

Abstract
Objectives:
Obstructive sleep apnea is associated with insulin resistance, glucose intolerance, and type 2 diabetes mellitus. Although several studies have suggested that intermittent hypoxia in obstructive sleep apnea may induce abnormalities in glucose homeostasis, it remains to be determined whether these abnormalities improve after discontinuation of the exposure. The objective of this study was to delineate the effects of intermittent hypoxia on glucose homeostasis, beta cell function, and liver glucose metabolism and to investigate whether the impairments improve after the hypoxic exposure is discontinued.
Interventions:
C57BL6/J mice were exposed to 14 days of intermittent hypoxia, 14 days of intermittent air, or 7 days of intermittent hypoxia followed by 7 days of intermittent air (recovery paradigm). Glucose and insulin tolerance tests were performed to estimate whole-body insulin sensitivity and calculate measures of beta cell function. Oxidative stress in pancreatic tissue and glucose output from isolated hepatocytes were also assessed.
Results:
Intermittent hypoxia increased fasting glucose levels and worsened glucose tolerance by 67% and 27%, respectively. Furthermore, intermittent hypoxia exposure was associated with impairments in insulin sensitivity and beta cell function, an increase in liver glycogen, higher hepatocyte glucose output, and an increase in oxidative stress in the pancreas. While fasting glucose levels and hepatic glucose output normalized after discontinuation of the hypoxic exposure, glucose intolerance, insulin resistance, and impairments in beta cell function persisted.
Conclusions:
Intermittent hypoxia induces insulin resistance, impairs beta cell function, enhances hepatocyte glucose output, and increases oxidative stress in the pancreas. Cessation of the hypoxic exposure does not fully reverse the observed changes in glucose metabolism.
Citation:
Polak J; Shimoda LA; Drager LF; Undem C; McHugh H; Polotsky VY; Punjabi NM. Intermittent hypoxia impairs glucose homeostasis in C57BL6/J mice: partial improvement with cessation of the exposure. SLEEP 2013;36(10):1483-1490.
Keywords: Glucose intolerance, insulin resistance, intermittent hypoxia, obstructive sleep apnea
INTRODUCTION
Obstructive sleep apnea is a prevalent sleep disorder affecting approximately 5-15% of middle-aged and older adults in the general population.1,2 Research over the past two decades has shown that untreated obstructive sleep apnea is associated with incident hypertension,3–5 cardiovascular disease,6,7 stroke,8–10 and all-cause mortality.11–14 A large body of observational evidence also indicates that obstructive sleep apnea is associated with glucose intolerance, insulin resistance, and type 2 diabetes mellitus.15,16 Clinical and epidemiological studies have revealed that the association between obstructive sleep apnea and impaired glucose homeostasis is independent of confounding factors such as age and central adiposity.15–19 A notable finding across many of the previous studies is that the severity of metabolic dysfunction is independently correlated with the degree of sleep related hypoxemia.20 Data from human and animal studies indicate that intermittent hypoxia has a fundamental role in impairing glucose homeostasis. Indeed, experimental work in several animal models, including genetically modified mice, has shown that acute and chronic intermittent hypoxia can lead to a variety of metabolic impairments including higher fasting glucose and insulin levels, impairments in whole-body insulin sensitivity, glucose intolerance, reduced beta cell function, and diminished glucose uptake in muscle.21–26 Moreover, healthy volunteers exposed to intermittent hypoxia for as little as 5 h exhibit decreased insulin sensitivity that is not accompanied by the expected compensatory increase in beta cell function.27 Despite these advancements, causal mechanisms mediating the association between obstructive sleep apnea and altered glucose metabolism have not been fully delineated. It is well established that fasting glycemia is primarily determined by hepatic glucose output whereas post-prandial glycemia is influenced by beta cell function, whole body insulin sensitivity, and modulation of hepatic glucose output.28,29 Although intermittent hypoxia can impair insulin sensitivity and beta cell function through oxidative stress and activation of the sympathetic nervous system, its effects on hepatic glucose metabolism remain largely unknown.
The repertoire of possible mechanisms provides a strong basis to speculate a causal association between intermittent hypoxia and altered glucose metabolism. However, studies evaluating whether treatment of obstructive sleep apnea with continuous positive airway pressure (CPAP) reverses the metabolic dysfunction are equivocal and largely inconclusive.15,16 The lack of a consistent and favorable response in glucose homeostasis to CPAP therapy may be due to methodological limitations such as small study samples and suboptimal adherence to CPAP therapy. Alternatively, it is possible that chronic exposure to intermittent hypoxia in obstructive sleep apnea irreversibly impairs glucose homeostasis, which would render CPAP therapy ineffective. To resolve the aforementioned issues, the current study investigated the effects of intermittent hypoxia on several key determinants of glucose homeostasis. Specifically, the aims of the current study were to examine whether intermittent hypoxia impairs glucose tolerance, whole-body insulin sensitivity, beta cell function, and hepatic glucose output. In addition, we also sought to investigate if the observed impairments improve with cessation of the hypoxic exposure. It was hypothesized that exposure to intermittent hypoxia would increase whole-body insulin resistance, induce beta cell dysfunction, and augment hepatic glucose output, which collectively would lead to fasting and postprandial hyperglycemia. Furthermore, it was hypothesized that these effects would be reversible after the hypoxic exposure was discontinued.
METHODS
Protocol for Intermittent Hypoxia
Adult C57BL6/J mice were housed in custom-modified cages connected with plastic tubing to a gas control delivery system regulating the flow of nitrogen, oxygen, and room air into cages as previously described.21,30 Programmable solenoid valves and flow regulators altered the composition of the gas with the cages so that during each cycle of hypoxia, the inspired fraction of oxygen (FiO2) was reduced from 21% to 6-7% over a period of 30 sec and rapidly returned to 21% during the following 30 sec. For the control arm, animals were exposed to alternating periods of room air gas (FiO2:21%), simulating a pattern comparable to intermittent hypoxia. On average, 60 episodes/h of oxygen desaturation were induced, mimicking oxyhemoglobin desaturations observed in severe obstructive sleep apnea.
All animals were housed at room temperature and subjected to a 12-h light/dark cycle. The following five exposure groups were used: (1) intermittent hypoxia for 7 days; (2) intermittent hypoxia for 14 days; (3) intermittent hypoxia for 7 days followed by air for 7 days (recovery paradigm); (4) intermittent air for 7 days; and (5) intermittent air for 14 days. Animals were exposed to hypoxia or control condition during the light phase for 12 h. Because intermittent hypoxia can induce weight loss, daily food intake in the control animals and during the recovery period was adjusted daily such that the body weight of mice in the control and recovery groups followed the same body weight trajectory as animals in the intermittent hypoxia group (see supplemental material). Animals were sacrificed on the last day of the exposures. Principles of laboratory animal care (National Institutes of Health publication no. 85-23, revised 1985) were followed and the study protocol was approved by the Animal Care and Use Committee of the Johns Hopkins University.
Intraperitoneal Glucose Tolerance Test and Insulin Tolerance Test
An intraperitoneal glucose tolerance test (GTT) was performed after a 5-h fast for each of the exposure paradigms (N = 10 per group). Following a glucose injection (1g/ kg glucose dissolved in saline to a concentration of 150 mg/ dL), glucose levels were measured at 0, 10, 20, 30, 60, 90, and 120 min after the glucose injection in tail-snip blood samples using a glucometer (Accu-Check Aviva, Roche, Indianapolis, IN, USA). An insulin tolerance test (ITT, N = 10 per exposure group) was similarly performed after a 2-h fast by measuring blood glucose levels at 0, 10, 20, 30, 40, 50, 60, 90, and 120 min after injecting 0.5 IU/kg of insulin (Humulin R, 100 U/ mL, Eli Lilly, Indianapolis, IN, USA). The GTT and ITT were performed in a distinct set of anesthetized animals in the fasted state without interruption in the exposure to the intermittent hypoxia or control conditions. Glucose-induced insulin secretion was assessed (N = 10 per exposure group) 30 min after a 1 g/kg glucose injection by measuring insulin levels in blood obtained by retro-orbital puncture using the Mouse Ultrasensitive EIA kit (Alpco, Salem, NH, USA). Fasting and glucose-induced insulin secretion was evaluated in a separate group of animals. Absorption of insulin from the peritoneal cavity was also assessed (N = 5 per exposure group) following a 2-h fasting period by measuring plasma radioactivity at 10, 30, and 60 min after the intraperitoneal injection of ∼1μCi of I125-radiolabeled insulin in 100 μL of saline (specific activity 378.8 μCi/μg, Perkin Elmer, Waltham, MA, USA). No significant differences were noted in insulin absorption across the different exposure groups (see supplemental material, Figure S1). All blood and plasma samples obtained during the GTT and ITT were frozen immediately and kept at -80°C until analysis. The homeostasis model assessment (HOMA) model was used to derive HOMA-IR (insulin·glucose/22.5) as a measure of insulin resistance and HOMA-β ([20·Insulin]/[Glucose-3.5]) as a measure of beta cell function.31
Determination of Liver Glycogen and Tissue Oxidative Stress
Liver, muscle (quadriceps), and pancreas were collected under isoflurane anesthesia, flash frozen in liquid nitrogen and stored at -80°C. Frozen tissue was mechanically homogenized in T-PER lysis buffer (Tissue Protein Extraction Reagent, Thermo Scientific, Waltham, MA, USA) containing complete protease inhibitor cocktail centrifuged at 16.1 relative centrifugal force for 10 min at 4°C and supernatants collected. All samples were assessed in duplicate and normalized to mg of protein as determined by bicinchoninic acid assay (BCA protein assay, Thermo Scientific, Waltham, MA, USA). Tissue lipid peroxidation (n = 5 per group) was determined using the thiobarbituric acid reactive substances (TBARS assay kit, Cayman Chemical Company, Ann Arbor, MI, USA). Glycogen content in liver (N = 5 per exposure group) was determined using the glycogen assay kit (Abcam, Cambridge, MA, USA) and normalized to liver weight.
Hepatocyte Isolation, Measurement of Glucose Output, and Gene Expression
Mouse hepatocytes were isolated from animals in all experimental groups using two-step portal vein perfusions under isoflurane anesthesia. Subsequently, to derive a measure of glycogenolysis, cells were washed and incubated for 4 h in glucose-free, phenol-free Dulbecco's Modified Eagle's Medium (DMEM) without glutamine and pyruvate (Sigma-Aldrich Co., St. Louis, MO, USA). Moreover, to derive a measure of total hepatocyte glucose output resulting from glycogenolysis and gluconeogenesis, cells were washed and incubated for 4 h in glucose-free, phenol-free DMEM supplemented with glutamine and pyruvate (Product No. G8540 and P2256, Sigma-Aldrich). Glucose secreted into the media was measured using a glucoseoxidase assay (GO Kit, Sigma-Aldrich) and normalized to mg protein. Details regarding messenger RNA extraction, primer sequences, and quantitative polymerase chain reaction (qPCR) protocol are described below.
Cultured cells isolated from animals (N = 4 for intermittent hypoxia and recovery groups, N = 3 for intermittent air group) were washed three times with 4 mL of glucose-free and phenol red-free DMEM medium. Total hepatocyte glucose output was determined in cells incubated for 4 h in glucose-free and phenol red-free DMEM medium supplemented with 0.11 g/L pyruvate and 0.584 g/L glutamine as substrates for gluconeogenesis. Glycogenolysis was determined in cells incubated for 4 h in glucose-free and phenol red-free DMEM medium without pyruvate and glutamine. The rate of gluconeogenesis was calculated as the difference in glucose output between the two incubations. Glucose in media was determined with glucoseoxidase methods (GO Kit). After completion of the exposure periods, cells were washed twice with 2 mL phosphate buffered saline (PBS 1X, Invitrogen, Grand Island, NY, USA) and lysed with TPER lysis buffer. Protein quantity was determined using the BCA protein assay (Thermo Scientific, Waltham, MA, USA) and used for data normalization.
cDNA Synthesis and qPCR
Liver samples (N = 5 per group) were flash frozen and stored at -80°C until analysis. Total RNA was extracted using the Trizol reagent (Life Technologies, Carlsbad, CA, USA). After assessing quality on an agarose gel (see supplemental material, Figure S2), cDNA was synthesized using the iScript cDNA Synthesis Kit (BioRad Life Science Research, Hercules, CA, USA) and qPCR was performed with iCycler instrument (BioRad Life Science Research) using the QuantiTect SYBR Green PCR Kit (Qiagen GmbH, Hilden, Germany). The following primers were used in reactions: phosphoenolpyruvate carboxykinase F: 5′-GGTGCATGAAAGGCCGCACC-3′ and R: 5′-ACCAATCTTGGCCAGCGGCG-3′, glucose 6-phosphatase F: 5′-CAACGCCCGTATTGGTGGGTCC-3′ and R: 5′-GGGACTTCCTGGTCCGGTCT-3′, cyclophyllin B F: 5′-CAGCAAGTTCCATCGTGTCA-3′ and R: 5′-GAAGCGCTCACCATAGATGC-3′. Expression levels of target genes in all groups were normalized to the expression of cyclophyllin B (a housekeeping gene) and expressed as the fold change over the expression level of the control group (2-ΔΔCT method). Differences in gene expression between groups were tested using the Pfafflalgorithm with the Relative Expression Software Tool (REST) v2.0.13 (Qiagen GmbH).
Statistical Analysis
Data are reported as means and standard error of the means (or medians with interquartile ranges). Intermittent hypoxia-induced changes in whole-body glucose metabolism were assessed by comparing the GTT and ITT-derived parameters between the intermittent hypoxia conditions (7- and 14-day exposure) and their respective control groups (7- and 14-day exposure). To determine whether cessation of intermittent hypoxia was associated with improvements in glucose homeostasis, the following three groups were compared: 14 days of intermittent hypoxia; 7 days of intermittent hypoxia followed by the recovery period with control condition; and 14 days of the control condition. Differences between groups in ITT and GTT glucose profiles were examined using analysis of variance (ANOVA). Between-group differences for all other variables were tested using one-way ANOVA with treatment group as the primary variable. Area under the glucose curve (AUC) during the ITT was calculated using the trapezoidal rule using the entire testing period of 120 min. Finally, the slope of the plasma glucose clearance after insulin injection during the initial 30 min of the ITT was determined and differences between exposure groups examined using ANOVA.
RESULTS
Fasting Glucose and Glucose Tolerance
Differences in fasting glucose levels or glucose tolerance were not observed between animals exposed to the control condition for 7 or 14 days. In contrast, intermittent hypoxia for 7 days increased fasting glucose levels by 51% (150 ± 6 mg/ dL versus 99 ± 5 mg/dL; P < 0.001). Extending the exposure duration to 14 days was not associated with further increase in fasting glucose levels (149 ± 5 md/dL versus 89 ± 5 mg/dL; P < 0.001). Glucose tolerance characterized by the GTT glucose profiles was not impaired after 7 days of intermittent hypoxia after accounting for differences in fasting glycemia. However, intermittent hypoxia exposure of 14 days did impair glucose tolerance and was associated with higher peak glucose levels (Figure 1A). The differences in GTT profiles between 7- and 14-day intermittent hypoxia exposure periods (P = 0.068) and the finding of a higher peak glucose values at 20 and 30 min during the GTT (Figure 1A) suggest that extending the duration of intermittent hypoxia from 7 to 14 days further deteriorates glucose tolerance. Finally, mice that were exposed to intermittent hypoxia for 7 days followed by a recovery period of 7 days revealed that fasting glucose and insulin levels returned to values similar to those observed in the control animals (Table 1). However, despite normalization of fasting glucose levels, glucose tolerance did not fully recover with cessation of the hypoxic exposure (Figure 1B).
Figure 1.
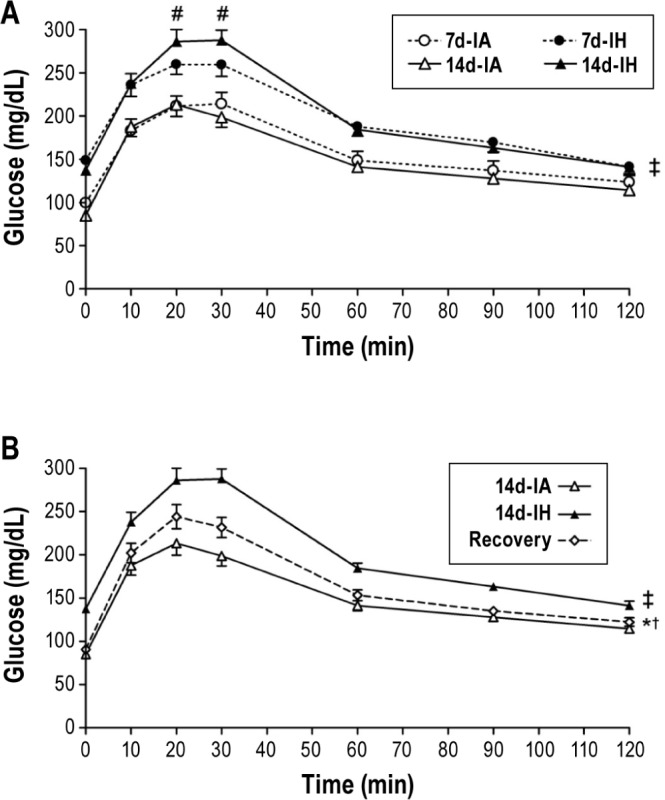
(A) Intraperitoneal glucose tolerance tests (GTT) after 7 and 14 days of intermittent hypoxia (7d-IH and 14d-IH) and intermittent air (7d-IA and 14d-IA, control conditions). Data were analyzed using an analysis of variance (ANOVA) of the GTT profiles assessing the interaction between exposure group (7d-IH or 14d-IH versus control) and time during the GTT. The GTT profile of the 7d-IH group is shifted upward relative to the 7d-IA group due to differences in fasting glucose levels. However, no significant interaction between exposure group and time was observed. ‡P < 0.001 for differences between 14d-IH and 14d-IA groups, #P = 0.048 for differences between 7d-IH and 14d-IH groups. (B) GTT profiles of 14d-IA, 14d-IH and recovery groups. Data were analyzed using an ANOVA of the GTT profiles investigating the interaction between exposure groups (14d-IA, 14d-IH, and recovery) and time of GTT. ‡P < 0.05 for differences between 14d-IH and 14d-IA groups, †P = 0.001 for differences between 14d-IA and recovery groups, *P < 0.013 for differences between 14d-IH and recovery groups.
Table 1.
Fasting and postchallenge glucose, insulin, and homeostasis model assessment values
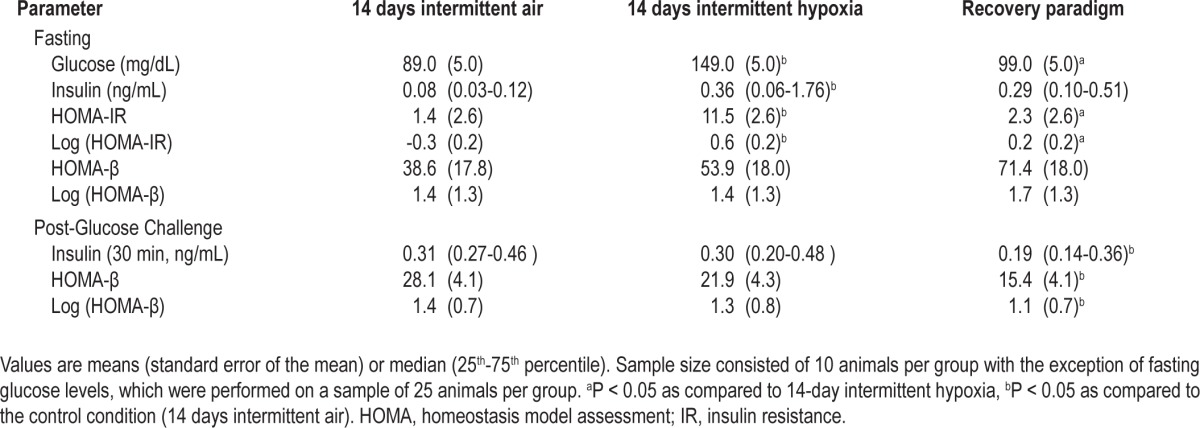
Whole-Body Insulin Sensitivity, Beta Cell Function, and Tissue Oxidative Stress
Figure 2A displays the glucose profiles after the insulin injection during the ITT. Because intermittent hypoxia led to fasting hyperglycemia, glucose profiles from the insulin tolerance test were normalized for fasting glucose values (Figure 2B). To quantify the differences in insulin sensitivity across experimental groups, AUC values for each of the glucose curves from the insulin tolerance test were determined (Figure 2C). Glucose profiles and the accompanying AUC values showed that 14 days of intermittent hypoxia diminishes insulin-induced glucose uptake. HOMA-IR, a surrogate marker of hepatic insulin resistance in the fasting state, increased after 14 days of intermittent hypoxia (1.4 ± 2.6 versus 11.5 ± 2.6, P = 0.012; Table 1). Animals subjected to 7 days of intermittent hypoxia followed by the recovery period also showed impairments in insulin sensitivity, as assessed by the glucose profiles and AUC values from the ITT (Figures 2A and 2C) as well as the HOMA-IR index (Table 1). Furthermore, analysis of the glucose trajectories during the ITT showed impaired insulin sensitivity after 14 days of intermittent hypoxia when compared to the control group (0.0231 ± 0.0014 versus 0.0117 ± 0.0001, respectively, P < 0.001) with evidence of partial improvement after 7 days of the recovery period (0.0173 ± 0.0014, from control P = 0.032). Despite cessation of the exposure for 7 days, insulin resistance (i.e., higher HOMA-IR values; Table 1) persisted, suggesting that intermittent hypoxia-induced impairment in insulin sensitivity does not fully recover.
Figure 2.
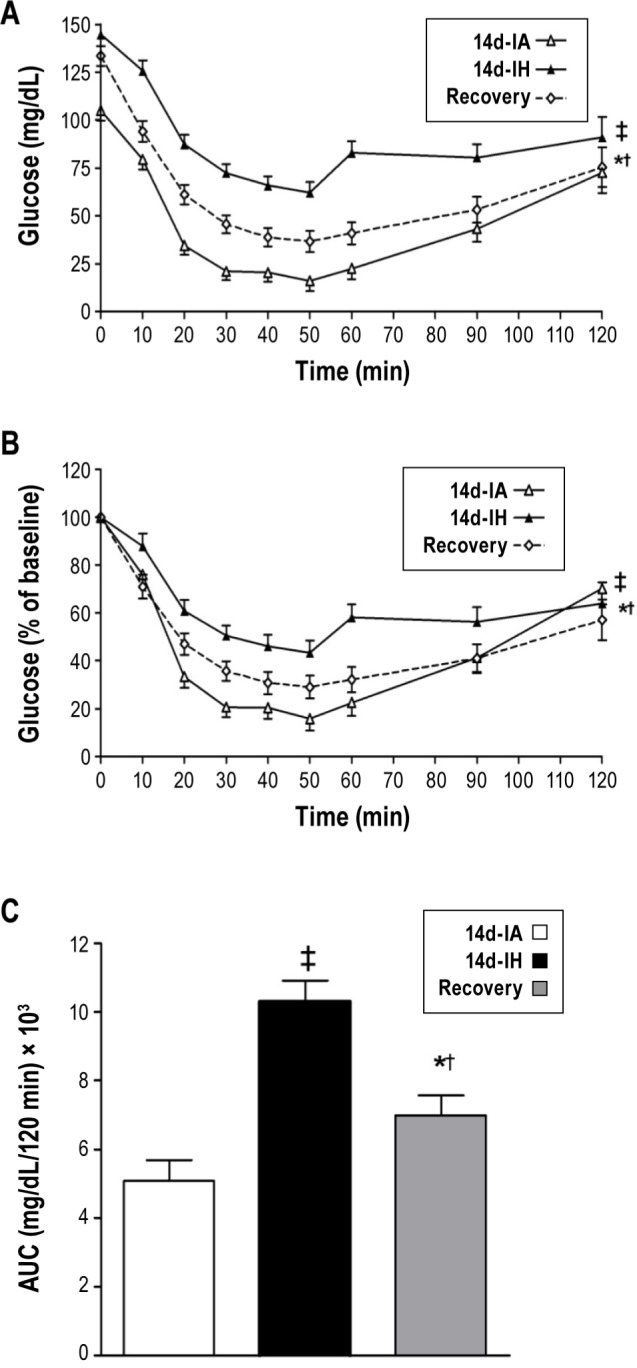
Insulin tolerance test-derived (A) absolute glucose levels; (B) normalized glucose levels (% of fasting glucose); and (C) area under the curve (AUC) of the normalized glucose curves after intermittent hypoxia (14d-IH), control condition (14d-IA), and recovery paradigm. ‡P = 0.028 for differences between 14d-IH and 14d-IA groups in Figure 2A (P = 0.011, for Figure 2B and P < 0.001 for Figure 2C), †P = 0.013 for differences between 14d-IA and recovery groups in Figure 2A (P = 0.015 for Figure 2B and P = 0.057, for Figure 2C), *P < 0.0013 for differences between 14d-IH and recovery groups in Figure 2A (P = 0.01 for Figure 2B).
Parameters of beta cell function revealed that 14 days of intermittent hypoxia was associated with impaired insulin secretion. Although fasting insulin levels were four-fold higher with intermittent hypoxia (Table 1), this compensatory increase was insufficient to normalize fasting glucose levels indicating an impaired beta cell response to fasting hyperglycemia. Moreover, in the fasting and postglucose challenge state, 14 days of intermittent hypoxia did not increase the HOMA-β index confirming impaired beta cell function (Table 1). Finally, animals subjected to the recovery paradigm showed no improvements in beta cell function after cessation of the hypoxic exposure. In fact, beta cell failure as demonstrated by a decreased HOMA-β and lower peak insulin levels was observed after the 7-day recovery period compared to the control group. Assessment of oxidative stress showed that TBARS levels in pancreatic tissue increased by 44% (TBARS: 0.66 ± 0.06 versus 0.95 ± 0.07 nmol/mg protein) and remained elevated even after discontinuation of the hypoxic exposure (Figure 3A). In contrast, markers of oxidative stress in muscle and liver tissue were unchanged with 14 days of intermittent hypoxia or the recovery paradigm (Figures 3B and 3C).
Figure 3.
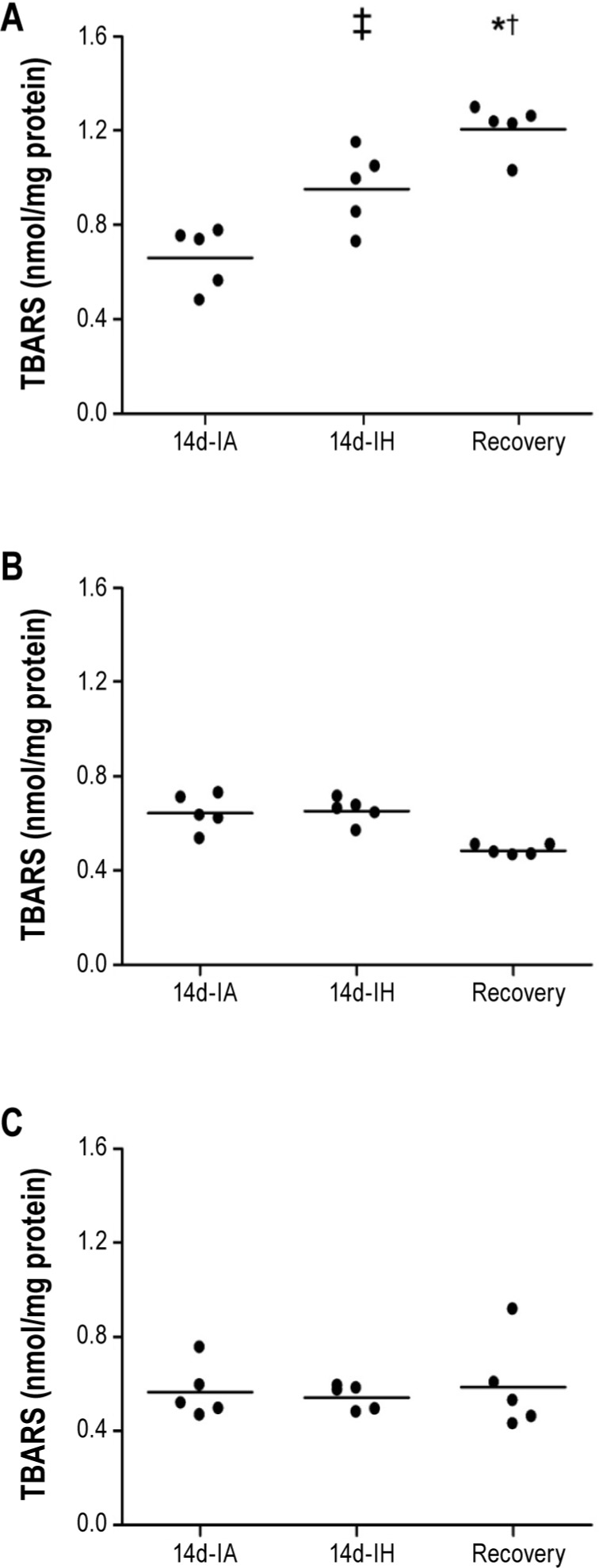
Thiobarbituric acid reactive substances (TBARS) in pancreas (A), liver (B), and muscle (C) after intermittent hypoxia (14-IH), control condition (14-IA), and recovery paradigm. Mean values were compared using one-way analysis of variance (ANOVA). ‡P = 0.005 for differences between 14d-IH and 14d-IA groups, †P < 0.001 for differences between 14d-IA and recovery groups, *P < 0.012 for differences between 14d-IH and recovery groups.
Liver Glucose Metabolism
To characterize the effects of intermittent hypoxia on liver glucose metabolism, hepatocytes were isolated from all experimental groups. Total glucose output (Figure 4A) was higher from hepatocytes isolated from mice exposed to intermittent hypoxia (0.103 ± 0.005 versus 0.160 ± 0.017 mg/mg protein/240 min; P < 0.021) compared to the control condition. Further characterization of glucose output demonstrated that gluconeogenesis, but not glycogenolysis, increased after 14 days of intermittent hypoxia (Figures 4B and 4C, respectively). Gene expression analysis revealed that phosphoenolpyruvate carboxykinase (PEPCK) and glucose 6-phosphatase, two key enzymes regulating gluconeogenesis, increased with 14 days of intermittent hypoxia. In mice exposed to 7 days of intermittent hypoxia followed by the recovery period, gluconeogenesis and glycogenolysis were comparable to the control group. However, a differential response in gene expression was noted. At the end of the recovery period, PEPCK mRNA levels were similar to the control group, whereas glucose 6-phosphatase levels remained elevated (Figure 4D). Finally, compared to the control condition, 14 days of intermittent hypoxia induced a 50-fold increase in liver glycogen stores that were correlated with the degree of fasting glycemia (Figures 4E and 4F, respectively).
Figure 4.

(A) Total glucose output from isolated hepatocytes. (B) Glucose output from gluconeogenesis. (C) Glucose output from glycogenolysis. (D) Phosphoenolpyruvate carboxykinase (PEPCK) and glucose 6-phosphatase messenger RNA levels. (E) Liver glycogen stores after intermittent hypoxia (14d-IH days), control condition (14d-IA days), and recovery paradigm. (F) Association between liver glycogen stores and fasting glucose levels. Mean values were compared using one-way analysis of variance (ANOVA). ‡P < 0.05 for differences between 14d-IH and 14d-IA groups (P = 0.021 for Figure 4A, P = 0.04 for Figure 4B, P = 0.004 for Figure 4C PEPCK, P = 0.011 for Figure 4D G6P, P = 0.004 for Figure 4E), †P < 0.05 for differences between 14d-IA and recovery groups (P = 0.004 for Figure 4D G6P), *P < 0.05 for differences between 14d-IH and recovery groups (P = 0.023 for Figure 4A, P = 0.001 for Figure 4B, P = 0.002 for Figure 4D PEPCK, P = 0.020 for Figure 4E).
DISCUSSION
The results of the current study demonstrate that exposure to intermittent hypoxia, a pathognomonic feature in obstructive sleep apnea, induces fasting hyperglycemia, glucose intolerance, insulin resistance, and beta cell dysfunction in mice. These alterations, which have a fundamental role in the development of type 2 diabetes, are accompanied by increased hepatic glycogen content and glucose output and higher levels of reactive oxygen species in pancreatic tissue. Cessation of the hypoxic exposure improved some, but not all, of the observed metabolic derangements. Specifically, glucose intolerance, insulin resistance, and pancreatic oxidative stress persisted despite cessation of the hypoxic exposure, whereas levels of fasting glucose and insulin returned to values comparable to those observed in the control animals.
The fact that intermittent hypoxia induced fasting hyperglycemia is consistent with previous studies21–26 demonstrating that short-term hypoxic exposure increases fasting glucose levels in nonobese mice—an effect that can be aggravated by diet-induced obesity.26 It is well established that glucose homeostasis in the fasting state depends primarily on hepatic glucose output.32 Previous work by Iiyori et al.22 has shown that exposure to intermittent hypoxia for 9 h can indeed increase hepatic glucose output, which then can lead to fasting hyperglycemia. While other studies have shown variable effects of intermittent hypoxia on glucose in the fasting state,33–36 the heterogeneity in the findings across these studies is likely attributed to differences in exposure duration, level of hypoxia, changes in body weight,36 fasting time prior to metabolic assessments, and variation across species.37 The current study extends previous observations by demonstrating that, at the cellular level, 14 days of exposure to intermittent hypoxia can increase hepatocyte glucose output. Our findings also motivate the hypothesis that alterations in hepatic gluconeogenesis may have a central role in the pathogenesis of fasting hyperglycemia with intermittent hypoxia. In fact, the observed 50-fold increase in liver glycogen content in conjunction with enhanced expression of glucose-6 phosphatase and PEPCK argue for gluconeogenesis as a central mechanism for intermittent hypoxia-induced fasting hyperglycemia. It is certainly also possible that hepatic insulin resistance and increased glycogenolysis can further augment the effects of intermittent hypoxia on fasting glycemia. Candidate pathways that can mediate the stimulation of gluconeo-genesis with intermittent hypoxia include activation of the sympathetic nervous system, increased circulating corticosteroids or leptin, and enhanced activity of hypoxia-inducible factors that, in turn, promote transcription of multiple enzymes involved in gluconeogenesis. Although further work will be required to elucidate the exact cellular mechanisms enhancing gluconeogenesis with intermittent hypoxia, our results demonstrate that availability of oxygen is fundamental for regulating cellular glucose metabolism.
It is well established that hepatic glucose output modulates fasting glycemia, whereas glucose levels after an intraperitoneal glucose challenge depend largely on the sensitivity of muscle and fat tissue to insulin and the ability of beta cells to secrete insulin in sufficient quantity to normalize plasma glucose. Previous studies have demonstrated that short-term intermittent hypoxia worsens insulin sensitivity and decreases muscle glucose utilization.21,22,38 Even though these derangements are critical in altering glucose homeostasis, overt glucose intolerance and hyperglycemia ensue if, and only if, beta cells fail to secrete the necessary amount of insulin to compensate for a given level of whole-body insulin resistance. Normal oxygen levels are obligatory for glucose-induced insulin secretion which rapidly deteriorates under conditions of sustained hypoxia.39,40 Because beta cells also exhibit high sensitivity to reactive oxygen species (ROS)-induced damage,25,41 the repetitive reoxygenation and increased production of ROS that occurs during intermittent hypoxia provides an ideal milieu for accentuating hypoxia-related functional impairment in beta cells. Indeed, our observation of high TBARS levels in pancreatic lysates of mice exposed to 14 days of intermittent hypoxia provides evidence that ROS production is increased by intermittent hypoxia. Furthermore, the sustained elevation in lipid peroxidation in the pancreas despite cessation of hypoxic exposure indicates a component of irreversible injury. Given the strong biological plausibility that intermittent hypoxia can have detrimental effects on glucose homeostasis, it is not surprising that previous studies,33–36,42 while in marked contrast to some of the findings reported here, have similar inferences regarding the effect of intermittent hypoxia on glucose intolerance, insulin resistance, and impaired insulin secretion.
There are two clinically relevant implications of the current study. First, significant controversy exists as to whether obstructive sleep apnea can lead to the development of insulin resistance, glucose intolerance, and type 2 diabetes.15,16 The findings of the current study, together with previous experimental work, indicate that intermittent hypoxia in obstructive sleep apnea may initiate a cascade of events (i.e., insulin resistance, beta cell dysfunction) that increase the predisposition for hyperglycemic states including type 2 diabetes. Second, a highly controversial issue in the existing literature has been the lack of consistent improvements in glucose metabolism in human studies after the initiation of therapy for obstructive sleep apnea.15 Inconsistencies across previous reports might be attributed to numerous methodological limitations. The current study provides an alternative explanation that perhaps some of the metabolic derangements induced by intermittent hypoxia, such as beta cell dysfunction, may not recover even though the inciting exposure is no longer present.
The current study has several important limitations that merit discussion. First, because metabolic testing was conducted with continued exposure to intermittent hypoxia, it is certainly possible that the observed changes in glucose homeostasis represent an acute effect that is mediated by the hypoxic stimulus immediately preceding the metabolic assessments. Nonetheless, it is important to recognize that small but statistically significant differences were observed in glucose tolerance when comparing the 7-day and 14-day intermittent hypoxia exposure period at the 20- and 30-min time points. Thus, it is more likely that the alterations in metabolic function are due to effects that have accumulated over the entire exposure period. Second, given that the GTT and the ITT circumvent the gastrointestinal tract, little insight is gained into the probable role of specific gut-derived hormones (i.e., incretins) in mediating the metabolic effects of intermittent hypoxia. Despite the demonstration that radiolabeled insulin was similarly absorbed with intermittent hypoxia and the control condition (see supplemental material), evaluating incretin profiles would delineate additional pathways through which intermittent hypoxia and obstructive sleep apnea impair glucose metabolism. Third, while the ITT provides information on whole-body insulin sensitivity, it does not characterize impairments in insulin action occurring in adipose tissue and liver. Fourth, the potential for recovery was evaluated after the hypoxic exposure was discontinued for only 7 days. It is certainly plausible that some of the metabolic parameters may require a longer recovery period to fully revert to the normal state. Prior work using animal models on the effects of intermittent hypoxia on glucose metabolism is notable for a wide range of exposure periods from 9 h22 (short-term) to several weeks36,37 (long term). Based on these previous reports, the 7- and 14-day exposures used in the current protocol would be considered of modest length. The decision to use the 7- and 14-day exposure duration was motivated, in part, by previous work from our laboratory demonstrating that such exposure durations are sufficient to induce abnormalities in glucose metabolism. An argument could also be made for a longer exposure period. However, the primary objective of this investigation was to probe the question of whether metabolic changes induced by intermittent hypoxia are reversible. Thus, the experiments described herein used an exposure duration that has been known to be associated with metabolic impairment. Given the findings of potential irreversibility, the next logical step would be to examine whether there is additional resolution as the recovery period is incrementally increased. Finally, it is important to recognize that the C57BL6/J lean mice are highly susceptible to the development of diabetes because of an acquired mutation in the nicotinamide nucleotide transhydrogenase gene.43 Additional research is needed to determine the generalizability of our findings to older and obese animals, as well as to translate these observations to obstructive sleep apnea.
In summary, intermittent hypoxia induces all of the features typical for type 2 diabetes, including fasting hyperglycemia, increased hepatic glucose output, insulin resistance, and beta cell dysfunction. While fasting glycemia normalizes after the hypoxic exposure is discontinued, insulin resistance and beta cell dysfunction persist. A better understanding of the factors involved in the development, progression, and reversibility of metabolic impairments is needed to ascertain whether early identification and treatment of obstructive sleep apnea curtails metabolic risk in those without preexisting diabetes or improves glycemic control in those with established type 2 diabetes.
DISCLOSURE STATEMENT
This was not an industry supported study. Dr. Punjabi (Johns Hopkins University) has received research grant support from ResMed for a multicenter clinical trial on the effects of CPAP therapy on glycemic control in patients with type 2 diabetes and obstructive sleep apnea. The other authors have indicated no financial conflicts of interest.
ACKNOWLEDGMENTS
The findings reported in this manuscript were derived from research supported by a grant from the National Institutes of Health (HL075078). Author contributions: conception and design (JP, LAS, and NMP), acquisition of data (JP, LFD, CU, and HM), analysis and interpretation of data (JP, LAS, VYP, and NMP) and manuscript preparation (JP, LAS, LFD, VYP, and NMP).
SUPPLEMENTAL MATERIAL
Effects of Intermittent Hypoxia on Body Weight
Given that intermittent hypoxia is associated with weight loss, daily food intake in the control animals and during the recovery period was adjusted daily such that the body weight of mice in the control and recovery groups followed the same body weight trajectory as animals in the intermittent hypoxia group. Table S1 describes the weights for animals in each of the exposure groups.
Assessment for interaction between exposure group and exposure time (before-after) on body weight showed no significant interaction in insulin tolerance test (ITT) or glucose tolerance test (GTT) groups (by ANOVA, F = 0.64 df = 2 and F = 0.154 df = 4, both P > 0.05 for ITT and GTT groups, respectively) which confirms that weight matching as performed was successful and animals in all groups lost comparable amount of body weight.
Hepatocyte Isolation Protocol
Mice were anesthetized with intraperitoneal injection of sodium pentobarbital (35 mg/kg) and fixed on a surgical plate. Midline laparotomy was performed and the portal vein was observed. Cranial and caudal ligatures were placed around the portal vein prior to making a small incision in it. Caudal ligature was tied, and catheter was inserted into the portal vein facing the liver hilus and fixed with the cranial ligature. Subsequently, catheter was attached to a peristaltic pump, the right heart ventricle was incised, and the liver perfused for 25-30 min with the full volume of 100 mL of Ca2+ free HBSS (HEPES Buffered Saline Solution: 7.59 g NaCl + 0.377g KCl + 0.244g MgCl2 +1.8g Dextrose + 4.76 HEPES [4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid] in 1 L of distilled water, pH = 7.4) supplemented with 1 uM EGTA preheated to 37°C. Immediately after this perfusion, a second perfusion with 50 mL of preheated (37°C) HBSS supplemented 10 μM CaCl2 and 40 ug/mL Liberase TM Enzyme (Roche, Indianapolis, IN, USA) was performed. Following the second perfusion, the liver was removed and minced with scissors in a Petri dish with prechilled DMEM media. Subsequently, the content of the Petri dish was filtered through a cell strainer into a 50-mL conical tube and centrifuged at 4°C for 2 min at the speed equivalent to 60 g. After centrifuging, supernatant was removed, cells were resuspended in fresh DMEM media, and the centrifuging step was repeated twice more. Following the last centrifuging step, viable cells were counted using the trypan blue exclusion method (isolations with < 75% viable were discarded). Cells were plated at the density of 500.000 cells/well in 3 mL DMEM (high glucose) + 10% fetal bovine serum (FBS) media in a six-well plate (BD Falcon, Franklin Lakes, New Jersey, USA). Unplated cells were washed after 4 h with fresh media and the six-well plate was incubated for additional 16-18 h in identical medium.
Mouse body weight across different experimental groups
Insulin absorption from the peritoneal cavity. Absorption of insulin from the peritoneal cavity was evaluated by measuring plasma radioactivity (A) or total blood radioactivity (B) at 10, 30, and 60 min after intraperitoneal injection of ∼1μCi of I125-radiolabeled insulin (Perkin Elmer, Waltham, MA, USA) using Cobra II Gamma Counter (Perkin Elmer). These data provide evidence that absorption of insulin from peritoneal cavity is not modified by exposure and thus, observed differences in ITT tests between groups can be attributed to the true pharmacological effect of insulin (rather than to a slower or decreased insulin absorption).
Quality control of total extracted RNA by 18S and 28S RNA bands. Total RNA (1μg) extracted from liver was electrophoretically separated on the agarose gel and visualized with ethidium-bromide. IA1-5 (14-day intermittent air), IH1-5 (14-day intermittent hypoxia), Rec1-5 (recovery paradigm). Note: sample IA1 was run on a separate gel. This gel provides information on extracted RNA quality as required by MIQE guidelines (The MIQE guidelines: Minimum Information for Publication of Quantitative Real-time PCR Experiments. Clin Chem 2009;55:611-22).
Western blot analysis of PEPCK protein expression in liver lysates. IH1-5 (14-day intermittent hypoxia), IA1-5 (14-day intermittent air), R (recovery paradigm). These findings support gene expression data presented in the main manuscript and demonstrate that besides enhanced gene expression, intracellular protein levels of PEPCK are also increased with 14d-IH and return to control levels with cessation of hypoxia. A blot of PEPCK and a loading control protein are shown. ‡P < 0.05 when compared to 14-day intermittent air exposure. *P < 0.05 when compared to 14-day intermittent hypoxic exposure.
REFERENCES
- 1.Young T, Palta M, Dempsey J, Skatrud J, Weber S, Badr S. The occurrence of sleep-disordered breathing among middle-aged adults. N Engl J Med. 1993;328(17):1230–5. doi: 10.1056/NEJM199304293281704. [DOI] [PubMed] [Google Scholar]
- 2.Young T, Peppard PE, Gottlieb DJ. Epidemiology of obstructive sleep apnea: a population health perspective. Am J Respir Crit Care Med. 2002;165(9):1217–39. doi: 10.1164/rccm.2109080. [DOI] [PubMed] [Google Scholar]
- 3.Peppard PE, Young T, Palta M, Skatrud J. Prospective study of the association between sleep-disordered breathing and hypertension. N Engl J Med. 2000;342(19):1378–84. doi: 10.1056/NEJM200005113421901. [DOI] [PubMed] [Google Scholar]
- 4.Cano-Pumarega I, Duran-Cantolla J, Aizpuru F, et al. Obstructive sleep apnea and systemic hypertension: longitudinal study in the general population: the Vitoria Sleep Cohort. Am J Respir Crit Care Med. 2011;184(11):1299–304. doi: 10.1164/rccm.201101-0130OC. [DOI] [PubMed] [Google Scholar]
- 5.Marin JM, Agusti A, Villar I, et al. Association between treated and untreated obstructive sleep apnea and risk of hypertension. JAMA. 2012;307(20):2169–76. doi: 10.1001/jama.2012.3418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gottlieb DJ, Yenokyan G, Newman AB, et al. Prospective study of obstructive sleep apnea and incident coronary heart disease and heart failure: the sleep heart health study. Circulation. 2010;122(4):352–60. doi: 10.1161/CIRCULATIONAHA.109.901801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yeboah J, Redline S, Johnson C, et al. Association between sleep apnea, snoring, incident cardiovascular events and all-cause mortality in an adult population: MESA. Atherosclerosis. 2011;219(2):963–8. doi: 10.1016/j.atherosclerosis.2011.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arzt M, Young T, Finn L, Skatrud JB, Bradley TD. Association of sleep-disordered breathing and the occurrence of stroke. Am J Respir Crit Care Med. 2005;172(11):1447–51. doi: 10.1164/rccm.200505-702OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Redline S, Yenokyan G, Gottlieb DJ, et al. Obstructive sleep apneahypopnea and incident stroke: the sleep heart health study. Am J Respir Crit Care Med. 2010;182(2):269–77. doi: 10.1164/rccm.200911-1746OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yaggi HK, Concato J, Kernan WN, Lichtman JH, Brass LM, Mohsenin V. Obstructive sleep apnea as a risk factor for stroke and death. N Engl J Med. 2005;353(19):2034–41. doi: 10.1056/NEJMoa043104. [DOI] [PubMed] [Google Scholar]
- 11.Marshall NS, Wong KK, Liu PY, Cullen SR, Knuiman MW, Grunstein RR. Sleep apnea as an independent risk factor for all-cause mortality: the Busselton Health Study. Sleep. 2008;31(8):1079–85. [PMC free article] [PubMed] [Google Scholar]
- 12.Young T, Finn L, Peppard PE, et al. Sleep disordered breathing and mortality: eighteen-year follow-up of the Wisconsin sleep cohort. Sleep. 2008;31(8):1071–8. [PMC free article] [PubMed] [Google Scholar]
- 13.Punjabi NM, Caffo BS, Goodwin JL, et al. Sleep-disordered breathing and mortality: a prospective cohort study. PLoS Med. 2009;6(8):e1000132. doi: 10.1371/journal.pmed.1000132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marshall NS, Delling L, Grunstein RR, et al. Self-reported sleep apnoea and mortality in patients from the Swedish Obese Subjects study. Eur Respir J. 2011;38(6):1349–54. doi: 10.1183/09031936.00022111. [DOI] [PubMed] [Google Scholar]
- 15.Punjabi NM. Do sleep disorders and associated treatments impact glucose metabolism? Drugs. 2009;69(Suppl 2):13–27. doi: 10.2165/11531150-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 16.Tasali E, Mokhlesi B, Van Cauter E. Obstructive sleep apnea and type 2 diabetes: interacting epidemics. Chest. 2008;133(2):496–506. doi: 10.1378/chest.07-0828. [DOI] [PubMed] [Google Scholar]
- 17.Strohl KP, Novak RD, Singer W, et al. Insulin levels, blood pressure and sleep apnea. Sleep. 1994;17(7):614–8. doi: 10.1093/sleep/17.7.614. [DOI] [PubMed] [Google Scholar]
- 18.Ip MS, Lam B, Ng MM, Lam WK, Tsang KW, Lam KS. Obstructive sleep apnea is independently associated with insulin resistance. Am J Respir Crit Care Med. 2002;165(5):670–6. doi: 10.1164/ajrccm.165.5.2103001. [DOI] [PubMed] [Google Scholar]
- 19.Punjabi NM, Sorkin JD, Katzel LI, Goldberg AP, Schwartz AR, Smith PL. Sleep-disordered breathing and insulin resistance in middle-aged and overweight men. Am J Respir Crit Care Med. 2002;165(5):677–82. doi: 10.1164/ajrccm.165.5.2104087. [DOI] [PubMed] [Google Scholar]
- 20.Punjabi NM, Shahar E, Redline S, Gottlieb DJ, Givelber R, Resnick HE. Sleep-disordered breathing, glucose intolerance, and insulin resistance: the Sleep Heart Health Study. Am J Epidemiol. 2004;160(6):521–30. doi: 10.1093/aje/kwh261. [DOI] [PubMed] [Google Scholar]
- 21.Polotsky VY, Li J, Punjabi NM, et al. Intermittent hypoxia increases insulin resistance in genetically obese mice. J Physiol. 2003;552(Pt 1):253–64. doi: 10.1113/jphysiol.2003.048173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iiyori N, Alonso LC, Li J, et al. Intermittent hypoxia causes insulin resistance in lean mice independent of autonomic activity. Am J Respir Crit Care Med. 2007;175(8):851–7. doi: 10.1164/rccm.200610-1527OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O'Donnell CP. Metabolic consequences of intermittent hypoxia. Adv Exp Med Biol. 2007;618:41–9. doi: 10.1007/978-0-387-75434-5_4. [DOI] [PubMed] [Google Scholar]
- 24.Yokoe T, Alonso LC, Romano LC, et al. Intermittent hypoxia reverses the diurnal glucose rhythm and causes pancreatic beta-cell replication in mice. J Physiol. 2008;586(3):899–911. doi: 10.1113/jphysiol.2007.143586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu J, Long YS, Gozal D, Epstein PN. Beta-cell death and proliferation after intermittent hypoxia: role of oxidative stress. Free Radic Biol Med. 2009;46(6):783–90. doi: 10.1016/j.freeradbiomed.2008.11.026. [DOI] [PubMed] [Google Scholar]
- 26.Drager LF, Li J, Reinke C, Bevans-Fonti S, Jun JC, Polotsky VY. Intermittent hypoxia exacerbates metabolic effects of diet-induced obesity. Obesity (Silver Spring) 2011;19(11):2167–74. doi: 10.1038/oby.2011.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Louis M, Punjabi NM. Effects of acute intermittent hypoxia on glucose metabolism in awake healthy volunteers. J Appl Physiol. 2009;106(5):1538–44. doi: 10.1152/japplphysiol.91523.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bonadonna RC, De Fronzo RA. Glucose metabolism in obesity and type 2 diabetes. Diabetes Metab. 1991;17(1 Pt 2):112–35. [PubMed] [Google Scholar]
- 29.Zierler K. Whole body glucose metabolism. Am J Physiol. 1999;276(3 Pt 1):E409–26. doi: 10.1152/ajpendo.1999.276.3.E409. [DOI] [PubMed] [Google Scholar]
- 30.Tagaito Y, Polotsky VY, Campen MJ, et al. A model of sleep-disordered breathing in the C57BL/6J mouse. J Appl Physiol. 2001;91(6):2758–66. doi: 10.1152/jappl.2001.91.6.2758. [DOI] [PubMed] [Google Scholar]
- 31.Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28(7):412–9. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- 32.Wahren J, Ekberg K. Splanchnic regulation of glucose production. Annu Rev Nutr. 2007;27:329–45. doi: 10.1146/annurev.nutr.27.061406.093806. [DOI] [PubMed] [Google Scholar]
- 33.Chiu LL, Chou SW, Cho YM, et al. Effect of prolonged intermittent hypoxia and exercise training on glucose tolerance and muscle GLUT4 protein expression in rats. J Biomed Sci. 2004;11(6):838–46. doi: 10.1007/BF02254369. [DOI] [PubMed] [Google Scholar]
- 34.Ling Q, Sailan W, Ran J, et al. The effect of intermittent hypoxia on bodyweight, serum glucose and cholesterol in obesity mice. Pak J Biol Sci. 2008;11(6):869–75. doi: 10.3923/pjbs.2008.869.875. [DOI] [PubMed] [Google Scholar]
- 35.Chen CY, Tsai YL, Kao CL, et al. Effect of mild intermittent hypoxia on glucose tolerance, muscle morphology and AMPK-PGC-1alpha signaling. Chin J Physiol. 2010;53(1):62–71. doi: 10.4077/cjp.2010.amk078. [DOI] [PubMed] [Google Scholar]
- 36.Carreras A, Kayali F, Zhang J, Hirotsu C, Wang Y, Gozal D. Metabolic effects of intermittent hypoxia in mice: steady versus high-frequency applied hypoxia daily during the rest period. Am J Physiol Regul Integr Comp Physiol. 2012;303(7):R700-9. doi: 10.1152/ajpregu.00258.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fenik VB, Singletary T, Branconi JL, Davies RO, Kubin L. Glucoregulatory consequences and cardiorespiratory parameters in rats exposed to chronic-intermittent hypoxia: effects of the duration of exposure and losartan. Front Neurol. 2012;3:51. doi: 10.3389/fneur.2012.00051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li J, Bosch-Marce M, Nanayakkara A, et al. Altered metabolic responses to intermittent hypoxia in mice with partial deficiency of hypoxiainducible factor-1alpha. Physiol Genomics. 2006;25(3):450–7. doi: 10.1152/physiolgenomics.00293.2005. [DOI] [PubMed] [Google Scholar]
- 39.Baum D, Griepp R, Porte D., Jr Glucose-induced insulin release during acute and chronic hypoxia. Am J Physiol. 1979;237(1):E45-50. doi: 10.1152/ajpendo.1979.237.1.E45. [DOI] [PubMed] [Google Scholar]
- 40.Dionne KE, Colton CK, Yarmush ML. Effect of hypoxia on insulin secretion by isolated rat and canine islets of Langerhans. Diabetes. 1993;42(1):12–21. doi: 10.2337/diab.42.1.12. [DOI] [PubMed] [Google Scholar]
- 41.Grankvist K, Marklund SL, Taljedal IB. CuZn-superoxide dismutase, Mnsuperoxide dismutase, catalase and glutathione peroxidase in pancreatic islets and other tissues in the mouse. Biochem J. 1981;199(2):393–8. doi: 10.1042/bj1990393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee EJ, Alonso LC, Stefanovski D, et al. Time-dependent changes in glucose and insulin regulation during intermittent hypoxia and continuous hypoxia. Eur J Appl Physiol. 2013;113(2):467–78. doi: 10.1007/s00421-012-2452-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Toye AA, Lippiat JD, Proks P, et al. A genetic and physiological study of impaired glucose homeostasis control in C57BL/6J mice. Diabetologia. 2005;48(4):675–86. doi: 10.1007/s00125-005-1680-z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Mouse body weight across different experimental groups
Insulin absorption from the peritoneal cavity. Absorption of insulin from the peritoneal cavity was evaluated by measuring plasma radioactivity (A) or total blood radioactivity (B) at 10, 30, and 60 min after intraperitoneal injection of ∼1μCi of I125-radiolabeled insulin (Perkin Elmer, Waltham, MA, USA) using Cobra II Gamma Counter (Perkin Elmer). These data provide evidence that absorption of insulin from peritoneal cavity is not modified by exposure and thus, observed differences in ITT tests between groups can be attributed to the true pharmacological effect of insulin (rather than to a slower or decreased insulin absorption).
Quality control of total extracted RNA by 18S and 28S RNA bands. Total RNA (1μg) extracted from liver was electrophoretically separated on the agarose gel and visualized with ethidium-bromide. IA1-5 (14-day intermittent air), IH1-5 (14-day intermittent hypoxia), Rec1-5 (recovery paradigm). Note: sample IA1 was run on a separate gel. This gel provides information on extracted RNA quality as required by MIQE guidelines (The MIQE guidelines: Minimum Information for Publication of Quantitative Real-time PCR Experiments. Clin Chem 2009;55:611-22).
Western blot analysis of PEPCK protein expression in liver lysates. IH1-5 (14-day intermittent hypoxia), IA1-5 (14-day intermittent air), R (recovery paradigm). These findings support gene expression data presented in the main manuscript and demonstrate that besides enhanced gene expression, intracellular protein levels of PEPCK are also increased with 14d-IH and return to control levels with cessation of hypoxia. A blot of PEPCK and a loading control protein are shown. ‡P < 0.05 when compared to 14-day intermittent air exposure. *P < 0.05 when compared to 14-day intermittent hypoxic exposure.