

Venous thromboembolism (VTE), including deep vein thrombosis (DVT) and its life-threatening complication, pulmonary embolism (PE), are among the most frequent causes of morbidity and mortality in developed countries. In the United States alone, the number of deaths due to VTE approaches 300,000 annually [1]. Blood flow restriction or stasis is considered a major factor driving DVT [2]. Regardless of its initial cause (bed-ridden position, long-haul flights, limb paralysis, etc.), delayed blood renewal in stasis is believed to produce limited oxygen supply to the vein walls (hypoxia), especially in the valvular sinus, which triggers thrombus development [2].
Certain circumstances may lead to an imbalance between tissue demand and actual oxygen supply. This can be caused by tissue requirement exceeding the capacity of oxygen delivery systems (e.g., physical exercise) or inadequate oxygen content in the ambient air. The latter case can be found in situations such as mountain climbing or commercial flights where the cabin is pressurized to a level corresponding to an altitude of 1.5 – 2.5 kilometers [3, 4]. Indeed, clinical studies have linked recent air travel of >4 hours to increased risk of developing DVT [5] and traveling patients with pro-thrombotic mutations predisposing to DVT are at even higher risk for thrombosis [6]. Moreover, an "absolute" risk for VTE in a ratio of 1 event per about 4,500 flights has been reported [7]. Even in healthy individuals, oxyhemoglobin saturation is decreased by 5 – 10% during flight, whereas in patients with chronic obstructive pulmonary disease a greater decrease has been reported [4].
In the majority of cases, hypoxia is followed by restoration of oxygen supply, i.e., reoxygenation. Hypoxia-reoxygenation (H/R), which is known to be harmful to tissues [8, 9], is proposed to be an exacerbating factor for thrombus development in veins although this causal relationship has not been directly proven. A crucial role of the reoxygenation phase in DVT can be suggested because thrombotic events frequently occur after landing [7, 10]. We report that exposure to 6% oxygen for 24 hours followed by 1–3 hours of reoxygenation in normal room air led to significantly increased thrombus prevalence in a mouse model of DVT. Eleven of 15 mice (73%) developed a thrombus after one hour stenosis of the inferior vena cava (IVC) as compared to 1 of 8 (13%) in control animals maintained in normoxic conditions (Figure A,B) (P<0.01). Weight and length of the thrombi from animals that underwent H/R significantly exceeded those in the control group. Thus, mice that were subjected to IVC stenosis after H/R demonstrated a clear pro-thrombotic phenotype.
Figure.
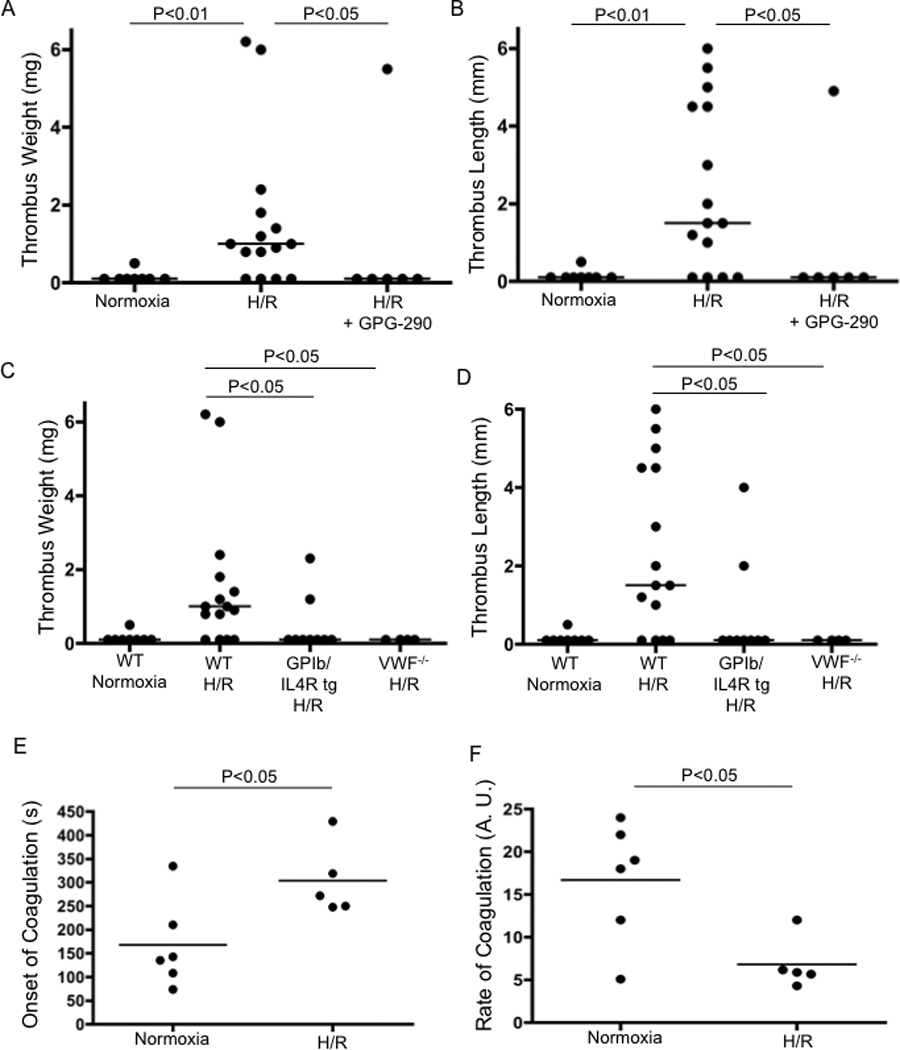
Hypoxia/Reoxygenation (H/R) promotes DVT. Mice subjected to H/R or normoxia followed by 1 h of IVC stenosis were euthanized and thrombi examined. Thrombus weight and length are shown in WT (normoxic), n=8; WT (H/R), n=15; WT (H/R + GPG-290), n=6; TgGP1bα/IL4R, n=9 and VWF−/− mice, n=4 (A–D). Onset and rate of coagulation were measured in WT mice exposed to normoxia (n=6) or H/R (n=5) (E, F).
Endothelial cells contain Weibel-Palade bodies (WPB) that store multiple thrombosis- and inflammation-related constituents, such as von Willebrand factor (VWF) and P-selectin [11–13] and hypoxia is a known inducer of WPB secretion [14]. von Willebrand factor is a large multimeric protein that mediates platelet adhesion and recruitment via its binding to receptors GPIbα and GPIIb-IIIa on the platelet surface [15]. Using a mouse model, we have recently demonstrated that release of WPB is a central event in the initiation of DVT [16]. This implies that DVT is most likely a thrombo-inflammatory pathology rather than a disease entirely based on defects of the hemostatic system. Liberation of VWF and platelet recruitment through the VWF/GPIbα axis was of particular importance. Therefore, we hypothesized that H/R would activate these pathways and thus promote DVT. Indeed, we confirmed that exposure to hypoxia for 24 hours followed by reoxygenation significantly elevated plasma VWF levels in our hypoxia model (data not shown). We therefore tested whether H/R exerts its pro-thrombotic effect through VWF. We administered GPG-290, a chimeric protein that prevents GPIbα interaction with VWF to wild-type (WT) mice as well as utilized mice that lack either the extracellular domain of GPIbα or VWF. Infusion of GPG-290 abolished the pro-thrombotic effect of H/R (Figure A,B). Mice lacking the extracellular domain of GPIbα and VWF−/− mice were protected against H/R-promoted DVT (22 and 0% of mice with a thrombus, respectively; thrombus analysis is shown in figure C,D). Thus, VWF released from WPBs and platelet binding through GPIbα are likely implicated in H/R-induced DVT.
It has been reported that hypoxia does not induce a pro-coagulant shift sufficient to explain thrombosis [17, 18]. In our model, we have confirmed these reports as the time to clotting onset was increased and clotting rate reduced in recalcified whole blood samples after H/R (Figure E,F), suggesting slower fibrin formation compared to normoxic samples.
Here, we demonstrate that H/R accelerates thrombosis in a mouse model of DVT. Interestingly, increased incidence of DVT under H/R conditions occurs despite decreased ex vivo blood clotting. The process is dependent on VWF-mediated interactions indicating that H/R enhances WPB secretion initiating rapid thrombosis in the stenosis model [16]. Many factors are proposed to lead to DVT in long flights, such as hypoxia in the cabin, dehydration and motionless limbs [4]. We demonstrate that hypoxia is at least partially responsible for this unfortunate side effect of travel.
Mice
Wild-type C57BL/6J mice purchased from the Jackson laboratory (Bar Harbor, ME, USA). GPIbα/IL4R mice [19] and VWF−/− mice [20] were also on C57BL/6J background. All experimental procedures involving mice were approved by the Animal Care and Use Committee of the Immune Disease Institute.
Hypoxia/Reoxygenation
Male mice, 7–8 weeks of age, were housed in a controlled atmosphere animal chamber (A-15274-P, Biospherix, Lacona, NY, USA) with 6.0 ± 0.2% oxygen for 24 h (hypoxia). Upon removal from the hypoxic chamber, mice were reoxygenated in normal room air for at least one hour and DVT surgery was performed during 1–3 hours of reoxygenation.
DVT model in mice
DVT was induced by applying stenosis to the inferior vena cava (IVC) as described [16]. Animals were re-opened after 1 h, thrombi were excised and their length and weight were measured. DVT on untreated WT (normoxic and H/R) groups was carried out in every experiment thus thrombus evaluation is combined in panels A–D.
Administration of GPG-290
GPG-290, a soluble chimeric GPIbα conjugated to the Fc fragment of human IgG was infused i.v. through the retroorbital plexus immediately after DVT surgery as previously described [16].
Whole blood recalcification analysis
The test was performed using the Sonoclot analyzer and non-activated clotting test kit (Sienco, CO) as described [21].
Statistics
Weight and length of thrombi were compared using the Mann-Whitney test. Thrombi prevalence between groups was compared by Fisher's exact test. Student's t-test was used for comparison of the blood clotting parameters. P<0.05 was considered statistically significant.
Acknowledgements
We thank Lesley Cowan for assistance with manuscript preparation and Ashish Bhandari for assistance with DVT surgery. This work was generously supported by National Heart, Lung and Blood Institute of the National Institutes of Health grants R01 HL041002 and RO1 HL095091 (to D.D.W.) and a National Research Service Award from the National Institutes of Neurological Disease and Stroke of the National Institutes of Health grant 1F32NS073245 (to G.L.S.).
Footnotes
Author Contributions
A.B. contributed to concept and design, analysis and/or interpretation, critical writing and final approval. G.L.S. contributed to concept and design, analysis and/or interpretation, critical writing and final approval. D. D. W contributed to concept and design, critical writing and final approval.
Disclosure of Conflicts of Interest: The authors state that they have no conflict of interest.
References
- 1.Heit JA. The epidemiology of venous thromboembolism in the community. Arterioscler Thromb Vasc Biol. 2008;28:370-2l. doi: 10.1161/ATVBAHA.108.162545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bovill EG, van der Vliet A. Venous valvular stasis-associated hypoxia and thrombosis: what is the link? Annu Rev Physiol. 2011;73:527-45l. doi: 10.1146/annurev-physiol-012110-142305. [DOI] [PubMed] [Google Scholar]
- 3.Gavish I, Brenner B. Air travel and the risk of thromboembolism. Intern Emerg Med. 2011;6:113–16l. doi: 10.1007/s11739-010-0474-6. [DOI] [PubMed] [Google Scholar]
- 4.Silverman D, Gendreau M. Medical issues associated with commercial flights. Lancet. 2009;373:2067–277l. doi: 10.1016/S0140-6736(09)60209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ferrari E, Chevallier T, Chapelier A, Baudouy M. Travel as a risk factor for venous thromboembolic disease: a case-control study. Chest. 1999;115:440–44l. doi: 10.1378/chest.115.2.440. [DOI] [PubMed] [Google Scholar]
- 6.Rosendaal FR. Air travel and thrombosis. Pathophysiol Haemost Thromb. 2002;32:341-2l. doi: 10.1159/000073594. [DOI] [PubMed] [Google Scholar]
- 7.Kuipers S, Cannegieter SC, Middeldorp S, Robyn L, Buller HR, Rosendaal FR. The absolute risk of venous thrombosis after air travel: a cohort study of 8,755 employees of international organisations. PLoS Med. 2007;4:e290l. doi: 10.1371/journal.pmed.0040290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li C, Jackson RM. Reactive species mechanisms of cellular hypoxiareoxygenation injury. Am J Physiol Cell Physiol. 2002;282:C227–C41l. doi: 10.1152/ajpcell.00112.2001. [DOI] [PubMed] [Google Scholar]
- 9.Wiles ME, Hechtman HB, Morel NM, Shepro D. Hypoxia reoxygenation-induced injury of cultured pulmonary microvessel endothelial cells. J Leukoc Biol. 1993;53:490-7l. doi: 10.1002/jlb.53.5.490. [DOI] [PubMed] [Google Scholar]
- 10.Kesteven P, Robinson B. Incidence of symptomatic thrombosis in a stable population of 650,000: travel and other risk factors. Aviat Space Environ Med. 2002;73:593-6l. [PubMed] [Google Scholar]
- 11.Wagner DD, Olmsted JB, Marder VJ. Immunolocalization of von Willebrand protein in Weibel-Palade bodies of human endothelial cells. J Cell Biol. 1982;95:355–60l. doi: 10.1083/jcb.95.1.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McEver RP, Beckstead JH, Moore KL, Marshall-Carlson L, Bainton DF. GMP-140, a platelet alpha-granule membrane protein, is also synthesized by vascular endothelial cells and is localized in Weibel-Palade bodies. J Clin Invest. 1989;84:92-9l. doi: 10.1172/JCI114175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bonfanti R, Furie BC, Furie B, Wagner DD. PADGEM (GMP140) is a component of Weibel-Palade bodies of human endothelial cells. Blood. 1989;73:1109–112l. [PubMed] [Google Scholar]
- 14.Pinsky DJ, Naka Y, Liao H, Oz MC, Wagner DD, Mayadas TN, Johnson RC, Hynes RO, Heath M, Lawson CA, Stern DM. Hypoxia-induced exocytosis of endothelial cell Weibel-Palade bodies. A mechanism for rapid neutrophil recruitment after cardiac preservation. J Clin Invest. 1996;97:493–500l. doi: 10.1172/JCI118440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bergmeier W, Chauhan AK, Wagner DD. Glycoprotein Ibalpha and von Willebrand factor in primary platelet adhesion and thrombus formation: lessons from mutant mice. Thromb Haemost. 2008;99:264–70l. doi: 10.1160/TH07-10-0638. [DOI] [PubMed] [Google Scholar]
- 16.Brill A, Fuchs TA, Chauhan AK, Yang JJ, De Meyer SF, Kollnberger M, Wakefield TW, Lammle B, Massberg S, Wagner DD. von Willebrand factor-mediated platelet adhesion is critical for deep vein thrombosis in mouse models. Blood. 2011;117:1400–147l. doi: 10.1182/blood-2010-05-287623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Toff WD, Jones CI, Ford I, Pearse RJ, Watson HG, Watt SJ, Ross JA, Gradwell DP, Batchelor AJ, Abrams KR, Meijers JC, Goodall AH, Greaves M. Effect of hypobaric hypoxia, simulating conditions during long-haul air travel, on coagulation, fibrinolysis, platelet function, and endothelial activation. Jama. 2006;295:2251–261l. doi: 10.1001/jama.295.19.2251. [DOI] [PubMed] [Google Scholar]
- 18.Hodkinson PD, Hunt BJ, Parmar K, Ernsting J. Is mild normobaric hypoxia a risk factor for venous thromboembolism? J Thromb Haemost. 2003;1:2131-3l. doi: 10.1046/j.1538-7836.2003.00407.x. [DOI] [PubMed] [Google Scholar]
- 19.Kanaji T, Russell S, Ware J. Amelioration of the macrothrombocytopenia associated with the murine Bernard-Soulier syndrome. Blood. 2002;100:2102–217l. doi: 10.1182/blood-2002-03-0997. [DOI] [PubMed] [Google Scholar]
- 20.Denis C, Methia N, Frenette PS, Rayburn H, Ullman-Cullere M, Hynes RO, Wagner DD. A mouse model of severe von Willebrand disease: defects in hemostasis and thrombosis. Proc Natl Acad Sci U S A. 1998;95:9524–959l. doi: 10.1073/pnas.95.16.9524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brill A, Yesilaltay A, De Meyer SF, Kisucka J, Fuchs TA, Kocher O, Krieger M, Wagner DD. Extrahepatic high-density lipoprotein receptor SR-BI and apoA-I protect against deep vein thrombosis in mice. Arterioscler Thromb Vasc Biol. 2012;32:1841–1871. doi: 10.1161/ATVBAHA.112.252130. [DOI] [PMC free article] [PubMed] [Google Scholar]