment with c-AUCB and L-NAME in noninduced rats did not prevent the increases in MAP and decreases in body weight as compared to L-NAME-treated noninduced rats. Similarly, as in I3C-induced rats, the combined RAS and NOS blockade abolished the increases in MAP and decreases in BW in noninduced rats (Fig. 1c and d).
As shown in Fig. 2a, untreated I3C-induced rats revealed pronounced albuminuria as compared with untreated noninduced rats (16.2 ± 2.1 vs. 0.40 ± 0.20 mg/24h, P < 0.05). Treatment with c-AUCB significantly attenuated the degree of albuminuria in I3C-induced rats. Administration of L-NAME further worsened the degree of albuminuria in I3C-induced rats and the combined treatment with c-AUCB and L-NAME did not prevent the increases in albuminuria in I3C-induced rats. In contrast, the combined RAS blockade and L-NAME treatment completely prevented increases in albuminuria in I3C-induced rats; thus, the level of albuminuria was similar to that observed in untreated noninduced rats (Fig. 2a).
FIGURE 2.
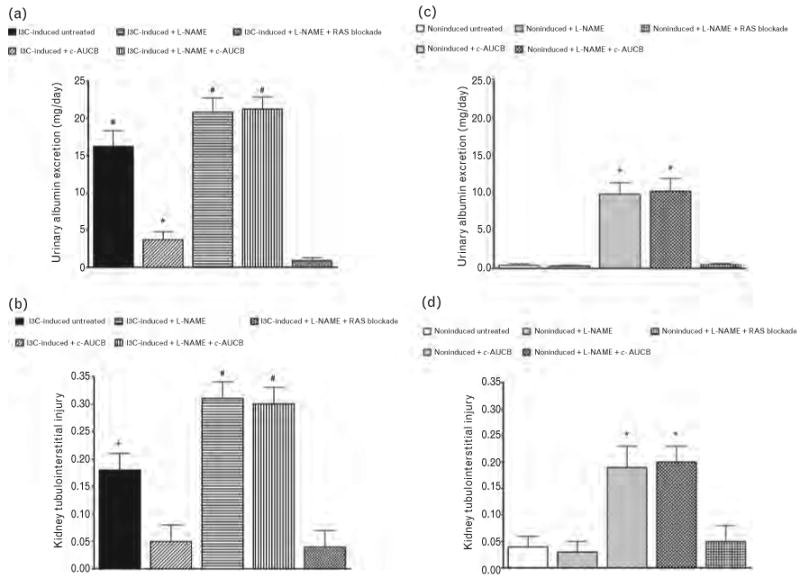
Urinary albumin excretion (a and c) and kidney tubulointerstitial injury (b and d) in indole-3-carbinol (I3C)-induced and noninduced Cyp1a1-Ren-2 transgenic rats and the effects of isolated cis-4-[4-(3-adamantan-1-yl-ureido)cyclohexyloxy]benzoic acid (c-AUCB) or Nω-nitro-l-arginine methyl ester (L-NAME) administration and either combined treatment with c-AUCB and L-NAME or combined RAS blockade and Nω-nitro-l-arginine methyl ester (L-NAME) treatment in these rats. *P < 0.05 vs. unmarked values. #P < 0.05 vs. all values.
As shown in Fig. 2c, treatment with c-AUCB in noninduced rats did not have any effects on albuminuria; however, the administration of L-NAME caused marked increases in albuminuria as compared to untreated noninduced rats (9.80 ± 1.50 vs. 0.40 ± 0.20mg/24h, P < 0.05) and this increase was unaffected by combined treatment with c-AUCB and L-NAME (Fig. 2c). Likewise in I3C-induced rats, the combined blockade of the RAS and of NOS completely prevented the increases in albuminuria in noninduced rats.
I3C-induced as well as noninduced rats exhibited a minimal degree of GSI (0.06 ± 0.02 and 0.05 ± 0.03), which was not significantly altered by any treatment protocol.
In contrast, as shown in Fig. 2b, untreated I3C-induced rats exhibited significantly stronger renal tubulointerstitial injury when compared with untreated noninduced rats (0.18 ± 0.03 vs. 0.04 ± 0.02, P < 0.05). The treatment with c-AUCB abolished the increases in kidney tubulointerstitial injury in I3C-induced rats. Administration of L-NAME significantly exacerbated the degree of kidney tubulointerstitial injury in I3C-induced rats and the treatment with c-AUCB did not prevent the augmented kidney tubulointerstitial injury in I3C-induced rats. In contrast, the combined inhibition of the RAS and NOS normalized the degree of kidney tubulointerstitial injury to levels observed in untreated noninduced rats. Treatment with c-AUCB in noninduced rats did not alter the degree of kidney tubulointerstitial injury; however, the administration of L-NAME caused marked increases in the level of kidney tubulointerstitial injury as compared with untreated noninduced rats (0.19 ± 0.03 vs. 0.03 ± 0.02, P < 0.05) and this increase was unaffected by combined treatment with c-AUCB and L-NAME. Likewise in I3C-induced rats with administration of L-NAME, the RAS blockade completely prevented increases in kidney tubulointerstitial injury in noninduced rats (Fig. 2d).
Tubulointerstitial changes of renal parenchyma of either noninduced or I3C-induced rats exposed to different treatment protocol are shown in supplemental Figures 1–5 (please see the online Data Supplement, http://links.lww.com/HJH/A214)
As summarized in Table 1, untreated I3C-induced rats showed significantly higher urinary NOX excretion than untreated noninduced rats. Treatment with c-AUCB significantly decreased urinary NOX excretion in I3C-induced rats and did not significantly change it in noninduced rats. Administration of L-NAME resulted in marked reduction of urinary NOX excretion to similar levels in I3C-induced and in noninduced rats. Combined administration of either c-AUCB with L-NAME or RAS blockade with L-NAME did not significantly alter urinary NOX excretion in I3C-induced or noninduced rats. As shown in Table 1, there were no significant differences in urinary UISO TBARS and H2O2 excretion between I3C-induced and noninduced rats; it was unaffected by any treatment protocol.
TABLE 1. Data from metabolic cages studies.
| Group name | UNOxV (μmol/day) | IISOV (ng/day) | UTBARSV (μmol/day) | UH2O2V (μmol/day) |
|---|---|---|---|---|
| I3C-induced + untreated | 19.68 ± 1.44# | 21.74 ± 2.45 | 0.24 ± 0.06 | 0.46 ± 0.09 |
| I3C-induced + c-AUCB | 9.88 ± 0.91* | 19.86 ± 1.74 | 0.32 ± 0.07 | 0.37 ± 0.07 |
| I3C-induced + L-NAME | 4.09 ± 0.99 | 21.72 ± 1.24 | 0.27 ± 0.05 | 0.41 ± 0.05 |
| I3C-induced + L-NAME + c-AUCB | 4.14 ± 0.56 | 22.14 ± 1.96 | 0.25 ± 0.04 | 0.43 ± 0.05 |
| I3C-induced + L-NAME + RAS blockade | 3.89 ± 1.01 | 20.72 ± 2.71 | 0.31 ± 0.06 | 0.39 ± 0.07 |
| Noninduced + untreated | 11.98 ± 0.91* | 25.14 ± 2.02 | 0.27 ± 0.04 | 0.43 ± 0.06 |
| Noninduced + c-AUCB | 10.42 ± 0.96* | 20.81 ± 1.96 | 0.32 ± 0.06 | 0.47 ± 0.09 |
| Noninduced + L-NAME | 4.12 ± 0.56 | 22.42 ± 1.76 | 0.29 ± 0.05 | 0.38 ± 0.06 |
| Noninduced + L-NAME + c-AUCB | 4.86 ± 0.64 | 19.86 ± 2.64 | 0.31 ± 0.06 | 0.42 ± 0.06 |
| Noninduced + L-NAME + RAS blockade | 5.04 ± 0.86 | 25.17 ± 2.89 | 0.28 ± 0.07 | 0.43 ± 0.07 |
c-AUCB, treatment with the soluble epoxide hydrolase inhibitor; I3C, administration of indole-3-carbinol; IISOV, urinary 8-isoprostane excretion; L-NAME, treatment with nitric oxide synthase inhibitor; RAS, renin–angiotensin system; UH2O2V, urinary hydrogen peroxide excretion; UNOxV, urinary nitrate/nitrite excretion; UTBARSV, urinary thiobarbituric acid-reactive substances excretion.
P < 0.05 vs. unmarked values.
P < 0.05 vs. all values.
Figures 3 and 4 summarize the availability of biologically active epoxygenase metabolites when expressed either as biologically active EETs or as almost biologically inactive DHETEs or as the ratio of EETs/DHETEs. As shown in Fig. 4, the ratio in untreated I3C-induced rats was significantly lower than in untreated noninduced rats (1.98 ± 0.25 vs. 3.74 ± 0.19, P < 0.05). The treatment with c-AUCB alone or the combined treatment with c-AUCB and L-NAME significantly increased this ratio in I3C-induced as well as in noninduced rats (Fig. 4a and b). However, these increases in the EETs/DHETEs ratio were markedly higher in I3C-induced rats than in noninduced rats. In addition, in I3C-induced rats the increases in the EETs/DHETEs ratios were the results of combination of significant increases of concentrations of EETs (698 ± 24 vs. 729 ± 29 and 804 ± 28 ng/g of tissue, P < 0.05 when compared with untreated I3C-induced vs. c-AUCB-treated I3C-induced rats) and significant decreases of concentrations of DHETEs (353 ± 21 vs. 209 ± 14 and 205 ± 16 ng/g of tissue, P < 0.05) (Fig. 3a and b). In contrast, in noninduced rats the increases in the EETs/DHETEs ratios were the results of significant increases of concentrations of EETs (824 ± 22 vs. 927 ± 28 and 919 ± 28 ng/g of tissue, P < 0.05, when compared with untreated noninduced rats vs. c-AUCB-treated noninduced rats) (Fig. 3c). Treatment with c-AUCB in noninduced rats exhibited a tendency to reduce kidney DHETEs concentrations when compared with untreated noninduced rats, but it did not reach statistical significance (192 ± 21 and 187 ± 15 vs. 219 ± 19ng/g of tissue) (Fig. 3d). The administration of L-NAME either alone or the combined RAS blockade and L-NAME treatment did not significantly change the ratio of EETs/DHETEs in I3C-induced as well as in noninduced rats (Fig. 4).
FIGURE 3.
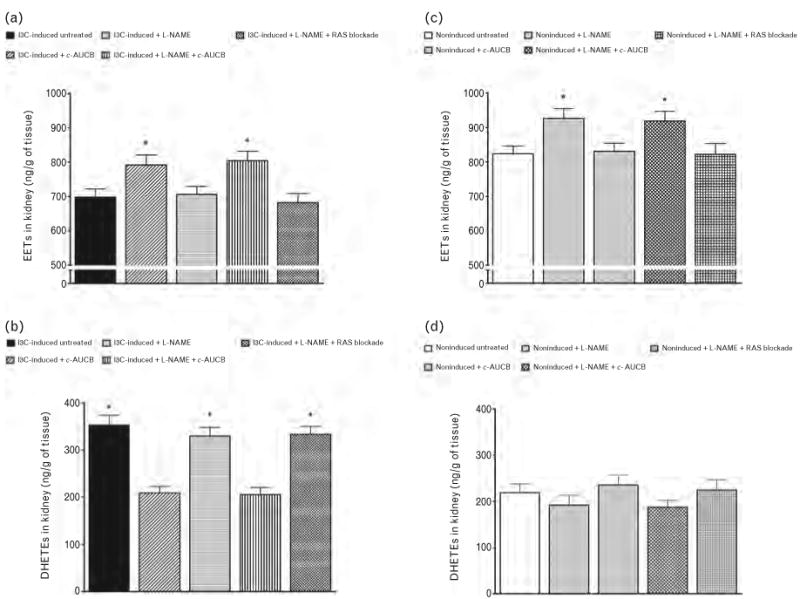
Epoxyeicosatrienoic acids (EETs) (a and c) and dihydroxyeicosatrienoic acids (DHETEs) (b and d) in kidney in indole-3-carbinol (I3C)-induced and noninduced Cyp1a1-Ren-2 transgenic rats and the effects of isolated cis-4-[4-(3-adamantan-1-yl-ureido)cyclohexyloxy]benzoic acid (c-AUCB) or Nω-nitro-l-arginine methyl ester (L-NAME) administration and either combined treatment with c-AUCB and L-NAME or combined RAS blockade and L-NAME treatment in these rats. *P < 0.05 vs. unmarked values.
FIGURE 4.
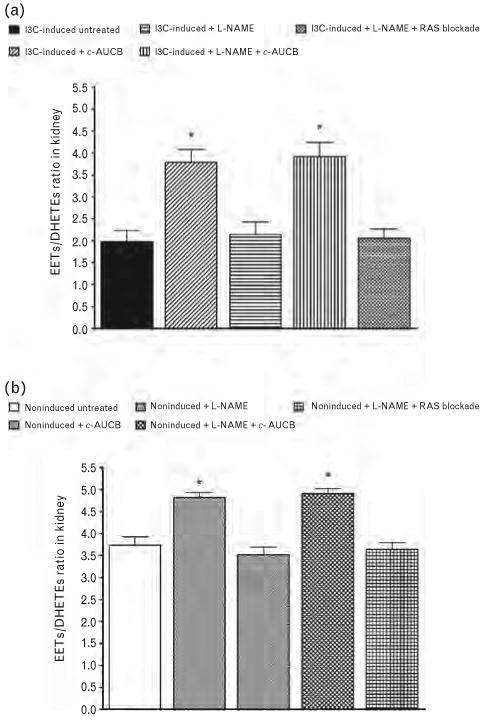
EETs/DHETEs ratio (a and b) in kidney in indole-3-carbinol (I3C)-induced and noninduced Cyp1a1-Ren-2 transgenic rats and the effects of isolated cis-4-[4-(3-adamantan-1-yl-ureido)cyclohexyloxy]benzoic acid (c-AUCB) or Nω-nitro-l-arginine methyl ester (L-NAME) administration and either combined treatment with c-AUCB and L-NAME or combined RAS blockade and L-NAME treatment on this ratio in these rats. *P < 0.05 vs. unmarked values.
As shown in Fig. 5, plasma and kidney ANG II levels were significantly higher in untreated I3C-induced rats than in untreated noninduced rats (68 ± 5 vs. 25 ± 3 fmol/ml and 262 ± 14 vs. 86 ± 5fmol/g, respectively, P < 0.05 in each case). The treatment with c-AUCB did not significantly change plasma and kidney ANG II concentrations in I3C-induced as well as in noninduced rats. Administration of L-NAME alone or combined treatment of L-NAME either with c-AUCB or RAS blockade resulted in significant decreases in plasma and kidney ANG II levels in I3C-induced as well as in noninduced rats.
FIGURE 5.
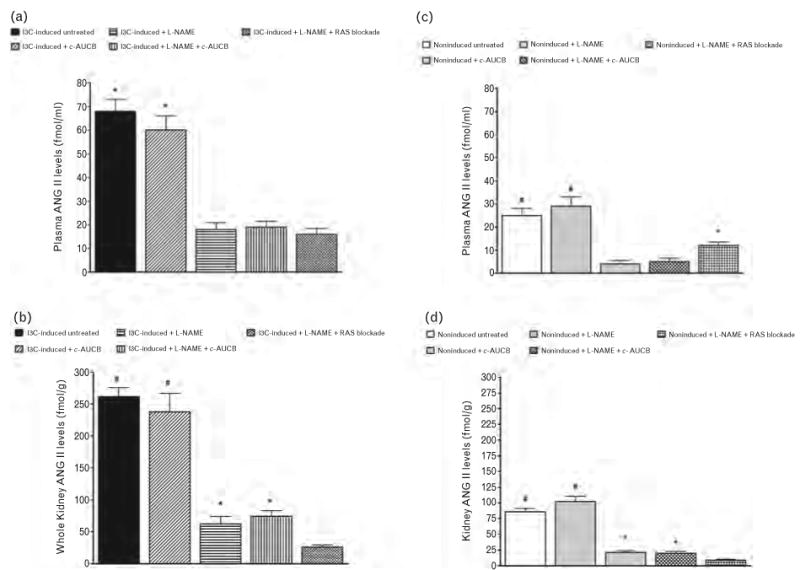
Plasma (a and c) and kidney (b and d) angiotensin II levels in indole-3-carbinol (I3C)-induced and noninduced Cyp1a1-Ren-2 transgenic rats and effects of isolated cis-4-[4-(3-adamantan-1-yl-ureido)cyclohexyloxy]benzoic acid (c-AUCB) or Nω-nitro-l-arginine methyl ester (L-NAME) administration and either combined treatment with c-AUCB and L-NAME or combined RAS blockade and L-NAME treatment in these rats. *P < 0.05 vs. unmarked values. #P < 0.05 vs. all values.
Series 2: effects on renal hemodynamics and excretory function
Table 2 summarizes the values (average from two clearance periods) of MAP, renal hemodynamics and electrolyte excretion. It is seen that untreated I3C-induced rats revealed significantly lower renal blood flow (RBF) as compared to untreated noninduced rats (P < 0.05). Treatment with c-AUCB prevented decreases in RBF in I3C-induced rats, but did not significantly change RBF in noninduced rats. Administration of L-NAME resulted in a striking reduction in RBF in I3C-induced as well as in noninduced rats. The combined treatment with c-AUCB and L-NAME did not prevent the decreases in RBF in I3C-induced and noninduced rats. The combined RAS blockade and L-NAME treatment slightly attenuated the decreases in RBF in I3C-induced and in noninduced rats as compared with animals that were administered L-NAME alone (P < 0.05 in both cases).
TABLE 2. Basal values of mean arterial pressure, renal function and electrolyte excretion.
| Group name | MAP (mmHg) | GFR (ml/min per g) | RBF (ml/min per g) | UNaV (μmol/min per g) | FENa (%) | V (μl/min per g) |
|---|---|---|---|---|---|---|
| I3C-induced + untreated | 162 ± 3@ | 1.45 ± 0.27* | 6.03 ± 0.34# | 1.48 ± 0.25@ | 1.32 ± 0.22@ | 13.51 ± 1.48@ |
| I3C-induced + c-AUCB | 134 ± 3* | 1.99 ± 0.19* | 9.24 ± 0.62@ | 1.91 ± 0.27@ | 2.04 ± 0.29@ | 12.18 ± 1.81@ |
| I3C-induced + L-NAME | 163 ± 4@ | 0.69 ± 0.11 | 2.61 ± 0.17 | 0.29 ± 0.09* | 0.25 ± 0.07* | 3.82 ± 0.34* |
| I3C-induced + L-NAME + c-AUCB | 161 ± 3@ | 0.61 ± 0.14 | 2.09 ± 0.24 | 0.35 ± 0.06* | 0.33 ± 0.04* | 4.11 ± 0.29* |
| I3C-induced + L-NAME + RAS blockade | 109 ± 4 | 0.74 ± 0.14 | 3.69 ± 0.23* | 0.66 ± 0.05# | 0.71 ± 0.06# | 6.52 ± 0.27 |
| Noninduced + untreated | 111 ± 3 | 1.89 ± 0.17* | 8.02 ± 0.41# | 0.43 ± 0.06* | 0.39 ± 0.08* | 4.98 ± 0.36* |
| Noninduced + c-AUCB | 112 ± 3 | 2.21 ± 0.21* | 7.22 ± 0.53# | 0.47 ± 0.05* | 0.44 ± 0.09* | 4.66 ± 0.42* |
| Noninduced + L-NAME | 136 ± 4* | 0.71 ± 0.19 | 2.37 ± 0.16 | 0.09 ± 0.02 | 0.11 ± 0.03 | 2.54 ± 0.22 |
| Noninduced + L-NAME + c-AUCB | 135 ± 5* | 0.65 ± 0.11 | 1.99 ± 0.21 | 0.14 ± 0.04 | 0.16 ± 0.05 | 2.63 ± 0.21 |
| Noninduced + L-NAME + RAS blockade | 110 ± 4 | 0.73 ± 0.14 | 3.71 ± 0.28* | 0.29 ± 0.04* | 0.33 ± 0.06* | 3.61 ± 0.18* |
c-AUCB, treatment with the soluble epoxide hydrolase inhibitor; FENa, fractional sodium excretion; GFR, glomerular filtration rate; I3C, administration of indole-3-carbinol; L-NAME, treatment with nitric oxide synthase inhibitor; MAP, mean arterial pressure; RAS, renin-angiotensin system; RBF, renal blood flow; UNaV, absolute sodium excretion; V, urine flow.
P < 0.05 vs. unmarked values.
P < 0.05 vs. marked values.
P < 0.05 vs. all values.
It is shown that untreated I3C-induced rats exhibited a tendency to lower glomerular filtration rate (GFR) when compared with untreated noninduced rats but this did not reach statistical significance. Treatment with c-AUCB abolished this tendency to decreases in GFR, but did not alter GFR in noninduced rats. Administration of L-NAME caused marked decreases in GFR in I3C-induced and in noninduced rats. Neither the combined treatment with c-AUCB and L-NAME nor the combined RAS blockade and L-NAME treatment did attenuate the decreases in GFR in I3C-induced or in noninduced rats as compared to animals that were administered L-NAME alone.
It is demonstrated that untreated I3C-induced rats showed significantly higher absolute and fractional sodium excretion and urine flow than untreated noninduced rats (P < 0.05 in all cases). Administration of L-NAME elicited profound decreases in absolute and fractional sodium excretion and in urine flow in I3C-induced and in noninduced rats (P < 0.05 in all cases). The combined treatment with c-AUCB and L-NAME did not prevent the decreases in absolute and fractional sodium excretion and urine flow in I3C-induced or noninduced rats. The combined RAS blockade and L-NAME treatment attenuated the decreases in absolute and fractional sodium excretion and urine flow in I3C-induced and in noninduced rats.
Discussion
The first and most important finding of the present study shows that the antihypertensive and renoprotective actions of chronic treatment with the sEH inhibitor c-AUCB on the development of malignant hypertension induced by IC3 in Cyp1a1-Ren-2 transgenic rats were completely abolished by the concomitant administration of the NOS inhibitor L-NAME. This lack of antihypertensive and renoprotective effects was present even if the combined treatment was, similar to previous studies, associated with significant increases in the availability of biologically active epoxygenase metabolites assessed as the ratio of EETs/DHETEs. Our previous [8,9] and present results show that untreated I3C-induced Cyp1a1-Ren-2 transgenic rats exhibit a reduced intrarenal availability of biologically active epoxygenase metabolites. In addition, our current data show that the increase in the intrarenal availability of EETs is markedly higher in I3C-induced Cyp1a1-Ren-2 transgenic rats than in noninduced rats and this increase in the EETs/DHETEs ratio was not altered by simultaneous administration of L-NAME. These data strongly suggest that the absence of antihypertensive and renoprotective actions in I3C-induced Cyp1a1-Ren-2 transgenic rats treated simultaneously with c-AUCB and L-NAME cannot be ascribed to a deficiency of the bioavailability of EETs.
Moreover, our data demonstrate that untreated I3C-induced Cyp1a1-Ren-2 transgenic rats have markedly higher urinary NOX excretion than untreated noninduced rats. These data indicate that untreated I3C-induced Cyp1a1-Ren-2 transgenic rats exhibit, similar to other ANG II-dependent model of hypertension, an increased intrarenal NOS activity that counteracts the vasoconstrictor influences of elevated circulating and intrarenal ANG II levels to sustain renal hemodynamics [24,28,29]. This notion is further supported by findings made by Patterson et al.[30] who found that neuronal NOS-derived NO exerts a pronounced renal vasodilator influence after induction of hypertension in Cyp1a1-Ren-2 transgenic rats. Furthermore, we found that although chronic treatment with the sEH inhibitor c-AUCB in I3C-induced Cyp1a1-Ren-2 transgenic rats exhibited antihypertensive actions, they resulted in a significant reduction of urinary NOX excretion as compared to untreated I3C-induced Cyp1a1-Ren-2 transgenic rats. These data question the original hypothesis that BP-lowering actions of sEH inhibition are mediated via increased availability of NO [11] and rather indicate that the decreased intrarenal NO activity is the consequence of diminished shear stress, which is known as a potent stimulus for NO synthesis and release [31,32], as the result of reduced BP in c-AUCB-treated Cyp1a1-Ren-2 transgenic rats.
L-NAME administration either alone or in combination with c-AUCB or with ACEI and AT1 receptor antagonist resulted in I3C-induced as well as in noninduced Cyp1a1-Ren-2 transgenic rats in a substantial reduction in urinary NOX excretion. These data suggest that the lack of antihypertensive effects of the combined c-AUCB and L-NAME treatment in I3C-induced Cyp1a1-Ren-2 transgenic rats can be attributed to a markedly reduced endogenous NO bioavailability. In contrast, as there were no significant differences in urinary UISO, TBARS and H2O2 excretion in any of the experimental groups, these results indicate that acute alterations in the production of reactive oxygen species do not play a critical role in the pathophysiology of early stages of malignant hypertension induced in Cyp1a1-Ren-2 transgenic rats.
Taken together, these findings indicate that the antihypertensive and renoprotective effects of chronic sEH inhibition by c-AUCB in this model of ANG II-dependent malignant hypertension are rather critically dependent on the endogenous NO bioavailability than on direct effects of increased availability of EETs. This is a critically important finding of our present study as it provides an important explanation for the results of studies performed during the last three decades which revealed that EETs exhibit important biological effects on the regulation of renal tubular transport of sodium and on the regulation of vascular tone [1–3]. At the kidney level it has been shown that EETs inhibit sodium reabsorption in the renal proximal tubule by blocking the sodium-hydrogen exchanger [33] and also decrease sodium reabsorption in the cortical collecting duct by blocking the epithelial sodium channels [34]. Most of the available evidence indicates that the EETs' antihypertensive properties are associated with their action on sodium excretion [3]. It has been demonstrated that either an absolute intrarenal deficiency of EETs or an inability to properly increase intrarenal EET levels are involved in the pathophysiology of certain forms of ANG II-dependent and salt-sensitive hypertension [3,4,35,36].
Taken together, based on previous studies from different investigators and also on the results of our own recent studies it has been proposed and so far generally accepted that the BP-lowering effects of chronic sEH inhibition are dependent on the direct vasodilatory actions of EETs and their direct influence on renal tubular transport of sodium [3,5,6,8–10]. However, it is obvious from our present study and from a recent study performed by Hercule et al.[11] that this generally accepted concept must be seriously questioned. These data rather indicate that the antihypertensive actions of sEH inhibition are dependent on an interaction of endogenous bioavailability of both EETs and NO. Nevertheless, future studies are needed to delineate the specific tubular transport mechanism(s) involved. Such experiments, however, are beyond the scope of the present study.
The second major finding that is of special interest is our observation that L-NAME administration in I3C-induced as well as in noninduced Cyp1a1-Ren-2 transgenic rats was associated with marked reduction of plasma and kidney ANG II levels. Therefore, the critical important issue of our present finding is related to the question: what are the underlying mechanism(s) responsible for the suppression of elevated plasma and kidney ANG II levels in L-NAME treated I3C-inducedas well as in noninduced Cyp1a1-Ren-2 transgenic rats?
A possible explanation could be, first, that these findings indicate that L-NAME-treated I3C-induced as well as noninduced Cyp1a1-Ren-2 transgenic rats possess preserved negative feedback baroreceptor mechanisms for renin secretion and ANG II levels [37,38]. However, against this notion argue our results from I3C-induced Cyp1a1-Ren-2 transgenic rats without L-NAME treatment because they, despite a similar level of hypertension did not exhibit any sign of suppression of ANG II levels.
A second possible explanation, and in our view the more likely one, is related to the role of NO in renin secretion. On the basis of currently available relevant data, it appears that NO serves as a stimulatory, permissive factor in renin secretion [37]. It is, therefore, conceivable that the administration of L-NAME resulted in a suppression of renin release and consequently a suppression of ANG II levels in Cyp1a1-Ren-2 transgenic rats. However, it is important to note that the relationship between BP and paracrine autacoids, such as NO, in the control of the renin system is particularly complex [37,39]. Therefore, it is obvious that additional studies, which are far beyond the scope and aim of the present study, will be necessary to address this issue more conclusively.
Our data regarding plasma and kidney ANG II concentrations are of critical importance because the majority of previous studies elucidating the pathophysiological mechanism(s) responsible for the development of hypertension in L-NAME-treated rats have postulated that decreased NO bioavailability results in unopposed activity of the RAS. This would then be inappropriately activated and would critically contribute to the development and maintenance of hypertension in this model of hypertension [40–42]. It has also been proposed that chronic NOS inhibition by L-NAME administration even activates the systemic and tissue RAS. However, these conclusions were drawn from studies evaluating the role of the RAS indirectly by employing pharmacological blockade of the RAS [43]. They are only few studies in either renin levels, the rate-limiting enzyme of the RAS, or ANG II concentrations, which are the main effector of the RAS, were measured directly and inconsistent results were obtained. It has been reported that the activity of the RAS is either increased [40,44,45], unchanged [19,46], or decreased [47] in response to chronic NOS inhibition. These contradictory findings suggest that the concept of unopposed or even increased RAS activity as one of the critical mechanisms responsible for the development of hypertension and hypertension-induced end-organ damage during chronic NOS inhibition is more complicated than has been considered so far. Our current findings that even if ANG II levels were substantially reduced in L-NAME-treated rats, the RAS blockade prevented the increases in BP and revealed important renoprotective actions indicate that the RAS still exhibits an important role in the pathophysiology of hypertension and hypertension-associated renal damage. This notion is also supported by findings made by Verhagen et al.[19] who demonstrated that the sensitivity to the detrimental effects of endogenous ANG II on the development of renal injury is increased during chronic NOS inhibition, despite unaltered or even suppressed intrarenal ANG II levels. However, our findings that plasma and kidney ANG II levels are suppressed after 3 days NOS inhibition can be considered as an appropriate physiological response to the elevated BP and renal perfusion pressure in L-NAME-treated animals. These findings indicate that the suppression of ANG II levels is a physiological compensation that at least partially compensates the absence of vasodilatory and natriuretic actions of NO in L-NAME-treated rats. Given the importance of the activation of the RAS in the pathophysiology of malignant forms of hypertension, especially in Cyp1a1-Ren-2 transgenic rats [14–17], one could predict that without this compensatory suppression of ANG II levels, the severity of hypertension and renal dysfunction would be markedly stronger in this form of malignant hypertension.
Of particular interest are our findings that even if pharmacological blockade of the RAS system in L-NAME-treated rats markedly reduced ANG II levels (e.g. kidney ANG II concentrations were even substantially lower than observed in untreated noninduced rats) and abolished BP increases in I3C-inducedaswellasnoninduced Cyp1a1-Ren-2 transgenic rats it did not prevent decreases in renal hemodynamics. These findings support the notion that intrarenal NO not only counteracts vasoconstrictor actions of ANG II [28,29], but physiologically exerts by itself pronounced renal vasodilator actions to maintain renal hemodynamics [31,32].
In summary, our present findings demonstrate that the antihypertensive and renoprotective actions of chronic sEH inhibition in Cyp1a1-Ren-2 transgenic rats with malignant hypertension are completely abolished by simultaneous administration of the NOS inhibitor L-NAME. These findings indicate that the antihypertensive actions of sEH inhibition in this ANG II-dependent malignant form of hypertension are dependent on the interactions of endogenous bioavailability EETs and NO and suggest that NO plays a permissive role for the mediating the antihypertensive and renoprotective actions of chronic sEH inhibition.
Reviewers' Summary Evaluations
Referee 1: Epoxyeicosatrienoic acids (EETs) seem to have a beneficial effect in the regulation of cardiovascular and renal function. Recent studies shown that the inhibition of soluble epoxide hydrolase (sEH) can prevent EETs degradation. The present study examined whether the antihypertensive and renoprotective actions of sEH inhibition were related to an interaction between increased bioavailability of EETs and NO in a rat model of angiotensin-dependent malignant hypertension. The design of the protocol was appropriate, the experiments were carefully performed and the results clearly presented. The results obtained from the present study provide evidence on some new mechanisms for the antihypertensive action of sEH inhibition.
Referee 2: This comprehensive study dissects the role of NO in the antihypertensive and renoprotective action of soluble epoxide hydrolase inhibition in the inducible mouse renin transgenic rat model. Overall the findings suggest that NO plays a permissive role and mediates the antihypertensive and renoprotective actions of chronic sEH inhibition.
Acknowledgments
Previous presentations: None.
This study was supported by grants No. NT/11230–4 and NT/12171–5 awarded by the Internal Grant Agency of the Ministry of Health to M.B and Z.H. L Č. is a recipient of grant No. MŠMT-Kontakt LH 11116 from the Ministry of Education, Youth and Sports for the support of the international research cooperation and this study was also supported by this grant.
H.J.K. was supported by grants from the German Research Foundation (Deutsche Forschungsgemeinschaft; DFG), Bonn (Kra 436/14–2 and 436 TSE 113/57/0–1) and the German Academic Exchange Service (Deutscher Akademischer Austauschdienst; DAAD), Bonn-Prague University partnership. This study also received financial support from the European Commission by the Operational Program Prague – Competitiveness; project ‘CEVKOON’ (#CZ.2.16/3.1.00/22126). A.S. is supported by grant No. P303/10/P170 awarded by Czech Science Foundation (GACR) and she is also a recipient of Marie Curie Fellowship from the European Commission Program PEOPLE (IRG 247847). J.D.I. is an Established Investigator of the American Heart Association and these studies were supported by NIH grants HL59699 and DK38226. S.H.H. was supported in part by the Howard Hughes Foundation and a fellowship from the NIEHS Supported Basic Research Program. The partial support was provided by NIEHS grant R01 ES02710, R01 Es013933 and P42 Es013933 and NIH grant R01 HL059699 awarded to B.D.H. B.D.H. is a George and Judy Marcus Senior Fellow of the American Asthma Foundation.
Abbreviations
- ACEI
angiotensin-converting enzyme inhibitor
- ANG II
angiotensin II
- AT1
receptors for angiotensin II, type 1
- BP
blood pressure
- c-AUCB
cis-4-[4-(3-adamantan-1-yl-ureido)-cyclohexyl-oxy]-benzoic acid
- CYP
cytochrome P-450
- DHETEs
dihydroxyeicosatrienoic acids
- EETs
epoxyeicosatrienoic acids
- GFR
glomerular filtration rate
- GSI
glomerulosclerosis index
- I3C
indole-3-carbinol
- L-NAME
Nω-nitro-l-arginine methyl ester
- MAP
mean arterial pressure
- NO
nitric oxide
- NOS
nitric oxide synthase
- NOX
nitrate/nitrite
- RAS
renin–angiotensin system
- RBF
renal blood flow
- sEH
soluble epoxide hydrolase
- UISO
8-isoprostane
Footnotes
Conflicts of interest: There are no conflicts of interest.
References
- 1.Campbell WB, Fleming I. Epoxyeicosatrienoic acids and endothelium-dependent response. Pfugers Arch. 2010;459:881–895. doi: 10.1007/s00424-010-0804-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roman RJ. P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol Rev. 2002;82:131–185. doi: 10.1152/physrev.00021.2001. [DOI] [PubMed] [Google Scholar]
- 3.Imig JD. Epoxides and soluble epoxide hydrolase in cardiovascular physiology. Physiol Rev. 2012;92:101–130. doi: 10.1152/physrev.00021.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li J, Carroll MA, Chander PN, Falck JR, Sangras B, Stier CT. Soluble epoxide hydrolase inhibitor, AUDA, prevents early salt-sensitive hypertension. Front Biosci. 2008;13:3480–3487. doi: 10.2741/2942. [DOI] [PubMed] [Google Scholar]
- 5.Huang H, Morisseau C, Wang JF, Yang T, Falck JR, Hammock BD, Wang MH. Increasing or stabilizing renal epoxyeicosatrienoic acid production attenuates abnormal renal function and hypertension in obese rats. Am J Physiol. 2007;293:F342–F349. doi: 10.1152/ajprenal.00004.2007. [DOI] [PubMed] [Google Scholar]
- 6.Sporkova A, Kopkan L, Varcabová A, Husková Z, Hwang SH, Hammock BD, et al. Role of cytochrome P450 metabolites in the regulation of renal function and blood pressure in 2-kidney, 1-clip hypertensive rats. Am J Physiol. 2011;300:R1468–R1475. doi: 10.1152/ajpregu.00215.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neckář J, Kopkan L, Husková Z, Kolář F, Papoušek F, Kramer HJ, et al. Inhibition of soluble epoxide hydrolase by cis-4-[4-(3-adamantan-I-ylureido)cyclohexyl-oxy]benzoic acid exhibits antihypertensive and cardioprotective actions in transgenic rats with angiotensin II-dependent hypertension. Clin Sci. 2012;122:513–525. doi: 10.1042/CS20110622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Honetschlägerová Z, Husková Z, Vaňourková Z, Sporková A, Kramer HJ, Hwang SH, et al. Renal mechanisms contributing to the antihypertensive action of soluble epoxide hydrolase inhibition in Ren-2 transgenic rats with inducible hypertension. J Physiol. 2011;589:207–219. doi: 10.1113/jphysiol.2010.199505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Honetschlägerová Z, Sporková A, Kopkan L, Husková Z, Hwang SH, Hammock BD, et al. Inhibition of soluble epoxide hydrolyse improves the impaired pressure-natriuresis relationship and attenuates the development of hypertension and hypertension-associated end-organ damage in Cyp1a1-Ren-2 transgenic rats. J Hypertens. 2011;29:1590–1601. doi: 10.1097/HJH.0b013e328349062f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Capdevila JH, Falck JR, Imig JD. Role of the cytochrome P450 arachidonic acid monooxygenases in the control of systemic blood pressure and experimental hypertension. Kidney Int. 2007;72:683–689. doi: 10.1038/sj.ki.5002394. [DOI] [PubMed] [Google Scholar]
- 11.Hercule HC, Schunck WH, Gross V, Seringer J, Leung FP, Weldon SM, et al. Interaction between P450 eicosanoids and nitric oxide in the control of arterial tone in mice. Aterioscler Thromb Vasc Biol. 2009;29:54–60. doi: 10.1161/ATVBAHA.108.171298. [DOI] [PubMed] [Google Scholar]
- 12.Kantachuvesiri S, Fleming S, Peters J, Peters B, Brooker G, Lammie AG, et al. Controlled hypertension, a transgenic toggle switch reveals differential mechanisms underlying vascular disease. J Biol Chem. 2001;276:36727–36733. doi: 10.1074/jbc.M103296200. [DOI] [PubMed] [Google Scholar]
- 13.Vaňourková Z, Kramer HJ, Husková Z, Vaněčková I, Opočenský M, Čertíková Chábová V, et al. AT1 receptor blockade is superior to conventional triple therapy in protecting against end-organ damage Cyp1a1-Ren-2 transgenic rats with inducible hypertension. J Hypertens. 2006;24:2465–2472. doi: 10.1097/01.hjh.0000251909.00923.22. [DOI] [PubMed] [Google Scholar]
- 14.Husková Z, Vaňourková Z, Erbanová M, Thumová M, Opočenský M, Mullins JJ, et al. Inappropriately high circulating and intrarenal angiotensin II levels during dietary salt loading exacerbate hypertension in Cyp1a1-Ren-2 transgenic rats. J Hypertens. 2010;28:495–509. doi: 10.1097/HJH.0b013e3283345d69. [DOI] [PubMed] [Google Scholar]
- 15.Mitchell KD, Bagatell SJ, Miller CS, Mouton CR, Seth DM, Mullins JJ. Genetic clamping of renin gene expression induces hypertension and elevation of intrarenal II levels of graded severity in Cyp1a1-Ren2 transgenic rats. JRAAS. 2006;7:74–86. doi: 10.3317/jraas.2006.013. [DOI] [PubMed] [Google Scholar]
- 16.Erbanová M, Thumová M, Husková Z, Vaněčková I, Vaňourková Z, Mullins JJ, et al. Impairment of the autoregulation of renal hemodynamics and of the pressure-natriuresis relationship precedes the development of hypertension in Cyp1a1-Ren-2 transgenic rats. J Hypertens. 2009;27:575–586. doi: 10.1097/hjh.0b013e32831cbd5a. [DOI] [PubMed] [Google Scholar]
- 17.Howard CG, Mitchell KD. Renal functional response to selective intrarenal renin inhibition in Cyp1a1-Ren-2 transgenic rats with ANG II-dependent malignant hypertension. Am J Physiol. 2012;302:F52–F59. doi: 10.1152/ajprenal.00187.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zicha J, Kuneš J, Vranková S, Jendeková L, Dobešová Z, Pintérová M, Pecháňová O. Influence of pertussion toxin pretreatment on the development of L-NAME-induced hypertension. Physiol Res. 2009;58:751–755. doi: 10.33549/physiolres.931898. [DOI] [PubMed] [Google Scholar]
- 19.Verhagen AMG, Braam B, Boer P, Grone HJ, Koomans HA, Joles JA. Losartan-sensitive renal damage caused by chronic NOS inhibition does not involve increased renal angiotensin II concentrations. Kidney Int. 1999;56:222–231. doi: 10.1046/j.1523-1755.1999.00542.x. [DOI] [PubMed] [Google Scholar]
- 20.Kujal P, Chabova VC, Vernerova Z, Walkowska A, Kompanowska-Jezierska E, Sadowski J, et al. Similar renoprotection after renin-angiotensin-dependent and -independent antihypertensive therapy in 5/6-nephrectomized Ren-2 transgenic rats: are there blood pressure-independent effects? Clin Exp Pharmacol Physiol. 2010;37:1159–1169. doi: 10.1111/j.1440-1681.2010.05453.x. [DOI] [PubMed] [Google Scholar]
- 21.Kurtz TW, Griffin KA, Bidani AK, Davisson RL, Hall JE. Recommendations for blood pressure measurements in humans and experimental animals. Part 2: blood pressure measurements in experimental animals. Hypertension. 2005;45:299–310. doi: 10.1161/01.HYP.0000150857.39919.cb. [DOI] [PubMed] [Google Scholar]
- 22.Sasser JM, Moningka NC, Tsarova T, Baylis C. Nebivolol does not protect against 5/6 ablation/infarction induced chronic kidney disease in rats – comparison with angiotensin II receptor blockade. Life Sci. 2012;91:54–63. doi: 10.1016/j.lfs.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kopkan L, Hess A, Husková Z, Červenka L, Navar LG, Majid DSW. High-salt intake enhances superoxide activity in eNOS knockout mice leading to the development of salt sensitivity. Am J Physiol. 2010;299:F656–F663. doi: 10.1152/ajprenal.00047.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kopkan L, Husková Z, Vaňourková Z, Thumová M, Škaroupková P, Červenka L, Majid DSW. Superoxide and its interaction with nitric oxide modulates renal function in prehypertensive Ren-2 transgenic rats. J Hypertens. 2007;25:2257–2265. doi: 10.1097/HJH.0b013e3282efb195. [DOI] [PubMed] [Google Scholar]
- 25.Čertíková Chábová V, Walkowska A, Kompanowska-Jezierska E, Sadowski J, Kujal P, Vernerová Z, et al. Combined inhibition of 20-hydroxyeicosatetraenoic acid formation and epoxyeicosatrienoic acids degradation attenuates hypertension and hypertension-induced end-organ damage in Ren-2 transgenic rats. Clin Sci. 2010;118:617–632. doi: 10.1042/CS20090459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakano Y, Hirano T, Uehara K, Nishibayashi S, Hattori K, Aihara M, Yamada Y. New rat model induced by antiglomerular basement membrane antibody shows severe glomerular adhesion in early stage and quickly progresss to end-stage renal failure. Pathol Int. 2008;58:361–370. doi: 10.1111/j.1440-1827.2008.02237.x. [DOI] [PubMed] [Google Scholar]
- 27.Vaněčková I, Kujal P, Husková Z, Vaňourková Z, Vernerová Z, Čertíková Chábová V, et al. Effects of combined endothelin A receptor and renin-angiotensin system blockade on the course of end-organ damage in 5/6 nephrectomized Ren-2 hypertensive rats. Kidney Blood Press Res. 2012;35:382–392. doi: 10.1159/000336823. [DOI] [PubMed] [Google Scholar]
- 28.Chin SY, Wang CT, Majid DSW, Navar LG. Renoprotective effects of nitric oxide in angiotensin II-induced hypertension in the rat. Am J Physiol. 1998;274:F876–F882. doi: 10.1152/ajprenal.1998.274.5.F876. [DOI] [PubMed] [Google Scholar]
- 29.Navar LG, Ichihara A, Chin SY, Imig JD. Nitric oxide-angiotensin II interactions in angiotensin II-dependent hypertension. Acta Physiol Scand. 2000;168:139–147. doi: 10.1046/j.1365-201x.2000.00630.x. [DOI] [PubMed] [Google Scholar]
- 30.Patterson ME, Mullins JJ, Mitchell KD. Renoprotective effects of neuronal NOS-derived nitric oxide and cyclooxygenase-2 metabolites in transgenic rats with inducible malignant hypertension. Am J Physiol. 2008;294:F205–F211. doi: 10.1152/ajprenal.00150.2007. [DOI] [PubMed] [Google Scholar]
- 31.Wilcox CS. L-Arginine-nitrix oxide pathway. In: Seldin DW, Giebisch G, editors. The kidney: physiology and pathophysiology. Philadelphia: Lippincott Williams & Wilkins; 2000. pp. 849–872. [Google Scholar]
- 32.Kone BC. Nitric oxide synthesis in the kidney:isoforms, biosynthesis, and functions in health. Semin Nephrol. 2004;24:299–315. doi: 10.1016/j.semnephrol.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 33.Madhun ZT, Goldthwait DA, McKay D, Hopfer U, Douglas JG. An epoxygenase metabolite of arachidonic acid mediates angiotensin II-induced rises in cytosolic calcium in rabbit proximal tubule epithelial cells. J Clin Invest. 1991;88:456–461. doi: 10.1172/JCI115325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sakairi Y, Jacobson HR, Noland DT, Capdevila JH, Falck JR, Breyer MD. 5,6-EET inhibits ion transport in collecting duct by stimulating endogenous prostaglandin synthesis. Am J Physiol. 1995;268:F931–F939. doi: 10.1152/ajprenal.1995.268.5.F931. [DOI] [PubMed] [Google Scholar]
- 35.Zhao X, Yamamoto T, Newman JW, Kim IH, Watanabe T, Hammock BD, et al. Soluble epoxide hydrolase inhibition protects the kidney from hypertension-induced damage. J Am Soc Nephrol. 2004;15:1244–1253. [PubMed] [Google Scholar]
- 36.Imig JD, Zhao X, Zaharis CZ, Olearczyk JJ, Pollock DM, Newman JW, et al. An orally active epoxide hydrolase inhibitor lowers blood pressure and provides renal protection in salt-sensitive hypertension. Hypertension. 2005;46:975–981. doi: 10.1161/01.HYP.0000176237.74820.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Castrop H, Höcherl K, Kurtz A, Schweda F, Todorov V, Wagner C. Physiology of kidney renin. Physiol Rev. 2010;90:607–673. doi: 10.1152/physrev.00011.2009. [DOI] [PubMed] [Google Scholar]
- 38.Hall JE, Brands MW. The renin-angiotensin-aldosterone system: renal mechanisms and circulatory homeostasis. In: Seldin DW, Giebisch G, editors. The kidney: physiology and pathophysiology. Philadelphia: Lippincott Williams & Wilkins; 2000. pp. 1009–1046. [Google Scholar]
- 39.Schweda F, Kurtz A. Regulation of renin release by local and systemic factors. Rev Physiol Biochem Pharmacol. 2011;161:1–44. doi: 10.1007/112_2008_1. [DOI] [PubMed] [Google Scholar]
- 40.Zatz R, Baylis C. Chronic nitric oxide inhibition model six years on. Hypertension. 1998;32:958–964. doi: 10.1161/01.hyp.32.6.958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Torok J. Participation of nitric oxide in different models of experimental hypertension. Physiol Res. 2008;57:813–825. doi: 10.33549/physiolres.931581. [DOI] [PubMed] [Google Scholar]
- 42.Kopkan L, Červenka L. Renal interactions of renin-angiotensin system, nitric oxide and superoxide anion: implications in the pathophysiology of salt-sensitivity and hypertension. Physiol Res. 2009;58(Suppl 2):S55–S57. doi: 10.33549/physiolres.931917. [DOI] [PubMed] [Google Scholar]
- 43.Zhou X, Frohlich ED. Differential effects of antihypertensive drugs on renal and glomerular hemodynamics and injury in the chronic nitrixoxide-suppressed rat. Am J Nephrol. 2005;25:138–152. doi: 10.1159/000085358. [DOI] [PubMed] [Google Scholar]
- 44.Ribeiro MO, Antune E, De Nucci G, Lovisolo SM, Zatz R. Chronic inhibition of nitric oxide synthesis: A new model of arterial hypertension. Hypertension. 1992;20:298–303. doi: 10.1161/01.hyp.20.3.298. [DOI] [PubMed] [Google Scholar]
- 45.Salazar FJ, Pinilla JM, López F, Romero JC, Quesada T. Renal effects of prolonged synthesis inhibition of endothelium-derived nitric oxide. Hypertension. 1992;20:113–117. doi: 10.1161/01.hyp.20.1.113. [DOI] [PubMed] [Google Scholar]
- 46.Jover B, Herizi A, Ventre F, Dupont M, Mimran A. Sodium and angiotensin in hypertension induced by long-term nitric oxide blockade. Hypertension. 1993;21:944–948. doi: 10.1161/01.hyp.21.6.944. [DOI] [PubMed] [Google Scholar]
- 47.Navarro-Cid J, Sanchez A, Sáiz J, Ruilope LM, García-Estaň J, Romero JC, et al. Hormonal, renal and metabolic alterations during hypertension induced by crhonic inhibition of NO in rats. Am J Physiol. 1994;267:R1516–R1521. doi: 10.1152/ajpregu.1994.267.6.R1516. [DOI] [PubMed] [Google Scholar]