

Abstract
The cytokine responses characterizing the inflammatory bowel diseases (IBDs) are the key pathophysiologic elements that govern the initiation, evolution and, ultimately, the resolution of these forms of inflammation. Studies over the last two decades now provide a detailed (but not yet complete) picture of the nature of these responses. The first tier of cytokine responses are governed by the T cell differentiation patterns dominating the disease. Thus, in Crohn’s disease, the major cytokines arise from Th1 and Th17 CD4+ T cell differentiation and consist of IFN-γ and IL-17/IL-22 generated by these types of differentiation. The relative importance of these cytokines to Crohn’s inflammation is still unclear, although evidence is mounting that IFN-γ is primus inter pare (first among equals). In contrast, in ulcerative colitis a Th2-like differentiation process is paramount which results in the expansion of NKT cells producing IL-13 (and perhaps IL-5). These disease-specific cytokine patterns give rise to a second tier of cytokines that span the Th1/Th17–Th2 divide and act as upstream facilitators and downstream mediators of inflammation. These cytokines include the well-known TNF-α, IL-1β, IL-6 triumphirate as well as a more recently studied cytokine known as TL1A. In this review, we will explore this cytokine landscape with the view of providing an understanding of how recent and future anti-cytokine therapies actually function.
Keywords: Crohn’s disease, ulcerative colitis, cytokines, IL-12, IL-23, IFN-g, IL-17, IL-22, TL1A
Introduction
In the past two decades, studies of the cytokines driving the inflammatory bowel diseases and other forms of mucosal inflammation has borne ample fruit, both in providing us with major insights into the mechanism of these diseases and in pointing us in the direction of new therapies. In this review we will focus on the main cytokine responses in Crohn’s disease (CD) and ulcerative colitis (UC) that initiate and sustain inflammation, leaving the task of discussing the regulatory cytokines that oppose such inflammation to other reviewers.
T cell Differentiation Pathways and Gut Inflammation
With the discovery in the late 1980’s that T helper (Th) cells differentiate into T helper cell Th1 and Th2 cells1 producing different sets of cytokines it was quickly established that CD differed from ulcerative colitis UC in that CD seemed to be a Th1 cytokine-mediated disease characterized by increased production of interferon(IFN)-γ whereas UC seemed to be a Th2 cytokine-mediated disease characterized by increased production of interleukin(IL)-5 production and normal IFN-γ production2,3. One caveat, however, was that production of the signature cytokine of the Th2 response(IL-4), was not increased in UC and it was thus clear that the latter was a “Th2-like” disease rather than a fully Th2 disease (See Figure 1).
Figure 1. The Basic Dichotomy of Cytokine Function In IBD.

Whereas Crohn’s disease and ulcerative colitis are both forms of inflammatory bowel disease (IBD) and exhibit overlapping genetic profiles, these disease are characterized by very different T cell responses. Crohn’s disease is driven by a Th1/Th17 response in which IL-12 and IL-23 cytokines play key roles. Ulcerative colitis, in contrast is driven by a Th2-like response in which NKT cells producing IL-13 (and IL-5) is the major response. In addition, in both forms of IBD cytokines that have functions both “upstream” and “downstream” of these basic cytokines also play important inflammatory roles.
Support for the above concepts came from studies of several murine models of IBD resembling CD, particularly trinitrobenzene sulfonic acid-induced colitis (TNBS)-colitis and cell transfer-induced colitis which showed that inflammation was reversed by treatment with anti-IL-12p40–an antibody directed against a cytokine initially identified as IL-12, the master cytokine driving the Th1 response4,5. These findings, along with the fact that patients with CD exhibited increased lamina propria IL-12 production as compared to controls,6-8 ultimately became the basis for the development of a humanized anti-IL-12p40 antibody for treatment of patients with CD. When such an antibody became available it was shown in clinical trials that anti-IL-12p40 had a level of therapeutic efficacy similar to that of anti-TNF-α and, moreover, could induce remission in patients with anti-TNF-α resistance9,10. These results not only formed the basis of a new therapy for CD, they also represented incontrovertible evidence that a cytokine containing a p40 chain played a major pathogenic role in this disease.
Parallel studies of UC to be discussed at greater length below, verified that UC was a Th2-like disease in that it was associated with increased IL-13 production (but not IL-4 production) by NKT cells, rather than by conventional T cells11. Thus, as in the case of CD, analysis of the pattern of T cell differentiation was predictive of the basic cytokines causing UC-type inflammation.
The Th17 Response in CD Pathogenesis
The concept that CD was an IL-12-driven Th1 inflammation did not remain unchallenged for long: about the time anti-IL-12p40 was shown to be effective in the treatment of CD, a new set of cytokines, the Th17 cytokines (IL-17 and IL-23), were shown to function as effectors in various autoimmune disease models12-15. Among the latter was the cell-transfer colitis model in which it was shown that the development of colonic inflammation was apparently more dependent on IL-23 than IL-1216,17. The idea that a Th17 response rather than a Th1 response was the major engine of inflammation in CD did not contradict the previously observed effect of anti-IL-12p40 in experimental and human CD because both IL-12 and IL-23 are heterodimers of which one chain is p4012; thus, anti-IL-12p40 can neutralize both IL-12 and IL-23.
To fully understand how Th17 T cell responses are involved in experimental colitis or CD it is well to take a moment to review certain salient features of Th17 immunobiology. First, IL-12 has a very different relation to IFN-γ than does IL-23 to IL-17. IL-12 is the major inducer of Th1 cells producing IFN-γ via direct interaction with IL-12 receptors on undifferentiated T cells18; in contrast, TGF-β and IL-6 (or IL-21) are the major inducers of Th17 cells producing IL-17 (and IL-22 as well) and the function of IL-23 is to interact with already differentiated Th17 cells (now expressing an IL-23 receptor) to cause “stabilization” and/or expansion of Th17 cells19-22. There is evidence that differentiation of Th17 cells in the absence of IL-23 leads to Th17 cells producing IL-10, an anti-inflammatory cytokine, which are therefore poor inducers of inflammation23. In addition, it has recently been shown that Th17 cells induced in the presence of IL-1β have a have a unique mRNA profile and an increased capacity to induce inflammation24. Thus, not all Th17 are “equal.”
Second, since Th17 cells usually require TGF-β for differentiation, a cytokine also involved in regulatory T cell differentiation, it is not surprising that Th17 cells and regulatory T cells have a “ying-yang” relationship wherein the development of one type of cell is reciprocal to the development of the other type of cell. This is seen in the fact that induction of Foxp3 expression, the signature protein of regulatory T cells, inhibits RORγt function, the main IL-17 transcription factor. Similarly, the induction of Th17 cells inhibits Foxp3 expression25,26. Another manifestation of the relationship between Th17 cells and regulatory T cells is that the latter cells produce large amounts of TGF-β and thus can induce naïve CD4+ cells to become IL-17-producing cells in an inflammatory milieu that contains IL-6 or can themselves be converted into Th17 cells under these circumstances27-30. This “plasticity” of IL-17-producing cells and regulatory T cells indicates that Th17 responses in IBD may morph into regulatory responses (and vice versa) depending on the character of the inflammation.
A third and final feature of Th17 development is that induction of IL-17 gives rise to IL-17-producing T cells as well as cells producing both IL-17 and IFN-γ; in addition, there is evidence that in a milieu lacking TGF-β, IL-12 and IL-23 tend to act on cells initially producing IL-17 to become cells producing IFN-γ31-33. This capacity of at least some Th17 T cells to produce IFN-γ is consonant with the heterogeneity of Thi17 cells mentioned above and may facilitate their pathologic potential.
The First Wave of Th17 Studies
The initial studies assessing the importance of Th17 responses in experimental colitis and CD utilized the aforementioned cell-transfer colitis model. This model consists of inflammation developing in immunodeficient mice (either RAG-deficient or severe combined immune-deficient (SCID) mice) following adaptive transfer of naïve CD4+ T cells (CD45RBhigh T cells) that develop into pro-inflammatory effector cells in the absence of a mature (CD45RBlow T cells) that contain regulatory T cells34,35. To put these studies into prospective one should be aware that anti-IFN-γ administration leads to complete amelioration of cell-transfer colitis and transfer of CD45RBhigh (colitis-inducing) cells unable to synthesize IFN-γ because they lack a key factor (T-bet) necessary for IFN-γ production, does not initiate colitis36,37. In addition, the percentage of cells in the inflamed lamina propria of this model producing IFN-γ alone is 15-25 times higher than the percentage of cells producing IL-17 alone and 10 times higher than those producing both IL-17 and IFN-γ38,39. These facts strongly suggest that the dominant effector cell driving cell-transfer colitis is an IFN-γ-producing cell that is likely originating mainly from a Th1 response not a Th17 response. Nevertheless, certain influential studies have appeared presenting compelling evidence that a Th17 response, is, in fact, the key effector cell response in this colitis model.
In the first of three such studies, it was shown that RAG-deficient mice also deficient in IL-23p19 (and thus deficient in IL-23) did not develop colitis upon transfer of naïve T cells whereas the same mice also deficient in IL-12p35 (and thus deficient in IL-12) did develop colitis upon transfer16,40. Correspondingly, pro-inflammatory cytokine production (TNF-α and IFN-γ) was greatly reduced in the IL-23p19-deficient mice whereas it was only moderately reduced in the IL-12p35-deficient mice. Interestingly, while the IL-12p35-deficient mice exhibited greatly increased levels of IL-17, they also exhibited increased levels of IFN-γ. This suggests that the inflammation in IL-12p35-deficient mice may have been due, as least in part, to the aforementioned IFN-γ component of the Th17 response which in the absence of the p35 chain comprising part of the inhibitory cytokine, IL-35, is exaggerated41. In a recent update of these findings, it was shown that transfer of naive T cells lacking the IL-23 receptor also failed to generate colitis 42. In addition, signaling through this receptor enhanced intestinal T cell proliferation and the accumulation of IL-17-producing cells, especially those producing both IL-17 and IFN-γ, which, again, could be a particularly pro-inflammatory Th17 cell subpopulation. Finally, the lack of T cell expansion in mice reconstituted by IL-23R-deficient T cells could be overcome by co-transfer of T cells bearing IL-23R, indicating that a factor produced by cells bearing the receptor can drive cell proliferation of cells lacking the receptor. Overall then, cell-transfer colitis was shown in studies of both IL-23-deficient mice and IL-23R-deficient mice to exhibit greatly diminished colitis.
In the second study SCID mice were initially transferred antigen-specific (i.e., flagellin-specific) T cells, i.e., memory T cells rather than naïve T cells, as in the usual cell-transfer colitis study. Interestingly, in this case the lamina propria of the inflamed colon contained a 5:1 preponderance of IL-17-producing cells vs. IFN-γ-producing cells, quite the opposite of what is seen with the transfer of naïve cells, 43. This could conceivably be explained by the fact that memory cells are more responsive to IL-23 than to IL-1212. Next, SCID mice were transferred previously polarized Th1 or Th17 flagellin-specific T cells producing mainly IFN-γ and IL-17 respectively. It was found that the mice transferred Th17 cells developed severe colitis whereas the mice transferred Th1 cells developed little if any colitis; furthermore, treatment of the Th17-reconstituted mice with anti-IL-23p19 either prevented colitis or reduced already-established colitis. These studies thus suggest that Th17 cells but not Th1 cells mediate inflammation in a modified cell-transfer colitis model. Two points about these results require further consideration. First, the inflamed colons of mice transferred Th17 cells and exhibiting colitis contained cells producing large amounts of IFN-γ even though at the time of cell administration they were producing mainly IL-17. Thus, the mice reconstituted with Th17 cells could conceivably have had inflammation driven by both IL-17 and IFN-γ. Second, the lack of inflammation in the mice transferred cells is discordant with earlier cell-transfer colitis studies already discussed in which colitis was shown to be reversed by treatment with anti-IFN-γ36; this could conceivably be explained by the fact that a memory rather than a naïve population was transferred to the SCID mice and that this memory cell population was subject to inhibition by regulatory T cells.
In the third study of the role of IL-17 in transfer-colitis, it was shown that while transfer of T cells lacking the capacity to produce any individual Th17 cytokine (IL-17A, IL-17F or IL-22) to Rag1-deficient mice led to full-blown colitis, transfer of T cells from an RORγt-deficient mice, i.e., mice lacking a transcription factor necessary for production of all of these Th17 cytokines, fail to develop colitis. Thus, while IL-17A and IL-17F are redundant, at least one member of the IL-17 cytokine family was required for the occurrence of colitis44. Further confirmatory studies showed that Rag1-deficient mice that fail to develop colitis upon transfer of RORγt-deficient cells, do develop colitis if administered exogenous IL-17A44. One possible discrepancy in these data was that transfer of RORγt-deficient T cells also led to a great reduction in the production of IFN-γ, suggesting that it was really the IFN-γ produced during a Th17 response that was mediating the inflammation. However, this possibility seemed to be at least partly negated by the fact that Rag1-deficient mice transferred IL-17F-deficient T cells and thus manifesting colitis due to IL-17A alone exhibit decreased colitis when treated with anti-IL-17A44.
Th17 Responses are Essential, but not Necessarily as Effector Cell Responses
The various studies discussed above appear to offer definitive evidence that at least one major type of experimental colitis, cell-transfer colitis, requires a Th17 response to support the development of colonic inflammation. However, before we accept this conclusion we need to consider studies probing the impact of Th17 responses on regulatory T cell responses. In an initial study of this question it was shown that the transfer of naïve T cells to immunodeficient (RAG1-deficient) mice also lacking IL-23p19 (i.e., mice able to mount a Th1 but not a Th17 response) only develop colitis if they have a concomitant IL-10 or TGF-β deficiency and thus cannot mount a regulatory T cell response45. In addition, IL-23p19 deficient mice exhibit increased numbers of regulatory T cells (Foxp3+ T cells) in the colon and transfer of naïve T cells into RAG1-deficient/IL-23p19-deficient mice that also lack Foxp3+ T cells develop colitis in spite of the absence of IL-23p19 (and the ability to mount a Th17 response). Similar findings were found in a second study, in which it was shown that Rag1-deficient mice reconstituted with IL-23R-deficient T cells also exhibit increased numbers of Foxp3+ T cells in the colon, which was then shown to be due to the lack of a negative effect of IL-23 on these cells42. These findings strongly suggest that IL-23 suppresses regulatory T cell development and thus introduce the possibility that mice that lack IL-23 fail to develop colitis, not because they cannot produce a key effector cytokine (IL-17) but rather because they have a dominant regulatory T cell response. In the same vein, the inability of transferred polarized Th1 cells to cause colitis could be due to the fact that in the absence of Th17 cells the Th1 cells are subject to suppression by regulatory T cells.
Additional questions about the role of Th17 responses in colitis models are raised by more recent studies of cell-transfer colitis as well as studies of TNBS-colitis and dextran sodium sulfate (DSS)-colitis. In these cell-transfer colitis studies it was found that transfer of T cell populations from IL-17-deficient mice to immunodeficient mice led to earlier onset of colitis and higher levels of IFN-γ production than transfer of cell populations from wild type mice46. These findings could be explained by the fact that Th1 cells bear IL-17 receptors and IL-17 signaling via these receptors inhibit Th1 differentiation by suppressing expression of T-bet, a factor necessary for Th1 T cell development46. Similarly, in the studies of TNBS-colitis and DSS-colitis it was shown that while IL-17 responses accompany IFN-γ responses in both models, induction of the colitis in IL-23p19-deficient mice, led to more severe inflammation than in wild type mice47,48. Thus, both in the cell-transfer model and in the TNBS/DSS-colitis models, lack of cells producing IL-17 led to more not less disease and the latter seemed to be playing a regulatory role rather than an effector role in the inflammation.
In summary, it now seems clear that while the Th17 response has the potential to be a pro-inflammatory response when present in sufficient numbers (as shown in the studies of the RORγt-deficient/RAG-1-deficient mice discussed above), it is more likely to be functioning mainly as a regulatory response in experimental colitis models where the number of Th17 cells may be limited. (See Figure 2). Support for this thesis is not only vested in the studies discussed above but also in the fact that IL-22, a Th17 cytokine generated under the same conditions that lead to the production of IL-17 has been shown to have anti-inflammatory effects in colitis models: IL-22 deficiency is associated with more severe colitis49,50 and IL-22 has been shown to induce Stat3 in epithelial cells and to thus promote epithelial integrity in the face of inflammation51. Finally, it is relevant to point out that the development of a relatively small Th17 response in the absence of a Th1 response does not necessarily lead to a CD-like inflammation. This is seen in recent studies of pig-tailed macaque monkeys who manifest decreased epithelial cell barrier function, entry of bacteria components into the lamina propria and increased numbers of lamina propria IL-17+ T cells in the absence of the transmural inflammation and granuloma formation characteristic of CD52.
Figure 2. Regulatory Mechanism Initiated by the Th17 Response.
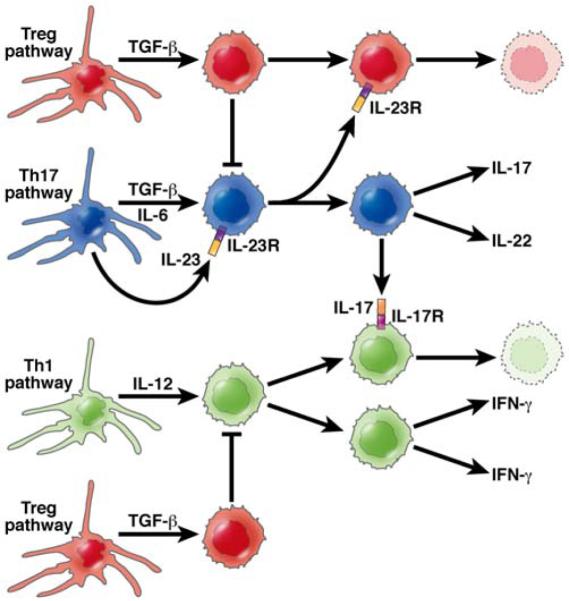
The role of the Th17 response in CD is under intense investigation. It is gradually emerging that in this disease this response may be more important in the regulation of the inflammation than in its induction. In this diagram the Th17 response (depicted by the cells shown in blue) generate cells producing IL-17 and IL-22, the latter a cytokine clearly known to ameliorate gut inflammation. This response depends on the production of IL-23 which also inhibits regulatory T cell (Treg) generation (depicted by the cells shown in red) and thus counter-acts the inhibitory effect on Tregs on both Th1 (IFN-γ) and Th17 (IL-17) pro-inflammatory responses. In contrast, IL-17 interaction with IFN-γ-producing cells (depicted by the cells shown in green) via an IL-17 receptor inhibits generation IFN-γ producing cells. Thus, the Th17 response is both permissive and inhibitory of the Th1 response, probably at different phases of the inflammatory cycle.
The Concept of Inflammatory Microdomains
So far in our discussion of experimental colitis we have focused mainly on data from the cell-transfer model of colitis. However, it is not prudent to rely too heavily on this model as a mirror of CD pathogenesis given the fact that colitis in this model develops under the very special conditions that obtain in an initially lymphopenic host. In addition, genetic studies of Crohn’s disease strongly suggest that unlike the situation in cell-transfer colitis, the T cell response directly or indirectly reflects a genetically determined tendency to hyper-respond to one or several microbial components in the commensal microflora53. For these reasons, an experimental colitis induced by TNP-modified antigens in the gut lumen, TNBS-colitis, may in some ways better approximate CD because in this model the mucosal immune response to a strong antigenic stimulus (TNP-modified protein) has been shown to be accentuated by a genetically determined hyper-responsiveness to LPS and perhaps other TLR ligands54. Thus, this model simulates the above mentioned hyper-responsiveness.
Recently, it has been shown that a chronic form of TNBS-colitis can be induced in BALB/c mice by administration of weekly intra-rectal instillation of relatively low doses of TNBS55. This form of TNBS-colitis is characterized by inflammation that is at once less intense and more prolonged than that in the acute model and thus allows detailed assessment of the evolution of the response. Of particular interest to the present discussion, the cytokine profile of mice with chronic TNBS-colitis proved to be unexpectedly dynamic: in the first few weeks of the colitis one sees a “pure” Th1 response characterized by high levels of both IL-12p40 and IFN-γ but no increases in the level of IL-17; by three weeks this response subsides, only to be replaced by the gradual increase in IL-23p19 production accompanied soon afterward by an increase in TGF-β and IL-17 production; this Th17 response reaches a sustained plateau by 40-45 days and then subsides after 70 days. Several other cytokines making a delayed appearance, in this case shortly after the onset of the IL-23p19 response, includes IL-25 and IL-13; these cytokines (along with TGF-β) are critical to the development of fibrosis in this model as well as its ultimate resolution despite continued TNBS administration. The remarkable aspect about the progression in cytokine responses in chronic TNBS-colitis is that the Th1 and Th17 responses are separate in time, suggesting that intestinal inflammations in humans may ordinarily be characterized by sequential responses that change the “mix” of Th1 and Th17 cells as the lesions mature. This concept is in keeping with the counter-regulatory character of Th1 and Th17 differentiation illustrated by the tendency of IL-17 to inhibit IFN-γ-producing T cells and vice versa31,33. In addition it is consonant with a recent parabiosis and co-transfer studies showing that Th1 and Th17 are in competition and down-regulate one another39. On these bases, it is attractive to speculate that Crohn’s disease inflammation consists of multiple microenvironments, each reflecting different stages of a cytokine response cycle. (See Figure 3).
Figure 3. Inflammatory Micro-Environments Characterizing CD.

There is considerable evidence that Th1 and Th17 responses are in an uneasy state of co-existence (See Figure 2). In this figure the concept is put-forward that such co-existence is minimized by the fact that Crohn’s inflammation consists of innumerable micro-environments, each exhibiting a progression of inflammatory patterns. In the initial and most intense phase of the inflammation, Th1 responses predominate; at this point, production of IL-23 in a nascent Th17 response inhibits Treg generation and feeds the inflammation. In a later phase a mixed T cell response prevails in which the Th1 response is still predominant but is now moderated by a Th17 response producing both IL-17 (which inhibits IFN-γ T cells) and IL-22.
Th1 and Th17 Cytokine Responses in Crohn’s Disease
Our evolving knowledge of cytokine production in Crohn’s disease is not unlike that in experimental models of colonic inflammation. Thus, as in the latter case, early studies pointed to the presence of an underlying Th1 response characterized by lamina propria T cells that produced increased amounts of IFN-γ and lamina propria antigen-presenting cells that produced increased amounts of IL-12p706. These early findings were corroborated by later studies showing that lamina propria T cells expressed increased amounts of IL-12/IFN-γ-induced factors such as Stat4, T-bet and IL-12Rβ237,56. Later, with the discovery that Th17 responses might drive experimental colitis, IL-17 producing cells were sought in the lamina propria of IBD patients. Indeed, in one study elevations were found in both Crohn’s Disease and ulcerative colitis (but to higher extent in the former than in the latter) and in another study IL-17R mRNA was actually higher in ulcerative colitis than in Crohn’s disease, although in the latter disease the level correlated with areas of disease activity57-59. More importantly, however, it was the discovery in 2006 that single nucleotide polymorphism in the IL-23R gene was associated with decreased susceptibility to both forms of IBD that really concentrated attention on the potential role of Th17 responses in these diseases60.
Two studies have recently appeared that address the importance of the Th17 response in CD vis a vis, the Th1 response. In the first attention was focused on the function of patient mesenteric node (MLN) T cells and dendritic cells, i.e., cell populations considered to reflect the responses occurring in the lamina propria. In initial studies it was shown the CD4+ T cells from CD MLN produced increased amounts of both IFN-γ and IL-17 compared to UC MLN and in this case the IL-17 levels in UC were barely elevated. Importantly, T cell IFN-γ production level was some 40-fold greater than the IL-17 production level61. Thus, as in the case of cell-transfer colitis, the IFN-γ response is far greater than the IL-17 response and one cannot assume the former was originating from Th17 cells producing both IL-17 and IFN-γ. In further studies the types of dendritic cells were analyzed and only minor differences were found among patients and controls; however, functional studies of one of the DC types (mature DCs) assessing their ability to induce cytokine in allogeneic T cells showed that the induction of cells producing IFN-γ was vastly greater than those producing IL-1761. Taken together, these studies strongly suggest that, from a quantitative point of view, the Th1 response in Crohn’s Disease is far greater than the Th17 response and thus that the Th17 response is not likely to be the major source of effector cytokines.
In a second study in which a somewhat different picture emerged, evidence was first presented showing that the surface marker CD161 identified T cells in the peripheral blood that in the resting state produced IL-17 and IL-22, but relatively little IFN-γ; furthermore, these cells expressed the Th17-associated receptor, IL-23R62. The authors thus concluded that CD161 is a surrogate marker for Th17 cells. In further studies in which cells from CD and control peripheral circulation and lamina propria were analyzed, it was shown that CD peripheral cells contained a higher percentage of IL-23R+ cells and exhibited increased IL-23-induced IL-17 and IFN-γ production compared to control cells. In addition, while CD lamina propria harbored increased numbers of CD161+ cells, the frequency of CD161 expression among CD4+ cells was high in both patient and control tissue (on the order of 80%). This may be due to the fact that CD161 appears on activated cells and in the activating milieu of the lamina propria, CD161 may be associated with both Th1 and Th17 cells. Thus, while these studies suggest that Th17 cells are in fact increased in the circulation and lamina propria of Crohn’s disease as compared to controls they don’t provide clear-cut information about the frequency of these cells vs. the frequency of Th1 cells in patient intestinal tissues.
Overall, studies of Crohn’s disease relating to the question of the relative importance of the Th1 and Th17 response to inflammation are in agreement with the findings in murine models in that they suggest that while Th17 responses occur in human disease and potentially play some role in the inflammatory process, the Th1 response is quantitatively greater and thus more likely to be the driving force of the inflammation. This conclusion is consonant with the known pathologic effects of IFN-γ in the intestine, the relation of Th1 responses to granulomatous disease and the Th1 provenance of extra-intestinal lesions63,40. In addition, it is consonant with a the results of a recent blinded clinical trial of anti-IL-17 in patients with Crohn’s disease which showed than anti-IL-17 had no therapeutic effect (Personal communication W. Hueber , Inhibition of IL-17A by Secukinumab is ineffective for Crohn’s disease, to be presented ECCO 2011). Finally, with respect to the protective role of the IL-23R polymorphism, it should be borne in mind that this polymorphism does not necessarily relate to the magnitude of the Th17 response, since we have seen that it also can control the regulatory function of Foxp3 T cells45; it thus might lead to less disease because it is associated with more negative regulation than with less pro-inflammatory Th17 activity.
Cytokine Responses in Ulcerative Colitis
As mentioned at the outset of this review, the cytokines driving ulcerative colitis were identified as having Th2-like characteristics in the initial studies attempting to place it within the Th1/Th2 spectrum. These consisted of lamina propria cells producing increased amounts of IL-5 in the conspicuous absence of increased amounts of IL-4, the more defining Th2 cytokine; hence, the descriptive phrase, Th2-like3. Also absent was any hint of an increased IFN-γ response, thus ruling out the presence of a Th1-driven inflammation. More recently, several studies have appeared showing that IL-17 levels were increased in ulcerative colitis compared to controls, but in the most reliable studies in which protein rather than mRNA was measured this increase was found to be far less than that found in Crohn’s disease61.
In an attempt to gain insight into the origin of this kind of inflammation investigators studied several colitis models driven by a Th2 response, such as TCRα chain deficiency and late-phase IL-10 deficiency64. However, these models differed from ulcerative colitis by the fact that the dominant cytokine abnormality was an elevated IL-4 response, which is not observed in UC. A breakthrough came when it was discovered that intra-rectal administration of oxazolone, an agent that like TNBS binds to self-proteins and renders them immunogenic, causes an intense but short-lived colitis that exhibits characteristics of ulcerative colitis65. The latter consisted of a superficial inflammation associated with micro-ulcerations of the epithelium, edema of the bowel wall and an inflammatory infiltrate containing granulocytes. In addition, the inflammation was most intense in the distal half of the colon, as frequently found in ulcerative colitis.
Initial cytokine analysis of oxazolone-colitis (oxa-colitis) revealed the presence of cells producing greatly increased amounts of IL-4 but not IFN-γ and increased amounts of TGF-β, a cytokine whose production is favored by Th2 conditions. Furthermore, treatment of oxa-colitis with anti-IL-4 prevented disease whereas as treatment with anti-IL-12p40 exacerbated disease. Thus, there could be no question that oxa-colitis, in contrast to TNBS-colitis, fit into the Th2 T cell spectrum. It should be noted that these studies of oxa-colitis was conducted with SJL/J mice, a strain which proved to be more susceptible than C57BL/6 to oxa-colitis, as was the case for TNBS-colitis.
In a second study of oxa-colitis, mice were pre-sensitized to oxazolone by skin painting and could thus be induced with a lower intra-rectal dose of oxazolone that caused a less intense and more persistent inflammation66. This modified oxa-colitis model revealed that the cytokine response, given more time, evolved from an initial IL-4 response lasting only several days into a more prolonged IL-13 response. In addition, the latter was shown to emanate from CD4+ NKT cells and not from conventional CD4+ T cells. Indeed, these studies proved that IL-13 and NKT cells are the driving force of oxa-colitis in that treatment of the latter with an IL-13 blocker such as IL-13Rα2-Fc or with antibodies that delete NKT cells, leads to amelioration of the inflammation. In addition, mice with CD1-deficiency that lack the ability to present antigens to NKT cells as well as mice with Jα281-deficiency that lack the invariant T cell receptors (TCRs) that NKT cells usually use to recognize antigen were also unable to support oxa-colitis. Finally, stimulation of lamina propria T cells with CD1-expressing antigen-presenting cells loaded with a-galactosyl-ceramide, the canonical antigen that has been shown to stimulate NKT cells expressing the “invariant” TCR associated with NKT cells, led to high level production of Th2 cytokines, including IL-13. Taken together, these data offered definitive proof that oxa-colitis was due to NKT cells producing IL-13.
The above studies of oxa-colitis set the stage for studies of ulcerative colitis, which would determine if an NKT cell/IL-13 abnormality also was present in this disease. Initial studies showed that stimulated lamina propria T cells from UC patients did indeed produce high levels of IL-13 but not IL-4 (or IFN-γ) whereas cells from CD patients produced only slightly increased amounts of IL-13 (compared to controls)11. Similarly, flow cytometric studies revealed that UC patients had high levels of lamina propria CD4+ T cells producing IL-13 whereas CD patients had only minor increases in such cells. In further studies, lamina propria T cells from UC patients were shown to produce IL-13 when stimulated with B cells stably transfected with CD1d, i.e., cells which preferentially activate NKT cells. It was noted, however, that stimulation of UC lamina propria cells with a-galactosyl-ceramide did not lead to activation of NKT cells as determined by up-regulation of Vα and Vβ chain expression. It was thus evident that UC lamina propria harbored increased numbers of IL-13-producing NKT cells but that these cells were not typical NKT cells bearing invariant T cell receptors (capable of responding to a-galactosyl-ceramide).
In a final series of studies, the potential of either NKT cells and/or IL-13 to mediate tissue damage was evaluated. It was found that purified lamina propria CD4+ T cells were cytotoxic to HT-29 epithelial cells pre-stimulated with LPS to up-regulate CD1d and that such cytotoxicity was augmented by IL-13. Furthermore, it was observed that IL-13 (but not IL-4) lowers the electrical resistance of HT-29 epithelial cell monolayers by inducing epithelial cell apoptosis and increasing expression of pore-forming tight-junction protein claudin-267. Interestingly, the latter was also observed in patients with ulcerative colitis. These studies suggest that the presence of NKT cells in the lamina propria of ulcerative colitis can lead to ulceration and inflammation by NKT cell-mediated cytotoxicity; in addition, such tissue injury is abetted by IL-13, which augments NKT cell cytotoxicity and has effects on epithelial cell integrity (See Figure 4).
Figure 4. The Pathogenesis of Ulcerative Colitis.
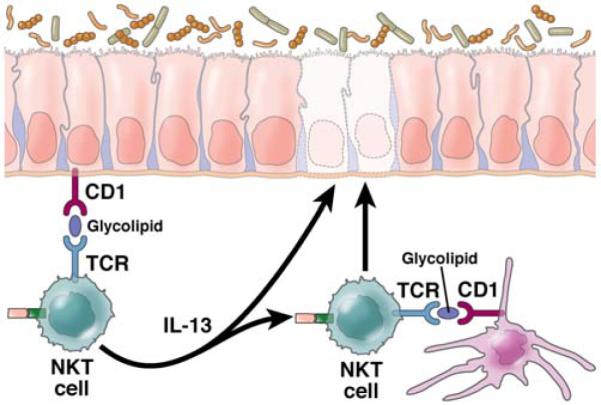
Inflammation in ulcerative colitis is initiated by glycolipid antigen(s) rising from either the microbiota or epithelial cells acted upon by microbiota. These antigens are presented to NKT cells in the context of CD1 on the surface of epithelial cells or dendritic cells and the NKT cells so stimulated act as effector cells mediating disease. Resultant epithelial cell damage and ulcer formation is caused by the cytotoxic activity of the NKT cells which recognize epithelial cells bearing antigen-loaded CD1 as targets or by IL-13 which has been shown to cause epithelial cell apoptosis and loss of epithelial barrier function; in addition, IL-13 enhances NKT cell cytotoxity.
The above studies of ulcerative colitis provide a basic framework with which to understand the immunopathogenesis of UC; nevertheless, they leave certain basic questions unanswered including the identification of the glycolipid antigen or antigens that stimulate UC NKT cells and the mechanism by which IL-13 stimulates NKT cell cytotoxic activity. In addition, in that they focus on IL-13 as a major factor in UC, they fail to explain why a polymorphism of the gene encoding the IL-23 receptor would result in protection from the development of UC (as well as CD), although one possibility here is that since IL-23R is found on NKT cells the polymorphism may be affecting NKT cell activity68. Finally, it is important to mention that the above studies offer no formal proof that either IL-13 or NKT cells (or both) are pathologic factors in UC as they manifestly are in oxa-colitis. Nothing short of clinical studies in which the efficacy of IL-13 or NKT cell inhibitors is tested under blinded conditions will suffice to establish this possibility. This said, one recently published clinical study does offer promising correlative evidence that IL-13 is a pathologic factor in UC. This consisted of a study of a group of patients with UC given IFN-β therapy in which it was found that significant response to this therapy was clearly correlated with decreased IL-13 production by lamina propria T cells69.
Effector Cytokines “Bridging” the Th1/Th17/Th2 Spectrum – TL1A
The cytokines discussed so far are those that are clearly located somewhere on the Th1/Th17/Th2 spectrum and that are mainly responsible for the distinctive type of inflammation characterizing the form of IBD with which they are associated. There are, however, a well known group of additional cytokines, such as TNF-α, IL-1β and IL-6, that are more promiscuous in their function in that they are associated with both forms of IBD to a lesser or greater degree53,64. These cytokines generally arise secondary to the primary Th1/Th17 or Th2-like response as a result of stimulation of innate immune cells (macrophages, epithelial cells, mast cells, etc), which are drawn into the inflammatory milieu. In addition, each of these cytokines activate NF-κB and the MAP kinases and thereby induce various “downstream” pro-inflammatory effects that are the immediate precursors of tissue and organ pathology in IBD. It is important to bear in mind, however, that these cytokines are also involved in important “upstream” roles. IL-6 and possibly IL-1β, for instance, are essential for initial induction of Th17 responses19,70. In addition, TNF-α enhances production of IL-12, a function which may account for the particular (but not exclusive) association of this cytokine with Th1 responses.
TL1A (Tumor Necrosis Factor-Like Ligand) a cytokine more recently shown to contribute to intestinal inflammation, also belongs to the category of cytokines that bridge the T cell differentiation spectrum. This cytokine is secreted by both antigen-presenting cells and T cells (as well as by endothelial cells) and signals through DR3, a TNF-family receptor that is found primarily on T cells; in addition, it is induced in antigen-presenting cells by TLR ligands and FcR cross-linking and in T cells by TCR stimulation71. The relevance to TL1A to colitis is shown by the fact that exogenous administration of TL1A to mice with DSS-colitis upregulated both Th1 and Th17 responses of T cells extracted from the inflamed colonic tissue72. Furthermore, administration of anti-TLA1 at least partially ameliorated DSS-colitis and in a subsequent study this antibody (or a DR3 blocker (DR3-Fc)) completely prevented the development of TNBS-colitis73,74.
In recent studies of DR3-deficient mice it was found TL1A-DR3 interactions co-stimulate cells subject to conventional TCR stimulation but is nevertheless not required for T cell differentiation or indeed for host-defense against toxoplasma infection75. However, it is required for the full expression of inflammation in experimental allergic encephalopathy (EAE) and experimental asthma. The increased role of DR3 signaling in these latter situations is facilitated by increased TL1A expression induced by innate factors such as TLR stimulation and Fc stimulation71,75. Recent studies of the function of TL1A in mice bearing a TL1A transgene expressed in T cells indicate that TL1A enhances baseline T cell and B cell activation by TCR stimulation and can induce spontaneous inflammation of the small bowel, most evident in the terminal ileum. Surprisingly, this inflammation was accompanied by greatly increased IL-13 synthesis which proved to be driving the inflammation since the latter was prevented by anti-IL-13 administration74. Yet another new set of findings concerning TL1A is that while TL1A inhibits the induction of new Foxp3+ regulatory T cells, it may induce expansion of existing Foxp3+ regulatory T cells76. Overall, these studies in mice suggest that TL1A is a co-stimulatory cytokine that optimizes both Th1 and Th17 responses characterizing murine models of inflammation and in addition can induce its own unique form of inflammation (See Figure 5).
Figure 5. TL1A Function.

TL1A is a TNF-family member that acts exclusively via DR3. It’s pro-inflammatory function bridges the Th1/Th17-Th2 dichotomy in that it enhances both types of T cell responses. As shown in this Figure, Th1 or Th17 differentiation results in T cells that express DR3 (the TL1A receptor) and are then subject to TL1A co-stimulation, the latter necessary for optimal proliferative and cytokine responses. Th2 cells are co-stimulated in a similar manner (not shown). TL1A has other effects such as those of Tregs that are not depicted here which may also contribute to inflammation.
Studies of TL1A in IBD are consistent with the above mouse studies. First, elevated TL1A levels have been noted in both CD and UC indicating that TL1A is not associated with a particular form of T cell differentiation71,72,77,78. Second, lamina propria CD14+ macrophages in CD produce increased amounts of TL1A and the latter enhance allo-antigen-induced T cell production of both IFN-γ and IL-17, but had little effect on its own72; thus, as in the mouse studies TL1A appears to have an enhancing effect rather than a primary effect on inflammatory responses. Finally, it has been shown that polymorphisms in the TL1A gene are associated with increased risk for Crohn’s disease, indicating that the above enhancing effects are not trivial and may in fact be necessary for disease expression in some patients79.
Anti-cytokine Agents Likely to be Useful in the Treatment of IBD
The review of the cytokine responses causing experimental and human intestinal inflammation presented above offers some guideposts relating to the type of new anti-cytokine therapy likely to be of use in the future treatment of CD and UC. Anti-TNF-α therapy has been and is likely to continue to be a major form of therapy for IBD. However, anti-TNF-α therapy is ineffective in about 50% of initially treated CD patients and becomes ineffective in another 50% of patients over time; thus, the need for additional anti-cytokine therapies is clear80. The reason for such treatment failure is presently unknown despite considerable study, but most likely relates to the fact that anti-TNF-α is either primarily or secondarily unable to cause apoptosis of T cells in many patients81,82.
Anti-IL-12p40 is a major new line of therapy which was proven to have therapeutic efficacy in initial phase 1 and 2 clinical trials9,10. However, in the most recent trial involving the largest group of patients, it had significant, but rather unspectacular effects at least during the induction phase and only marginal effects during the maintenance phase83. It should be noted, however, that in this latter study, doses of antibody administered during induction were considerably separated in time and it may be necessary to provide a more concentrated dosing schedule to see clear-cut therapeutic effects. Although anti-IL-12p40 has the potential to neutralize both Th1 and Th17 responses its ability to neutralize the former may be its most important function assuming that, as discussed above, the main function of the Th17 response is to restrain the regulatory T cells that would otherwise inhibit the Th1 response. For this reason, anti-IL-23p19 is, theoretically, likely to be a less effective anti-inflammatory agent and anti-IL12p40, particular if it inhibits the anti-inflammatory effects of IL-22 and the down-regulatory effects of IL-17 on the Th1 response49,50. Another anti-Th1 agent, anti-IFN-γ, has already be subjected to trial and has been found to have little effectiveness as has anti-IL1784. This may be due to the fact that only anti-cytokine agents that cause apoptosis of effector elements, such as anti-IL-12p40 and anti-TNF-α, are likely to have major therapeutic benefit. In the absence of this quality, a therapeutic anti-cytokine (such as anti-IFN-γ) must accomplish the daunting task of constantly neutralizing newly produced inflammatory cytokines85.
Several years ago anti-IL-6 was proposed as a useful anti-inflammatory agent in IBD and other autoimmune diseases and indeed some success in the treatment of rheumatoid arthritis with this agent has been reported86,87. This form of anti-cytokine therapy may owe its effectiveness to the fact that antibody binding to IL-6 interferes with the ability of this cytokine to bind to soluble IL-6 and to thus engage in “trans-signaling” via insertion of the IL-6/IL-6 receptor into the cell which ordinarily deliver an anti-apoptotic signal to the cell. Thus, anti-IL-6 prevents apoptosis of effector cells and prolongs inflammation88.
Finally, anti-TL1A has been proposed as a useful anti-cytokine therapy owing to the ability of this anti-cytokine to potentially block the ability of TL1A to enhance proliferation of both Th1 and Th2 effector cells72-77. However, since TL1A has no effect on T cells in the absence of a primary stimulus, anti-TL1A is most likely to work in tandem with other forms of anti-cytokine therapy.
Conclusion
This analysis of the cytokine responses mediating intestinal inflammation in IBD calls attention to the multi-layered and complex nature of these responses. Nevertheless, that point with ever more clarity to a rational anti-cytokine therapy of IBD which holds great promise to providing an effective approach to long-term control of IBD inflammation.
Supplementary Material
Biographies


Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors state that there are no conflicts of interest.
References
(FIRST 50 IN PRINT; REST ONLINE ONLY AS SUPP. FILE)
- 1.Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol. 1986;136:2348–2357. [PubMed] [Google Scholar]
- 2.Braese E, Braegger CP, Corrigan CJ, Walker-Smith JA, MacDonald TT. Interleukin-2- and interferon-gamma-secreting T cells in normal and diseased human intestinal mucosa. Immunology. 1993;78:127–131. [PMC free article] [PubMed] [Google Scholar]
- 3.Fuss IJ, Neurath M, Boirivant M, Klein JS, de la Motte C, Strong SA, Fiocchi C, Strober W. Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn’s disease LP cells manifest increased secretion of IFN-gamma, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. J Immunol. 1996;157:1261–1270. [PubMed] [Google Scholar]
- 4.Neurath MF, Fuss I, Kelsall BL, Stüber E, Strober W. Antibodies to interleukin 12 abrogate established experimental colitis in mice. J Exp Med. 1995;182:1291–1290. doi: 10.1084/jem.182.5.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu Z, Beboes K, Heremans H, Overbergh L, Mathieu C, Rutgeerts P, Ceuppens JL. Role of interleukin-12 in the induction of mucosal inflammation and abrogation of regulatory T cell function in chronic experimental colitis. Eur J Immunol. 2001;31:1550–1560. doi: 10.1002/1521-4141(200105)31:5<1550::AID-IMMU1550>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 6.Fuss IJ, Becker C, Yang Z, Groden C, Hornung RL, Heller F, Neurath MF, Strober W, Mannon PJ. Both IL_12p70 and IL-23 are synthesized during active Crohn’s disease and are down-regualted by treatment with anti-IL_12 p40 monoclonal antibody. Inflamm Bowel Dis. 2006;12:9–15. doi: 10.1097/01.mib.0000194183.92671.b6. [DOI] [PubMed] [Google Scholar]
- 7.Hart AL, Al-Hassi HO, Rigby RJ, Bell SJ, Emmanuel AV, Knight SC, Kamm MA, Stagg AJ. Characteristics of intestinal dendritic cells in inflammatory bowel diseases. Gastroenterology. 2005;129:50–65. doi: 10.1053/j.gastro.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 8.Matsuaka K, Inoue N, Sato T, Okamoto S, Hisamatsu T, Kishi Y, Sakuraba A, Hitotsumatsu O, Ogata H, Koganei K, Fukushima T, Kanai T, Watanabe M, Ishii H, Hibi T. T-bet upregulation and subsequent interleukin 12 stimulation are essential for induction of Th1 mediated Immunopathology in Crohn’s disease. Gut. 2004;53:1303–1308. doi: 10.1136/gut.2003.024190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mannon PJ, Fuss IJ, Elson CO, Sandborn WJ, Present D, Dolin B, Goodman N, Groden C, Hornung RL, Quezado M, Yang Z, Neurath MF, Salfeld J, Veldman GM, Schwertschlag U, Strober W, Anti-IL-12 Crohn’s Disease Study Group Anti-interleukin-12 antibody for active Crohn’s disease. N Engl J Med. 2004;351:2069–2079. doi: 10.1056/NEJMoa033402. [DOI] [PubMed] [Google Scholar]
- 10.Sandborn WJ, Feagan BG, Fedorak RN, Scherl E, Fleisher MR, Katz S, Johann J, Blank M, Rutgeers P, Uslekinumab Crohn’s Disease Study Group A randomized trial of Ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with moderate-to-severe Crohn’s disease. Gastroenterology. 2008;135:1130–1141. doi: 10.1053/j.gastro.2008.07.014. [DOI] [PubMed] [Google Scholar]
- 11.Fuss IJ, Heller F, Boirivant M, Leon F, Yosida M, Fichtner-Feigl S, Yang Z, Exley M, Kitani A, Blumberg RS, Mannon P, Strober W. Nonclassical CD1d-restricted NK T cells that produce IL-13 characterize an atypical Th2 response in ulcerative colitis. J Clin Invest. 2004;113:1490–1497. doi: 10.1172/JCI19836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oppman B, Lesley R, Blom B, Timans JC, Xu Y, Hunte B, Vega F, Yu N, Wang J, Singh K, Zonin F, Valsberg E, Churakova T, Liu M, Gorman D, Wanger J, Zurawski S, Liu Y, Arams JS, Moore KW, Rennick D, de Waal-Maleft R, Hannum C, BAzan JF, Kastelein RA. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000;13:715–725. doi: 10.1016/s1074-7613(00)00070-4. [DOI] [PubMed] [Google Scholar]
- 13.Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem. 2003;278:1910–1914. doi: 10.1074/jbc.M207577200. [DOI] [PubMed] [Google Scholar]
- 14.Yen D, Cheung J, Scheerens H, Poulet F, McClanahan T, McKenzie B, Kleinschek MA, Owyang A, Mattson J, Blumenschein W, Murphy E, Sathe M, Cua DJ, Kastelein RA, Rennick D. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J Clin Invest. 2006;116:1310–1316. doi: 10.1172/JCI21404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen Y, Langrish CL, McKenzie B, Joyce-Shaikh B, Stumhofer JS, McClanahan T, Blumenschein W, Churakovsa T, Low J, Presta L, Hunter CA, Kastelein RA, Cua DJ. Anti-IL-23 therapy inhibits multiple inflammatory pathways and ameliorates autoimmune encephalomyelitis. J Clin Invest. 2006;116:1317–1326. doi: 10.1172/JCI25308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hue S, Ahern P, Buonocore S, Kullberg MC, Cua DJ, McKenzie BS, Powrie F, Maloy KJ. Interleukin-23 drives innate and T cell-mediated intestinal inflammation. J Exp Med. 2006;203:2473–2483. doi: 10.1084/jem.20061099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kullberg MC, Jankovic D, Feng CG, Hue S, Gorelick PL, McKenzie BS, Cua DJ, Powrie F, Cheever AW, Maloy KJ, Sher A. IL-23 plays a key role in Helicobacter hepaticus-induced T cell-dependent colitis. J Exp Med. 2006;203:2485–2494. doi: 10.1084/jem.20061082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trinchieri G. Interleukin-12: a Proinflammatory cytokine with immunoregulatory functions that bridge innate resistance and antigen-specific adaptive immunity. Ann Rev Immunol. 1995;13:251–276. doi: 10.1146/annurev.iy.13.040195.001343. [DOI] [PubMed] [Google Scholar]
- 19.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 20.McGeachy MJ, Chen Y, Tato CM, Laurence A, Joyce-Shaik B, Blumenschein WM, McClanahan TK, O’Shea JJ, Cua DJ. The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17-producing effector T helper cells in vivo. Nat Immunol. 2009;10:314–324. doi: 10.1038/ni.1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ivanov II, Zhou L, Littman DR. Transcriptional regulation of Th17 cell differentiation. Semin Immunol. 2007;19:409–417. doi: 10.1016/j.smim.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bettelli E, Oukka M, Kuchroo VK. T(H)-17 cells in the circle of immunity and autoimmunity. Nat Immunol. 2007;8:345–350. doi: 10.1038/ni0407-345. [DOI] [PubMed] [Google Scholar]
- 23.McGeachey MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, Cua DJ. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol. 2007;8:1390–1397. doi: 10.1038/ni1539. [DOI] [PubMed] [Google Scholar]
- 24.Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE, Ramos HL, Wei L, Davidson TS, Bouladoux N, Grainger JR, Chen Q, Kanno Y, Watford WT, Sun HW, Eberl G, Shevach EM, Belkaid Y, Cua DJ, Chen W, O’shea JJ. Generation of pathogenic T(H)17 cells in the absence of TGF-β signaling. Nature. 2010;467:967–971. doi: 10.1038/nature09447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou L, Littman DR. Transcriptional regulatory networks in Th17 cell differentiation. Curr Opin Immunol. 2009;21:146–152. doi: 10.1016/j.coi.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang XO, Pappu BP, Nurieva R, Akimzhanov A, Kang HS, Chung Y, Ma L, Shah B, Panopoulos AD, Schluns KS, Watowich SS, Tian Q, Jetten AM, Dong C. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity. 2008;28:29–39. doi: 10.1016/j.immuni.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu L, Kitani A, Fuss I, Strober W. Cutting edge: regulatory T cells induce CD4+CD25-Foxp3-T cells or are self-induced to become Th17 cells in the absence of exogenous TGF-beta. J Immunol. 2007;178:6725–6729. doi: 10.4049/jimmunol.178.11.6725. [DOI] [PubMed] [Google Scholar]
- 28.Zhou L, Lopes JE, Chong MM, Ivanov II, Min R, Victoria GD, Shen Y, Du J, Rubtsov YP, Rudensky AY, Ziegler SF, Littman DR. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature. 2008;453:236–240. doi: 10.1038/nature06878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koenen JH, Smeets RL, Vink PM, van Rijssen E, Boots AM, Joosten I. Human CD25highFoxp3pos regulatory T cells differentiate into IL-17-producing cells. Blood. 2008;112:2340–2352. doi: 10.1182/blood-2008-01-133967. [DOI] [PubMed] [Google Scholar]
- 30.Yang XO, Nrurieva R, Martinez GJ, Kang HS, Chung Y, Pappu BP, Shah B, Chang SH, Schluns KS, Watowich SS, Feng XH, Jetten AM, Dong C. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity. 2008;29:44–56. doi: 10.1016/j.immuni.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 32.Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 33.Lee YK, Turner H, Maynard CL, Oliver JR, Chen D, Elson CO, Weaver CT. Late developmental plasticity in the T helper 17 lineage. Immunity. 2009;30:92–107. doi: 10.1016/j.immuni.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Powrie F, Correa-Oliveira R, Mauze S, Coffman RL. Regulatory interactions between CD45RBhigh and CD45RBlow CD4+ T cells are important for the balance between protective and pathogenic cell-mediated immunity. J Exp Med. 1994;179:589–600. doi: 10.1084/jem.179.2.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Powrie F, Mason D. OX-22 high CD4+ T cells induce wasting disease with multiple organ pathology: prevention by the OX-22 low subset. J Exp Med. 1990;172:1701–1708. doi: 10.1084/jem.172.6.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Powrie F, Leach MW, Mauze S, Menon S, Caddie LB, Coffman RL. Inhibition of Th1 responses prevents inflammatory bowel disease in scid mice reconstituted with CD45RBhi CD4+ T cells. Immunity. 1994;1:553–562. doi: 10.1016/1074-7613(94)90045-0. [DOI] [PubMed] [Google Scholar]
- 37.Neurath MF, Weigmann B, Finotto S, Glickman J, Nieuwenhuis E, Iijima H, Mizoguchi A, Mizoguchi E, Mudler J, Galle PR, Bhan A, Autschbach F, Sullivan BM, Szabo SJ, Glimcher LH, Blumberg RS. The Transcription factor T-bet regulates mucosal T cell activation in experimental colitis and Crohn’s disease. J Exp Med. 2002;195:1129–1143. doi: 10.1084/jem.20011956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Griseri T, Asquith M, Thompson C, Powrie F. OX40 is required for regulatory T cell-mediated control of colitis. JEM. 2010;207:699–709. doi: 10.1084/jem.20091618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mikami Y, Kanai T, Sujino T, Ono Y, Hayashi A, Okazawa A, Kamada N, Matsuoka K, Hisamatsu T, Okamoto S, Takaishi H, Inoue N, Ogata H, Hibi T. Competition between colitogenic Th1 and Th17 cells contributes to the amelioration of colitis. Eur J Immunol. 2010;40:2409–2422. doi: 10.1002/eji.201040379. [DOI] [PubMed] [Google Scholar]
- 40.Uhlig HH, McKenzie BS, Huie S, Thompson C, Joyce-Shaikh B, Stepankova R, Robinson N, Buonocore S, Tlaskalova-Hogenova H, Cua DJ, Powrie F. Differential activity of IL-12 and IL-23 in mucosal and systemic innate immune pathology. Immunity. 2006;25:309–318. doi: 10.1016/j.immuni.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 41.Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, Cross R, Sehy D, Blumberg RS, Vignali DA. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature. 2007;450:566–569. doi: 10.1038/nature06306. [DOI] [PubMed] [Google Scholar]
- 42.Ahern PP, Schiering C, Buonocore S, McGeachy MJ, Cua DJ, Maloy KJ, Powrie F. Interleukin-23 drives intestinal inflammation through direct activity on T cells. Immunity. 2010;33:279–288. doi: 10.1016/j.immuni.2010.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Elson CO, Cong Y, Weaver CT, Schoeb TR, McClanahan TK, Fick RB, Kastelein RA. Monoclonal anti-interleukin 23 reverses active colitis in a T cell-mediated model in mice. Gastroenterology. 2007;132:2359–2370. doi: 10.1053/j.gastro.2007.03.104. [DOI] [PubMed] [Google Scholar]
- 44.Leppkes M, Becker C, Ivanov II, Hirth S, Wirtz S, Neufert C, Pouly S, Murphy AJ, Valenzuela DM, Yancopoulos GD, BEcher B, Littman DR, Neurath MF. RORgamma-expressing Th17 cells induce murine chronic intestinal inflammation via redundant effects of IL-17A and IL-17F. Gastroenterology. 2009;136:256–267. doi: 10.1053/j.gastro.2008.10.018. [DOI] [PubMed] [Google Scholar]
- 45.Izcue A, Hue S, Buonocore S, Arancibia-Cárcamo CV, Ahern PP, Iwakura Y, Maloy KJ, Powrie F. Interleukin-23 restrains regulatory T cell activity to drive T cell-dependent colitis. Immunity. 2008;28:559–570. doi: 10.1016/j.immuni.2008.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.O’Connor O, Jr, Kamanaka M, Booth C, Town T, Nakae S, Iwakura Y, Kolls JK, Flavell RA. A protective function for interleukin 17A in T cell-mediated intestinal inflammation. Nature Immunology. 2009;10:603–609. doi: 10.1038/ni.1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang XO, Chang SH, Park H, Nurieva R, Shah B, Acero L, Wang YH, Schluns KS, Broaddus RR, Zhu Z, Dong C. Regulation of inflammatory responses by IL-17F. J Exp Med. 2008;205:1063–1075. doi: 10.1084/jem.20071978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Becker C, Domhoff H, Neufert C, Fantini MC, Wirtz S, Huebner S, Nikolaev A, Lehr HA, Murpha AJ, Valenzeula DM, Yancopoulos GD, Galle PR, Karow M, Neurath MF. Cutting edge: IL-23 cross-regulates IL-12 production in T cell-dependent experimental colitis. J Immunol. 177:2760–2764. doi: 10.4049/jimmunol.177.5.2760. 206. [DOI] [PubMed] [Google Scholar]
- 49.Sugimoto K, Ogawa A, Mizoguchi E, Shimomura Y, Andoh A, Bhan AK, Blumberg RS, Xavier RJ, Mizoguchi A. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J Clin Invest. 2008;118:534–544. doi: 10.1172/JCI33194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Stevens S, Flavell RA. Innate and adaptive interleukin-22 protects mice from inflammatory bowel disease. Immunity. 2008;29:947–957. doi: 10.1016/j.immuni.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.