

Abstract
A major concern in treating premature infants with birth-associated head trauma is the rapid determination of reliable biomarkers of neuroinflammation. To this end a chip-based immunoaffinity CE device has been applied to determine the concentrations of inflammation-associated chemokines in samples of cerebral spinal fluid collected from such subjects. The chip utilizes replaceable immunoaffinity disks, to which reactive antibody fragments (FAb) of 6 anti-chemokine-specific antibodies were immobilized. Following injection of a sample into the device, the analytes were captured by the immobilized FAbs, labelled in-situ with a red laser dye, chemically released and separated by CE. Each resolved peak was measured on-line by laser-induced fluorescence detection and the results compared to standard curves produced by running known chemokine standards through the immunoaffinity system. The complete processing of a sample took 10 min with separation of all 6 analytes being achieved in less than 2 min. The system compared well to commercial ELISA, analysis of the results by linear regression demonstrating r2 values in the range of 0.903 – 0.978 and intra- and inter-assay coefficients of variance (CV) of the migration times and the measured peak areas being less than 2.3% and 5%, respectively. Application of the system to analysis of cerebrospinal fluid from head traumatized babies clearly indicated the group with mild trauma versus those with severe injury. Additionally, CE analysis demonstrated that the severe trauma group could be divided into individuals with good and poor prognosis, which correlated with the clinical finding for each patient.
Keywords: Chip-based immunoaffinity capillary electrophoresis, Head trauma, Preterm babies, Chemokines, Miniaturization
1. Introduction
Traumatic head injury during birth is a major concern in preterm babies often causing the onset of cranial inflammation which can lead to localized or more extensive tissue injury [1, 2]. Therefore it is important that assessment of the pathophysiological mediators be performed quickly in order to start effective therapy. Clinical assessment of central nervous system (CNS) pro-inflammatory mediators, such as cytokines and chemokines, can be a useful tool for when assessing the outcome of such injury [3, 4]. The chemokines monocyte chemoattractive protein-1 (CCL2) and interleukin-8 (CXCL8) are associated with the activation of phagocytic cells and their migration into local tissue. Both chemokines play important roles in the initiation of an inflammatory response and several reports have demonstrated that elevated concentrations of the both chemokines can be detected in the cerebrospinal fluid (CSF) of head trauma patients [5-8]. However, prolonged inflammatory responses are associated with the influx of both phagocytic cells and lymphocytes, the presence of the latter often leading to severe tissue injury [9]. Recently, interest has focused on the role of B cells as antigen presenting cells to T cells in neuroinflammation and assessment of B cell-attracting chemokines, such as macrophage inflammatory protein-3 (CCL19), secondary lymphoid-tissue chemokine (CCL21), stromal cell derived factor-1 (CXCL12) and B cell attracting chemokine-1 (CXCL13), have been shown to influence the migration of B cells into brain tissue [10]. These chemokines have been detected in CNS tissue and CSF of patients with clinically diagnosed neuroinflammation [8, 10-12]. CXCL13 has been shown to play a role in the formation of ectopic lymphoid tissue in the CNS with elevated concentrations in the CSF of patients with multiple sclerosis and neuroinflammation [8, 13]. Kowarik and colleagues demonstrated that CXCL13 was elevated in patients with neuroinflammation and concluded that this chemokine was responsible for the recruitment of B-cells into the central nervous system [10]. CXCL13, together with CXCL12 are chemoattractive for B cells, T cells and monocytes and both can be elevated in inflammatory diseases of the CNS [10]. The chemokines, CCL19 and CCL21 are chemoattractive for B cells, certain subsets of T cells and mature dendritic antigen-presenting cells [10]. Additionally, CCL19 has been shown to be elevated in the CSF of patients with neuroinflammation and multiple sclerosis [14]. Pro-inflammatory mediator assessment in the CSF of infants has been performed by a number of different immunoassays including ELISA [10, 15-17] and multiplexed bead assays [17-19]. Although both of these techniques are capable of analysing multiple samples in the same run, they do require at least 3 h to perform the analyses and often require reasonably large sample volumes (circa 50-100 μL).
Considering the importance of rapid diagnosis and treatment in pre-term babies with traumatic head injury at birth, there is a need for analytical tests capable of rapidly detecting and measuring biomarkers of inflammation. It has been reported that concentrations of inflammatory biomarkers in peripheral blood does not reflect the situation in-situ [18, 20]. CSF is a good source of CNS-associated fluid and although, somewhat difficult to collect, it is often readily available in pre-term infants with birth-associated head trauma where CSF samples are used to clinically assess the baby’s status. However, this fluid has several drawbacks such has minimal sample, low analyte abundance, and high viscosity. The sample size can be overcome by the application of CE coupled with selective immunoaffinity extraction, which has been successfully used to measure cytokines in a number of biological fluids including plasma [21], urine [21], dried blood spots [22], sweat [23] and CSF [6]. The advantage of immunoaffinity CE over other immunoassays is that several analytes can be measured during the same run and the electrophoretic separation following the immunoaffinity extraction lessens the possibility of false positives [24]. The small sample requirement (less than 1 μL) plus the increased sensitivity when laser-induced fluorescence (LIF) detection is used, makes immunoaffinity CE an ideal candidate for preterm baby CSF analysis. Further, the introduction of chip-based CE further speeds the analytical time due to the short length of the separation channel and the increased sensitivity afforded by the integrated LIF detector.
In the present communication, a chip-based CE system with an integrated immunoaffinity pre-analytical selective phase and an integrated LIF detector has been used for the rapid analysis of six inflammation-associated chemokines in CSF samples taken from preterm babies during clinical assessment of birth-associated head injury.
2. Materials and Methods
2.1 Reagents
Recombinant human chemokines (CCL2, CCL19, CCL21, CXCL8, CXCL12, and CXCL-13) and their corresponding biotinylated polyclonal anti-chemokine antibodies were obtained from R & D Systems (Minneapolis, MN, USA) and reconstituted to stock solutions of 1μg/mL in 0.1 M phosphate buffer, pH 7.4.ELISA kits, specific for each chemokine, were obtained from R & D Systems. Neutravidin and carbonyldiimidazole and ImmunoPure IgG FAb preparation kit were purchased from Pierce Biotechnology (Rockford, IL, USA). Liquid artificial human CSF was obtained from Tocris Bioscience (R & D Systems – product No. 3525, composed of 150mM Na, 3mM K, 1.4mM Ca, 0.8mM Mg, 1mM P and 155mM Cl in sterile distilled water). Octylphenoxy)polyethoxyethanol (Igepal CA-630) a non-ionic detergent, used to minimalize protein adherence to the chip channels when sampling “sticky” biological fluids such as CSF, was obtained from Sigma-Aldrich, St Louis, MO, USA). All other chemicals were purchased from Acros Chemicals (Fisher Scientific, Pittsburgh, PA, USA). Prior to use, all solutions were passed through 0.2-μm nitrocellulose filters (Millipore, Bedford, MA, USA) to remove particulate matter.
2.2. Instrumentation
The immunoaffinity extraction and analyses were performed with a slight modification to the procedure previously described [25, 26]. Briefly, a Micralyne μTK microfluidic electrophoresis system (Micralyne, Edmonton, Alberta Canada) consisting of four dual-channel 6 kV power boards attached to four platinum electrodes, a custom-designed chip stage that accepted standard CHT8050 CE chips from Micronit Microfluidics BV (Enschede, The Netherlands) and an integrated 635 nm, 8mW red diode laser (Micralyne) was used. Detection was achieved by focusing the integrated epiillumination microscope onto the chip channel via a 10× magnification objective and passing the light to the built-in Hamamatsu H5773-03 photomultiplier tube coupled to a 16-bit data acquisition board.
The CHT8050 chip (Micronit Microfluidics BV)with a double T injection configuration, a basic cruciform pattern with 50-μm wide × 20-μm deep semicircular channels ending in a 2-mm diameter × 0.1-mm deep port and a separation channel length of 80-mm was chosen over the previously described CHT3550 chip [26]. The new chip had a separation channel over twice the length of the previous chip enabling all six chemokines of interest to be resolved. Ports were labelled 1-4 in a clockwise fashion starting at the anodal port of the separation channel (Figure 1A). Fluids were introduced into port 2 via a Harvard “11” syringe pump (Harvard Apparatus, Holliston, MA, USA) connected to the chip via an automatic injection valve (Upchurch Scientific, Oak Harbor, WA, USA) fitted with a calibrated 500nL loop. Control of the entire system was achieved by a PC, running Microsoft Windows XP with a compiled LabView interface (National Instruments, Austin, TX, USA). Fluid and electrical connections to the chip were achieved using either normal or modified Upchurch F-121H nuts (Upchurch Scientific) attached to a laboratory-built poly-ether-ether-ketone cover plate. The modified F-121H nuts were fitted with platinum wire electrodes glued into the bore of the nut, thus allowing the nut to be used for both fluidic transfers and electrophoresis. The modified nuts were placed in ports 2 and 4.
Figure 1.
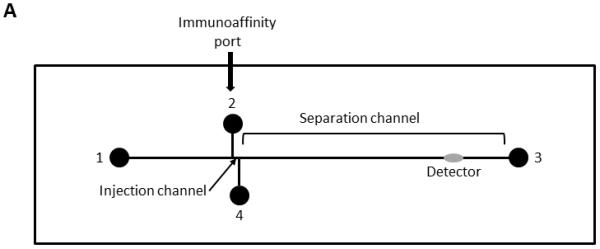
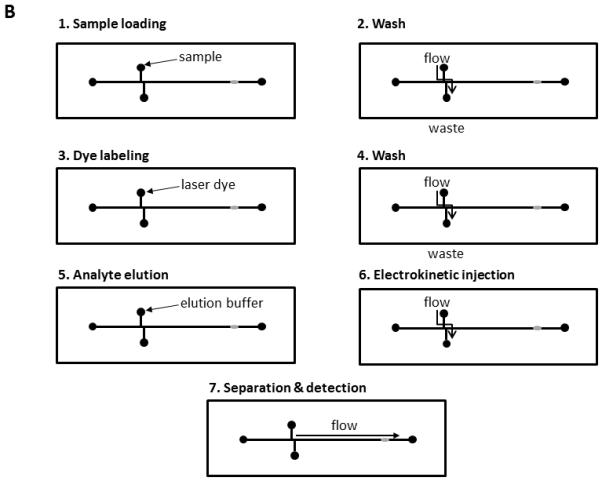
Diagrammatic representation of the layout of the immunoaffinity chip and the basic steps in performing the assay. A. Diagram indicating the major components of the chip. B. Diagram of the steps used to run an immunoaffinity CE assay. Step 1: the sample is loaded into the immunoaffinity port. Step 2: Unbound materials are washed out of the port to waste. Step 3: The FAb bound chemokines are labelled with a laser dye. Step 4: The unbound dye is washed out of the port to waste. Step 5: The dye-labelled chemokines are released from the immobilized FAb by disruption of the antibody/antigen binding. This is achieved using an acid elution solution. Step 6: The released chemokines are electrokinetically injected into the injection channel. Step 7: the chemokines are electrophoretically separated and detected by the on-line LIF detector.
2.3. Immunoaffinity disks
The immunoaffinity capture and isolation of the analytes was performed on a disposable insert as previously described [26]. Briefly, 100 2-mm diameter disks were punched from AP40 glass fibre filters (Millipore). This was achieved by using a laboratory-built hollow steel punch (sharpened at the cutting end) and a small hammer. The disks were placed in a 10 mL glass beaker and soaked in 5 mL of 10% v/v aqueous 3-aminopropyl-triethoxysilane for 10 min, after which the beaker and its contents were heated at 100°C for 60 min and cooled to 22°C (room temperature). The fluid was removed by suction and the beaker filled with 5 mL of fresh silane solution and reheated to 100°C. This process was repeated four times before the silane solution was removed by suction and the disks placed into 5 mL of 10mM hydrochloric acid, incubated for 60 min at 100 °C, washed twice in 10 mL of distilled water (the water being removed by gentle decantation). No mechanical stirring or shaking was used during any of the steps. Finally, the disks were incubated in a 5 mL solution of 1mg/mL carbonyldiimidazole dissolved in formamide for 6 h at 22°C on a rotary shaker. Prior to immobilization of the antibody, the disks were washed five times in 5 mL of formamide and then placed into 5 mL of a 1 μg/mL solution of Neutravidin dissolved in 100mM phosphate buffer, pH 7.4. The disks were incubated for 6 h at 22°C on a rotary plate mixer and the excess or non-bound Neutravidin removed by washing 20 times in 5 mL of 100mM phosphate buffer, pH 7.4. Following each wash, the fluid was gently removed by suction prior to adding a fresh wash; care was taken not to allow the disks to become dry. Excess carbonyldiimidazole side chains were blocked by a further incubation in a 5 mL solution of 200mM Tris-HCl buffer, pH 9.0 for 10 min at 22°C on a rotary shaker. Prior to attachment, each anti-chemokine antibody was reduced to FAb fragments using the Pierce IgG FAb fragmentation kit according to the manufacturer’s instructions. Attachment of the biotinylated FAbs was achieved by incubating the disks in a 5 mL solution of 100mM phosphate buffer, pH 7.4 containing 10ng of each of the anti-chemokine FAbs. Considering that the concentrations of the chemokines were unknown in the patient samples, the immunoaffinity disks were loaded with approximately 50-fold excess antibody as previously experimentally determined to be optimal amounts for analyte capture [6]. Free biotin sites were blocked by incubating with a 5 mL solution of 1mg/mL biotin for 30 min at 22°C. The disks were washed in 1 mL of 100mM phosphate buffer, pH 7.4 and stored at 4°C until required. Generally, throughout this process, we only checked the efficiency of the FAb binding step as we considered this the most important step. Following processing, the disks underwent a small degree of shrinkage and were found to be 1.8-mm ±0.1-mm and a single disk could easily be placed into port 2 or the immunoaffinity port of the chip (2-mm) in order to perform the assay.
Routinely, the immunoaffinity disks were produced in batches of 100 and tested for uniformity and specificity by running five randomly selected disks against a standard solution containing a mixture of all six chemokines (each chemokine at a concentration of 100pg/mL).
2.4. Patient groups
CSF was routinely obtained by the hospital staff from preterm infants during their initial clinical assessment at the Neonatal Intensive Care unit of The George Washington University Hospital, Washington, DC, USA. A 5 μL sample of each patient was donated for the present study and the remainder volume was used for clinical assessment of the subject. The infants were divided into 3 groups, based upon the clinical and clinical pathological assessment - group A which was composed of 20 individuals with severe head trauma that occurred during difficult physical birth, group B which consisted of 22 individuals that received mild forceps trauma during birth, and group C which consisted of 15 individuals from whom CSF was taken but no pathological or clinical abnormalities were found. In this study, group C was determined to be “normal” controls. Consent to use the samples were obtained from all subjects and no name indicators were assigned to any samples as required by the hospital institutional review board. Measurement of the total protein content of each sample was performed by direct spectrophotometry at 280/260 nm using a NanoDrop ND-100 micro-spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA) using a molar absorption coefficient of ten as recommended by Pierce Biotechnology [27]. Each sample was then adjusted to a protein concentration of 1 mg/mL in 100mM phosphate buffer/Igepal, pH 7.4 prior to analysis.
2.5. Preparation of Standards
Standards of 10, 50, 100, 250, and 500pg/mL were prepared for each chemokine by dissolving the stock solution of each chemokine (1 μg/mL) in 0.1M phosphate buffer, pH 7.4. These standard solutions were also used to spike the artificial CSF fluid to make final solutions containing a mixture of 10, 50, 100, 250, and 500-pg/mL of each cytokine.
2.6. Immunoaffinity measurement of chemokines in CSF
Basically, the analytical process was accomplished using a LabView interface, which controlled both the syringe pump and the CE system; this included sample injection, affinity port washing, analyte labelling, and elution. Figure 1B is a schematic of the basic procedure for the immunoaffinity CE system. After placing the immunoaffinity disk into port 2, all the chip channels were flushed by pumping 2 mL of 25mM phosphate containing 0.1% Igepal CA-630, pH 7.4 through the entire channel system at a flow rate of 2 mL/min for 1 min. A 500nL sample was injected into port 2 at a flow rate of 0.1 mL/min for 30 s and allowed to interact for 4 min, during which the immobilized FAbs reacted with and bound their reactive analytes from the sample (4 min was found experimentally to be the ideal time for optimal antibody-antigen reactions to take place using these anti-chemokine antibodies). The pump was then programmed to flush 1 mL of the phosphate/Igepal buffer through the immunoaffinity port at a flow rate of 1 mL/min. This fluid exited through port 4 to waste, thus removing the unbound materials from the immunoaffinity port before adding 2μL a 1-mg/mL solution of AlexaFluor 633 laser dye (Molecular Probes/Invitrogen, Eugene, OR, USA), a dye excited at 633nm and optimally suited to the output of the laser used in the electrophoresis system. The dye was added at a flow rate of 8 μL/min for 15s and allowed to interact with the FAb-bound analytes for 2 min (2 min incubation was found experimentally to be the ideal time for optimal analyte labelling in this system). The non-reacted dye was flushed from the immunoaffinity port by pumping 1 mL of phosphate/Igepal buffer at a flow rate of 1 mL/min through ports 2 and 4 for 1 min. This removed unreacted dye from the immunoaffinity port prior to elution, separation and detection of the captured chemokines. The labelled chemokines were recovered by introducing 500nL of elution buffer (phosphate/Igepal/HCl buffer, pH 1.0) into port 2 at a flow rate of 1 mL/min for 30 s. The elution buffer was allowed to interact with the immobilized immune complex for 2 min, during which time the acidic buffer disrupted the FAb/analyte complex thus releasing the bound analyte for analysis by electrophoresis. This incubation time was determined experimentally by spectrophotometrically measuring the amount of analyte released from an immunoaffinity disk over time. The eluted materials were transported into the injection channel by applying a 4 kV potential between ports 2 and 4 for 30 s. Separation was performed by applying a 6 kV potential between ports 1 and 3 for 2 min. All analyses were performed at 22°C and the concentrations of the separated chemokines were calculated by comparing the peak areas against peak area calibration curves constructed by running known amounts of each chemokine through the system.
2.7. Validation
Split samples of both spiked artificial CSF and patient samples were diluted 1:4 in order to provide enough material to perform parallel assays and run by both immunoaffinity chip electrophoresis and standard commercially-available ELISA, which were run according to the manufacturer’s instructions.
3. Results and Discussion
3.1 Characteristics of the system
A complete assay, including injection, analyte capture, labelling, recovery and separation with detection of the resolved peaks, could be performed in approximately 10 min with separation of the analytes being performed in 2 min. These parameters are in keeping with work on chip-based immunoaffinity CE as previously published by our group [25, 26]. Figure 2 illustrates a typical electropherogram produced by running a mixture containing 100 pg/mL of each of the six chemokines through the system. As illustrated in figure 2, the 6 chemokines eluted in the following order with migration times given in brackets - CXCL8 (88.1±0.2s), CXCL12 (89.7±0.3s), CCL2 (91.5±0.3s), CXCL13 (92.2±0.2s), CCL19 (96.2±0.3s) and CCL21 (112.5±0.4s).
Figure 2.
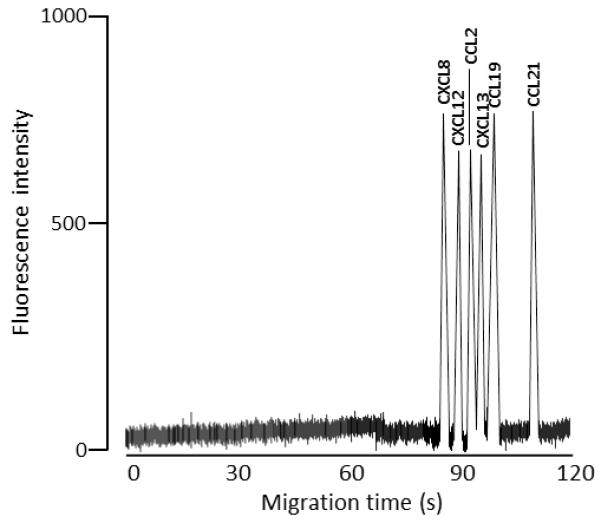
Typical electropherogram showing the separation of the six chemokines in artificial human CSF. Chemokines were separated according to the conditions described in section 2.6. Peaks represent these concentrations of the different chemokines: 97.8 pg of CXCL8, 97.9 pg of CXCL12, 86.5 pg of CCL2, 97.9 pg of CXCL13, 97.5pg of CCL19, and 98.4 pg of CCL21.
ELISA, using anti-mouse detection reagents (R & D Systems), was used on randomly selected immunoaffinity disks to determine the amounts of FAb bound to the disk. These studies demonstrated that each disk could bind approximately 4.6ng of the anti-chemokine antibody mixture. A laboratory-built reverse ELISA, using labelled chemokines (R & D Systems), demonstrated that the six different antibodies were bound in approximately equal parts. The LOD of the disks was calculated at 1.3 – 1.8 pg/mL as determined by analysing dilutions of each chemokine until no further signal could be detected. A similar procedure was employed to determine the saturation of the disks. Increasing amounts of each chemokine was applied to the disk until no increase in signal could be detected – the average saturation point was calculated to be between 2.7 and 2.8ng/mL. Table 1 summarizes the recovery efficiency of the disks when analysing spiked artificial CSF at concentrations of 10, 50, 100, 250, and 500pg/mL. The values are calculated by comparing the peak areas of the different chemokines against calibration curves constructed from the peak areas of known standards run under identical conditions. Intra- and inter-assay CVs were calculated from five repeat analyses of a sample of artificial CSF, spiked with a mixture containing 100 pg/mL of all six chemokines, performed on either the same day or on five different days, using the same immunoaffinity disk. Intra-assay CV’s were calculated at 4.21%, 3.65%, 3.15%, 4.11%, 4.25%, and 4.02% for CCL2, CCL19, CCL20, CXCL8, CXCL12 and CXCL13, respectively. Inter-assays CV’s were calculated at 4.61%, 3.99%, 3.54%, 4.66%, 4.57%, and 4.33% for CCL2, CCl19, CCL20, CXCL8, CXCL12 and CXCL13, respectively.
Table 1.
Recovery of chemokines from spiked artifical CSF
| Amount added (pg) |
Amount recovered (pg/mL)* |
|||||
|---|---|---|---|---|---|---|
|
| ||||||
| CCL2 | CCL19 | CCL21 | CXCL8 | CXCL12 | CXCL13 | |
| 10 | 8.8 | 9.7 | 9.1 | 9.6 | 9.4 | 9.8 |
| 50 | 47.3 | 48.9 | 47.9 | 48.8 | 48.1 | 49.8 |
| 100 | 86.5 | 97.5 | 98.4 | 97.8 | 97.9 | 97.9 |
| 250 | 246.1 | 247.6 | 248.1 | 248.6 | 248.3 | 248.7 |
| 500 | 491.2 | 496.4 | 497.7 | 498.2 | 496.5 | 498.6 |
The recovered amount was calculated by comparing the peak areas of the different chemokines to a calibration curve constructed by running chemokine standards through the immunoaffinity system.
Generally, it was found that an immunoaffinity disk could be regenerated 10-15 times before detectable loss of activity could be detected. Every batch of immunoaffinity disks were subjected to quality control testing by randomly selecting 5 disks and running them against a mixture of spiked CSF containing 100 pg/mL of each chemokine. This ensured batch-to-batch uniformity; a batch of disks being discarded when a 5-6% loss of activity was discovered. This criterion was also applied to any disk during its active life.
3.2 Comparison with commercial immunoassays
When the immunoaffinity disks were compared to commercially available ELISA kits, a high degree of agreement was seen when recovery was compared; the agreement between the two assays demonstrated r2 values of 0.915 for CCL2, 0.966 for CCL19, 0.936 for CCL21, 0.985 for CXC8, 0.903 for CXCL12, and 0.978 for CXCL13 when analyzed by the least-squares linear regression module of the Prism 4 software program (GraphPad Software, San Diego, CA, USA). Comparison of detection limits between the two assays differed as the LOD of the ELISA kits was found to be 5 pg/mL, while that of the immunoaffinity CE assay was 0.5 pg/mL. This added sensitivity aids in the detection of trace analytes, such as chemokines, in complex biological fluids. Additionally, the electrophoretic separation of the isolated analytes aids in the detection of contaminating components, while the immunoextraction and the characteristic migration time also lessens the possibility of false positives [24]. Both of these characteristics are advantages over conventional immunoassays.
3.3 Measurement of chemokine concentrations in CSF of the patient groups
Analysis of the individual patients in each of the three patient groups showed distinct differences in the chemokine pattern. Individuals in group A demonstrated the most chemokines detectable at concentrations greater than the normal range (8-12pg/mL). In this group the most elevated chemokines were CXCL13 (1600 – 2341pg/mL), CCL19 (521 – 680pg/mL), CXCL8 (277 – 358pg/mL) and CCL21 (116-180pg/mL) with CCL2 and CXCL12 within normal range. Individuals in group B demonstrated elevated concentrations of CCL2 (305 – 369pg/mL), CCL19 (160 – 194pg/mL) and CXCL8 (135 – 170pg/mL) with CCL21, CXCL12, and CXCL13 within normal range. Group C patients all demonstrated concentrations of all six chemokines within the normal range (8-12pg/mL). Figure 3 shows representative electropherogram patterns seen in the three groups. The representative sample from group A (figure 3A) shows chemokine concentrations of CXCL8 – 346 pg/mL; CXCL12 – 21 pg/mL; CCL2 – 15 pg/mL; CXCL13 – 230 pg/mL; CCL19 – 669 pg/mL; and CCL21 – 169 pg/mL. The sample from group B (figure 3B) demonstrates chemokine concentrations of CXCL8 – 169 pg/mL; CXCL12 – 10 pg/mL; CCL2 – 365 pg/mL; CXCL13 – 16 pg/mL; CCL19 – 191 pg/mL; and CCL21 – 8 pg/mL. The representative sample from group c (figure 3C) demonstrates chemokine concentrations of CXCL8 – 10 pg/mL; CXCL12 – 6 pg/mL; CCL2 – 11 pg/mL; CXCL13 – 12 pg/mL; CCL19 – 10 pg/mL; and CCL21 – 10 pg/mL.
Figure 3.
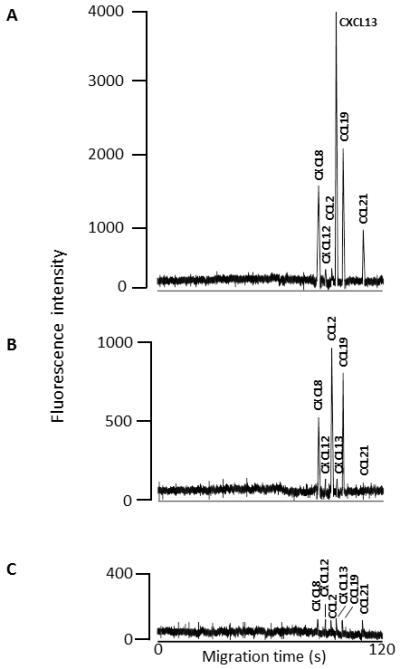
Representative electropherograms from selected patients showing the different chemokine patterns in the three patient groups. The electrophoretic conditions are as described in section 2.6. A. Electrophoretic pattern obtained from running a sample from a patient demonstrating the electrophoretic pattern seen in patients from group A; B. Pattern obtained by running a sample from a patient demonstrating the electrophoretic pattern seen in patients from group B; C. Baseline pattern obtained by running a sample from a patient in group C. All electropherograms were recorded using the same detector scale and settings.
Detection of chemokines in the CSF of infants with traumatic head injury has been reported by others [5, 8, 10, 17]. Whalen et al [5] reported that concentrations of CXCL8 in the CSF of children with traumatic brain injury as measured by ELISA could be used as an indicator of injury and outcome. However, our findings are comparable to those reported by Kowarik et al [10], who employed ELISA to study chemokines in the CSF from patients with various neurological diseases, including neuroinflammation and demonstrated that CCL2, CCL19, CXCL8, and CXC13 were important chemokines in brain injury and were responsible for the infiltration of lymphoid cells into the site of injury. These authors concluded that CXCL13 was one of the most important chemokines for assessing neuroinflammation, however, our studies show that CXCL8, CCL19 and CCL21 in addition to CXCL13 are present in infants with head trauma and using the immunoaffinity CE assay, we were able to detect varying concentrations of CCL21, which has been reported as undetectable in CSF when using ELISA [10]. Buttram et al [17] and Stein et al [18] both used a multiplex bead immunoassay to measure CXCL8 in patients with head trauma and concluded the CXCL8 was associated with a poor prognosis. In a similar fashion, our present study demonstrated that the presence of CXCL8, a neutrophil attractant, was present in the group A subjects and that in the treatment study (see below) that this chemokine persisted the longest during the treatment regime.
Further examination of the data generated by the analysis of the individual patients in all three groups indicated that the subjects in group A could be divided into two further groups on the basis of the presence of different chemokines and were labelled A1 and A2 accordingly. The electropherograms from individual patients in group A1 exhibited elevated concentrations of CXCL13 and CXCL8, while those of group A2 exhibited elevated concentrations of chemokines CCL19, CCL21 and CXCL8 (Figure 4). Examination of the clinical information regarding the status of the patients in group A indicated that clinically the group could also be divided into two groups based on the clinical chemistry profile and physical examination by a neurologist. According to the clinical chemical and physical assessment, the subjects in group A1 showed poor clinical progress with prolonged indications of head trauma, poor responses to treatment and in three cases, death. The subjects in group A2, however, although they initially showed clinical signs of severe head trauma and inflammation, appeared to respond well to treatment.
Figure 4.
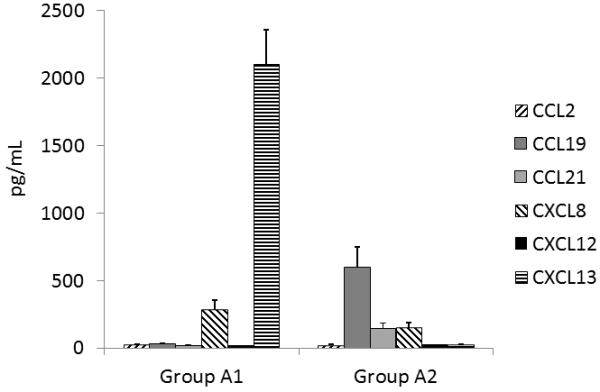
Graph illustrating the distribution of the six chemokines in the two subgroups of patient group A. The error bars represent the mean ± SEM.
A study of chemokine behaviour during non-steroid anti-inflammatory treatment was performed on samples from 6 subjects in group A2. CSF samples were taken from all subjects at weekly intervals and subjected to immunoaffinity CE analysis. Initially, analysis of the chemokine concentrations in the individual patients, demonstrated chemokine concentrations of 579 ± 61 pg/mL for CCL19, 150 ± 39 pg/mL for CCL21, and 170 ± 41 pg/mL for CXCL8 but following one week of therapy the concentrations of CCL19 and CCL21 fell to 312 ± 28 pg/mL and 70 ± 15 pg/mL, respectively while CXC8 remained at approximately its original value (166 ± 30 pg/mL). A further week of treatment reduced both CCL19 and CCL21 to 106 ± 18 pg/mL and 42 ± 8 pg/mL respectively and a slight drop in CXC8 (138 ± 11 pg/mL). Week 3 of the treatment reduced CCL19 to 15± 4 0pg/mL, CCL21 to 28 ± 10 pg/mL and CXC8 to 77 ± 18 pg/mL. The fourth week reduced all 3 chemokines to within normal range (8-12pg/mL). This data is summarized in figure 5. These findings coincided with the clinical findings and the clinical situation – all 6 infants returning to a “normal” status by week 4 post-treatment.
Figure 5.
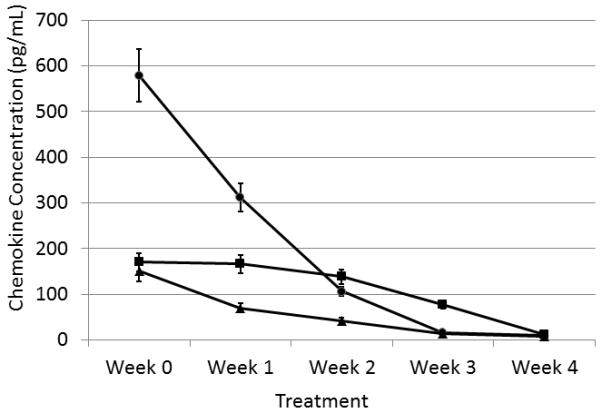
Graph illustrating the concentrations of chemokines in the treated subjects from group A2 over time. The solid squares represent the concentrations of CCL19; the solid triangles the concentrations of CCL21; and the solid circles represent the concentrations of CXCL8. The error bars represent the mean ± SEM.
The ability of the immunoaffinity CE system to distinguish between subjects with good clinical outcome and those that were not so fortunate together with the speed of analysis and the small sample size requirement makes this system a good approach for the clinical laboratory diagnosis of preterm head injury associated with neuroinflammation. Further the electrophoresis unit plus a laptop computer can easily be placed on a laboratory cart, thus making the unit portable and certainly available for installation in a room in a paediatric emergency room. The speed of analysis is also an advantage as a chemokine assessment can be performed in less time than conventions clinical chemistry and much faster than convention ELISA or Luminex bead assays. Considering that both analytical and treatment speed is the essential in head trauma cases, especially in new-borns, the immunoaffinity CE offers an ideal instrument. The minimal sample requirement is also a great asset when working with pre-term babies, who are often small with little bodily fluids. The immunoaffinity CE required only 0.5 μL of sample and is well suited to analysing single samples. The development of better designed chips (lab-on-a-chip) and more efficient detection systems such as chemiluminescence and mass spectrometry plus the further miniaturization of the entire instrument further enhances the possibilities of this approach to its establishment in neonatal intensive care units.
Chip-based CE systems are an excellent platform for developing rapid clinical tests especially in critical situations, where the speed of the analysis can be critical. In this communication, an immunoaffinity CE device was applied to the rapid analysis of CSF samples from preterm babies with traumatic head injury and demonstrated that the resolution of 6 inflammation-associated chemokines could be achieved in 10 minutes with a CE separation and on-line detection being performed in circa 2 min. Pre-analysis immunoaffinity extraction is becoming very popular with applications in HLPC [28], GC-MS [29], HPLC-MS [30] and MS [31]. In the analysis of CSF, the antibody pre-selection of the analyte of interest greatly reduced the matrix effect of the viscose sample, allowing the unreacted materials to be removed before the CE separation. In miniature systems, such as chip-based CE, antibody capture with removal of unbound materials prior to analysis greatly reduces non-specific interference in the relatively short separation channel. Further, it was found that the acidic conditions used to recover the bound analytes, from the immunoaffinity insert, greatly reduced protein adherence to the channel wall.
4. Conclusions
A chip-based CE system has been employed to isolate and measure 6 pro-inflammatory chemokines from the CSF of infants with birth-associated head trauma. The isolation of the chemokines from the complex biological fluid was achieved by incorporating an immobilized antibody immunoextraction unit into the CE system. This unit contained reactive fragments (FAb) of 6 anti-chemokine antibodies immobilized onto a glass fiber disk, which was used as a solid-phase extraction unit to specifically isolate the chemokines of interest. The isolated chemokines were then electrophoretically separated and their concentrations measured by on-line LIF detection, following labelling of the isolated chemokines with a laser dye. This system successfully analysed three patients groups, distinguishing between the subjects with neuroinflammation and the normal controls. Further, analysis of the chemokine patterns could predict the future outcome of the traumatized patients and additionally monitor chemokine concentrations during anti-inflammation therapy. The results were comparable to those reported using conventional immunoassays but immunoaffinity CE chip was more reliable, faster and utilized less sample – an important aspect when studying new-born infants.
Acknowledgements
This work was supported by the intramural research program of the National Institutes of Health.
Abbreviations
- CCL2
monocyte chemoattraction protein-1
- CCL19
macrophage inflammatory protein 3 beta
- CCL21
secondary lymphoid tissue chemokine
- CXCL8
interleukin-8
- CXCL12
stromal cell-derived factor-1
- CXCL13
B cell attracting chemokine-1
Footnotes
Conflict of interest statement.
Both authors declare that they have no conflict of interest.
References
- 1.Degos V, Favrais G, Kaindl AM, Peineau S, Guerrot AM, Verney C, Gressens PJ. Neural. Transm. 2010;117:1009–1017. doi: 10.1007/s00702-010-0411-x. [DOI] [PubMed] [Google Scholar]
- 2.Berger I, Peleg O, Ofek-Shlomai N. Isr. Med. Assoc. J. 2012;14:318–323. [PubMed] [Google Scholar]
- 3.Kaindl AM, Favrais G, Gressens PJ. Child. Neurol. 2009;24:1112–1118. doi: 10.1177/0883073809337920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Malaeb S, Dammann O. J. Child. Neurol. 2009;24:1119–1126. doi: 10.1177/0883073809338066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Whalen MJ, Carlos TM, Kochanek PM, Wisniewski SR, Bell MJ, Clark RS, DeKosky ST, Marion DW, Adelson PD. Crit. Care Med. 2000;28:929–934. doi: 10.1097/00003246-200004000-00003. [DOI] [PubMed] [Google Scholar]
- 6.Phillips TM. Electrophoresis. 2004;25:1652–1659. doi: 10.1002/elps.200305873. [DOI] [PubMed] [Google Scholar]
- 7.Stefini R, Catenacci E, Piva S, Sozzani S, Valerio A, Bergomi R, Cenzato M, Mortini P, Latronico N. J. Neurosurg. 2008;108:958–962. doi: 10.3171/JNS/2008/108/5/0958. [DOI] [PubMed] [Google Scholar]
- 8.Szmydynger-Chodobska J, Strazielle N, Gandy JR, Keefe TH, Zink BJ, Ghersi-Egea JF, Chodobski A. J. Cereb. Blood Flow Metab. 2012;32:93–104. doi: 10.1038/jcbfm.2011.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brandes M, Legler DF, Spoerri B, Schaerli P, Moser B. Int. Immunol. 2000;12:1285–1292. doi: 10.1093/intimm/12.9.1285. [DOI] [PubMed] [Google Scholar]
- 10.Kowarik MC, Cepok S, Sellner J, Grummel V, Weber MS, Korn T, Berthele A, Hemmer B. J. Neuroinflammation. 2012;9:93–103. doi: 10.1186/1742-2094-9-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aloisi F, Columba-Cabezas S, Franciotta D, Rosicarelli B, Magliozzi R, Reynolds R, Ambrosini E, Coccia E, Salvetti M, Serafini B. J. Neuroimmunol. 2008;198:106–112. doi: 10.1016/j.jneuroim.2008.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chakraborty S, Kaushik DK, Gupta M, Basu A. J. Neurosci. Res. 2010;88:1615–1631. doi: 10.1002/jnr.22343. [DOI] [PubMed] [Google Scholar]
- 13.Khademi M, Kockum I, Andersson ML, Iacobaeus E, Brundin L, Sellebjerg F, Hillert J, Piehl F, Olsson T. Mult. Scler. 2011;17:335–343. doi: 10.1177/1352458510389102. [DOI] [PubMed] [Google Scholar]
- 14.Krumbholz M, Theil D, Steinmeyer F, Cepok S, Hemmer B, Hofbauer M, Farina C, Derfuss T, Junker A, Arzberger T, Sinicina I, Hartle C, Newcombe J, Hohlfeld R, Meinl E. J. Neuroimmunol. 2007;190:72–79. doi: 10.1016/j.jneuroim.2007.07.024. [DOI] [PubMed] [Google Scholar]
- 15.Chiaretti A, Genovese O, Aloe L, Antonelli A, Piastra M, Polidori G, Di Rocco C. Childs Nerv. Syst. 2005;21:185–193. doi: 10.1007/s00381-004-1032-1. [DOI] [PubMed] [Google Scholar]
- 16.Zajkowska J, Moniuszko-Malinowska A, Pancewicz SA, Muszyńska-Mazur A, Kondrusik M, Grygorczuk S, Swierzbińska-Pijanowska R, Dunaj J, Czupryna P. Adv. Med. Sci. 2011;56:311–317. doi: 10.2478/v10039-011-0033-z. [DOI] [PubMed] [Google Scholar]
- 17.Buttram SD, Wisniewski SR, Jackson EK, Adelson PD, Feldman K, Bayir H, Berger RP, Clark RS, Kochanek PM. J. Neurotrauma. 2007;24:1707–1717. doi: 10.1089/neu.2007.0349. [DOI] [PubMed] [Google Scholar]
- 18.Stein DM, Lindell A, Murdock KR, Kufera JA, Menaker J, Keledjian K, Bochicchio GV, Aarabi B, Scalea TM. J. Trauma. 2011;70:1096–1103. doi: 10.1097/TA.0b013e318216930d. [DOI] [PubMed] [Google Scholar]
- 19.Tateishi T, Yamasaki R, Tanaka M, Matsushita T, Kikuchi H, Isobe N, Ohyagi Y, Kira J. J. Neuroimmunol. 2010;222:76–81. doi: 10.1016/j.jneuroim.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 20.Ellison VJ, Mocatta TJ, Winterbourn CC, Darlow BA, Volpe JJ, Inder TE. Pediatr. Res. 2005;57:282–286. doi: 10.1203/01.PDR.0000148286.53572.95. [DOI] [PubMed] [Google Scholar]
- 21.Phillips TM, Dickens BF. Electrophoresis. 1998;19:2991–2996. doi: 10.1002/elps.1150191632. [DOI] [PubMed] [Google Scholar]
- 22.Nelson KB, Grether JK, Dambrosia JM, Walsh E, Kohler S, Satyanarayana G, Nelson PG, Dickens BF, Phillips TM. Pediatr. Res. 2003;53:600–607. doi: 10.1203/01.PDR.0000056802.22454.AB. [DOI] [PubMed] [Google Scholar]
- 23.Marques-Deak A, Cizza G, Eskandari F, Torvik S, Christie IC, Sternberg EM, Phillips TM. J. Immunol. Methods. 2006;315:99–109. doi: 10.1016/j.jim.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 24.Guzman NA, Phillips TM. Electrophoresis. 2011;32:1565–1578. doi: 10.1002/elps.201000700. [DOI] [PubMed] [Google Scholar]
- 25.Phillips TM, Wellner EF. Electrophoresis. 2007;28:3041–3048. doi: 10.1002/elps.200700193. [DOI] [PubMed] [Google Scholar]
- 26.Phillips TM, Wellner EF. Electrophoresis. 2009;30:2307–2312. doi: 10.1002/elps.200900095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pierce Biotechnology. Tech Tip 6, Thermo Scientific; Rockford, IL: 2008. [Google Scholar]
- 28.Hage DS, Anguizola JA, Bi C, Li R, Matsuda R, Papastavros E, Pfaunmiller E, Vargas J, Zheng X. J. Pharm. Biomed. Anal. 2012;69:93–105. doi: 10.1016/j.jpba.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang W, Wang H, Wang J, Li X, Jiang H, Shen J. J. AOAC. Int. 2006;89:1677–1681. [PubMed] [Google Scholar]
- 30.Liu L, Jin H, Sun L, Ma S, Lin R. Phytochem. Anal. 2012;23:469–476. doi: 10.1002/pca.2343. [DOI] [PubMed] [Google Scholar]
- 31.Moser AC, Hage DS. Bioanalysis. 2010;2:769–790. doi: 10.4155/bio.10.31. [DOI] [PMC free article] [PubMed] [Google Scholar]