

Abstract
Background
Clinical studies report that scopolamine, an acetylcholine muscarinic receptor antagonist, produces rapid antidepressant effects in depressed patients, but the mechanisms underlying the therapeutic response have not been determined. The present study examines the role of the mammalian target of rapamycin complex 1 (mTORC1) and synaptogenesis, which have been implicated in the rapid actions of NMDA receptor antagonists.
Methods
The influence of scopolamine on mTORC1 signaling was determined by analysis of the phosphorylated and activated forms of mTORC1 signaling proteins in the prefrontal cortex (PFC). The numbers and function of spine synapses were analyzed by whole cell patch clamp recording and 2-photon image analysis of PFC neurons. The actions of scopolamine were examined in the forced swim test in the absence or presence of selective mTORC1 and AMPA receptor inhibitors.
Results
The results demonstrate that a single, low dose of scopolamine rapidly increases mTORC1 signaling and the number and function of spine synapses in layer V pyramidal neurons in the PFC. Scopolamine administration also produces an antidepressant response in the forced swim test that is blocked by pretreatment with the mTORC1 inhibitor or by a glutamate AMPA receptor antagonist.
Conclusions
Taken together, the results demonstrate that the antidepressant actions of scopolamine require mTORC1 signaling and are associated with increased glutamate transmission, and synaptogenesis, similar to NMDA receptor antagonists. These findings provide novel targets for safer and more efficacious rapid acting antidepressant agents.
Keywords: Depression, acetylcholine, GABA, glutamate, ketamine, synaptic plasticity
Depressive illness affects over 15 percent of the population and results in enormous personal and socioeconomic consequences (1). Moreover, currently available medications that have been designed to block the reuptake or breakdown of monoamines have significant limitations, including low response rates (approximately one third of patients achieve remission with the first prescribed antidepressant) and a therapeutic time lag of weeks to months (2). Clinical studies have identified two different drug classes with rapid antidepressant actions in depressed patients: ketamine, a non-competitive glutamate N-methyl-D-aspartate (NMDA) receptor antagonist (3–5); and scopolamine, a nonselective acetylcholine muscarinic receptor antagonist (6–9). The ability of these agents to produce a rapid response by mechanisms completely different than currently available agents represents a paradigm shift in the field of depression.
Recent work has begun to elucidate the molecular and cellular mechanisms underlying the actions of NMDA receptor antagonists. These studies demonstrate that ketamine rapidly stimulates the mammalian target of rapamycin (mTOR) complex 1 (mTORC1) and increases synapogenesis in a rapamycin sensitive manner in rat prefrontal cortex (PFC) (10, 11). Similar effects have been reported for NMDA antagonists that are selective for the NR2B receptor (10) consistent with clinical reports (12). The mTORC1 pathway has been implicated in activity-dependent synaptic plasticity and is localized in neuronal dendrites and spines where it controls the synthesis of proteins that are required for new synapse formation (13). Conversely, a possible role for reduced mTORC1 signaling and synaptogenesis in the pathogenesis of depression is supported by postmortem studies demonstrating that levels of mTORC1 signaling and synaptic proteins are decreased in the PFC of depressed subjects (14). Stimulation of mTORC1-mediated synaptogenesis could represent a mechanism for rapid reversal of the behavioral and synaptic deficits that are caused by exposure to stress and have been implicated in depression (11).
The current study was undertaken to determine if scopolamine rapidly increases mTORC1 signaling and synaptogenesis in the PFC, and if the behavioral actions of this muscarinic receptor antagonist require stimulation of mTORC1 signaling. The results demonstrate that scopolamine rapidly activates mTORC1 signaling, increases the number and function of spine synapses in the PFC, and produces mTORC1 dependent behavioral effects, similar to NMDA receptor antagonists.
MATERIALS AND METHODS
Animals
Male Sprague–Dawley rats weighing 175–250 g were pair-housed and maintained in standard conditions with a 12-h light/dark cycle and ad libitum access to food and water. Animal use and procedures were in accordance with the National Institutes of Health guidelines and approved by the Yale University Animal Care and Use Committees.
Drug Administration and Surgical Procedure
Animals received a single acute injection of vehicle, scopolamine (i.p.) or the preferential M1 selective antagonist telenzepine (s.c.). Tissue was collected from separate groups of animals for molecular or electrophysiological studies, and separate cohorts were also used in behavioral paradigms or microdialysis experiments as described below. For experiments involving central administration of rapamycin, rats were implanted with intracerebral ventricular (i.c.v.) guide cannula under Nembutal anesthesia (i.p. 55 mg/kg) as previously reported (15, 16). After recovery for 7 d, rapamycin (0.2 nmol in 2 µl), or a vehicle (DMSO) was delivered at the rate of 0.25 µl/min 30 minutes before scopolamine injections. This dose of rapamycin is based on previous reports demonstrating effective and selective inhibition of the mTORC1 signaling (15, 16).
Immunoblotting
For analysis of mTORC1 signaling synaptoneurosomes were prepared and western blotting for the phosphorylated forms of mTORC1 signaling proteins, as well as upstream kinases was conducted as previously described (16). The primary antibodies used for both phosphorylated and total proteins were: phospho-mTORC1 (Ser2448), mTORC1, Total p70 S6 kinase (S6K) (Thr389), phospho-S6K, total extracellular-signal regulated kinase (ERK), phospho-ERK (Thr202/Tyr204), total protein kinase B (PKB or Akt), phospho-Akt (all from Cell Signaling, Boston, MA), GluR1 (Abcam, Cambridge, MA), and GAPDH (Advanced Immunochemical, Long Beach, CA). Levels of immunoreactive bands were quantified by densitometry using NIH Image J software and normalized to the control group for each protein.
Brain Slice Preparation and Electrophysiological Recordings
Brain slices were prepared as previously described (16, 17). Briefly, one day after scopolamine treatment, rats were anesthetized (chloral hydrate, 400 mg/kg, i.p.) and brains removed. Coronal slices 400 µm thick were cut from a block of tissue containing the mPFC, placed in a submerged recording chamber at 32 °C in standard ACSF (pH 7.35). There was recovery period of 1–2 hr before recording.
Pyramidal neurons in layer V were patched under visual control using a microscope (60× IR lens; Olympus, Center Valley Pennsylvania) with infrared differential interference contrast microscopy (IR/DIC). The pipette solution contained the following: 115 mM K gluconate, 5 mM KCl, 2 mM MgCl2, 2 mM Mg-ATP, 2 mM Na2ATP, 10 mM Na2-phosphocreatine, 0.4 mM Na2GTP, and 10 mM Hepes, pH 7.33. Neurobiotin (0.3%) was added to the pipette solution to mark cells for later processing and imaging.
Whole-cell recordings were made with an Axoclamp-2B amplifier (Molecular Devices, Sunnyvale, California). The output signal was low-pass-filtered at 3 KHz and digitized at 15 kHz; data were acquired by pClamp 9.2/Digidata 1320 software (Molecular Devices). Series resistance, which was monitored throughout the experiment, was usually between 4 and 8 MΩ. To minimize series resistance errors, cells were discarded if series resistance rose above 10 MΩ. Postsynaptic currents were studied in the continuous single-electrode voltage-clamp mode (3000 Hz low-pass filter) clamped near resting potential (75 mV ± 5 mV). Known concentrations of drugs in ACSF were applied through a stopcock arrangement (~4 ml/min) to reach the slice within 7–10 s.
Spine Density Analysis
After completion of recording, slices were transferred to 4% paraformaldehyde (0.1 M phosphate buffer) and stored overnight at 4°C. Slices were then processed with streptavidin conjugated to the fluorophore Alexa 594 (1:1000) for visualization of labeled cells; this procedure enabled visualization of even the most distant spines in the apical tuft. Labeled neurons within layer V of anterior cingulate and prelimbic mPFC were imaged with a two-photon Ti:sapphire laser scanning system (810 nanometers; Mai Tai, Spectra Physics, Mountain View, California) coupled to direct detection Radiance 2000 BioRad laser scanner (Zeiss Micromaging, Thornwood, New York) mounted on a Olympus BX50WI microscope, using a 60× (0.9 numerical aperture) water-immersion objective. For spine density analysis, Z-stacks usually consisted of 2–5 scans at high zoom at 1-µm steps in the z axis. Spine density was sampled in two zones: (i) tips of tuft branches as they approach the pial membrane and (ii) proximal tuft dendrites distal to the bifurcation of the apical shaft; previous studies had shown that, in contrast to basilar dendrites, the distal tuft in layer V pyramidal cells is especially sensitive to chronic stress (18). Spine density and diameter were analyzed by automated Neuroleucida software (Autoneuron/Autospine; MBF Bioscience, Williston VT). Density results were expressed in terms of spine density per 10 µm.
Behavioral analysis: Forced Swim Test (FST)
Behavioral responses in the FST were conducted as previously described (16). 24 hr prior to the treatment with scopolamine, rats were placed for 15 min in a clear cylinder with water (24±1C, 45cm depth). Rats were administered scopolamine, and 24 hr later were tested for immobilization in the FST. In the blocking studies, rapamycin was administered (0.2 nmol in 2 µl, i.c.v.) or NBQX was administered (10 mg/kg, i.p.) 30 min prior to scopolamine. Video recorded sessions were scored for total immobility time during the first 5min by a blinded experimenter. Brains were collected and cannula placement was determined by histology; rats with incorrect placement were excluded. Immobility values were analyzed using a one-way ANOVA with LSD post-hoc tests as appropriate. Significance was determined at P<0.05 and data were plotted as total seconds immobile.
Microdialysis and glutamate measurement
Rats were anaesthetized with pentobarbital (55 mg/kg, i.p.) and cannula guides CMA / 11 (Harvard Apparatus, Holliston, MA) were stereotaxically implanted in the prefrontal cortex (AP: + 3.5 mm and L: +0.7mm from bregma; DV: 2 mm from dura). After handling daily for 7 days to reduce variability and decrease injection stress the influence of scopolamine and ketamine administration on glutamate levels in freely moving rats was determined. Microdialysis probes (CMA / 11, Harvard Apparatus; 2 mm length of the microdialysis membrane; molecular weight cut-off, 20 kDa) were lowered through the guide cannula so that the tip of the dialysing membrane reached DV: 4 mm from dura). Perfusion solution (Harvard Apparatus) was applied at a constant flow rate (1 µL/min) using a micropump (Harvard Apparatus). Effluents were collected every 10 min and were immediately frozen at −80°C. Sample collection started 90 min after the onset of perfusion to achieve stable conditions, and then collected every 10 min for 60 min before the injection of either ketamine (10 mg/kg, i.p.) or scopolamine (25 µg/kg, i.p.). Sampling was carried out for an additional 120 min period (12 samples). Animals were then injected with saline and sample collection was carried out for another 60 min (6 samples).
Glutamate was measured in the microdialysis effluents by ultraperformance liquid chromatography combined with tandem mass spectrometry (UPLC/MS/MS). Briefly, 6 µL of microdialysis effluent was processed using the EZfaast kit with U-13C-glutamate (Cat # CLM 1800, Cambridge Isotope Laboratories, Andover, MA) added to the internal standard solution. After drying with nitrogen, the derivatized sample was reconstituted with 50 µL of 50% methanol. An aliquot of 5 µL reconstituted sample was injected onto a reverse-phase microbore column using a Waters ACQUITY autosampler/solvent manager (Waters Corporation, Milford, MA). The column (ACQUITY UPLC HSS T3, 1.8 µm particle diameter, 2.1 mm inner diameter, 100 mm length; Waters Corporation) was kept at 55 °C. The sample was separated using the following gradient at a flow of 0.5 mL/min: 0 – 4.5 min: 45 – 35% A (2 mmol/L ammonium acetate and 0.1% formic acid in water), 55 – 65 % B (2 mmol/L ammonium acetate and 0.1% formic acid in methanol); 4.5 – 4.75 min: 35 – 0% A; 4.75 – 5.25 min: 0% A; 5.25 – 5.75 min: 0 – 45% A; 5.75 – 6 min: 45% A. The column effluent was then analyzed by multiple reaction monitoring (MRM) in a Waters/Micromass Quattro Micro tandem mass spectrometer (Waters) after positive ionization by electrospray (ES+). The following mass spectrometry parameters were used: capillary 0.5kV, cone 15–16V, source 120°C, desolvation 350°C, cone gas flow 40 L/hr, desolvation gas flow 850 L/hr, collision 13–14eV, dwell time 0.1s, inter scan delay 0.05s At these conditions, glutamate and U-13C glutamate eluted at 3.9 min as the 318>172 and 323>177 transitions respectively. One blank, five calibrators and two quality control (QC) samples were processed in parallel with the microdialysis samples and included in each run. The area under the curve of glutamate divided by the area under the curve of standard-13C glutamate was used for quantitation. The concentration of glutamate was determined by comparison with a 5-point calibrator curve for the amino acid. The between-run coefficient of variation (CV) of quality control samples measured over a period of several months was <10% for glutamate at 2.5 µM. The limit of quantitation (i.e. the concentration where the between-run CV is 20%) was approximately 0.25 µM.
RESULTS
Muscarinic receptor antagonists rapidly increase mTORC1 signaling
Based on previous studies demonstrating that the rapid acting antidepressant effects of NMDA antagonists (i.e., ketamine) require mTORC1, we hypothesized that scopolamine would also increase mTORC1 signaling and related upstream signaling proteins ERK and Akt (19). A dose of 25 µg/kg was chosen based on preliminary studies indicating that low doses of scopolamine within this range increased mTORC1 signaling, while higher doses (100 µg/kg) had no effect (not shown). This is consistent with the low dose of scopolamine that is used for the treatment of depressed patients (4 µg/kg) (6, 7). Scopolamine administration resulted in a small but significant induction of phosphor-mTOR, phospho-Akt, and phospho-S6K 1 hr after treatment, but no effect on phospho-ERK (Figure 1a–b). Levels of phospho-S6K were also significantly increased 2 hr after scopolamine (Figure 1a–b).
Figure 1. Muscarinic receptor antagonist administration increases mTORC1 signaling in rat frontal cortex.
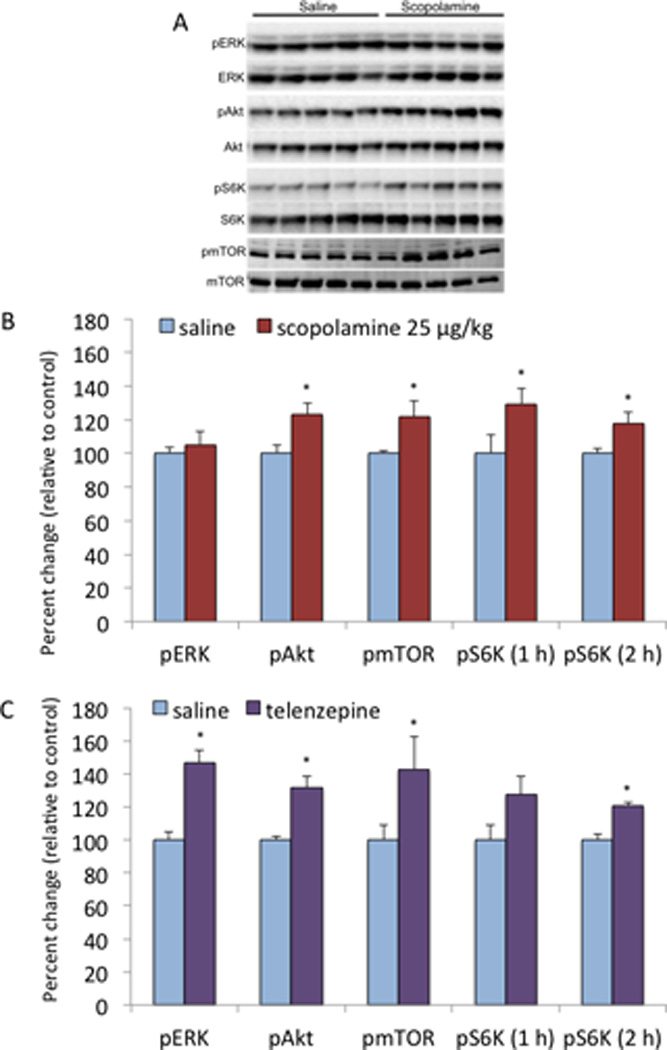
Rats were administered scopolamine (25 µg/kg, i.p.) or telenzapine (3 mg/kg, s.c.) and levels of mTORC1 signaling were determined 1 or 2 hr as indicated. Levels of the phosphorylated and activated forms of mTORC1, ERK, Akt, S6K were determined by western blot analysis. Levels of total mTORC1 and GAPDH were determined to control for loading differences. (a) Representative western blot images for each signaling protein are shown. (b) Results were quantified and are the mean ± SEM, percent of control (n = 6 animals; *P < 0.05, compared to control, Student’s t-test).
We also examined another muscarinic receptor antagonist, telenzepine, that has limited selectivity for the M1 receptor subtype (~6 fold higher affinity for M1 vs. M3) (20). A dose of 3 mg/kg was chosen based on preliminary studies demonstrating that doses of 3 to 10 mg/kg were effective. Telenzepine administration significantly increased levels of phospho-mTOR, as well as phospho-Akt and phospho-ERK 1 hr after administration (Figure 1c). There was a trend for increased levels of phospho-S6K at this time point, that were significantly higher 2 hr after telenzepine administration, indicating a short time lag for induction of this downstream kinase that regulates mRNA translation and synaptic protein synthesis.
Scopolamine rapidly increases the number and function of spine synapses in PFC neurons
Our previous studies demonstrate that ketamine administration also rapidly increases the number and function of spine synapses in the PFC (10), raising the possibility that scopolamine increases spine density and function. A 24 hr time point was chosen because previous studies report anecdotal evidence of an antidepressant therapeutic response to scopolamine (7) and because ketamine-induction of spine number was determined at 24 hr (10). Pyramidal neurons in layer V were visualized in slices of PFC and whole cell recordings were conducted as previously described (16, 17). Scopolamine administration significantly increased the frequency of 5-HT or hypocretin-induced excitatory postsynaptic currents (EPSCs) by ~50 to 75 percent; EPSC amplitude was also increased, as shown by a rightward shift in the cumulative fraction figures (Figure 2a–c).
Figure 2. Scopolamine administration rapidly increases EPSC responses in PFC layer V pyramidal cells.

Rats were administered scopolamine and 24 hr later slices of PFC were prepared for whole-cell recordings followed by neurobiotin labeling. (a) Sample whole cell voltage-clamp traces of 5-HT and hypocretin-induced EPSCs in slices from saline control or scopolamine treated rats (24 hr post drug treatment). (b) Frequency of 5-HT- and hypocretin-induced EPSCs (*p < 0.05 and **p < 0.01 Student’s t-test) (c) Cumulative probability distributions showing that scopolamine administration exposure increases the amplitude of EPSCs (P < 0.001 for 5-HT, Kolmogorov-Smirnov-test z value =3.65; p < 0.001 for Hcrt, Kolmogrov-Smirnov-test z value = 3.83) (n = 18 neurons for 5-HT group; n = 15 neurons for Hcrt group).
While recording, the neurons were filled passively with neurobiotin, and two-photon laser imaging was used for analysis of spine density and morphology. Scopolamine administration significantly increased the density of spines in the distal segments of layer V neurons in the PFC, and caused a non significant increase in proximal dendrites (Figure 3a, b). Scopolamine also produced a small but significant increase in spine head diameter in both the distal and proximal segments of these neurons (Figure 3c, d). The increase in spine number and diameter are consistent with increased synaptic function (i.e., increased frequency and amplitude, respectively, Figure 2) of the inputs to these neurons.
Figure 3. Scopolamine administration increases spine density and maturation in PFC layer V pyramidal cells.

After whole-cell recordings slices were subjected to post hoc two-photon microscopy image of the neurobiotin-labeled layer V pyramidal cells. (a) Representative images are shown of high magnification Z-stack projections of distal and proximal segments of the layer V pyramidal cell apical tuft dendrites (Scale: 10 µm). (b) The density of spines was analyzed using automated software (MBF Bioscience, Neurolucida V10/Autospine) and the results are the mean ± SEM (12 cells from 5 rats for control and 14 cells from 6 rats for scopolamine). (c, d) Quantification of distal and proximal spine head diameter, and cumulative fraction curves; note the increase in population of large diameter, mushroom spines in the scopolamine group as compared to the saline controls *p < 0.05; **p < 0.01; Student’s t-test).
Antidepressant response to scopolamine in the FST requires mTORC1 signaling and glutamate receptor activation
The antidepressant actions of scopolamine and telenzepine were examined 24 hr after scopolamine administration in the FST to be consistent with the synaptogenic studies, as well as previous reports of ketamine (10). Scopolamine administration at a dose of 25 µg/kg or telenzepine at a dose of 3 mg/kg significantly decreased immobility in the FST, indicative of an antidepressant response (Figure 4a). There was also a trend for a response to a higher dose of scopolamine tested (50 µg/kg), although this effect was not significant and the lower dose was used for subsequent studies. In our studies to test the role of mTORC1 signaling, we found that pretreatment with rapamycin (i.c.v.) completely blocked the antidepressant effect of scopolamine in the FST (Figure 4b). Our previous studies have demonstrated that a single injection of rapamycin (i.c.v.) alone at this dose has no effect on immobility in the FST (10). Scopolamine administration did not influence distance traveled in an open field, indicating that the effects in the FST are not due to a generalized effect on locomotor activity (saline 39.52±11.91 m; scopolamine 39.54±11.93 m).
Figure 4. Behavioral actions of scopolamine in the FST require mTORC1 signaling and glutamate-AMPA receptors.

Rats were administered scopolamine or telenzapine at the doses indicated and immobility in the FST was determined 24 hr later. (a) Both scopolamine and telenzapine significantly decreased immobility in the FST at the same doses that increased mTORC1 signaling and synaptogenesis. Values represent mean ± SEM (n = 3 per group (Sco) and n=6 per group (Tel); *P < 0.05 compared to control. (b) Pretreatment with the selective mTORC1 inhibitor rapamycin (0.2 nmol, i.c.v., 30 min before scopolamine) completely blocked the antidepressant effects of scopolamine (25 µg/kg, i.p.). Values represent mean ± SEM (n = 7 per group; *P < 0.05 compared to scopolamine alone (ANOVA). (c) Pretreatment with the selective AMPA receptor antagonist NBQX (10 mg/kg, i.p. 30 min before scopolamine) completely blocked the antidepressant effects of scopolamine (25 µg/kg, i.p.). Values represent mean ± SEM (n = 9; *P < 0.05 compared to control by ANOVA.
The rapid antidepressant actions of ketamine are reported to be dependent on glutamate transmission, and are blocked by pretreatment with an antagonist of the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor (10, 21). Similarly, in the current study we found that pretreatment with the AMPA receptor antagonist NBQX also completely blocked the antidepressant effects of scopolamine on immobility in the FST (Figure 4c).
The influence of systemic injection of scopolamine on extracellular glutamate levels in the PFC was measured by microdialysis in freely moving animals. As a positive control, we also tested ketamine, which is reported to increase extracellular glutamate in the PFC (Moghaddam et al., 1997). Administration of scopolamine (25 µg/kg) or ketamine (10 mg/kg) significantly increased extracellular glutamate levels (repeated measures ANOVA, P < 0.05) (Supplement: Figure S1). The mean increases for scopolamine and ketamine were 145.5 ± 23.8 and 144.3 ± 28.1 (mean ± S.E.M.), respectively. The induction of glutamate was rapid (first 10 min) and transient. There was no significant difference between groups, although ketamine produced slightly higher levels of extracellular glutamate than scopolamine over the 90 min period following drug administration. There was no effect of saline injection administered at 120 min (Figure 5), consistent with our previous experiments showing that post-operative handling and analgesic drug injections (i.e., after dialysis probe surgery) reduce glutamate release due to the injection-stress.
Figure 5. Model depicting the cellular mechanisms for the synaptogenic actions of scopolamine.

Scopolamine rapidly increases the number, maturation, and function of spine synapses in pyramidal neurons in the medial PFC. This could occur via increased glutamate transmission, or a glutamate burst resulting in a long-term potentiation (LTP) like synaptogenic effect. The induction of glutamate could occur via blockade of muscarinic receptors located on GABAergic interneurons, acetylcholine (Ach) terminals, or even glutamate terminals. In addition, postsynaptic muscarinic receptors that couple to protein kinase C (PKC) can produce long-term depression (LTD), and blockade of these receptors could enhance the synaptogenic response to glutamate. Scopolamine also increases mTORC1-p70S6K (S6K) signaling resulting in increased translation of synaptic proteins, including AMPA receptors, which are required for the expansion and stabilization of spines. The antidepressant behavioral responses to scopolamine require activation of the mTORC1 pathway (demonstrated by blockade with rapamycin). The actions of ketamine, another rapid acting antidepressant, require BDNF-TrkB-Akt signaling, and we hypothesize that scopolamine-induction of mTORC1 signaling also requires release of this neurotrophic factor, although this has not yet been tested. The model also shows that depression relapse, as well as stress, are associated with a reduction in spine synapses, due to failure of synaptic homeostasis. This could result from genetic mutations or environmental factors such as sustained stress, and decreased expression of BDNF.
DISCUSSION
The results demonstrate that administration of scopolamine, a muscarinic receptor antagonist with rapid antidepressant actions in humans, leads to fast activation of mTORC1 signaling and increased synaptogenesis in layer V pyramidal neurons of the PFC. These effects are similar to the synaptogenic actions of ketamine (10, 11) and together these findings indicate that mTORC1 signaling and synaptogenesis are key factors underlying the response to rapid acting antidepressants.
The results demonstrate that scopolamine rapidly increases the phosphophorylated and activated forms of mTOR and S6K, two canonical mTORC1 complex 1 signaling proteins (19). Scopolamine also significantly increased phospho-Akt, an upstream kinase that is known to be involved in activation of the mTORC1 signaling pathways (19). As with ketamine (10), higher doses were ineffective. In addition, the results demonstrate that telenzepine, another muscarinic receptor antagonist with modest selectivity for M1 receptors (~6 fold for M1 vs. M3), significantly increased phosphorylated mTOR and S6K, as well as the ERK and Akt. The reason for the more robust effects of telenzepine, particularly on ERK, could be related to the selectivity of this agent (20), although further studies with more selective M1 antagonists will be required to test this hypothesis. Together the results demonstrate that muscarinic receptor blockade results in activation of mTORC1 signaling, similar to the actions of NMDA receptor blockade.
Scopolamine administration also increased the number and function of spine synapses in layer V pyramidal neurons of the PFC. The increased frequency and amplitude of 5-HT and hypocretin-induced EPSCs indicates an increase of cortical-cortical (5-HT) and thalamocortical (hypocretin) synaptic inputs, respectively (17). Increased spine number was most apparent in the distal tuft dendrites of layer V neurons, with a non-significant trend in the proximal tuft. Similarly, there was an increase in the population of large-diameter mushroom spines in the distal tuft but only a marginal albeit significant change proximally. These changes in spine density and diameter are less robust than previously observed after one dose of ketamine (10). It is possible that greater effects would be observed after 2 or 3 doses of scopolamine, as reported for the antidepressant actions of repeated scopolamine dosing in depressed patients (6, 7). In any case, the overall increase in the total and large diameter spines correlates with significant increases in the frequency and amplitude, respectively, of 5-HT- and hypocretin-induced EPSCs.
The influences of scopolamine on antidepressant behavior and the role of mTORC1 signaling were examined in the FST model by pretreatment with the selective mTORC1 inhibitor, rapamycin. A single dose of scopolamine or telenzepine significantly decreased immobility in the FST, consistent with previous reports demonstrating antidepressant actions of muscarinic receptor antagonists (25, 26). The behavioral actions of ketamine are dependent on mTORC1 signaling and glutamate AMPA transmission (10, 21, 27). Here we find that rapamycin pretreatment completely blocked the effects of scopolamine in the FST, similar to rapamycin-blockade of ketamine (10, 27). In addition, pretreatment with the AMPA receptor antagonist NBQX completely blocked the antidepressant effects of scopolamine in the FST, demonstrating a requirement for glutamate-AMPA activity. Recent studies also demonstrate that mGlu2/3 receptor antagonists, which increase glutamate transmission via blockade of synaptic autoreceptors, also produce antidepressant responses that require mTORC1 signaling (27, 28). Further studies will be required to demonstrate the mTORC1 signaling is also required for the synaptogenic actions of scopolamine, and to determine if scopolamine is capable of producing a rapid response in chronic stress models that require chronic administration of a conventional antidepressant. However, the current findings clearly demonstrate that mTORC1 signaling and AMPA receptor activation are required for the behavioral actions of scopolamine in the FST, and importantly show a link with the mechanisms underlying the actions of ketamine.
The results also demonstrate that scopolamine rapidly increases extracellular glutamate levels in medial PFC, determined by microdialysis. Application of scopolamine via the dialysis probe into the striatum is also reported to increase extracellular glutamate in this region (29). Although the exact cellular mechanisms are unknown, electrophysiology studies demonstrate that muscarinic receptors can influence pyramidal neuron excitability via effects on multiple receptor subtypes and processes (30). This includes inhibition of muscarinic receptors on GABAergic interneurons, which would result in disinhibition of glutamate transmission. Consistent with this possibility, M1 agonist incubation increases GABA overflow in striatum (31). A similar mechanism is thought to underlie ketamine-induction of extracellular glutamate in the PFC (32, 33). Based on these findings it is possible that transient cortical disinhibition represents a common feature of both ketamine and scopolamine (Figure 5). The transient nature of this effect may be critical as sustained induction of glutamate could produce toxic, damaging effects. Additional studies will be required to test the hypothesis that blockade of GABAergic interneurons underlies the induction of glutamate transmission and the rapid antidepressant actions of scopolamine, as well as ketamine.
Muscarinic receptors can also influence synaptic activity and function via additional mechanisms. Muscarinic agonists produce long-term depression at postsynaptic sites via M1 activation of protein kinase C or via activation of hippocalcin leading to AMPA and/or NMDA receptor internalization (34, 35), and M1 receptor blockade could enhance glutamate transmission via inhibition of these mechanisms (Figure 5). Presynaptic muscarinic receptors also influence the release of glutamate, as well as acetylcholine (30). Muscarinic receptor antagonists could influence glutamate release via one or more of these mechanisms and thereby contribute to a burst of glutamate transmission that results in transient long-term potentiation like effects and increased synaptogenesis (Figure 5).
The synaptogenic and behavioral actions of scopolamine, like ketamine (10), are observed after 24 hr, consistent with anecdotal reports of the rapid antidepressant actions of scopolamine (6–9). Together the results support the hypothesis that the induction of synaptogenesis is a delayed response to scopolamine-activation of mTORC1 signaling and the resulting increase in levels of synaptic proteins (Figure 6). Scopolamine-induction of spines synapses would be expected to block or reverse the effects of chronic stress and depression, which are known to decrease the number and function of spines on PFC neurons (36–40). These effects could also lead to reversal of the decreased volume of the PFC in depressed patients (41, 42). Together, these findings raise the possibility that scopolamine, like ketamine (11), causes a functional reconnection of PFC neurons that could reinstate its ability to modulate amygdala control of emotion and mood (43).
The ability of muscarinic receptor antagonists, as well as ketamine, to rapidly increase synaptogenesis provides further evidence for the role of synaptic plasticity in the treatment of depression. The results of the present study demonstrate that another pharmacological agent reported to have rapid acting antidepressant effects in depressed patients can rapidly stimulate the formation and function of new spines/synapses. Brain imaging studies that directly or indirectly measure synaptogenesis and/or glutamate transmission will be required to directly test this hypothesis in depressed patients.
In summary, the results provide direct evidence that muscarinic receptor antagonists increase mTORC1 signaling and synaptogenesis in the PFC and produce rapamycin sensitive antidepressant behavioral responses. The results provide novel targets for development of next-generation fast-acting antidepressant medications, including selective muscarinic receptor antagonists that could potentially be more efficacious and have fewer side effects than scopolamine. Studies to address this issue are currently underway, although many of the selective agents must be infused directly into the brain because of lack of brain penetration. This question is also being addressed with mice with a null mutation of selected muscarinic receptor subtypes to identify the key receptors that mediate the actions of scopolamine.
Supplementary Material
Acknowledgement
This work is supported by United States Public Health Service Grants MH45481 (RSD) and 2 P01 MH25642 (RSD), R01 MH081211 (GS), the Connecticut Mental Health Center (RSD).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Financial Disclosures
The authors reported no biomedical financial interests or potential conflicts of interest.
References
- 1.Kessler RC, Berglund P, Demler O, Jin R, Koretz D, Merikangas KR, et al. The epidemiology of major depressive disorder: results from the National Comorbidity Survey Replication (NCS-R) JAMA. 2003;289:3095–3105. doi: 10.1001/jama.289.23.3095. [DOI] [PubMed] [Google Scholar]
- 2.Trivedi MH, Rush AJ, Wisniewski SR, Nierenberg AA, Warden D, Ritz L, et al. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry. 2006;163:28–40. doi: 10.1176/appi.ajp.163.1.28. [DOI] [PubMed] [Google Scholar]
- 3.Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, et al. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. 2000;47:351–354. doi: 10.1016/s0006-3223(99)00230-9. [DOI] [PubMed] [Google Scholar]
- 4.Zarate CA, Jr, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, et al. A randomized trial of an N-methyl-D-aspartate antagonist in treatmentresistant major depression. Arch Gen Psychiatry. 2006;63:856–864. doi: 10.1001/archpsyc.63.8.856. [DOI] [PubMed] [Google Scholar]
- 5.Liebrenz M, Borgeat A, Leisinger R, Stohler R. Intravenous ketamine therapy in a patient with a treatment-resistant major depression. Swiss Med Wkly. 2007;137:234–236. doi: 10.4414/smw.2007.11852. [DOI] [PubMed] [Google Scholar]
- 6.Drevets W, Furey ML. Replication of scopolamine's antidepressant efficacy in major depressive disorder: A randomized, placebo-controlled clinical trial. Biol Psych. 2010;67:432–438. doi: 10.1016/j.biopsych.2009.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Furey M, Drevets WC. Antidepressant efficacy of the antimuscarinic drug scopolamine: a randomized, placebo-controlled clinical trial. Arch Gen Psych. 2006;63:1121–1129. doi: 10.1001/archpsyc.63.10.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Furey M, Khanna A, Hoffman EM, Drevets WC. Scopolamine produces larger antidepressant and antianxiety effects in women than in men. Neuropsychopharmacol. 2010;35:2479–2488. doi: 10.1038/npp.2010.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Furey M, Nugent AC, Speer AM, Luckenbaugh DA, Hoffman EM, Frankel E, Drevets WC, Zarate CA., Jr Baseline mood-state measures as predictors of antidepressant response to scopolamine. Psychiatry Res. 2012 doi: 10.1016/j.psychres.2012.01.003. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li N, Lee BY, Liu RJ, Banasr M, Dwyer J, Iwata M, Li XY, Aghajanian G, Duman RS. mTORC1-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 2010;329:959–964. doi: 10.1126/science.1190287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li N, Liu R-J, Dwyer J, Banasr M, Lee B, Son J, Li X-Y, Aghajanian G, Duman RS. Glutamate N-methyl-D-aspartate receptor antagonists rapidly reverse behavioral and synaptic deficits caused by chronic stress exposure. Biol Psych. 2011;69:754–761. doi: 10.1016/j.biopsych.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Preskorn S, Baker B, Omo K, Kolluri S, Menniti FS, Landen JW. A placebo-controlled trial of the NR2B subunit specific NMDA antagonist CP-101,606 plus paroxetine for treatment resistant depression (TRD) APA. 2007 [Google Scholar]
- 13.Hoeffer CA, Klann E. mTORC1 signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci. 2010;33:67–75. doi: 10.1016/j.tins.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jernigan C, Goswami DB, Austin MC, Iyo AH, Chandran A, Stockmeier CA, Karolewicz B. The mTORC1 signaling pathway in the prefrontal cortex is compromised in major depressive disorder. Prog Neuro-Psychopharmacol & Biol Psych. 2011;35:1774–1779. doi: 10.1016/j.pnpbp.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cota D, Proulx K, Smith KA, Kozma SC, Thomas G, Woods SC, et al. Hypothalamic mTORC1 signaling regulates food intake. Science. 2006;312:927–930. doi: 10.1126/science.1124147. [DOI] [PubMed] [Google Scholar]
- 16.Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, Iwata M, et al. mTORC1- dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 2010;329:959–964. doi: 10.1126/science.1190287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu RJ, Aghajanian GK. Stress blunts serotonin- and hypocretin-evoked EPSCs in prefrontal cortex: role of corticosterone-mediated apical dendritic atrophy. Proc Natl Acad Sci U S A. 2008;105:359–364. doi: 10.1073/pnas.0706679105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu R-J, Aghajanian GK. Stress blunts serotonin- and hypocretinevoked EPSCs in prefrontal cortex: role of corticosterone-mediated apical dendritic atrophy. PNAS. 2008;105:359–364. doi: 10.1073/pnas.0706679105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoeffer C, Klann E. mTORC1 signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci. 2010;33:67–75. doi: 10.1016/j.tins.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Doods H, Mathy MJ, Davidesko D, van Charldorp KJ, de Jonge A, van Zwieten PA. Selectivity of muscarinic antagonists in radioligand and in vivo experiments for the putative M1, M2 and M3 receptors. J Pharm Exp Ther. 1987;242:257–262. [PubMed] [Google Scholar]
- 21.Maeng S, Zarate CA, Jr, Du J, Schloesser RJ, McCammon J, Chen G, Manji HK. Cellular mechanisms underlying the antidepressant effects of ketamine: role of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors. Biol Psych. 2008a;63:349–352. doi: 10.1016/j.biopsych.2007.05.028. [DOI] [PubMed] [Google Scholar]
- 22.Berman R, Cappiello A, Anand A, et al. Antidepressant effects of ketamine in depressed patients. Biol Psychiat. 2000;47:351–354. doi: 10.1016/s0006-3223(99)00230-9. [DOI] [PubMed] [Google Scholar]
- 23.Machado-Vieira R, Ibrahim L, Henter ID, Zarate CA., Jr Novel glutamatergic agents for major depressive disorder and bipolar disorder. Pharmacol Biochem Behav. 2012;100:678–687. doi: 10.1016/j.pbb.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zarate CJ, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, Charney DS, Manji HK. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psych. 2006b;63:856–864. doi: 10.1001/archpsyc.63.8.856. [DOI] [PubMed] [Google Scholar]
- 25.Borsini F, Lecci A, Sessarego A, Frassine R, Meli A. Discovery of antidepressant activity by forced swimming test may depend on pre-exposure of rats to a stressful situation. Psychopharmacol. 1989;97:183–188. doi: 10.1007/BF00442247. [DOI] [PubMed] [Google Scholar]
- 26.Kitada Y, Miyauchi T, Satoh A, Satoh S. Effects of antidepressants in the rat forced swimming test. Eur J Pharmacol. 1981;72:145–152. doi: 10.1016/0014-2999(81)90269-7. [DOI] [PubMed] [Google Scholar]
- 27.Koike H, Lijima M, Chaki S. Involvement of the mammalian target of rapamycin signaling in the antidepressant-like effect of group II metabotropic glutamate receptor antagonists. Neuropharmacol. 2011;61:1419–1423. doi: 10.1016/j.neuropharm.2011.08.034. [DOI] [PubMed] [Google Scholar]
- 28.Dwyer J, Lepack AE, Duman RS. mTORC1 activation is required for the antidepressants effects of mGluR2/3 blockade. 2011 doi: 10.1017/S1461145711001702. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rawls S, McGinty JF. Muscarinic receptors regulate extracellular glutamate levels in the rat striatum: an in vivo microdialysis study. J Pharmacol Exp Ther. 1998;286:91–98. [PubMed] [Google Scholar]
- 30.Amar M, Lucas-Meunier E, Baux G, Fossler P. Blockade of different muscarinic receptor subtypes changes the equilibrium between excitation and inhibition in rat visual cortes. Neurosci. 2010;169:1610–1620. doi: 10.1016/j.neuroscience.2010.06.019. [DOI] [PubMed] [Google Scholar]
- 31.Harsing L, Zigmond MJ. Postsynaptic integration of cholinergic and dopaminergic signals on medium-sized GABAergic projection neurons in the neostriatum. Br Res Bill. 1998;45:607–613. doi: 10.1016/s0361-9230(97)00460-7. [DOI] [PubMed] [Google Scholar]
- 32.Homayoun H, Moghaddam B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J Neurosci. 2007;27:11496–11500. doi: 10.1523/JNEUROSCI.2213-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moghaddam B, Adams B, Verma A, Daly D. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J Neurosci. 1997;17:2912–2917. doi: 10.1523/JNEUROSCI.17-08-02921.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Caruana D, Warburton EC, Bashir ZI. Induction of Activity-Dependent LTD Requires Muscarinic Receptor Activation in Medial Prefrontal Cortex. J Neurosci. 2011;31:18464–18478. doi: 10.1523/JNEUROSCI.4719-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jo J, Son GH, Winters BL, Kim MJ, Whitcomb DJ, Dickinson BA, Lee YB, Futai K, Amici M, Sheng M, Collingridge GL, Cho K. Muscarinic receptors induce LTD of NMDAR EPSCs via a mechanism involving hippocalcin. Nat Neurosci. 2010;13:1216–1224. doi: 10.1038/nn.2636. [DOI] [PubMed] [Google Scholar]
- 36.Radley JJ, Rocher AB, Janssen WG, Hof PR, McEwen BS, Morrison JH. Reversibility of apical dendritic retraction in the rat medial prefrontal cortex following repeated stress. Exp Neurol. 2005;196:199–203. doi: 10.1016/j.expneurol.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 37.Radley JJ, Morrison JH. Repeated stress and structural plasticity in the brain. Ageing Res Rev. 2005;4:271–287. doi: 10.1016/j.arr.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 38.Liston C, Miller MM, Goldwater DS, Radley JJ, Rocher AB, Hof PR, et al. Stress-induced alterations in prefrontal cortical dendritic morphology predict selective impairments in perceptual attentional set-shifting. J Neurosci. 2006;26:7870–7874. doi: 10.1523/JNEUROSCI.1184-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Radley JJ, Rocher AB, Miller M, Janssen WG, Liston C, Hof PR, et al. Repeated stress induces dendritic spine loss in the rat medial prefrontal cortex. Cereb Cortex. 2006;16:313–320. doi: 10.1093/cercor/bhi104. [DOI] [PubMed] [Google Scholar]
- 40.Goldwater DS, Pavlides C, Hunter RG, Bloss EB, Hof PR, McEwen BS, et al. Structural and functional alterations to rat medial prefrontal cortex following chronic restraint stress and recovery. Neuroscience. 2009;164:798–808. doi: 10.1016/j.neuroscience.2009.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bremner J, Vythilingam M, Vermetten E, Nazeer A, Adil J, Khan S, Staib LH, Charney DS. Reduced volume of orbitofrontal cortex in major depression. Biol Psychiat. 2002;51:273–279. doi: 10.1016/s0006-3223(01)01336-1. [DOI] [PubMed] [Google Scholar]
- 42.Drevets WC, Price JL, Simpson JR, Todd RD, Reich T, Vannier M, Raichle ME. Subgenual prefrontal cortex abnormalities in mood disorders. Nature. 1997;386:824–827. doi: 10.1038/386824a0. [DOI] [PubMed] [Google Scholar]
- 43.Milad M, Quirk GJ. Fear extinction as a model for translational neuroscience: ten years of progress. Annu Rev Psychol. 2012;63:129–151. doi: 10.1146/annurev.psych.121208.131631. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.