

Abstract
Here we present a biomimetic strategy towards nanoparticle design for controlled immune response through encapsulation of conserved internal influenza proteins on the interior of virus like particles (VLPs) to direct CD8+ cytotoxic T cell protection. Programmed encapsulation and sequestration of the conserved nucleoprotein (NP) from influenza on the interior of a VLP, derived from the bacteriophage P22, results in a vaccine that provides multi-strain protection against 100 times lethal doses of influenza in an NP specific CD8+ T cell-dependent manner. VLP assembly and encapsulation of the immunogenic NP cargo protein is the result of a genetically programmed self-assembly making this strategy amendable to the quick production of vaccines to rapidly emerging pathogens. Addition of adjuvants or targeting molecules were not required for eliciting the protective response.
Keywords: virus like particle, VLP, P22, nucleoprotein, influenza, CD8, biomimetic
Protein cage architectures have emerged as important platforms for both soft and hard materials and there is significant interest in their biomedical application.1-4 Protein cages have been used to encapsulate inorganic nanomaterials either through directed nucleation and mineralization5-9 or by a directed self-assembly templated around a preformed nanomaterial.10-15 Small molecule and polymer attachment to a wide range of protein cages have been successful in making designer hybrid materials with great potential as MR imaging agents or drug delivery systems.16-23 In addition, the directed encapsulation of gene products (proteins, polypeptides) inside viral capsids has developed as a novel method for investigating the effects of protein crowding,24 the creation of novel nanoreactors,25-28 and the delivery of gene products for medical applications.29 While significant work is emerging on the use of protein cage nanomaterials in biomedical applications, lingering concerns over the immune response to these materials in vivo remains. However, biomedical applications exist where induction of specific immune responses is desired in the creation of new immune responsive therapeutic materials for combating major human health problems and disease.4, 30 In particular, and the focus of this work, is the persistent difficulty in producing materials that will effect broad range protection against influenza. We specifically utilize a novel strategy for biomimetic display of antigens within a virus like particle (VLP) that is generalizable to nearly any gene product (protein), easily produced, and amenable to rapid modification and production, with the ability to produce new antigenic VLPs in quick response to emerging pathogens, as discussed in detail below.
VLPs, a class of protein cage architectures, have found significant utility in the development of biomaterials31 and towards biomedical materials development, particularly in the areas of therapeutic delivery, imaging, and vaccine development.3 These multimeric protein assemblies, derived from viral capsids, exhibit complex architectures with coat proteins assembled around a hollow interior space. They are non-infectious because they are assembled without incorporating any genetic material on the interior. Several VLP vaccines are in clinical use and there are many undergoing preclinical trials, some of which target influenza.32-37 These vaccines have largely utilized the coat proteins of the VLP as the agent for inducing an immune response that provides protection. In addition, much of the work on VLP presentation of antigens has primarily focused on the exterior display of proteins/peptides from antigens targeting the generation of neutralizing antibody responses via a major histocompatibility complex type II (MHCII) presentation.32-37 However, VLPs present a rich biomaterial architecture and platform for displaying antigens in a spatially controlled manner, either on the exterior and/or on the interior, which could be exploited to direct specific immune response pathways. For instance, display of an antigen exclusively on the interior would first require degradation of the VLP to expose the antigen to the immune system, mimicking the display of internal antigens of pathogens which stimulate CD8+ T cell responses and aid in the clearance of infected cells. Alternatively, display of antigens on the exterior directly exposes the antigen to the immune system and might generate neutralizing antibodies, as mentioned above, which would act to prevent infection. The ability to specifically engineer VLPs to present antigens to the immune system so as to drive a particular immunogenic response is a potentially useful strategy for constructing new vaccines and other therapeutic agents.
The use of nanoparticle architectures to harbor antigens to elicit protective immune responses has been shown to be effective at generating protective immune responses.30, 38, 39 Kasturi et al. synthesized synthetic polymer nanoparticles loaded with hemagglutinin (HA) from the avian influenza H5N1 virus and Toll-like receptor ligands that afforded protection against lethal avian and swine influenza virus strains in mice and induced robust immunity against H1N1 influenza in rhesus macaques.40 While utilizing synthetic nanoparticles can provide rapid synthesis of new vaccines, the genetic control provided by VLP platforms, such as the P22 bacteriophage system described here, utilizes knowledge of the structure and location of antigens at a molecular level not available in other synthetic systems. In addition, the use of VLPs can allow for rapid synthesis of new vaccine candidates by expression of complete vaccine constructs via heterologous expression, providing added economic and sustainability advantages. For instance, we have shown that the VLP derived from the bacteriophage P22 from Salmonella typhimurium can be used to encapsulate a wide range of protein cargoes in vivo by co-expression the capsid coat protein (CP) with genetic fusion of the cargo protein and the scaffold protein (SP), which directs capsid assembly and is incorporated onto the interior of the assembled VLP.41-43 P22 VLPs encapsulating cargo in this manner produces up to 200 mg P22 VLPs/L media when expressed in E. coli and requires relatively little processing to obtain pure native structured P22 VLPs. Utilizing organisms for the genetic fabrication of nanoparticle vaccines eliminates the need for synthesizing components, in the case of synthetic nanoparticles, and the use of organic solvents for their synthesis. Furthermore, the use of a P22 VLP based approach would allow new vaccines to be produced against rapidly emerging pathogen variants, such as influenza, by standard molecular biology techniques to mirror the changes in the pathogens, with the ability to produce the new vaccine variants in a matter of weeks (currently requires ∼6 months to begin production of new vaccines). These properties make the P22 VLP a unique platform for constructing biomaterials with controlled immunogenic characteristics.
We have previously found that the P22 VLP alone provides non-specific protection from lethal doses of pathogens such as influenza and MRSA.44 These results suggested that P22 could be an effective biomimetic platform for encapsulating immunogenic antigens in order to elicit and direct desired protective immune responses. Recent reports have highlighted the urgent need for new vaccines for the prevention of influenza45, 46 and our previous results showing non-specific protection against influenza by P22 fueled our investigation of constructing P22 VLPs for directing protective immune responses against influenza presented here. The ability of influenza virus to thwart current vaccine strategies is a major problem for human health resulting in over 41,000 deaths annually in the United States alone and even greater numbers in years of pandemic outbreaks.47 Prevention of yearly seasonal outbreaks has primarily focused on developing vaccines which induce neutralizing antibodies directed at viral glycoproteins, particularly HA, to prevent cell infection.48, 49 Vaccines against seasonal influenza are generated by monitoring virus strains circulating in the human population every year and generating a trivalent vaccine (composed of three strains) to the most prevalent strains observed from the surveillance.50 However, the antigenic sites of HA are prone to continuous amino acid mutation leading to antigenic drift. In addition, reassortment of viral gene segments between various influenza viruses of human and zoological origin, known as antigenic shift, leads to the emergence of new influenza strains not accounted for in the current vaccine design.50 It has become apparent that vaccines for certain viruses and intracellular bacteria should induce protective CD8+ T cell responses to be most effective. Attenuated pathogen vaccines have been used because they induce strong oligo-clonal CD8+ T cell responses but infection after vaccination of immunocompromised individuals, occurrence of revertant mutations in the vaccine strain, or recombination with other pathogens in infected patients are possible dangers with attenuated vaccines.51 In the case of a new rapidly emerging virus, attenuation of the virus into a safe vaccine strain takes much time to develop and, therefore, synthetic vaccines that induce protective CD8+ T cell responses have also been pursued. These include peptide-loaded dendritic cells52 and DNA vaccination,53 which are effective approaches but are not necessarily practical. Incorporation of CD8+ T cell epitopes into liposomes54 has been shown to be effective in inducing CD8+ T cell-dependent immunity but requires the inclusion of strong adjuvants. VLPs with adjuvants such as CpG,55, 56 or dendritic cell targeting molecules57 have also been shown to induce protective CD8+ T cell responses. However, there is a significant need for a simple yet effective vaccine delivery system that induces strong CD8+ T cell-dependent immunity locally in the lungs without the need for adjuvants which could damage the lungs.
For eliciting a broadly protective CD8+ T cell response to influenza, we identified the conserved nucleoprotein (NP) as an antigen for encapsulation within the P22 VLP. The large size of NP (500 amino acids) would present a major challenge for encapsulation within alternative VLP systems and allows us to test the fidelity of the P22 VLP system. NP is conserved across multiple serotypes of influenza A strain viruses, which is the leading cause of seasonal influenza infection in humans, and between the three different strains.58 NP is responsible for the binding and packaging of the viral genome, hence it is located on the inside of the mature influenza virus. Because NP is an internal protein it induces an immunogenic response different from that of HA, which is external and produces neutralizing antibodies. Exposure of the immune system to influenza NP requires uptake and degradation of the virus for presentation of NP. Immunization with NP has been shown to provide broad serotype protection that elicits a virus-specific CD8+ CTL response, which has been suggested to contribute to heterosubtypic immunity.49, 59, 60 However, current vaccine strategies do not exploit NP for providing broad strain protection, perhaps due in part to the low yields from recombinant expression of NP (personal correspondence with T. D. Randall). Commercial sources of NP are not widely available, and NP stocks purchased previously by the authors from commercial sources were found to be of very poor quality. Developing a bioinspired vaccine utilizing NP or conserved NP epitopes, where NP proteins are presented to the immune system as it is in the natural influenza virus (i.e. encapsulated inside a viral capsid), that can elicit a specific CD8+ CTL response could lead to a universal protective vaccine against influenza. In addition, antigen encapsulation within the P22 VLP could be utilized as a method for readily obtaining NP, in a delivery ready form, by heterologous expression. Here we present results examining our strategy of utilizing P22 as a platform for antigenic protein display by encapsulating NP constructs on the interior of the VLP and show the ability of the NP-P22 nanoparticles to elicit protective CD8+ CTL responses, that are NP specific, against multiple serotypes of strain A influenza (Figure 1).
Figure 1. Schematic representations of the expression and in vivo encapsulation of the nucleoprotein through programmed self-assembly of the P22 VLP and the biomimetic display in order to elicit nucleoprotein-specific CD8+T cell response.
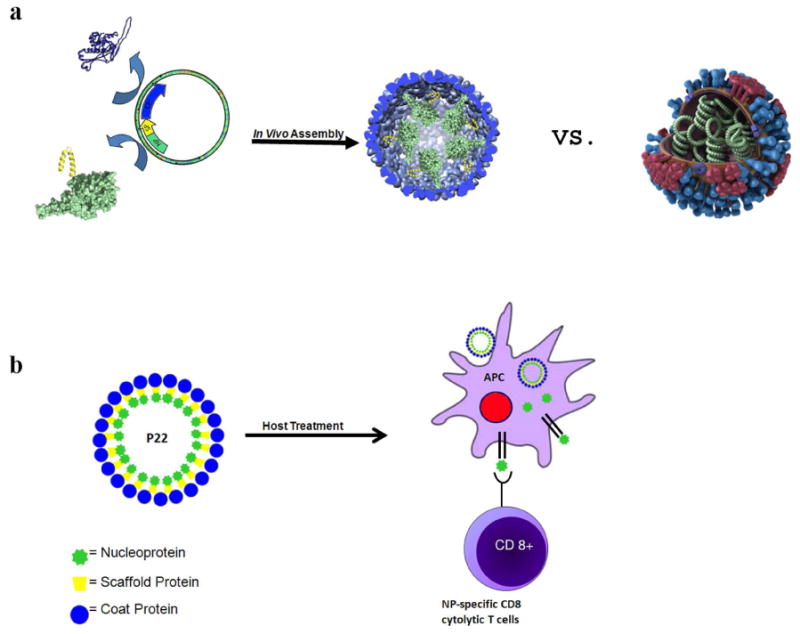
a) Nucleoprotein (NP; green) fusion with the scaffold protein (SP; yellow) is co-expressed with the coat protein (blue), resulting in assembly of the NP-P22 VLP. A model of the natural influenza virus (made available by the Center for Disease Control) is shown illustrating the display of NP (green), neuraminidase (red), hemagglutinin (blue), and M2 ion channels (purple) to highlight the biomimetic design of the NP-P22. b) Treatment of a host (immunization) with NP-P22, due to its biomimetic display of NP, is expected to be processed by the pathway that generates CD8+ T cells specific for NP.
Results and Discussion
Using our previously demonstrated methodology whereby a desired cargo can be encapsulated by genetically fusing it to the N terminus of a truncated P22 scaffold protein (SP), which directs the self-assembly of the P22 capsid when co-expressed with the coat protein (CP), we examined the construction of NP-P22 VLP nanoparticles (Figure 1). An initial construct consisting of the first third of the nucleoprotein (amino acids 1-163 (NP163)) genetically fused to truncated SP (NP163-SP) was co-expressed with the CP from a pETDuet-1 expression vector containing the two genes. The self-assembled P22 capsid was subsequently purified (Figures S1-S2) using a slightly modified method to those previously reported, whereby the mature P22 VLPs encapsulating NP was purified by ultracentrifugation over a cesium chloride gradient. Overall the modified method requires only two ultracentrifugation spins after obtaining the cell lysis supernatant, providing an efficient means for generating potential vaccines based on the P22 platform. Wild type recombinant P22 capsid and the NP163-P22 material exhibited nearly identical biophysical characteristics. Analysis of NP163-P22 by SDS-PAGE showed bands corresponding to NP163-SP (37.5 kDa) and CP (46.7 kDa) (Figure 2a) as expected. Direct visualization of the particles by transmission electron microscopy (TEM) verified that NP163-SP P22 samples were assembled into structures consistent with the size and morphology of native P22 and showed evidence for dense packing of protein on the interior (Figure 2b). Analysis of NP163-P22 by HPLC-SEC coupled to multi-angle and quasi elastic light scattering detectors (Figure 2c and Tables S1 and S2) revealed a molecular weight (Mw) of 28.0 ± 0.13 MDa, whereas empty P22 (no scaffold protein; data not shown) had a Mw of 19.1± 0.05 MDa (as compared to the expected Mw of ∼19.7 MDa). The difference in Mw, 8.9 MDa, corresponds to an estimated loading of 222 ± 3.5 NP163 per capsid, an extremely large number of packaged proteins per VLP. Light scattering revealed an average radius of gyration (Rg) of 25.5 nm and average hydrodynamic radius (RH) of 28.7 nm. The Rg/RH ratio (0.89) aligns well with a solid sphere model61, 62 and is consistent with highly dense packaging of NP-SP fusion proteins inside the P22, whereas empty P22 has been observed to give Rg/RH values closer to 1 (0.95) as expected for a hollow sphere model with infinitely thin walls.61, 62
Figure 2. Purification and characterization of the nucleoprotein encapsulated P22 VLP constructs (NP163-P22 and NP-P22).
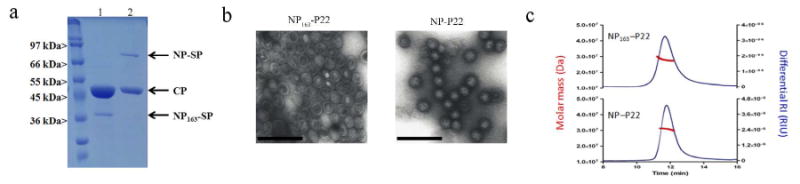
a) SDS-PAGE analysis of purified NP163-P22 (lane 1) and NP-P22 (lane 2) showing the coat protein (CP) and nucleoprotein-scaffold protein fusion constructs. b) Characterization of NP163-P22 (left) and NP-P22 (right) VLPs by TEM reveal homogeneous particles after purification by cesium chloride gradient ultracentrifugation. The scale bars represent 200 nm. c) Representative SEC elution profile monitored by absorbance and light scattering showing analysis of the molar mass across the elution peak of NP163-P22 (top) and NP-P22 (bottom).
The ability of the NP163-P22 to elicit a protective immune response was examined by immunization of mice with NP163-P22, P22 or PBS control, and subsequent challenge with lethal doses of influenza A PR8 (H1N1) followed by challenge with influenza A X-31 (H3N2). Because the P22 itself elicits a protective immune response44 we administered 100 (PR8) and 50 (X-31) times the lethal doses to mice in order to determine whether protection was associated with the encapsulated NP. Mice treated with NP163-SP P22 showed protection against both challenges of influenza, with 80% survival over the course of the experiment (after both challenges), whereas mice treated with P22 or control groups given buffer solution (PBS) did not survive the first challenge (Figure 3a). Only the NP163-P22 immunized mice and a new group of naïve mice were left for the X-31 challenge and all of the naïve mice died by 7 days after challenge, whereas all of the NP163-P22 immunized mice survived. To determine the role of CD8+ T cells in the protection, some of the NP163-P22 and P22 immunized mice were depleted of CD8+ T cells before the challenges. Only 1 out of 5 CD8+ T cell-depleted mice immunized with NP163-P22 survived the first dose of influenza (Figure 3a), indicating that the NP163-P22-induced protection was CD8+ T cell-dependent. Body weight data showed that the mice in all groups were infected, as indicated by the initial loss in body weight, however, the NP163-P22 treated mice were the only group that recovered after the initial body weight loss (Figure 3b). The body weight response observed for NP163-P22 treated mice is consistent with an expected CD8+ CTL protective response elicited by NP, which aids in clearance of influenza and recovery starting at day 7 but does not prevent infection as indicated by the loss in body weight after infection. That the NP163-P22-immunized mice were resistant to both H1N1 and H3N2 influenza viruses is consistent with the fact that NP is well conserved across influenza strains and responses to NP are cross protective to influenza strains.
Figure 3. Nasal immunization with NP163-P22 protects mice against 100× the lethal challenge with influenza.
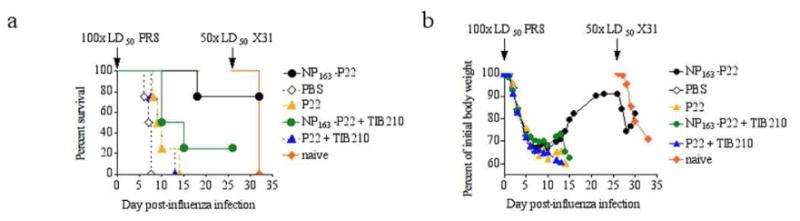
a) Survival of mice infected with 100× LD50 of PR8 influenza 26 days after completion of immunization and re-challenged with 50× LD50 of X31 influenza 20 days later. CD8+ T cell-depleted mice are identified with addition of TIB210 (+TIB210, green and blue traces). b) Body weights of infected mice were monitored daily. Decrease in body weight is presented as a percentage of initial body weight (at the time of challenge). Groups where 50% or more mice succumbed to the initial infection with PR8 influenza were excluded from further data collection. Results depict the average of 5 mice per group.
Major histocompatibility complex class I (MHCI) tetramers containing NP366-374 have previously been developed that are fluorescently labeled, which allows staining of CD8+ T cells that are specific for NP366-374.63 Since the NP163-P22 does not contain this epitope, we constructed a fusion protein consisting of the full length NP (NP) and SP (see Supporting Information for DNA and protein sequences) utilizing the same expression vector system. Expression, purification (Figures S1 and S2), and characterization of NP-P22 was carried out as was done for NP163-P22 with bands of the expected size for NP-SP (75.5 kDa) and CP (46.7 kDa) observed by SDS-PAGE and TEM results (observed 53. 5 ± 2.5 nm average diameter) consistent with those observed for NP163-P22 and native P22 (Figure 2). Analysis of NP-P22 by HPLC-SEC coupled to multiangle and quasi-elastic light scattering detectors (Figure 3c) showed an Mw of 30.6 ± 0.41 MDa. The difference in Mw between NP-P22 and empty P22 (see above), 10.9 MDa, corresponds to an estimated loading of 145 ± 5.5 NPs per capsid, still a very large number of encapsulated proteins, but less than observed for the smaller truncated NP163. The results in packaging between the two constructs agree well with other results we have observed in our lab, that smaller proteins/protein fragments are incorporated in higher numbers due to the limited volume that can be filled by the cargo proteins (unpublished observations). This has important implications for future design, as truncation of antigenic proteins to a minimal antigenic epitope will allow greater copy numbers to be incorporated allowing reduced dose requirements to elicit the same response as constructs containing fewer overall numbers of these epitopes. Light scattering revealed an average radius of gyration (Rg) of 22.6 nm and average hydrodynamic radius (RH) of 27 nm. Again, the Rg/RH ratio (0.84) aligns well with a solid sphere model61, 62 and is consistent with highly dense packaging of NP-SP fusion proteins inside the P22 and is smaller than the value observed for the empty P22 (0.95), which fits the empty thin walled hollow sphere model.
Mice immunized with the full length NP-P22, P22 or PBS were challenged with 50 times the lethal doses of PR8 and X-31 influenza strains as above. Over the course of the two challenges, 90% of the NP-P22-immunized mice survived, whereas only 70% of the CD8+ T cell-depleted NP-P22-immunized mice survived, and 40% of the P22-immunized mice survived, and none of the PBS treated mice survived (Figure 4a), again indicating the efficacy of NP-P22 immunization against various strains of influenza and the CD8+ T cell dependence. Interestingly, at the lower dose of PR8 used in this experiment, the partial protective ability of P22 is apparent, and the initial better survival of mice dosed with P22 and depleted of CD8+ T cells is likely due to reduced lung damage resulting from delayed response by repopulating CD8+ T cells. The apparent discrepancy in the level of protection against influenza infection granted by P22 in figures 3a and 4a is likely a result of different infection doses used in these experiments (100× LD50 PR8 in experiment in Figure 3a and 50× LD50 of PR8 in Figure 4a). As mentioned above, all of the P22 constructs were purified from E. coli expression system and thus contain lipopolysaccharide (LPS), a well-known immune adjuvant.64 However, we have not seen any survival differences between mice immunized with LPS-free or LPS-containing proteins (data not shown). To determine whether NP-P22 treatment induced an NP-specific CD8+ T cell response, cells in bronchoalveolar lavage fluid (BALF) of mice 7 days after influenza challenge and after receiving the various immunizing agents, were stained using fluorophore-linked MHCI tetramers that recognize NP-specific T cell receptors63 and fluorophore labeled antibodies specific for CD8+ T cells and were analyzed by flow cytometry (Figures 4b, S4 and S5). Figure 4b shows that the BALF of NP-P22-treated mice had significantly higher expression rates of NP-specific CD8+ T cells than either P22 immunized or PBS controls. These results confirm that the NP-P22 immunization causes an augmented NP-specific CD8+ T cell response upon influenza challenge, consistent with the biomimetic design of the NP-P22.
Figure 4. Immunization of mice with NP-P22 induces significant NP-specific CD8+ T cell responses following influenza infection.
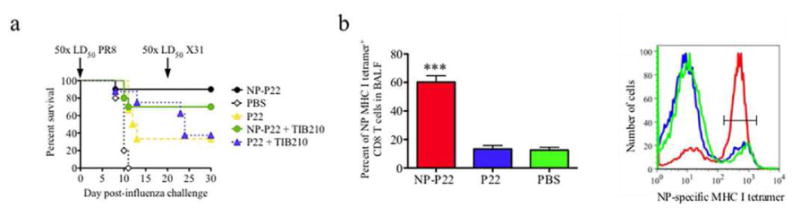
a) Survival of mice infected with 50× LD50 of PR8 influenza 26 days after completion of immunization and re-challenged with 50× LD50 of X31 influenza 20 days later. CD8+ T cell-depleted mice are identified with addition of TIB210 (+TIB210, green and blue traces). Survival curves show average survival of 8-10 mice per group. b) Flow cytometry analysis of cells isolated from BALF to determine the percent of NP-specific CD8+ T cells from mice immunized with NP-P22 (red), P22 (green), or PBS (blue). Bar graph (left) shows average percentage of tetramer+ cells from 5 mice per group and a representative histogram (right) shows the number of cells stained with NP-specific tetramer.
Depletion of CD8+ T cells in mice immunized with NP163-P22 increased mortality over 60% when compared to non-depleted mice immunized with NP163-P22 (80% survival of NP163-P22 immunized mice and 20% of the mice immunized with NP163-P22 and depleted of CD8 T cells; Figure 3a). In contrast to these mice, depletion of CD8+ T cells from mice immunized with NP-P22 resulted in only 20% increase in mice mortality (Figure 4a). To determine whether differences in induction of antibodies accounted for this difference in survival, we determined the levels of NP-specific IgG antibodies in serum of mice immunized with NP163-P22, NP-P22, P22 or PBS. None of the groups of mice had detectable levels of NP-, P22- or PR8-specific antibodies prior (day 0) to immunization (Figure 5a-c). On day 15 and 28 post-immunization, only mice that received NP-P22, but neither NP163-P22 nor P22 immunization induced production of NP-specific antibodies (Figure 5a). At day 35 after immunization (5 days after influenza challenge), all groups of mice showed similar titers of antibodies to both NP protein and PR8 membrane preparation (Figures 5 a,c). Interestingly, when the mice were challenged with a 2× LD50 of PR8, all of the PBS-treated mice lost weight rapidly (Figure 5D) and had died by about 10 days but none of the mice immunized with either P22 or one of the NP-P22 constructs died. Thus at this low dose of PR8 the protective effects of P22 only are apparent. However, mice immunized with the NP163-P22 lost more body weight than mice immunized with either P22 or NP-P22. The reason for the greater body weight loss in the NP163-P22-immunized mice could have been the result of less NP-specific antibody production in these mice (Figure 4a) and a greater reliance on CD8+ T cell-dependent protection as compared to NP-P22-immunized mice (Figures 3a and 4a)‥ NP-P22, NP163-P22 and P22-immunized mice all produced P22-specific antibodies, which suggest that antibodies to P22 were not instrumental in NP-P22-mediated protection from influenza.
Figure 5. Immunization with NP-P22 but not NP163-P22 induces NP-specific antibody responses prior to challenge with influenza.
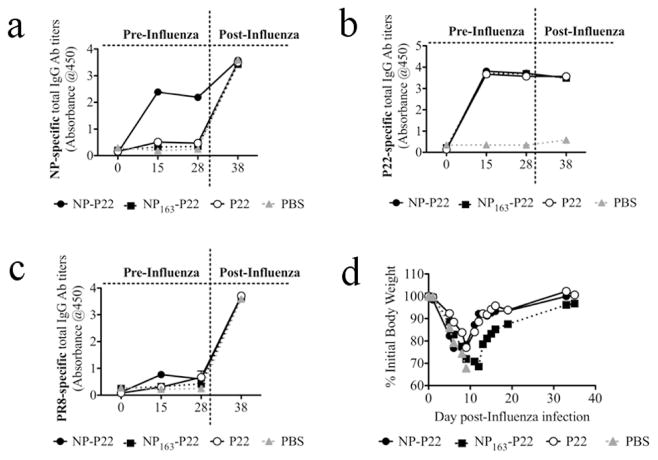
Mice were intranasally immunized daily for 5 days with 100 ug of designated protein. At 30 days after initial immunization mice were challenged with 2× LD50 of PR8 influenza. NP- (a), P22- (b), and PR8 membrane preparation-specific IgG antibody titers (c) were measured by ELISA in serum of mice immunized with NP-P22, NP163-P22 and P22 before and after influenza infection. d) Body weights of influenza-infected mice were monitored daily. Decrease in body weight is presented as a percentage of initial body weight (at the time of challenge). Groups where 50% or more mice succumbed to the infection with PR8 influenza (PBS-dosed mice) were excluded from further data collection. The average of 5 mice per group is depicted.
Conclusions
The results presented here provide strong evidence for constructing VLP based vaccines by encapsulation of highly conserved, non-surface exposed, immunogenic proteins via biomimetic display on the VLP interior. The results show the robust nature of the P22 system to incorporate large complete antigenic protein, which is often a limiting factor in other VLP systems. P22 encapsulating NP (truncated and full length constructs) elicited a strong protective immune response in mice against challenge with both H1N1 and H3N2 influenza viruses, without the addition of adjuvants. The NP-P22-induced protection was dependent, at least in part, on a NP-specific CD8+ T lymphocyte response, as we had predicted in our design, which was not found for control mice. Interestingly, we also observed that protective NP-specific antibodies were generated by the full length NP-P22 construct but not the truncated NP construct, leading to reduced protection in the truncated construct. Further studies are underway to determine a complete picture of the intricacies of the immune response generated by NP-P22 and provide a guide towards optimizing desired immune responses.
Materials and Methods
The NP gene, provided by Dr. Troy D. Randall in the pTrcHis vector, was amplified using the primers 5′-AAAAAAAAACCATGGCGCTCGAGGCGAGCCAGGGCACCA AACGTAG-3′ and 5′- AAAAAAAAAGGATCCGTCGACGTTATCGTATTCTTCCGC GTTATCGCC-3′ by PCR. NP163 or NP were incorporated into a pETDuet-1 assembler vector, containing the truncated scaffold protein (residues 141-303, SP141) 42 (multiple cloning site 1; placed in with BamHI/SacI) and P22 coat protein (multiple cloning site 2; placed in with NdeI/XhoI), utilizing NcoI and BamHI restriction sites. Incorporation of NP required removal of an internal BamHI (Supplementary Information) in the gene, utilized for incorporation of NP163, by PCR site specific mutagensis using the primers 5′-GTGCGTACCGGCATGGACCCGCGTATGT-3′ and 5′-ACATACGCGGGTCCATGC CGGTACGCAC-3′. Protein expression and purification was carried out as described previously for P22,41 but with a final purification step by ultracentrifugation at 38,000 rpm (TH-641 rotor; Thermo WX Ultra 80 ultracentrifuge) over a cesium chloride gradient (0.26-0.54 g CsCl/mL in PBS) for 2 hours replacing SEC purification. Band fractions from the cesium chloride gradient (Figures S2 and S3) were removed by pipet and analyzed by SDS-PAGE, TEM, and light scattering and all calculations performed as described previously.43 Experimental details of nanoparticle preparation and administration, TIB210 CD8+ T cell depletion, influenza infection, NP specific MHCI tetramer and FACS analysis, and flow cytometry of mouse studies was performed as previously described.44, 63, 65 Mice were lightly anesthetized with oxygen delivered isofluorane, USP (Piramal; Bethlehem, PA) and were intranasally dosed with 50 μl of sterile PBS or P22 containing 100 μg of protein. This dosing procedure was repeated 5 times (once daily for a total of 5 days). Infections with PR8 influenza strain were performed 26 days after last treatment dose. For infection with influenza, mice were lightly anesthetized with oxygen delivered isofluorane, USP and were intranasally dosed with 50 μl of virus preparations. The influenza virus strains A/PR8/8/34 (PR8; H1N1) was obtained from the Trudeau Institute, Saranac Lake, NY, USA. The PR8 influenza inoculum contained 1500 PFU.
Supplementary Material
Acknowledgments
This work was supported by grants from NIH (R01 EB012027), NIH/NIAID (R56AI089458), and the Idea Network for Biomedical Research Excellence (INBRE) (P20GM103500). We would like to thank Dr. Troy D Randall for the NP vector.
Footnotes
Supporting Information Available: DNA and protein sequences, supplementary Figures S1-S5 and Tables S1 and S2. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Uchida M, Klem MT, Allen M, Suci P, Flenniken M, Gillitzer E, Varpness Z, Liepold LO, Young M, Douglas T. Biological Containers: Protein Cages as Multifunctional Nanoplatforms. Adv Mater. 2007;19:1025–1042. [Google Scholar]
- 2.Shen L, Bao N, Zhou Z, Prevelige PE, Gupta A. Materials Design Using Genetically Engineered Proteins. J Mater Chem. 2011;21:18868–18876. [Google Scholar]
- 3.Yildiz I, Shukla S, Steinmetz NF. Applications of Viral Nanoparticles in Medicine. Curr Opin Biotechnol. 2011;22:901–908. doi: 10.1016/j.copbio.2011.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee LA, Wang Q. Adaptations of Nanoscale Viruses and Other Protein Cages for Medical Applications. Nanomedicine. 2006;2:137–149. doi: 10.1016/j.nano.2006.07.009. [DOI] [PubMed] [Google Scholar]
- 5.Reichhardt C, Uchida M, O'Neil A, Li R, Prevelige PE, Douglas T. Templated Assembly of Organic-Inorganic Materials Using the Core Shell Structure of the P22 Bacteriophage. Chem Commun. 2011;47:6326–6328. doi: 10.1039/c1cc11215e. [DOI] [PubMed] [Google Scholar]
- 6.Douglas T, Strable E, Willits D, Aitouchen A, Libera M, Young M. Protein Engineering of a Viral Cage for Constrained Nanomaterials Synthesis. Adv Mater. 2002;14:415-+. [Google Scholar]
- 7.Klem MT, Young M, Douglas T. Biomimetic Synthesis of Beta-TiO2 inside a Viral Capsid. J Mater Chem. 2008;18:3821–3823. [Google Scholar]
- 8.Allen M, Willits D, Young M, Douglas T. Constrained Synthesis of Cobalt Oxide Nanomaterials in the 12-Subunit Protein Cage from Listeria Innocua. Inorg Chem. 2003;42:6300–6305. doi: 10.1021/ic0343657. [DOI] [PubMed] [Google Scholar]
- 9.Flenniken ML, Willits DA, Brumfield S, Young MJ, Douglas T. The Small Heat Shock Protein Cage from Methanococcus Jannaschii Is a Versatile Nanoscale Platform for Genetic and Chemical Modification. Nano Lett. 2003;3:1573–1576. [Google Scholar]
- 10.Douglas T, Young M. Host-Guest Encapsulation of Materials by Assembled Virus Protein Cages. Nature. 1998;393:152–155. [Google Scholar]
- 11.Chen C, Daniel MC, Quinkert ZT, De M, Stein B, Bowman VD, Chipman PR, Rotello VM, Kao CC, Dragnea B. Nanoparticle-Templated Assembly of Viral Protein Cages. Nano Lett. 2006;6:611–615. doi: 10.1021/nl0600878. [DOI] [PubMed] [Google Scholar]
- 12.Aniagyei SE, DuFort C, Kao CC, Dragnea B. Self-Assembly Approaches to Nanomaterial Encapsulation in Viral Protein Cages. J Mater Chem. 2008;18:3763–3774. doi: 10.1039/b805874c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang X, Bronstein LM, Retrum J, Dufort C, Tsvetkova I, Aniagyei S, Stein B, Stucky G, McKenna B, Remmes N. Self-Assembled Virus-Like Particles with Magnetic Cores. Nano Lett. 2007;7:2407–2416. doi: 10.1021/nl071083l. [DOI] [PubMed] [Google Scholar]
- 14.Daniel MC, Tsvetkova IB, Quinkert ZT, Murali A, De M, Rotello VM, Kao CC, Dragnea B. Role of Surface Charge Density in Nanoparticle-Templated Assembly of Bromovirus Protein Cages. ACS Nano. 2010;4:3853–3860. doi: 10.1021/nn1005073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zheng B, Zettsu N, Fukuta M, Uenuma M, Hashimoto T, Gamo K, Uraoka Y, Yamashita I, Watanabe H. Versatile Protein-Based Bifunctional Nano-Systems (Encapsulation and Directed Assembly): Selective Nanoscale Positioning of Gold Nanoparticle-Viral Protein Hybrids. Chem Phys Lett. 2011;506:76–80. [Google Scholar]
- 16.Francis MB. Synthetically Modified Viral Capsids as Targeted Delivery Vehicles for Therapeutic Cargo. Biopolymers. 2011;96:417–417. [Google Scholar]
- 17.Stephanopoulos N, Tong GJ, Hsiao SC, Francis MB. Dual-Surface Modified Virus Capsids for Targeted Delivery of Photodynamic Agents to Cancer Cells. ACS Nano. 2010;4:6014–6020. doi: 10.1021/nn1014769. [DOI] [PubMed] [Google Scholar]
- 18.Wu W, Hsiao SC, Carrico ZM, Francis MB. Genome-Free Viral Capsids as Multivalent Carriers for Taxol Delivery. Angew Chem, Int Ed Engl. 2009;48:9493–9497. doi: 10.1002/anie.200902426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tong GJ, Hsiao SC, Carrico ZM, Francis MB. Viral Capsid DNA Aptamer Conjugates as Multivalent Cell-Targeting Vehicles. J Am Chem Soc. 2009;131:11174–11178. doi: 10.1021/ja903857f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garimella PD, Datta A, Romanini DW, Raymond KN, Francis MB. Multivalent, High-Relaxivity Mri Contrast Agents Using Rigid Cysteine-Reactive Gadolinium Complexes. J Am Chem Soc. 2011;133:14704–14709. doi: 10.1021/ja204516p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lucon J, Qazi S, Uchida M, Bledwell G, LaFrance B, Prevelige P, Douglas T. Using the Interior of the P22 Capsid for Site Specific Initiation of Atom Transfer Radical Polymerization with Tremendously Increased Cargo Loading. Nature Chem. 2012;4:781–788. doi: 10.1038/nchem.1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liepold LO, Abedin MJ, Buckhouse ED, Frank JA, Young MJ, Douglas T. Supramolecular Protein Cage Composite Mr Contrast Agents with Extremely Efficient Relaxivity Properties. Nano Lett. 2009;9:4520–4526. doi: 10.1021/nl902884p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Franzen S, Lommel SA. Targeting Cancer with ‘Smart Bombs’: Equipping Plant Virus Nanoparticles for a ‘Seek and Destroy’mission. Nanomedicine. 2009;4:575–588. doi: 10.2217/nnm.09.23. [DOI] [PubMed] [Google Scholar]
- 24.Minten IJ, Claessen VI, Blank K, Rowan AE, Nolte RJM, Cornelissen JJLM. Catalytic Capsids: The Art of Confinement. Chem Sci. 2011;2:358–362. [Google Scholar]
- 25.Comellas-Aragones M, Engelkamp H, Claessen VI, Sommerdijk NAJM, Rowan AE, Christianen PCM, Maan JC, Verduin BJM, Cornelissen JJLM, Nolte RJM. A Virus-Based Single-Enzyme Nanoreactor. Nat Nanotechnol. 2007;2:635–639. doi: 10.1038/nnano.2007.299. [DOI] [PubMed] [Google Scholar]
- 26.Fiedler JD, Brown SD, Lau JL, Finn MG. Rna-Directed Packaging of Enzymes within Virus-Like Particles. Angew Chem, Int Ed Engl. 2010;49:9648–9651. doi: 10.1002/anie.201005243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Inoue T, Kawano MA, Takahashi RU, Tsukarnoto H, Enornoto T, Imai T, Kataoka K, Handa H. Engineering of Sv40-Based Nano-Capsules for Delivery of Heterologous Proteins as Fusions with the Minor Capsid Proteins Vp2/3. J Biotechnol. 2008;134:181–192. doi: 10.1016/j.jbiotec.2007.12.006. [DOI] [PubMed] [Google Scholar]
- 28.de la Escosura A, Nolte RJM, Cornelissen JJLM. Viruses and Protein Cages as Nanocontainers and Nanoreactors. J Mater Chem. 2009;19:2274–2278. [Google Scholar]
- 29.Schott JW, Galla M, Godinho T, Baum C, Schambach A. Viral and Non-Viral Approaches for Transient Delivery of Mrna and Proteins. Curr Gene Ther. 2011;11:382–398. doi: 10.2174/156652311797415872. [DOI] [PubMed] [Google Scholar]
- 30.Moon JJ, Huang B, Irvine DJ. Engineering Nano- and Microparticles to Tune Immunity. Adv Mater. 2012;24:3724–3746. doi: 10.1002/adma.201200446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Douglas T, Young M. Viruses: Making Friends with Old Foes. Science. 2006;312:873–875. doi: 10.1126/science.1123223. [DOI] [PubMed] [Google Scholar]
- 32.Bachmann MF, Jennings GT. Vaccine Delivery: A Matter of Size, Geometry, Kinetics and Molecular Patterns. Nat Rev Immunol. 2010;10:787–796. doi: 10.1038/nri2868. [DOI] [PubMed] [Google Scholar]
- 33.Dormitzer PR, Tsai TF, Del Giudice G. New Technologies for Influenza Vaccines. Hum Vaccines Immunother. 2012;8:45–58. doi: 10.4161/hv.8.1.18859. [DOI] [PubMed] [Google Scholar]
- 34.Schneider-Ohrum K, Ross TM. Virus-Like Particles for Antigen Delivery at Mucosal Surfaces. Curr Top Microbiol. 2012;354:53–73. doi: 10.1007/82_2011_135. [DOI] [PubMed] [Google Scholar]
- 35.Grgacic EVL, Anderson DA. Virus-Like Particles: Passport to Immune Recognition. Methods. 2006;40:60–65. doi: 10.1016/j.ymeth.2006.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kang SM, Song JM, Quan FS, Compans RW. Influenza Vaccines Based on Virus-Like Particles. Virus Res. 2009;143:140–146. doi: 10.1016/j.virusres.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Plummer EM, Manchester M. Viral Nanoparticles and Virus-Like Particles: Platforms for Contemporary Vaccine Design. Wiley Interdiscip Rev: Nanomed Nanobiotechnol. 2011;3:174–196. doi: 10.1002/wnan.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Akagi T, Baba M, Akashi M. Biodegradable Nanoparticles as Vaccine Adjuvants and Delivery Systems: Regulation of Immune Responses by Nanoparticle-Based Vaccine. Adv Polym Sci. 2011:1–34. [Google Scholar]
- 39.McNeela E, Lavelle E. Recent Advances in Microparticle and Nanoparticle Delivery Vehicles for Mucosal Vaccination. Mucosal Vaccines. 2012:75–99. doi: 10.1007/82_2011_140. [DOI] [PubMed] [Google Scholar]
- 40.Kasturi SP, Skountzou I, Albrecht RA, Koutsonanos D, Hua T, Nakaya HI, Ravindran R, Stewart S, Alam M, Kwissa M, et al. Programming the Magnitude and Persistence of Antibody Responses with Innate Immunity. Nature. 2011;470:543–U136. doi: 10.1038/nature09737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Patterson DP, Schwarz B, El-Boubbou K, van der Oost J, Prevelige PE, Douglas T. Virus-Like Particle Nanoreactors: Programmed Encapsulation of the Thermostable Celb Glycosidase inside the P22 Capsid. Soft Matter. 2012;8:10158–10166. [Google Scholar]
- 42.O'Neil A, Reichhardt C, Johnson B, Prevelige PE, Douglas T. Genetically Programmed In Vivo Packaging of Protein Cargo and Its Controlled Release from Bacteriophage P22. Angew Chem, Int Ed Engl. 2011;50:7425–7428. doi: 10.1002/anie.201102036. [DOI] [PubMed] [Google Scholar]
- 43.Patterson DP, Prevelige PE, Douglas T. Nanoreactors by Programmed Enzyme Encapsulation inside the Capsid of the Bacteriophage P22. ACS Nano. 2012;6:5000–5009. doi: 10.1021/nn300545z. [DOI] [PubMed] [Google Scholar]
- 44.Rynda-Apple A, Dobrinen E, McAlpine M, Read A, Harmsen A, Richert LE, Calverley M, Pallister K, Voyich J, Wiley JA. Virus-Like Particle-Induced Protection against Mrsa Pneumonia Is Dependent on Il-13 and Enhancement of Phagocyte Function. Am J Pathol. 2012;181:196–210. doi: 10.1016/j.ajpath.2012.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Imai M, Watanabe T, Hatta M, Das SC, Ozawa M, Shinya K, Zhong G, Hanson A, Katsura H, Watanabe S. Experimental Adaptation of an Influenza H5 Ha Confers Respiratory Droplet Transmission to a Reassortant H5 Ha/H1n1 Virus in Ferrets. Nature. 2012;486:420–428. doi: 10.1038/nature10831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Herfst S, Schrauwen EJA, Linster M, Chutinimitkul S, de Wit E, Munster VJ, Sorrell EM, Bestebroer TM, Burke DF, Smith DJ. Airborne Transmission of Influenza a/H5n1 Virus between Ferrets. Science. 2012;336:1534–1541. doi: 10.1126/science.1213362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Beigel JH. Concise Definitive Review: Influenza. Crit Care Med. 2008;36:2660–2666. doi: 10.1097/CCM.0b013e318180b039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rappuoli R, Del Giudice G. Influenza Vaccines for the Future. 2nd. p xiv. Springer; Basel: 2010. p. 446 p. [Google Scholar]
- 49.Hillaire MLB, Osterhaus ADME, Rimmelzwaan GF. Induction of Virus-Specific Cytotoxic T Lymphocytes as a Basis for the Development of Broadly Protective Influenza Vaccines. J Biomed Biotechnol. 2011;2011:1–12. doi: 10.1155/2011/939860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kaur K, Sullivan M, Wilson PC. Targeting B Cell Responses in Universal Influenza Vaccine Design. Trends Immunol. 2011;32:524–531. doi: 10.1016/j.it.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zaiss DMW, Boog CJP, van Eden W, Sijts AJAM. Considerations in the Design of Vaccines That Induce Cd8 T Cell Mediated Immunity. Vaccine. 2010;28:7716–7722. doi: 10.1016/j.vaccine.2010.08.101. [DOI] [PubMed] [Google Scholar]
- 52.Nestle FO, Alijagic S, Gilliet M, Sun YS, Grabbe S, Dummer R, Burg G, Schadendorf D. Vaccination of Melanoma Patients with Peptide- or Tumor Lysate-Pulsed Dendritic Cells. Nat Med. 1998;4:328–332. doi: 10.1038/nm0398-328. [DOI] [PubMed] [Google Scholar]
- 53.Fissolo N, Riedl P, Reimann J, Schirmbeck R. DNA Vaccines Prime Cd8(+) T Cell Responses to Epitopes of Viral Antigens Produced from Overlapping Reading Frames of a Single Coding Sequence. Eur J Immunol. 2005;35:117–127. doi: 10.1002/eji.200425608. [DOI] [PubMed] [Google Scholar]
- 54.Engler OB, Schwendener RA, Dai WJ, Wolk B, Pichler W, Moradpour D, Brunner T, Cerny A. A Liposomal Peptide Vaccine Inducing Cd8(+) T Cells in Hla-A2.1 Transgenic Mice, Which Recognise Human Cells Encoding Hepatitis C Virus (Hcv) Proteins. Vaccine. 2004;23:58–68. doi: 10.1016/j.vaccine.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 55.Storni T, Ruedl C, Schwarz K, Schwendener RA, Renner WA, Bachmann MF. Nonmethylated Cg Motifs Packaged into Virus-Like Particles Induce Protective Cytotoxic T Cell Responses in the Absence of Systemic Side Effects. J Immunol. 2004;172:1777–1785. doi: 10.4049/jimmunol.172.3.1777. [DOI] [PubMed] [Google Scholar]
- 56.Schwarz K, Meijerink E, Speiser DE, Tissot AC, Cielens I, Renhof R, Dishlers A, Pumpens P, Bachmann MF. Efficient Homologous Prime-Boost Strategies for T Cell Vaccination Based on Virus-Like Particles. Eur J Immunol. 2005;35:816–21. doi: 10.1002/eji.200425755. [DOI] [PubMed] [Google Scholar]
- 57.Bonifaz LC, Bonnyay DP, Charalambous A, Darguste DI, Fujii SI, Soares H, Brimnes MK, Moltedo B, Moran TM, Steinman RM. In Vivo Targeting of Antigens to Maturing Dendritic Cells Via the Dec-205 Receptor Improves T Cell Vaccination. J Exp Med. 2004;199:815–824. doi: 10.1084/jem.20032220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Portela A, Digard P. The Influenza Virus Nucleoprotein: A Multifunctional Rna-Binding Protein Pivotal to Virus Replication. J Gen Virol. 2002;83:723–734. doi: 10.1099/0022-1317-83-4-723. [DOI] [PubMed] [Google Scholar]
- 59.Epstein SL, Kong WP, Misplon JA, Lo CY, Tumpey TM, Xu L, Nabel GJ. Protection against Multiple Influenza a Subtypes by Vaccination with Highly Conserved Nucleoprotein. Vaccine. 2005;23:5404–5410. doi: 10.1016/j.vaccine.2005.04.047. [DOI] [PubMed] [Google Scholar]
- 60.Flynn KJ, Belz GT, Altman JD, Ahmed R, Woodland DL, Doherty PC. Virus-Specific Cd8(+) T Cells in Primary and Secondary Influenza Pneumonia. Immunity. 1998;8:683–691. doi: 10.1016/s1074-7613(00)80573-7. [DOI] [PubMed] [Google Scholar]
- 61.Egelhaaf SU, Schurtenberger P. Shape Transformations in the Lecithin Bile-Salt System - from Cylinders to Vesicles. J Phys Chem. 1994;98:8560–8573. [Google Scholar]
- 62.Hotz J, Meier W. Vesicle-Templated Polymer Hollow Spheres. Langmuir. 1998;14:1031–1036. [Google Scholar]
- 63.Wiley JA, Hogan RJ, Woodland DL, Harmsen AG. Antigen-Specific Cd8(+) T Cells Persist in the Upper Respiratory Tract Following Influenza Virus Infection. J Immunol. 2001;167:3293–3299. doi: 10.4049/jimmunol.167.6.3293. [DOI] [PubMed] [Google Scholar]
- 64.Seppälä I, Mäkelä O. Adjuvant Effect of Bacterial Lps and/or Alum Precipitation in Responses to Polysaccharide and Protein Antigens. Immunology. 1984;53:827–836. [PMC free article] [PubMed] [Google Scholar]
- 65.Meissner N, Swain S, McInnerney K, Han S, Harmsen AG. Type-I Ifn Signaling Suppresses an Excessive Ifn-Γ Response and Thus Prevents Lung Damage and Chronic Inflammation During Pneumocystis (Pc) Clearance in Cd4 T Cell-Competent Mice. Am J Pathol. 2010;176:2806–2818. doi: 10.2353/ajpath.2010.091158. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.