

Abstract
A robust, high-throughput method has been developed to screen one-bead-one-compound peptide libraries to systematically profile the sequence specificity of protein kinases. Its ability to provide individual sequences of the preferred substrates permits the identification of sequence contextual effects and non-permissive residues. Application of the library method to kinases Pim1, MKK6, and Csk revealed that Pim1 and Csk are highly active toward peptide substrates and recognize specific sequence motifs, whereas MKK6 has little activity or sequence selectivity against peptide substrates. Pim1 recognizes peptide substrates of the consensus RXR(H/R)X(S/T); it accepts essentially any amino acid at the S/T-2 and S/T+1 positions, but strongly disfavors acidic residues (Asp or Glu) at the S/T-2 position and a proline residue at the S/T+1 position. The selected Csk substrates show strong sequence covariance and fall into two classes with the consensus sequences of (D/E)EPIYφXφ and (D/E)(E/D)S(E/D/I)YφXφ (where X is any amino acid and φ is a hydrophobic amino acid). Database searches and in vitro kinase assays identified phosphatase PTP-PEST as a Pim1 substrate and phosphatase SHP-1 as a potential Csk substrate. Our results demonstrate that the sequence specificity of protein kinases is defined not only by favorable interactions between permissive residue(s) on the substrate and their cognate binding site(s) on the kinase, but also by repulsive interactions between the kinase and non-permissive residue(s).
Approximately 30% of all mammalian proteins are phosphorylated during some point of their life time; more than 100000 phosphoserine (pS), phosphothreonine (pT), and phosphotyrosine (pY) sites have been identified by high- and low-throughput methods.1 In human, the phosphorylation events are carried out by 518 putative protein kinases.2 The large number of kinases and potential protein substrates necessitates tight control of the kinase activity to ensure fidelity of the phosphorylation events with respect to both the kinase and the substrate protein. It is now established that protein kinases utilize a number of mechanisms to differentiate their specific substrates from a large pool of other proteins. These include temporal expression of the kinase and/or substrate, localization of the kinase and/or substrate to subcellular structures, protein-protein interaction through the use of recruiting domains/surfaces or scaffolding proteins, and interactions between the kinase active site and the linear sequence motif surrounding the phosphorylatable residue (or the intrinsic sequence specificity of the kinase domain).3,4
For some protein kinases (e.g., protein kinase A), the intrinsic sequence specificity of the kinase domain is the major determinant of their in vivo substrate specificity.3,4 Several methods have been developed to determine the sequence specificity of protein kinases and use the specificity profiles to predict their physiological substrates. Cantley and coworkers pioneered the use of oriented peptide libraries to profile kinase substrate specificity.5,6 Their method involved treatment of a combinatorial peptide library with a kinase of interest in solution, separation of the phosphorylated peptides by metal affinity chromatography, and Edman sequencing of the enriched peptide pool. A modification of this method (or positional scanning peptide libraries) was later introduced and used to define the sequence specificity of 61 yeast kinases.7–9 This method employs a series of 198 sublibraries, each of which contains a fixed amino acid at one position and random residues at all other positions. After kinase treatment in solution with 32P-labeled ATP, the peptides (which are all tagged with a C-terminal biotin during library synthesis) were captured by streptavidin-coated nitrocellulose paper and the amount of phosphorylation was determined by the radioactivity incorporated. These methods have been very useful for obtaining an overall preference of amino acids at a given position. A limitation of the above methods is that they do not provide individual sequences and therefore cannot detect any sequence contextual effect. In fact, data derived from positional scanning peptide libraries have led to the proposal that protein kinases do not exhibit sequence contextual effects.10 On the other hand, our studies of protein binding domains11–13 and protein phosphatases14,15 have demonstrated that it is not uncommon for the same active/binding site of a protein to recognize two or more different types of consensus sequences. There is a need for more robust library screening methods to reevaluate the contribution of sequence covariance to kinase specificity. Very recently, several investigators reported the use of peptide libraries derived from proteolytic digestion of bacterial or mammalian proteomes and identification of phosphorylated peptides by LC-MS/MS-based proteomics methods.16–18 These MS-based methods provide individual sequences and can potentially reveal sequence contextual effects. Their main drawback is that the peptide library has limited sequence space (especially when a bacterial proteome is used) and is biased towards abundant proteins. In addition, they are incompatible with posttranslationally modified amino acids (e.g., pS, pT, or pY) and false positives may result if a peptide is phosphorylated in the proteome but incompletely dephosphorylated by the in vitro phosphatase treatment. Finally, other investigators have attempted on-bead screening of one-bead-one-compound (OBOC) peptide libraries against protein kinases. Lam and co-workers treated OBOC peptide libraries with 32P-labeled ATP, isolated the resulting radioactive beads by autoradiography, and identified the kinase substrates by Edman sequencing.19 This method has not been widely used because isolation of positive beads by autoradiography and Edman sequencing are inconvenient and time-consuming. Lee et al. used anti-pY antibodies for on-bead detection of protein tyrosine kinase products;20 for protein serine/threonine kinases, they employed base-mediated β-elimination of the pS/pT residue and conjugate addition of a biotin tag to the resulting dehydroalanine residue.21 Unfortunately, binding of anti-pY antibodies to pY peptides exhibit substantial sequence selectivity of their own,22 whereas base treatment also causes β-elimination of Ser and Thr side chains resulting in false positives (Lee et al. excluded Ser and Thr from their libraries). Here we report a simple, robust method for on-bead screening of OBOC peptide libraries against protein serine/threonine as well as tyrosine kinases and its application to determine the specificity profiles of Pim1 kinase, mitogen-activated protein kinase kinase 6 (MKK6), and C-terminal Src kinase (Csk). Our study shows that protein kinases do exhibit sequence contextual effects and their substrate specificity is defined not only by the preferred residues, but also by the presence of non-permissive residues. Additionally, the specificity data allowed us to identify several new protein substrates of the kinases.
MATERIALS AND METHODS
Materials
Fmoc-protected L-amino acids were purchased from Advanced ChemTech (Louisville, KY), Peptides International (Louisville, KY), or Aapptec (Louisville, KY). O-Benzotriazole-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HBTU) and 1-hydroxybenzotriazole hydrate (HOBt) were from Aapptec. [γ-S]ATP and anti-Tyr(P) antibody (clone 4G10) were purchased from EMD Millipore (Billerica, MA). Pyruvate kinase type II, lactate dehydrogenase type II and HRV3C protease were purchased from Sigma-Aldrich (St. Louis, MO). Carboxytetramethylrhodamine succinimidyl ester (Rhodamine-NHS) was purchased from Pierce (Rockford, IL). All solvents and other chemical reagents were obtained from Sigma-Aldrich, Fisher Scientific (Pittsburgh, PA), or VWR (West Chester, PA) and were used without further purification unless noted otherwise. N-(9-Fluorenylmethoxycarbonyloxy)succinimide (Fmoc-OSu) was from Advanced ChemTech. Phenyl isothiocyanate (PITC) was purchased in 1-mL sealed ampoules from Sigma-Aldrich, and a freshly opened ampoule was used in each experiment. Six-histidine-tagged SCAND1 and CBX1 were purchased from ProSpec (East Brunswick, NJ).
Purification of WT and Mutant GST-Csk
Escherichia coli DH5α cells harboring prokaryotic expression vector pGEX-3x containing GST-tagged WT or mutant human Csk (kindly provided by P. A. Cole of Johns Hopkins University) were grown in LB medium containing ampicillin (75 μg/ml) at 37 °C until the OD600 reached ~0.6. Protein expression was induced by the addition of 0.1 mM of isopropyl-thio-β-D-galactoside (IPTG) for 4 h at 30 °C. Cells were harvested by centrifugation at 5000 rpm, 4 °C for 30 min, and the cell pellet was suspended in GST binding buffer (20 mM HEPES, pH 8.0, 150 mM NaCl, and 1 mM β-mercaptoethanol) and lysed by sonication in the presence of protease inhibitors (2 mM benzamidine, 1 mM phenylmethanesulfonyl fluoride (PMSF), 2 μg/mL pepstatin, 2 μg/mL leupeptin, 100 μg/mL soybean trypsin inhibitor). The lysate was centrifuged at 15000 rpm, 4 °C for 30 min and clear supernatant was loaded onto a glutathione-Sepharose column. After incubation at 4 °C for 30 min, the column was washed with 150 mL of GST binding buffer, and GST-Csk was eluted with GST elution buffer (20 mM HEPES, pH 8.0, 150 mM NaCl, 10 mM reduced glutathione, and 1 mM β-mercaptoethanol). The resulting GST-Csk solution was concentrated, mixed with an equal volume of 60% glycerol, and flash frozen and stored at −80 °C. The protein concentration was determined by Bradford assay, using bovine albumin serum as the standard.
Purification of GST-MKK6
Expression vector pGEX-2T containing GST-tagged human S207E/T211E MKK6 (kindly provided by D. Maly of University of Washington) was used to transform E. coli BL21(DE3) CodonPlus cells. The S207E and T211E mutations result in constitutively activated MKK6. The recombinant cells were grown in LB medium containing ampicillin (75 μg/mL) at 37 °C until the OD600 reached ~0.3. The culture was cooled to 20 °C and allowed to grow until OD600 reached ~0.8. Protein expression was induced by the addition of 0.2 mM IPTG and the cells were incubated overnight at 20 °C. The cells were harvested and GST-MKK6 was purified as described for GST-Csk.
Purification of Pim1
E. coli BL21 (DE3) CodonPlus cells harboring plasmid pLIC-SGC containing the human Pim1 gene and a N-terminal six-histidine tag (kindly provided by D. Maly of University of Washington) were grown in LB medium containing ampicillin (75 μg/ml) at 37 °C until OD600 reached ~0.5. The incubator temperature was adjusted to 30 °C and the cells were grown until OD600 reached ~0.8. Protein expression was induced by the addition of 0.1 mM IPTG for 4 h at 18 °C. The cells were harvested by centrifugation at 5000 rpm, 4 °C for 30 min and suspended in His-tag binding buffer (50 mM HEPES, pH 7.5, 500 mM NaCl, and 5% glycerol) and lysed by sonication in the presence of the protease inhibitor cocktail The crude lysate was centrifuged at 15000 rpm, 4 °C for 30 min and the clear supernatant was loaded onto a Nickel-NTA column. After 30 min incubation at 4 °C, the column was washed with 150 mL of His-tag wash buffer (50 mM HEPES, pH 7.5, 500 mM NaCl, 10 mM imidazole, 5% glycerol). The bound Pim1 protein was eluted with His-tag elution buffer (50 mM HEPES, pH 7.5, 500 mM NaCl, 150 mM imidazole, 5% glycerol) and concentrated to ~3 mg/mL. Dephosphorylation of Pim1 was carried out by incubation with 200 units (20 μL) of calf intestinal alkaline phosphatase overnight at 4 °C. The reaction mixture was loaded onto a Q-Sepharose column and washed with 10 mL of 50 mM HEPES, pH 7.5. The column was eluted with a linear gradient of 0–1 M NaCl in 50 mM HEPES, pH 7.5. Pim1 and phosphorylated Pim1 were separated, concentrated, flash frozen and stored at −80 °C.
Purification of PTP-PEST and S39A Mutant
Expression vector pGEX-6P-2 containing GST-tagged catalytic domain of human PTP-PEST (amino acids 5–300) was used to transform E. coli BL21(DE3) CodonPlus cells. Expression and purification of GST-PTP-PEST was carried out as previously described.23 GST-PTP-PEST S39A mutant was generated by QuikChange mutagenesis using 5′–CATGCGGTTAAGAAGATTGGCTACCAAATATAGAACAG as the primer. Expression and purification of GST-PTP-PEST S39A were carried out in the same manner as that of the wild type protein.48 Cleavage of the GST tag from GST-PTP-PEST was carried out by incubating the fusion protein with HRV3C protease (50:1 w/w protein/protease) overnight at 4 °C. Briefly, 100 μg of GST-PTP-PEST was incubated with 2 μg of HRV3C protease in 40 mM HEPES, pH 7.4, 120 mM NaCl, 2 mM DTT for 16 h at 4 °C with gentle agitation. The reaction mixture was used in kinase assays without further purification.
Synthesis of Peptide Libraries
Library I (X5ZX4NNBBRM-resin) was synthesized on 2 g of amino polyethylene glycol polyacrylamide (PEGA) resin (0.2 mmol/g, 300–500 μm in water). All manipulations were performed at room temperature unless otherwise noted. The invariant positions (NNBBRM) were synthesized with 4 equivalents of Fmoc-amino acids using HBTU/HOBt/N-methylmorpholine (NMM) as the coupling reagents, and the coupling was terminated after negative ninhydrin test. For the synthesis of the random positions, the resin was split into 20 equal portions and each was coupled with 5 equiv of a different Fmoc-amino acid/HBTU/HOBt/NMM for 1.5 h. To facilitate sequence determination by mass spectrometry, 5% (mol/mol) of CD3CO2D was added to the coupling reactions of Lys and Leu, whereas 5% CH3CD2CO2D was added to the coupling reaction of Nle.24 The resin bound library was washed with dichloromethane (DCM) and deprotected using modified reagent K (7.5% phenol, 5% water, 5% thioanisole, 2.5% ethanedithiol, 2.5% triisopropylsilane, 1.25% anisole in trifluoroacetic acid (TFA)) for 2 h. The library was washed extensively with TFA, DCM and DMF and stored in DMF at −20 °C until use. Library II (X1–5 YX6–12 YNBNBRM-resin) was similarly synthesized but with the following modification. After the synthesis of the 7 random positions, the resin was treated with 20% piperidine in DMF to remove the N-terminal Fmoc group, and the exposed N-terminal amine was capped with a 6:4 (mol/mol) mixture of Alloc-OSu and Fmoc-OSu. After removal of the Fmoc group, Fmoc-Tyr was coupled to the resin with HBTU/HOBt/NMM. The 5 partially randomized N-terminal residues were added in the same manner except that the resin was split into 3 or 4 equal portions according to the number of amino acids were used at each of the X1–5 positions.
Synthesis of Rhodmaine Label (Scheme 1)
Scheme 1.

Synthesis of tetramethylrhodamine labeling agent.
To a solution of rhodamine-NHS (10.5 mg, 0.02 mmol) in 500 μL DMF was added N-Boc-ethylenediamine (3.33 μL, 0.0202 mmol). The mixture was stirred overnight at room temperature and the solvent was removed by vacuum. TFA was added to the residue and the reaction mixture was stirred for 2 h at room temperature to yield rhodamine-ethyldiamine (TNR-NH2). After removal of TFA under reduced pressure, the amine was dissolved in 400 μL of anhydrous DMSO to give a 50 mM stock solution. To 78. μL of this stock solution was added N-succinimidyl 3-[2-pyridyldithio]-propionate (SPDP) (1.2 mg, 0.00384 mmol) and triethylamine (0.53 μL) and the reaction was allowed to proceed at room temperature, in the dark, for 2 h to yield 3-[2-pyridyldithio]propionyl rhodamine (TMR-S-S-Py). The crude reaction product was stored at −20 °C and used for library screening without further purification.
Library Screening
In a typical screening reaction, 100–200 mg of resin (100000–200000 beads) was placed in a plastic micro-BioSpin column (2 mL, Bio-Rad) and washed extensively with DMF, ddH2O and screening buffer. The resin was then transferred to a microcentrifuge tube using buffer and incubated with the kinase of interest. For Csk, the kinase reaction contained 3 μM Csk and 2 mM [γ-S]ATP in 60 mM Tris, pH 7.4, 2 mM MnCl2, 5 or 150 mM NaCl, 2 mM dithiothreitol (DTT), and 0.02% Tween-20. The reaction mixture was incubated for 20–24 h at 30 °C with mixing on a rotary shaker (200 rpm). The Pim1 screening reaction contained 3 μM Pim1 (or phosphorylated Pim1) and 2 mM [γ-S]ATP in 30 mM HEPES, pH 7.4, 30 mM MgCl2, 2 mM DTT, and 0.02% Tween-20 and was incubated for 6 h at 30 °C. For MKK6, the screening reaction contained 20 μM MKK6 and 2 mM [γ-S]ATP in 20 mM Tris, pH 7.4, 30 mM NaCl, 20 mM MgCl2, 1 mM EGTA, and 1 mM DTT and was incubated for 48 h at 30 °C. The kinase reaction was terminated by washing the resin extensively with screening buffer, water and 4 M guanidine-HCl (pH 7). The resin was incubated in 4 M guanidine-HCl for 45 min and washed extensively with water and labeling buffer [1:1 (v/v) 50 mM HEPES (pH 7.4) and N-methylpyrrolidone (NMP)]. The resin was then treated with 0.1 μM TMR-NH2 in labeling buffer (1.5 mL) for 1 h in the dark, followed by extensive washing with labeling buffer, DMF, water and selection solution (50 mM NaCl and 0.02% Tween-20 in water). The resin was transferred into a petri dish using the selection solution and fluorescent beads (false positives caused by nonspecific binding of dye) were isolated with a micropipette under a fluorescent microscope. The remaining non-fluorescent beads were transferred back the micro-BioSpin column and treated with 0.1 μM TMR-S-S-Py in labeling buffer (1.5 mL) for 1 h in the dark. The resin was washed, transferred to a petri dish, and fluorescent beads were isolated and transferred to another petri dish containing 50 mM tris(2-carboxyethyl)phosphine (TCEP) in selection solution. Beads that lost their fluorescence signals in the presence of TCEP were selected and sequenced by the partial Edman degradation-mass spectrometry (PED-MS) method.24
Synthesis of Selected Peptides
Individual peptides were synthesized on 100 mg of CLEAR-amide resin using standard Fmoc/HBTU/HOBt chemistry. Cleavage and deprotection of resin-bound peptides were carried out using modified reagent K at room temperature for 2 h. After evaporation of solvents, the mixture was triturated three times with 20 volumes of cold Et2O. The precipitate was collected and dried under vacuum. The crude peptides were purified by reversed-phase HPLC on a semi-preparative C18 column. The identity of each peptide was confirmed by MALDI-TOF mass spectrometric analysis.
Enzyme Coupled Kinase Assay
Csk and Pim1 activities against peptide substrates were determined using a previously described coupled assay.25 A typical reaction (total volume 150 μL) at room temperature contained 100 mM Tris, pH 7.4, 5 mM MnCl2 (or 20 mM MgCl2 for Pim1), 5 mM NaCl, 5 mM KCl, 10 mM DTT, 1 mM ATP, 1 mM phosphoenolpyruvate, 0.2 mM NADH, 5.5 units of pyruvate kinase, 4.5 units of lactate dehydrogenase, and 0–2400 μM peptide substrate. The reaction was initiated by the addition of the kinase (1–3 μM final concentration for GST-CSK or 40–1200 nM for im1) and monitored continuously on a UV-Vis spectrophotometer at 340 nm. The initial rates were calculated from the early regions of the reaction progress curves (<2 min) and fitted against the Michaelis-Menten equation V = Vmax • [S]/(KM + [S]) or the simplified equation V = kcat[E][S]/KM (when KM ≫ [S]) to give the kinetic constants kcat, KM, and/or kcat/KM.
Kinase Assay by HPLC
The MKK6 activity toward peptide substrates were too low to be reliably determined by the enzyme coupled assay. For HPLC-based assay, a typical reaction (total volume of 0.1 mL) contained 20 mM Tris, pH 7.5, 30 mM NaCl, 20 mM MgCl2, 1 mM EGTA, 1 mM DTT, 1 mM ATP, and 100–500 μM peptide substrate. The reaction was initiated by the addition of MKK6 (final concentration 10 μM) and allowed to proceed at room temperature for 40 h. The reaction was quenched by mixing with an equal volume of 0.05% TFA in water and the mixture was analyzed by reversed-phase HPLC using a C18 analytical column. The identities of the substrate and product were confirmed by MALDI-TOF mass spectrometry. The percentage of substrate-to-product conversion and initial velocity were determined by integration of the areas underneath the reactant and product peaks. Data fitting against the simplified Michaelis-Menten equation V = kcat[E][S]/KM (when KM ≫ [S]) gave the constant kcat/KM. For each substrate, a control reaction was carried out under the same conditions except that no ATP was added.
[γ-32P]ATP Assay with Protein Substrates
Phosphorylation of protein substrates by Pim1 were carried out using 100–300 nM Pim1 and 1–2 μM protein substrates. A typical reaction (total volume of 20 μl) contained 100 mM Tris, pH 7.4, 50 mM NaCl, 20 mM MgCl2, 1 mM DTT, 1 mM ATP (including 0.1 μCi/μl [γ-32P]ATP). The reaction was allowed to proceed at room temperature for 2 h and terminated by the addition of 20 μL of 2x SDS-PAGE loading buffer, followed by heating at 100 °C for 10 min. Phosphorylated proteins were separated by SDS-PAGE and visualized by phosphorimaging on a GE Typhoon Trio imager.
Kinase Assay by Western Blotting
SHP-1 (and mutants) phosphorylation by GST-Csk was carried out using 4 μM GST-Csk and 20–25 μM SHP-1. The reaction (total volume of 30 μL) contained 100 mM Tris, pH 7.4, 10 mM NaCl, 2 mM MnCl2, 1 mM DTT and 1 mM ATP. The reaction was allowed to proceed at room temperature and 15-μL aliquots were withdrawn and quenched at 30 min with 15 μL of 2x SDS-PAGE loading buffer. After heating for 10 min at 100 °C, the proteins were separated by SDS-PAGE, transferred to a nitrocellulose membrane and immunoblotted with anti-Tyr(P) antibody 4G10. The membrane was washed with 10 mM Tris, pH 7.4, 100 mM NaCl, 0.1% Tween-20 and signal development was carried out following the manufacturer’s recommendations.
RESULTS AND DISCUSSION
Peptide Library Design, Synthesis, and Screening
For a protein kinase of unknown specificity, we typically begin the specificity profiling with a generic kinase substrate library in the form of X5ZX4NNBBRM-resin (library I), where B is β-alanine, Z is Ser, Thr, and/or Tyr, and X is any of the 19 proteinogenic amino acids except for methionine [replaced by L-norleucine (Nle or M)] and cysteine. The inclusion of a fixed Ser, Thr, and/or Tyr ensures that each peptide contains at least one phosphorylatable residue, although phosphorylation may also take place at any of the randomized positions. The linker sequence, NNBBRM, permits selective peptide release (cleavage after Met by CNBr) and facilitates peptide sequencing by PED-MS (Arg provides a fixed positive charge and improves aqueous solubility).24 This library has a theoretical diversity of 199 or 2.6 × 1011, but the actual number of peptide sequences is limited by the amount of resin used. Library I was synthesized on 2 g of PEGA resin (300–500 μm in water, ~1 million beads/g) in the OBOC format and thus contained ~2 million different sequences. In the resulting library, each bead carried ~200 pmol of a unique peptide sequence. Our previous studies with protein-binding domains and other enzymes have shown that the specificity profile of a protein domain or enzyme can be unambiguously determined by sampling just a small fraction of the entire sequence space.11–15 It should be noted that the choice of PEGA or other resins that are permeable to relatively large proteins is crucial for successful on-bead screening reactions.26
Library screening involved treating a portion of the peptide library with a kinase of interest for a limited amount of time, so that only beads carrying the most efficient kinase substrates underwent partial phosphorylation (usually a few percent) while the rest of the beads had no to little reaction. To facilitate the identification of the phosphorylated (positive) beads, we performed the kinase reaction in the presence of adenosine 5′-O-(3-thio)triphosphate ([γ-S]ATP) instead of ATP, resulting in the transfer of a thiophosphoryl group to the positive beads (Figure 1A). Subsequently, a disulfide exchange reaction with a pyridyldithio-containing tetramethylrhodamine derivative (TMR-S-S-Py) resulted in covalent attachment of the rhodamine dye to the thiophosphorylated beads. The fluorescent beads (Figure 1B) were manually isolated from the library with a micropipette under a fluorescence microscope and individually sequenced by the PED-MS method.24 Most protein kinases accept [γ-S]ATP as substrate although substitution of [γ-S]ATP for ATP decreases the catalytic activity of kinases by 15–30 fold.27,28 Since the disulfide exchange reaction is specific and essentially quantitative, the fluorescence intensity on a positive bead directly reflects the amount of (thio)phosphorylation on that bead. The positive beads may be separated into “intensely colored” and “lightly colored” categories, corresponding to the most active and less efficient substrates, respectively. This screening method is applicable to both serine/threonine and tyrosine kinases. It also has a wide dynamic range; as described below, we were able to profile the specificity for kinases that have catalytic efficiency (kcat/KM) ranging from 0.1 to 106 M−1 s−1, kinases that are highly sequence specific (Pim1), and kinases that have little sequence selectivity (MKK6). This method provides individual peptide sequences, thus permitting the identification of not only amino acids that contribute positively to the kinase-substrate interaction (permissive residues), but also amino acids that negatively impact the kinase function (non-permissive residues), as well as any sequence covariance. Another useful feature of our method is that genuine positive beads and false positive beads (e.g., those caused by nonspecific binding of the fluorescent dye molecule) are readily differentiated by treating the fluorescent beads with tris(carboxyethyl)phosphine (TCEP). A true positive bead would undergo cleavage of the disulfide linkage and lose the red fluorescence, whereas a false positive bead would not be affected by the TCEP treatment.
Figure 1.
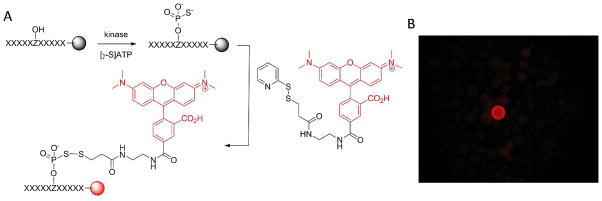
Screening peptide libraries for optimal kinase substrate motifs. (A) Reactions involved in the kinase screening procedure. Z, Ser, Thr, or Tyr. (B) Photograph of a portion of the library beads after the screening reactions (viewed under a fluorescence microscope).
Substrate Specificity of Pim1 Is Defined by both Preferred and Non-Permissive Residues
To validate the on-bead screening method, we first screened library Ia (where Z = Ser and Thr) against protein serine/threonine kinase Pim1. Pim1, along with Pim2 and Pim3, forms the Pim subfamily of Ca2+/calmodulin-dependent protein kinases. Pim1 is involved in cell proliferation and survival, as well as regulation of DNA transcription.29 Its substrate specificity has been investigated by several methods, including the positional scanning peptide library method and the MS-based proteomics method.17,30,31 Pim1 strongly prefers basic residues (especially Arg) at the S/T-3 and S/T-5 positions (relative to the phosphorylatable Ser/Thr, which is defined as position 0). It was also reported to have some preference for His and Arg at the S/T-2 position and Gly at the S/T+1 position.17,30 Screening of Pim1 against 100 mg of library I (~100,000 beads) produced ~500 fluorescent beads; 89 of the most intensely colored beads were individually sequenced to give 44 complete sequences (Supplementary Table S1). These sequences were aligned with respect to the putative phosphorylated Ser/Thr (position 0) on the basis of their similarity. Most of the peptides (40 out of 44 sequences) were apparently phosphorylated at the fixed Ser/Thr residue, while four sequences were phosphorylated at other positions. A plot of the aligned sequences in the Weblogo format shows that the overall specificity profile is very similar to that determined by other library screening methods17,30 and a profile generated with the phosphorylation motifs derived from established Pim1 protein substrates (Figure 2). The availability of individual sequences provided information on underrepresented amino acids at each position. For example, although amino acids of different physicochemical properties were selected at the S/T-2 position, none of the selected sequences contained an acidic residue at this position. Similarly, proline was not selected at the S/T+1 position. These data suggest that acidic and proline residues are disfavored (non-permissive residues) at these positions.
Figure 2.

Weblogo plots showing the specificity profile of Pim1. (A) Plot generated with the 44 sequences selected from library Ia. (B) Plot generated with 30 sequences derived from known Pim1 protein substrates.
A small panel of peptides were individually synthesized and tested against Pim1 in solution to confirm the library screening results (Table 1). Peptide 1 (Ac-KYRHPTNMYY-NH2) is one of the sequences selected from library Ia and has a kcat/KM value of 1.9 × 104 M−1s−1 toward Pim1. Substitution of the most preferred residues at S/T-5 (K→R) and S/T+1 positions (N→G) gave a consensus peptide Ac-RYRHPTGMYY-NH2 (Table 1, peptide 2), which is an excellent Pim1 substrate (kcat/KM value of 8.7 × 105 M−1s−1). As expected, replacement of Arg by Ala at either the S/T-5 or S/T-3 position greatly reduced the Pim1 activity (910- and 730-fold, respectively), while removal of both Arg side chains abolished the Pim1 activity (Table 1, peptides 2–5). Remarkably, replacement of His at position S/T-2 by an acidic residue (Asp) also reduced the Pim1 activity by 620-fold (Table 1, compare peptides 2 and 6), a magnitude similar to that caused by removal of the Arg residue at the S/T-5 or S/T-3 position. The co-crystal structure of Pim1 bound with peptide ARKRRRHPSGPPTA showed that the substrate-binding site of Pim1 contains a Glu residue (Glu-243), which interacts electrostatically with a His or Arg at the S/T-2 position of the peptide substrate.30 An acidic residue (e.g., Asp) at the S/T-2 position would result in repulsive interaction with Glu-243, which is likely responsible for the poor activity of Pim1 toward peptide 6. Similarly, substitution of proline for glycine at the S/T+1 position resulted in a 62-fold reduction in activity (Table 1, compare peptides 2 and 7). We also replaced the S/T+1 glycine with an Arg residue, which was not selected from the library, and found that the Arg residue only slightly decreased the Pim1 activity. Our results indicate that the substrate specificity of Pim1 (and likely other kinases as well) is determined not only by favorable interactions between the kinase active site and the side chains of preferred substrate residues, but also by unfavorable (repulsive or steric) interactions between the kinase and other regions of the substrate (hereafter referred to as non-permissive residues). The contribution of non-permissive residues to kinase specificity has only recently started to be recognized. Proline has been proposed as a non-permissive or “veto” residue at the S/T+1 position to insure orthogonal substrate specificity between proline-directed (e.g., cdk1) and other kinases (e.g., AGC and CAMK kinases).32 Examples of other non-permissive amino acids or at other substrate positions (other than the S/T+1 position) have been rare.33 Commonly, a kinase prefers amino acids of certain physicochemical property at a given position (e.g., hydrophobic or basic residues) and therefore disfavors amino acids of the opposite property at that position (e.g., hydrophilic or acidic residues). Pim1, on the other hand, accepts essentially any amino acid except for Asp and Glu at the S/T-2 position and, therefore, Asp and Glu act as genuine “veto” residues. The negative selectivity of Pim1 kinase has not previously been reported, although its disfavor for proline at the S/T+1 position is apparent from the primary data of a previous position scanning library study.30
Table 1.
Kinetic Properties of Pim1 toward Selected Peptides.
| Peptide No. | Sequencea | kcat (s−1) | KM (μM) | kcat/KM (M−1s−1) |
|---|---|---|---|---|
| 1 | KYRHPTNMYY | 7.8 ± 0.4 | 410 ± 43 | 19000 |
| 2 | RYRHPTGMYY | 14 ± 1 | 16 ± 2 | 870000 |
| 3 | AYRHPTGMYY | >800 | 960 | |
| 4 | RYAHPTGMYY | >600 | 1200 | |
| 5 | AYAHPTGMYY | ND | ND | ND |
| 6 | RYRDPTGMYY | 1.8 ± 0.1 | 1300 ± 53 | 1400 |
| 7 | RYRHPTPMYY | 2.9 ± 0.1 | 200 ± 26 | 14000 |
| 8 | RYRHPTRMYY | 7.5 ± 0.1 | 24 ± 2 | 310000 |
All peptides contain an N-terminal acetyl group and a C-terminal amide.
ND, no detectable activity.
MKK6 Has Low Sequence Selectivity and Intrinsic Catalytic Activity
MKK6 is an upstream regulator of p38 MAP kinase (MAPK) and involved in cell proliferation, apoptosis, and stress-induced cellular responses. As a dual-specificity kinase, it is known to phosphorylate both Thr-180 and Tyr-182 residues of the p38 MAPK activation motif.34 Other than that, little is known about its substrate specificity or whether it has other biological functions. Compared to Pim1, screening of MKK6 against library I was more problematic. High kinase concentration (up to 15 μM) and extended incubation time (up to 48 h) were necessary to produce beads of detectable fluorescence. Screening of 100 mg of library Ia gave 81 lightly fluorescent beads, which were sequenced to afford 67 sequences (Supplementary Table S2). Similar results were obtained when MKK6 was screened against library Ib, X5YX4NNBBRM-resin, which is similar to library Ia but contains a fixed Tyr in the middle and no Tyr at the random positions. A total of 61 sequences (44 complete sequences) were selected from 100 mg of library Ib (Supplementary Table S3). MKK6 does not exhibit strong sequence selectivity toward peptide substrates, other than a slight preference for hydrophobic residues from the −4 to +4 positions (Figure 3).
Figure 3.

Specificity profile of MKK6 toward Ser-/Thr- (A) and Tyr-containing peptides (B). The Weblogo plots were generated with the 67 and 44 sequences selected from libraries I a and Ib, respectively.
Five of the selected peptides, Ac-RRWRHFMNIYKFPP-NH2, Ac-RRWRDGIKIYNLLF-NH2, Ac-RRWRIFKFISAHNP-NH2, Ac-RRWRFVEGHTKAMF-NH2, and Ac-RRRYEILPTWNVY-NH2 (Table 2, peptides 9–13) were resynthesized and assayed against MKK6 in solution. Due to the low activity of MKK6 toward peptide substrates, the low solubility of the peptides, and interference from MKK6 autophosphorylation, the enzyme-coupled kinase assay could not reliably measure the MKK6 activity. We therefore monitored the kinase reaction by HPLC. The inclusion of a RRWR motif in peptides 9–13 improved their aqueous solubility and facilitated UV detection at 280 nm. Among the five peptides, only peptide 13 was active enough to allow its kcat/KM value accurately determined (0.11 M−1 s−1, KM >500 μM). Peptides 9 and 12 were active against MKK6 and the phosphorylated products were detected by MALDI-TOF MS, whereas peptides 10 and 11 did not show detectable activity. For comparison, we also tested two peptides corresponding to the in vivo MKK6 phosphorylation sites in p38 MAPK, Ac-WTDDEMT180GFVATR-NH2 and Ac-WTDDEMDGY182VATR-NH2 (Table 2, peptides 14 and 15). Peptide 14 had no detectable activity, whereas phosphorylation of peptide 15 could only be detected by MS (kcat/KM <0.1 M−1 s−1). The lack of sequence selectivity and the low intrinsic kinase activity of MKK6 toward peptide substrates indicate that substrate recruiting and/or allosteric activation is required for MKK6 to achieve high activity toward its physiological substrates. This is consistent with a previous observation that MKK6 exists as an autoinhibited dimer in the absence of substrates.35 Docking interaction between p38 and a related upstream kinase (MKK3) has been established and the docking peptide motif of MKK3 is also conserved in MKK6.36
Table 2.
Kinetic Properties of MKK6 toward Selected Peptides
| Peptide No. | Sequencea | Activity Detectable ? | kcat/KM (M−1 s−1) |
|---|---|---|---|
| 9 | RRWRHFMNIYKFPP | by MS | ND |
| 10 | RRWRDGIKIYNLLF | Not detectable | ND |
| 11 | RRWRIFKFISAHNP | Not detectable | ND |
| 12 | RRWRFVEGHTKAMF | by MS | ND |
| 13 | RRRYEILPTWNVY | by HPLC and MS | 0.11 |
| 14 | WTDDEMTGFVATR | Not detectable | ND |
| 15 | WTDDEMDGYVATR | by MS | ND |
All peptides contain an N-terminal acetyl group and a C-terminal amide.
ND, not determined.
Csk Recognizes Two Different Sequence Motifs
Csk is a protein tyrosine kinase that phosphorylates a C-terminal tyrosine of Src family kinases (SFKs), resulting in down-regulation of their kinase activities. Although generally thought to be specific for SFKs, Csk has been shown to phosphorylate a cell surface glycoprotein, platelet endothelial cell adhesion molecule-1 (PECAM-1; also termed CD31),37 suggesting that it may phosphorylate other yet unidentified substrates in vivo. Previous screening of an oriented peptide library gave a consensus sequence of EEEIpYFFF, which bears little resemblance to the C-terminal sequences of SFKs.28 Mutagenesis and crystallographic studies revealed that a docking interaction between Csk and Src kinase domains is critical for the high activity and specificity of Csk toward SFK substrates.38,39 However, it remains to be determined how much the local sequence surrounding the phosphorylation site contributes to Csk activity and/or specificity and whether Csk has physiological substrates other than SFKs and PECAM-1.
Csk was first screened against library Ib (X5YX4NNBBRM-resin). To minimize any interference from its SH2 domain, which may amplify the kinase reaction on beads containing SH2-binding sequences by recruiting more Csk to their surfaces, we performed library screening with a Csk mutant that harbors an R107A mutation in its SH2 domain. Screening of 50 mg of library Ib gave 23 complete sequences (Supplementary Table S4). Csk strongly prefers a Glu at the Y-3 position, an acidic residue at the Y-4 position, and acidic or hydrophobic residues at positions Y-5 and Y-1 (Figure 4a). It prefers a small residue, especially Pro, Ser and Gly at the Y-2 position. On the C-terminal side of Tyr, Csk has an overwhelming preference for hydrophobic residues (Nle, Val, Ile, Phe, and Pro) at the Y+1 and Y+3 positions and strong preference for acidic residues at the Y+2 position. The Y+4 position does not exhibit any obvious specificity. Given that Csk shows selectivity at 8 out of the 9 random positions, sampling a small fraction of the possible sequence space is unlikely to identify the most active Csk substrate. Therefore, we synthesized a more focused library, X1X2X3X4X5YX6–12NNBBRM-resin (library II), in which X1–5 featured the most preferred residues identified from library Ib (X1 = Asp, Glu, or Phe; X2 = Asn, Asp, or Glu; X3 = Asp, Glu, Ser, or Val; X4 = Gly, Pro, or Ser; and X5 = Asp, Glu, Ile, or Phe), whereas X7–12 were randomized with all 18 proteinogenic amino acids except for Tyr and Met (replaced by Nle). Screening of this library should reveal the relative fitness of the preferred N-terminal residues as well as the most optimal sequences on the C-terminal side of Tyr. Screening of a total of 150 mg of library II (in 2 separate experiments) produced 114 most reactive sequences (Supplementary Table S5). The data confirmed Csk’s preference for hydrophobic residues at the Y+1 and Y+3 positions and acidic residues at the Y+2 position. Again, Csk has no major selectivity at Y+4 to Y+7 positions, other than a small overrepresentation of acidic residues. On the N-terminal side of Tyr, the data from library II are largely consistent with those derived from library Ib, but also revealed several new features. First, Pro is strongly preferred at the Y-2 position, followed by Ser, and Gly was only occasionally selected. Second, essentially all of the selected Csk substrates (113/114) contained acidic residue(s) at either Y-3 or Y-4 position and frequently at both positions. Third, although Glu, Phe, Ile, and Asp were selected at the Y-1 position at similar frequency, there was strong sequence covariance between the Y-1 and Y-2 residues. When Pro was the Y-2 residue, the Y-1 residue was most frequently Ile and Phe (53 out of 85 sequences) (Supplementary Table S5). On the other hand, when Ser was the Y-2 residue, the Y-1 residue was mostly Asp and Glu (19 out of 24 sequences). In fact, EPIY and (E/D)S(E/D)Y were among the most frequently selected sequence motifs (Table S4 and S5).
Figure 4.

Weblogo plots showing the specificity profile of Csk. (A) Plot generated with 23 sequences selected from library Ib; (B) Plot generated with 114 sequences selected from library II.
To evaluate the screening results, we synthesized peptides 16–29 and determined their kinetic constants against GST-Csk in solution (Table 3). Peptide 16 (Ac-KKKKEEIYFFF-NH2) is the Csk consensus sequence previously determined by screening an oriented peptide library28 and used here for comparison. Peptide 17 (Ac-FEEIDYVSPW-NH2) is one of the sequences selected from library Ib, whereas peptide 18 (Ac-FEEPDYVEFI-NH2) corresponds to the consensus sequence of library Ib screening data. Peptides 19 (Ac-EEEPEYIEPDDDE-NH2) and 20 (Ac-FEEPEYIEPIDFE-NH2) represent the consensus sequences of library II screening data. Peptides 21–23, 26, and 28 were individual sequences selected from library II, while peptides 24, 25, 27, and 29 were synthetic variants of the above library II-derived peptides for the purpose of assessing the sequence covariance between the Y-2 and Y-1 residues.
Table 3.
Kinetic Properties of GST-Csk toward Selected Peptides
| Entry No. | Sequencea | kcat (s−1) | KM (μM) | kcat/KM (M−1 s−1) |
|---|---|---|---|---|
| 16 | KKKKEEIYFFF | ND | ND | 580 |
| 17 | FEEIDYVSPW | ND | ND | 390 |
| 18 | FEEPDYVEFI | ND | ND | 490 |
| 19 | EEEPEYIEPDDDE | ND | ND | 190 |
| 20 | FEEPEYIEPIDFE | ND | ND | 540 |
| 21 | EEEPEYAEIIVLP | ND | ND | 490 |
| 22 | DDEPEYIEFDAHN | ND | ND | 850 |
| 23 | DDEPIYAELADIT | 0.77 ± 0.10 | 120 ± 22 | 6300 |
| 24 | DDEPIYIEFDAHN | 0.72 ± 0.03 | 190 ± 19 | 3800 |
| 25 | FEEPIYIEPIDFE | 0.21 ± 0.01 | 150 ± 15 | 1400 |
| 26 | FEDSDYADIFIEE | ND | ND | 280 |
| 27 | FEDSIYADIFIEE | ND | ND | 320 |
| 28 | DDESEYINIPDGE | ND | ND | 130 |
| 29 | DDESIYINIPDGE | ND | ND | 180 |
All peptides contain an N-terminal acetyl group and a C-terminal amide.
ND, not determined.
Peptide 16, which was the most active peptide substrate previously known for Csk, has a kcat/KM value of 580 M−1s−1. It was previously reported that native Csk is 15-fold more active than GST-Csk and has a kcat/KM value of 6050 M−1s−1 toward peptide 16.28 Peptides 17 and 18 derived from the primary library (Ib) have slightly lower activity than peptide 16, with kcat/KM values of 390 and 490 M−1s−1, respectively. As expected, peptides derived from library II generally have higher activity toward Csk than those from library Ib (Table 3, compare peptide 17 versus 21–23). In particular, peptide 23 (Ac-DDEPIYAELADIT-NH2), which was from an intensely colored bead (Table S5), has a kcat/KM value of 6300 M−1s−1. Thus, peptide 23 is 11-fold more active than the previous consensus peptide (16) and its activity (which corresponds to a kcat/KM value of 9.5 × 104 M−1s−1for native Csk) approaches that of Csk protein substrates (2.0 × 105 and 3.7 × 105 M−1s−1 for Lck and Src respectively).40 It was previously reported that a Leu or Ile at the Y-1 position enhances Csk activity.28,41 To test the importance of the Ile residue, we replaced the Glu or Asp residue at the Y-1 position of peptides 20, 22, 26, and 28 by an Ile residue to generate peptides 25, 24, 27, and 29, respectively (Table 3). We found that substitution of Ile indeed increases the Csk activity by 2.6- and 4.5-fold for peptides 20 and 22, respectively, which both have a Pro as the Y-2 residue. Interestingly, the same substitution has a smaller effect on peptides 26 and 28 (1.1- and 1.4-fold, respectively), which contain a Ser at the Y-2 position. This is consistent with the frequent selection of EPIY-containing but not ESIY-containing sequences during library screening (Table S5). The more frequent selection of (D/E)S(D/E)Y than (D/E)S(I/F)Y sequences (despite the similar or slightly higher activity of the latter) is likely caused by the better aqueous solubility of the former, which rendered the resin-bound peptides more accessible to Csk.42 A further manifestation of the importance of sequence context for optimal Csk activity is the observation that peptides featuring the most frequently selected residues at every position (“consensus” peptides 19 and 20) actually have lower activity (kcat/KM values of 190 and 540 M−1s−1, respectively) than many of the individual sequences selected from the library. By contrast, the consensus peptide of Pim1 (Table 1, peptide 2) is much more active than the individual sequences selected from the peptide library.
Identification of Novel Pim1 Protein Substrates
A search of the PhosphoSite database (http://www.phosphosite.org) against the Pim1 consensus sequence, RXR(H/R)X(S/T), gave 149 potential protein substrates, 3 of which have previously been reported as in vivo and/or in vitro Pim1 substrates (Table S6). We selected two of the predicted substrates, SCAND1 and PTP-PEST, for further testing because they were available commercially (SCAND1) or already in this laboratory (PTP-PEST). SCAND1 is a transcription co-activator and contains a potential Pim1 phosphorylation site at Thr-173 (RIRRRT173DVRI).43 PTP-PEST is a non-receptor protein tyrosine phosphatase, which contains an optimal Pim1 motif at Ser-39 (RLRRLS39TKYR).44 For comparison, we also tested CBX1 and phosphatase PTP1B as potential Pim1 substrates. CBX1 is a component of the heterochromatin; its binding to histone H3 tails methylated at Lys-9 leads to epigenetic repression.45 It was predicted to be a less active Pim1 substrate, because its putative Pim1 site has two Lys residues at the S-5 and S-3 positions (KRKADS89DSED). PTP1B contains a sequence motif at Ser-50 (RYRDVS50PFDH) that matches the Pim1 consensus sequence with respect to the preferred Arg residues at the S-5 and S-3 positions but has non-permissive residues at the S-2 (Asp) and S+1 positions (Pro). We predicted PTP1B to be a poor substrate of Pim1. In vitro phosphorylation with [γ–32P]ATP followed by SDS-PAGE analysis showed that PTP-PEST, is indeed efficiently phosphorylated by Pim1 (Figure 5A). Kinetic analysis with an enzyme-coupled assay gave a kcat/KM value of 34000 M−1s−1 against the recombinant PTP-PEST protein. Mutation of Ser-39 into alanine greatly reduced the amount of PTP-PEST phosphorylation by Pim1 (Figure 5B), indicating that Ser-39 is indeed the primary site of phosphorylation by Pim1. The Ser-39 site of PTP-PEST was previously shown to be phosphorylated by PKA and PKC kinases and phosphorylation at this site downregulates its phosphatase activity by 2-fold.46 Similarly, SCAND1 was phosphorylated by Pim1 although less efficiently as compared to PTP-PEST (Figure 5A). As expected, CBX1 showed only weak phosphorylation, whereas phosphorylation of PTP1B by Pim1 was not detectable under the experimental conditions (Figure 5A).
Figure 5.
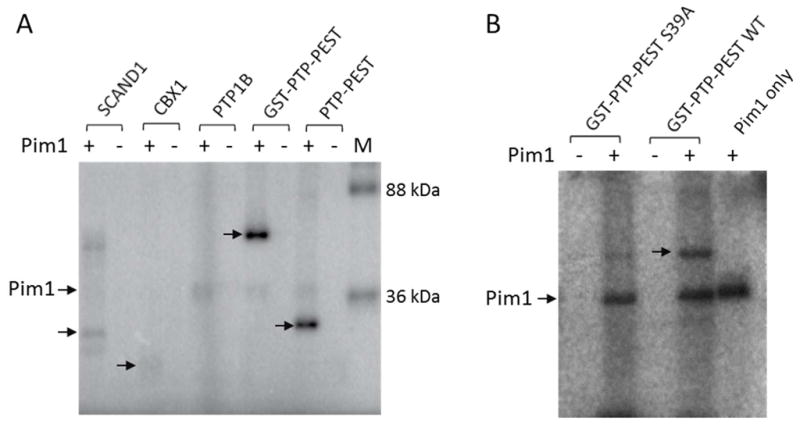
In vitro phosphorylation of protein substrates by Pim1. Indicated proteins (1–2 μM) were incubated with [γ-32P]ATP in the presence and absence of Pim1 (100 nM) for 2 h, followed by SDS-PAGE and phosphorimaging analysis. Arrows indicate the phosphorylated proteins. M, molecular weight markers.
Csk Phosphorylates Proteins other than the Src Family Kinases
The C-terminal tails of SFKs have sequences of EPQY (Src, Fgr, and Fyn), EGQY (Lck and Lyn), DSSY (Frk), and FTSY (Brk). PECAM-1, a non-SFK substrate of Csk,37 was recently reported to be phosphorylated by Csk at Tyr-663 (NSDVQY663TEVQV).47 These sequences (with the exception of Brk) are similar to the sequence motifs selected from our peptide library [EPIY, EPEY, (E/D)S(E/D)Y, EGEY, and EV(H/R)Y], suggesting that the local sequence surrounding the phosphorylation site plays an important role in defining the in vivo substrate specificity of Csk. Encouraged by this finding, we searched the PhosphoSite for potential Csk substrates using the consensus sequence motifs of Csk [(E/D)PIY and (D/E)S(D/E)Y] (Table S7). This search identified Tyr-536 (GQESEY536GNITY) of protein tyrosine phosphatase SHP-1 as a potential Csk substrate. Tyr-536 of SHP-1 is known to be phosphorylated in vivo48 and its phosphorylation activates the catalytic activity of SHP-1 by 8-fold.49 Interestingly, in vitro phosphorylation of recombinant SHP-1 by Csk showed that while the full-length SHP-1 (68 kDa) is a poor substrate of Csk, a 55-kDa proteolytic fragment apparently lacking the N-terminal SH2 domain(s) is efficiently phosphorylated (Figure 6). To confirm the phosphorylation of Tyr-536, we carried out the kinase reaction with two SHP-1 truncation mutants, SHP-1(ΔSH2/ΔC35) and SHP-1(ΔSH2/ΔC60), which are catalytically deficient due to an active-site mutation (C453S) and lack the two SH2 domains and the C-terminal 35 and 60 amino acids, respectively.50 The 40-kDa SHP-1(ΔSH2/ΔC35) fragment, which contains Tyr-536, is an efficient Csk substrate, whereas the 37-kDa SHP-1(ΔSH2/ΔC60) fragment (which does not contain Tyr-536) is not (Figure 6). We thus conclude that SHP-1(ΔSH2/ΔC35) and the 55 kDa fragment were phosphorylated by Csk at Tyr-536. Although the physiological relevance of this phosphorylation event remains to be determined, our data (together with the PECAM-1 finding) demonstrate that Csk is capable of efficient phosphorylation of protein substrates other than the SFKs. It is yet unclear why full-length SHP-1 is resistant to phosphorylation by Csk; however, the N-terminal SH2 domain and the C-terminus of SHP-1 have been shown to cooperatively regulate the catalytic activity of SHP-1.50 Our database search with the other Csk consensus sequence, (E/D)PIY, revealed several notable potential Csk substrate proteins including the CagA protein of Helicobacter pylori. CagA is a bacterial toxin which upon entering mammalian cells, undergoes tyrosine phosphorylation at multiple EPIYA motifs.51 Csk is recruited to some of the phosphorylated EPIYA sites via its SH2 domain, although the biological function of this recruitment is unclear.52 It is tempting to suggest that the Csk kinase domain may phosphorylate the other EPIY motifs on CagA.
Figure 6.
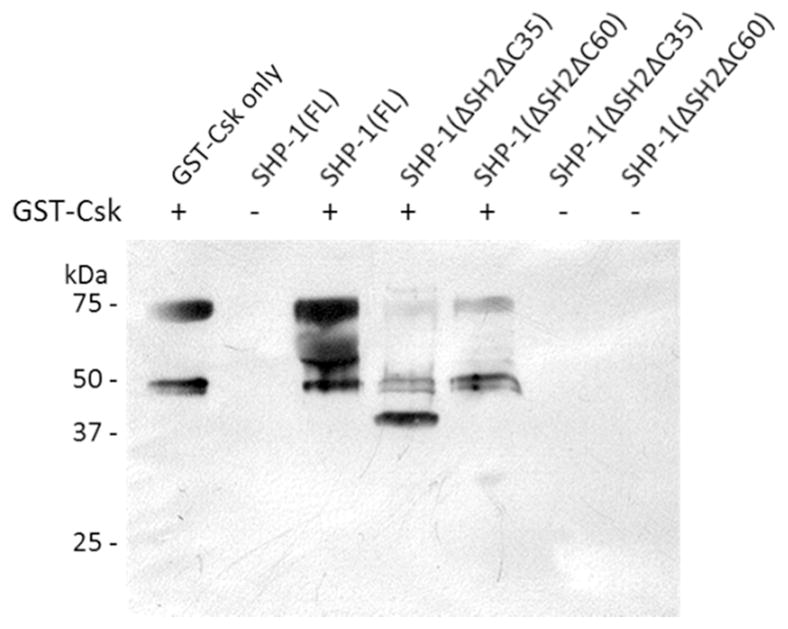
In vitro phosphorylation of full-length and truncation mutants of SHP-1 by GST-Csk. Full-length or truncated SHP-1 (20 μM) was treated with GST-Csk (4 μM) in the presence of 1.0 mM ATP for 30 min, followed by SDS-PAGE and immunoblotting with anti-pY antibody 4G10. All SHP-1 variants contained a C453S mutation in the PTP active site. The 75- and 50-kDa bands correspond to GST-Csk and Csk (from proteolysis of GST-Csk), respectively.
CONCLUSION
We have developed a simple but robust method to systematically profile the sequence specificity of protein kinases. Our method shares the desirable attributes of the previously reported kinase profiling methods (including the ability to provide information on individual substrate sequences and therefore ability to identify any sequence covariance and/or disfavored residues, compatibility with modified amino acids such as pS, pT, and pY,42 high-throughput capability, and relative ease of the screening protocol) while avoiding most of their drawbacks (e.g., the use of radioactivity or anti-pY antibody, which exhibits substantial sequence-dependence in binding to pY peptides,22 and sequence biases in the proteome-derived peptide libraries). The improved screening method allowed us to uncover two important properties of Pim1 and Csk kinases, both of which had previously been scrutinized by other library methods. One finding is the importance of non-permissive residues (perhaps as important as the preferred residues) in defining the specificity of kinases. A second finding is that kinase-peptide substrate interaction exhibits sequence contextual effect, i.e., a kinase may recognize more than one type of consensus motifs. The contribution of non-permissive residues and contextual effect to the sequence specificity of protein-binding domains and protein phosphatases has also recently been described.11–15 It appears that Nature uses a combination of preferred residues (favorable interaction), non-permissive residues (unfavorable interaction), and sequence context (interaction within the peptide ligand) as a general strategy to ensure high fidelity in protein-protein and enzyme-substrate interactions. This also highlights the importance of obtaining individual peptide ligands in any form of library screening. Our data suggest that in addition to the SFKs, Csk may phosphorylate other protein substrates in vivo. Finally, it should be noted that some kinases (e.g., MKK6) may have little intrinsic sequence specificity and their in vivo substrate specificity is likely dictated by protein-protein interactions and/or other mechanisms.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health grants (CA132855 and GM062820).
We thank Drs. Philip Cole and Dustin Maly for providing the kinase expression vectors and Drs. David Taggart, Zucai Suo, and Jane Jackman for help with the radioactive kinase assays.
ABBREVIATIONS
- PTP
protein tyrosine phosphatase
- PEGA
poly[acryloyl-bis(aminopropyl)polyethylene glycol]
- PED-MS
partial Edman degradation-mass spectrometry
- TCEP
tris(carboxyethyl)phosphine
- TMR
tetramethylrhodamine
- MALDI-TOF
matrix-assisted laser desorption/ionization-time of flight
- SFK
Src-family kinase
- Csk
C-terminal Src kinase
- MKK6
mitogen-activated protein kinase kinase 6
Footnotes
Supporting Information: Supplementary tables containing sequences selected from peptide libraries. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Hornbeck PV, Kornhauser JM, Tkachev S, Zhang B, Skrzypek E, Murray B, Latham V, Sullivan M. PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 2012;40:261–270. doi: 10.1093/nar/gkr1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 3.Ubersax JA, Ferrell JE., Jr Mechanisms of specificity in protein phosphorylation. Nat Rev Mol Cell Biol. 2007;8:530–541. doi: 10.1038/nrm2203. [DOI] [PubMed] [Google Scholar]
- 4.Zhu G, Liu Y, Shaw S. Protein kinase specificity. A strategic collaboration between kinase peptide specificity and substrate recruitment. Cell Cycle. 2005;4:52–56. doi: 10.4161/cc.4.1.1353. [DOI] [PubMed] [Google Scholar]
- 5.Songyang Z, Blechner S, Hoagland N, Hoekstra MF, Piwnica-Worms H, Cantley LC. Use of an oriented peptide library to determine the optimal substrates of protein kinases. Curr Biol. 1994;4:973–982. doi: 10.1016/s0960-9822(00)00221-9. [DOI] [PubMed] [Google Scholar]
- 6.Obata T, Yaffe MB, Leparc GG, Piro ET, Maegawa H, Kashiwagi A, Kikkawa R, Cantley LC. Peptide and protein library screening defines optimal substrate motifs for AKT/PKB. J Biol Chem. 2000;275:36108–36115. doi: 10.1074/jbc.M005497200. [DOI] [PubMed] [Google Scholar]
- 7.Hutti JE, Jarrell ET, Chang JD, Abbott DW, Storz P, Toker A, Cantley LC, Turk BE. A rapid method for determining protein kinase phosphorylation specificity. Nat Methods. 2004;1:27–29. doi: 10.1038/nmeth708. [DOI] [PubMed] [Google Scholar]
- 8.Fujii K, Zhu G, Liu Y, Hallam J, Chen L, Herrero J, Shaw S. Kinase peptide specificity: improved determination and relevance to protein phosphorylation. Proc Natl Acad Sci USA. 2004;101:13744–13749. doi: 10.1073/pnas.0401881101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mok J, Kim PM, Lam HY, Piccirillo S, Zhou X, Jeschke GR, Sheridan DL, Parker SA, Desai V, Jwa M, Cameroni E, Niu H, Good M, Remenyi A, Ma JL, Sheu YJ, Sassi HE, Sopko R, Chan CS, De Virgilio C, Hollingsworth NM, Lim WA, Stern DF, Stillman B, Andrews BJ, Gerstein MB, Snyder M, Turk BE. Deciphering protein kinase specificity through large-scale analysis of yeast phosphorylation site motifs. Science Signal. 2010;3:ra12. doi: 10.1126/scisignal.2000482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Joughin BA, Liu C, Lauffenburger DA, Hogue CW, Yaffe MB. Protein kinases display minimal interpositional dependence on substrate sequence: potential implications for the evolution of signaling networks. Philos Trans R Soc London: Ser B Biol Sci. 2012;367:2574–2583. doi: 10.1098/rstb.2012.0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sweeney MC, Wavreille A, Park J, Butchar JP, Tridandapani S, Pei D. Decoding protein-protein interactions through combinatorial chemistry: sequence specificity of SHP-1, SHP-2, and SHIP SH2 domains. Biochemistry. 2005;44:14932–14947. doi: 10.1021/bi051408h. [DOI] [PubMed] [Google Scholar]
- 12.Wavreille AS, Pei D. A chemical approach to the identification of tensin-binding proteins. ACS Chem Biol. 2007;2:109–118. doi: 10.1021/cb600433g. [DOI] [PubMed] [Google Scholar]
- 13.Chen X, Tan PH, Zhang Y, Pei D. On-bead screening of combinatorial libraries: reduction of nonspecific binding by decreasing surface ligand density. J Comb Chem. 2009;11:604–611. doi: 10.1021/cc9000168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luechapanichkul R, Chen X, Taha HA, Vyas S, Guan X, Freitas MA, Hadad CM, Pei D. Specificity profiling of dual specificity phosphatase vaccinia VH1-related (VHR) reveals two distinct substrate binding modes. J Biol Chem. 2013;288:6498–6510. doi: 10.1074/jbc.M112.449611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen X, Ren L, Kim S, Carpino N, Daniel JL, Kunapuli SP, Tsygankov AY, Pei D. Determination of the substrate specificity of protein-tyrosine phosphatase TULA-2 and identification of Syk as a TULA-2 substrate. J Biol Chem. 2010;285:31268–31276. doi: 10.1074/jbc.M110.114181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang SY, Tsai ML, Chen GY, Wu CJ, Chen SH. A systematic MS-based approach for identifying in vitro substrates of PKA and PKG in rat uteri. J Proteome Res. 2007;6:2674–2684. doi: 10.1021/pr070134c. [DOI] [PubMed] [Google Scholar]
- 17.Kettenbach AN, Wang T, Faherty BK, Madden DR, Knapp S, Bailey-Kellogg C, Gerber SA. Rapid determination of multiple linear kinase substrate motifs by mass spectrometry. Chem Biol. 2012;19:608–618. doi: 10.1016/j.chembiol.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chou MF, Prisic S, Lubner JM, Church GM, Husson RN, Schwartz D. Using bacteria to determine protein kinase specificity and predict target substrates. PloS One. 2012;7:e52747. doi: 10.1371/journal.pone.0052747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu J, Ma QN, Lam KS. Identifying Substrate Motifs of Protein Kinases by a Random Library Approach. Biochemistry. 1994;33:14825–14833. doi: 10.1021/bi00253a022. [DOI] [PubMed] [Google Scholar]
- 20.Kim YG, Shin DS, Kim EM, Park HY, Lee CS, Kim JH, Lee BS, Lee YS, Kim BG. High-throughput identification of substrate specificity for protein kinase by using an improved one-bead-one-compound library approach. Angew Chem Int Ed. 2007;46:5408–5411. doi: 10.1002/anie.200700195. [DOI] [PubMed] [Google Scholar]
- 21.Kim M, Shin DS, Kim J, Lee YS. Substrate screening of protein kinases: detection methods and combinatorial peptide libraries. Biopolymers. 2010;94:753–762. doi: 10.1002/bip.21506. [DOI] [PubMed] [Google Scholar]
- 22.Tinti M, Nardozza AP, Ferrari E, Sacco F, Corallino S, Castagnoli L, Cesareni G. The 4G10, pY20 and p-TYR-100 antibody specificity: profiling by peptide microarrays. Nat Biotechnol. 2012;29:571–577. doi: 10.1016/j.nbt.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 23.Yang Q, Co D, Sommercorn J, Tonks NK. Cloning and expression of PTP-PEST. A novel, human, nontransmembrane protein tyrosine phosphatase. J Biol Chem. 1993;268:6622–6628. [PubMed] [Google Scholar]
- 24.Thakkar A, Wavreille AS, Pei D. Traceless capping agent for peptide sequencing by partial edman degradation and mass spectrometry. Anal Chem. 2006;78:5935–5939. doi: 10.1021/ac0607414. [DOI] [PubMed] [Google Scholar]
- 25.Cole PA, Grace MR, Phillips RS, Burn P, Walsh CT. The role of the catalytic base in the protein tyrosine kinase Csk. J Biol Chem. 1995;270:22105–22108. doi: 10.1074/jbc.270.38.22105. [DOI] [PubMed] [Google Scholar]
- 26.Leon S, Quarrell R, Lowe G. Evaluation of resins for on-bead screening: a study of papain and chymotrypsin specificity using PEGA-bound combinatorial peptide libraries. Bioorg Med Chem Lett. 1998;8:2997–3002. doi: 10.1016/S0960-894X(98)00534-4. [DOI] [PubMed] [Google Scholar]
- 27.Allen JJ, Li M, Brinkworth CS, Paulson JL, Wang D, Hübner A, Chou WH, Davis RJ, Burlingame AL, Messing RO, Katayama CD, Hedrick SM, Shokat KM. A semisynthetic epitope for kinase substrates. Nat Methods. 2007;4:511–516. doi: 10.1038/nmeth1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu WQ, Zhou SY, Eck MJ, Cole PA. Peptide and protein phosphorylation by protein tyrosine kinase Csk: Insights into specificity and mechanism. Biochemistry. 1998;37:165–172. doi: 10.1021/bi9722960. [DOI] [PubMed] [Google Scholar]
- 29.Brault L, Gasser C, Bracher F, Huber K, Knapp S, Schwaller J. PIM serine/threonine kinases in the pathogenesis and therapy of hematologic malignancies and solid cancers. Haematologica. 1995;95:1004–1015. doi: 10.3324/haematol.2009.017079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bullock AN, Debreczeni J, Amos AL, Knapp S, Turk BE. Structure and substrate specificity of the Pim-1 kinase. J Biol Chem. 2005;280:41675–41682. doi: 10.1074/jbc.M510711200. [DOI] [PubMed] [Google Scholar]
- 31.Peng C, Knebel A, Morrice NA, Li X, Barringer K, Li J, Jakes S, Werneburg B, Wang L. Pim kinase substrate identification and specificity. J Biochem. 2007;141:353–362. doi: 10.1093/jb/mvm040. [DOI] [PubMed] [Google Scholar]
- 32.Zhu G, Fujii K, Belkina N, Liu Y, James M, Herrero J, Shaw S. Exceptional disfavor for proline at the P + 1 position among AGC and CAMK kinases establishes reciprocal specificity between them and the proline-directed kinases. J Biol Chem. 2005;280:10743–10748. doi: 10.1074/jbc.M413159200. [DOI] [PubMed] [Google Scholar]
- 33.Alexander J, Lim D, Joughin BA, Hegemann B, Hutchins JR, Ehrenberger T, Ivins F, Sessa F, Hudecz O, Nigg EA, Fry AM, Musacchio A, Stukenberg PT, Mechtler K, Peters JM, Smerdon SJ, Yaffe MB. Exceptional disfavor for proline at the P + 1 position among AGC and CAMK kinases establishes reciprocal specificity between them and the proline-directed kinases. Science Signal. 2011;4:ra42. doi: 10.1074/jbc.M413159200. [DOI] [PubMed] [Google Scholar]
- 34.Raingeaud J, Whitmarsh AJ, Barrett T, Dérijard B, Davis RJ. MKK3- and MKK6-regulated gene expression is mediated by the p38 mitogen-activated protein kinase signal transduction pathway. Mol Cell Biol. 1996;16:1247–1255. doi: 10.1128/mcb.16.3.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Min X, Akella R, He H, Humphreys JM, Tsutakawa SE, Lee SJ, Tainer JA, Cobb MH, Goldsmith EJ. The structure of the MAP2K MEK6 reveals an autoinhibitory dimer. Structure. 2009;17:96–104. doi: 10.1016/j.str.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chang CI, Xu BE, Akella R, Cobb MH, Goldsmith EJ. Crystal structures of MAP kinase p38 complexed to the docking sites on its nuclear substrate MEF2A and activator MKK3b. Mol Cell. 2002;9:1241–1249. doi: 10.1016/s1097-2765(02)00525-7. [DOI] [PubMed] [Google Scholar]
- 37.Cao MY, Huber M, Beauchemin N, Famiglietti J, Albelda SM, Veillette A. Regulation of mouse PECAM-1 tyrosine phosphorylation by the Src and Csk families of protein-tyrosine kinases. J Biol Chem. 1998;273:15765–15772. doi: 10.1074/jbc.273.25.15765. [DOI] [PubMed] [Google Scholar]
- 38.Lee S, Lin X, Nam NH, Parang K, Sun G. Determination of the substrate-docking site of protein tyrosine kinase C-terminal Src kinase. Proc Natl Acad Sci USA. 2003;100:14707–14712. doi: 10.1073/pnas.2534493100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Levinson NM, Seeliger MA, Cole PA, Kuriyan J. Structural basis for the recognition of c-Src by its inactivator Csk. Cell. 2008;134:124–134. doi: 10.1016/j.cell.2008.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang D, Huang XY, Cole PA. Molecular determinants for Csk-catalyzed tyrosine phosphorylation of the Src tail. Biochemistry. 2001;40:2004–2010. doi: 10.1021/bi002342n. [DOI] [PubMed] [Google Scholar]
- 41.Ruzzene M, Songyang Z, Marin O, Donella-Deana A, Brunati AM, Guerra B, Agostinis P, Cantley LC, Pinna LA. Sequence specificity of C-terminal Src kinase (CSK)--a comparison with Src-related kinases c-Fgr and Lyn. Eur J Biochem. 1997;246:433–439. doi: 10.1111/j.1432-1033.1997.t01-1-00433.x. [DOI] [PubMed] [Google Scholar]
- 42.Ren L, Chen X, Luechapanichkul R, Selner NG, Meyer TM, Wavreille A, Chan R, Iorio C, Zhou X, Neel BG, Pei D. Substrate specificity of protein tyrosine phosphatases 1B, RPTPα, SHP-1, and SHP-2. Biochemistry. 2011;50:2339–2356. doi: 10.1021/bi1014453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Edelstein LC, Collins T. The SCAN domain family of zinc finger transcription factors. Gene. 2005;359:1–17. doi: 10.1016/j.gene.2005.06.022. [DOI] [PubMed] [Google Scholar]
- 44.Veillette A, Rhee I, Souza CM, Davidson D. PEST family phosphatases in immunity, autoimmunity, and autoinflammatory disorders. Immunol Rev. 2009;228:312–324. doi: 10.1111/j.1600-065X.2008.00747.x. [DOI] [PubMed] [Google Scholar]
- 45.Verschure PJ, van der Kraan I, de Leeuw W, van der Vlag J, Carpenter AE, Belmont AS, van Driel R. In vivo HP1 targeting causes large-scale chromatin condensation and enhanced histone lysine methylation. Mol Cell Biol. 2005;25:4552–4564. doi: 10.1128/MCB.25.11.4552-4564.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Garton AJ, Tonks NK. PTP-PEST: a protein tyrosine phosphatase regulated by serine phosphorylation. EMBO J. 1994;13:3763–3771. doi: 10.1002/j.1460-2075.1994.tb06687.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tourdot BE, Brenner MK, Keough KC, Holyst T, Newman PJ, Newman DK. Immunoreceptor tyrosine-based inhibitory motif (ITIM)-mediated inhibitory signaling is regulated by sequential phosphorylation mediated by distinct nonreceptor tyrosine kinases: a case study involving PECAM-1. Biochemistry. 2013;52:2597–2608. doi: 10.1021/bi301461t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lorenz U, Ravichandran KS, Pei D, Walsh CT, Burakoff SJ, Neel BG. Lck-dependent tyrosyl phosphorylation of the phosphotyrosine phosphatase SH-PTP1 in murine T cells. Mol Cell Biol. 1994;14:1824–1834. doi: 10.1128/mcb.14.3.1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang Z, Shen K, Lu W, Cole PA. The role of C-terminal tyrosine phosphorylation in the regulation of SHP-1 explored via expressed protein ligation. J Biol Chem. 2003;278:4668–4674. doi: 10.1074/jbc.M210028200. [DOI] [PubMed] [Google Scholar]
- 50.Pei D, Lorenz U, Klingmüller U, Neel BG, Walsh CT. Intramolecular regulation of protein tyrosine phosphatase SH-PTP1: a new function for Src homology 2 domains. Biochemistry. 1994;33:15483–15493. doi: 10.1021/bi00255a030. [DOI] [PubMed] [Google Scholar]
- 51.Mueller D, Tegtmeyer N, Brandt S, Yamaoka Y, De Poire E, Sgouras D, Wessler S, Torres J, Smolka A, Backert S. c-Src and c-Abl kinases control hierarchic phosphorylation and function of the CagA effector protein in Western and East Asian Helicobacter pylori strains. J Clin Invest. 2012;122:1553–1566. doi: 10.1172/JCI61143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Naito M, Yamazaki T, Tsutsumi R, Higashi H, Onoe K, Yamazaki S, Azuma T, Hatakeyama M. Influence of EPIYA-repeat polymorphism on the phosphorylation-dependent biological activity of Helicobacter pylori CagA. Gastroenterology. 2006;130:1181–1190. doi: 10.1053/j.gastro.2005.12.038. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.