

Abstract
Aims/hypothesis
We sought to determine the impact of longstanding type 1 diabetes on haematopoietic stem/progenitor cell (HSC) number and function and to examine the impact of modulating glycoprotein (GP)130 receptor in these cells.
Methods
Wild-type, gp130−/− and GFP chimeric mice were treated with streptozotocin to induce type 1 diabetes. Bone marrow (BM)-derived cells were used for colony-formation assay, quantification of side population (SP) cells, examination of gene expression, nitric oxide measurement and migration studies. Endothelial progenitor cells (EPCs), a population of vascular precursors derived from HSCs, were compared in diabetic and control mice. Cytokines were measured in BM supernatant fractions by ELISA and protein array. Flow cytometry was performed on enzymatically dissociated retina from gfp+ chimeric mice and used to assess BM cell recruitment to the retina, kidney and blood.
Results
BM cells from the 12-month-diabetic mice showed reduced colony-forming ability, depletion of SP-HSCs with a proportional increase in SP-HSCs residing in hypoxic regions of BM, decreased EPC numbers, and reduced eNos (also known as Nos3) but increased iNos (also known as Nos2) and oxidative stress-related genes. BM supernatant fraction showed increased cytokines, GP130 ligands and monocyte/macrophage stimulating factor. Retina, kidney and peripheral blood showed increased numbers of CD11b+/CD45hi/CCR2+/Ly6Chi inflammatory monocytes. Diabetic gp130−/− mice were protected from development of diabetes-induced changes in their HSCs.
Conclusions/interpretation
The BM microenvironment of type 1 diabetic mice can lead to changes in haematopoiesis, with generation of more monocytes and fewer EPCs contributing to development of microvascular complications. Inhibition of GP130 activation may serve as a therapeutic strategy to improve the key aspects of this dysfunction.
Keywords: Bone marrow microenvironment, Endothelial progenitors, GP130 deletion, Haematopoietic stem cells, IL-1β and IL-10, MMPs, Monocytes, Side population cells, Type 1 diabetes
Introduction
Bone marrow (BM)-derived cells contribute to vascular repair and tissue perfusion, and their use has spawned the field of therapeutic revascularisation [1–4]. However, they can participate not only in physiological repair but also in pathological neovascularisation. BM-derived endothelial progenitor cell (EPC) number in the circulation is reduced in diabetic individuals with non-proliferative diabetic retinopathy [5], and in animal models of diabetes [6]. A reduction in circulating progenitor cell number marks the clinical onset of type 2 diabetes [7]. This loss of EPCs is implicated in the pathogenesis of the vasodegenerative phase of diabetic retinopathy and in the generalised poor angiogenesis, as seen in reduced wound healing in poorly controlled diabetic patients. In contrast, increased numbers of EPCs have been implicated in the ‘angiogenic paradox’ seen as increased neovascularisation in proliferative diabetic retinopathy [8].
In diabetic animals and humans, not only are the numbers of EPCs affected, but EPCs also become dysfunctional [9]. Diabetes affects their mobilisation [10], proliferation and homing, and these deficiencies contribute to development of vascular complications [11–13]. Increased levels of reactive oxygen species (ROS) [14], reduced bioavailable nitric oxide (NO), and increased levels of proinflammatory mediators are implicated in the pathogenesis of EPC reparative dysfunction in diabetes [15, 16].
The BM is also the source of myelomonocytic cells [17], which contribute to diabetes-associated inflammation [18] and which, when blocked, can prevent development of retinopathy [19, 20]. The increase in local cytokine levels [9], in particular IL-6 [21], IL-8 [22] and TNF-α [23], affects the function of white blood cells, such as monocytes, shifting them towards a ‘proinflammatory’ phenotype [9] while simultaneously inhibiting the proliferation of EPCs [23]. Moreover, increased production of the inflammatory factor, monocyte/macrophage colony-stimulating factor (M-CSF), by stromal cells shifts haematopoiesis towards generation of monocytes [24]. A series of key cytokines that bind to glycoprotein (GP)130, the receptor for IL-6/leukaemia inhibitory factor (LIF), have been implicated in diabetic vascular dysfunction [25]. In this study, we sought to determine the impact of long-standing type 1 diabetes on haematopoiesis and haematopoietic stem cell (HSC) function and specifically examined the impact of modulating GP130 receptor in these cells on their function.
Methods
Mouse model of type 1 diabetes
All animal studies were approved by the Institutional Animal Care and Use Committee at the University of Florida, University of Oklahoma, and Case Western University. C57BL/6 mice were purchased (Jackson Laboratory, Bar Harbor, ME, USA) and made diabetic by streptozotocin (STZ) injection as described [26]. Please refer to the electronic supplementary material (ESM) (Methods, section 1.1) for details.
Mouse gp130−/− model studies
Tissue-specific deletion of GP130 was accomplished using Tie2-cre transgenic mice, expressing cre recombinase in vascular endothelial cells and all BM-derived cells [27]. Tie2-cre mice were mated with GP130-floxed (gp130f/f) mice [28]. Mice were inbred to produce Tie2-cre/gp130f/f mice, with >95% deletion of GP130 in vascular endothelial cells and BM-derived cells [27]. Diabetic mice with blood glucose levels >13.8 mmol/l were used for the study.
GFP chimeric mice studies
GFP BM chimeric mice were generated as previously described [4]. Briefly, BM was harvested from the femur of gfp+ transgenic (homozygous) mice. From the total BM cells, the c-Kit+ Sca-1+ HSCs were enriched by flow cytometry using anti-c-Kit and anti-Sca-1 antibody (BD Biosciences, San Jose, CA, USA). Cell purity was assessed by flow cytometry. HSCs were then injected into lethally irradiated adult C57BL/6J mice in the retro-orbital sinus. After 1 month, successful engraftment was confirmed by flow cytometry analysis of blood from the recipient mice.
Colony-forming assay using Lineage− c-Kit+ Sca-1+ (LSK) cells
LSK cells were isolated according to the manufacturer’s instructions (STEMCELL Technologies, Vancouver, Canada) and were plated on methocult medium (STEMCELL Technologies). Colonies were assessed for colony forming units (CFU)-G (granulocyte colonies), CFU-M (macrophage colonies) and CFU-GM (granulocyte-macrophage colonies). Please refer to the ESM for detailed methods (Methods, section 1.2 to 1.5).
For generation of early endothelial progenitors (eEPCs), bone marrow mononuclear cells (BMMNCs) were isolated from diabetic and control mice, and equal numbers of cells were plated on fibronectin in the presence of 20 ng/ml granulocyte colony-stimulating factor (G-CSF) at day 0. After 2 days, non-adherent cells from fibronectin-coated dishes were replated on to new six-well dishes. At day 5–7, the number of eEPC colonies was determined manually.
To generate endothelial colony-forming cells (ECFCs), equal numbers of BMMNCs were plated on collagen in the presence of 20 ng/mlG-CSF. ECFCs were obtained after 3–4 weeks from adherent cells.
Measurement of fat and water content by proton magnetic resonance spectroscopy (MRS)
1H single-voxel spectroscopy of BM was performed at 11.1 T magnetic field strength (470 MHz) (Bruker Biospin, Billerica, MA, USA) using Paravision 3.0.2 software (Bruker) to determine BM fat vs water ratio in diabetic (n=16) vs non-diabetic (n=14) mice. Data analysis was performed using XWin-NMR 3.0.2 software (Bruker). The method has been described in detail in the ESM (Methods, section 1.6).
Studies with mouse progenitors
Real-time PCR was performed using a 7500 Fast Real Time System (ABI Biosystems, Foster City, CA, USA). The in vitro migration assay was carried out according to the manufacturer’s instructions (Chemicon International, Temecula, CA, USA), and NO generation was measured as previously described [29]. Please refer to the ESM for detailed methods (Methods 1.7 to 1.9) and information about the primers (ESM Table 1).
Studies with BM supernatant fraction
BM was flushed with PBS, and cells were removed by centrifugation. The supernatant fraction was concentrated with an ultracentrifugation filter unit (Millipore, Bedford, MA, USA) and analysed for cytokines and growth factors by ELISA (R&D Systems, Minneapolis, MN, USA) and a luminex bead-based immunoassay platform (Assaygate, Ijamsville, MD, USA). Data were plotted as pg or ng of analyte per total amount of protein. Activity of matrix metalloproteinase (MMP)-9 was measured as described previously [30]. Detailed methods are described in the ESM (Methods 1.10).
Höechst and pimonidazole binding in vivo followed by FACS analysis of anti-pimonidazole staining and side population (SP) analysis
Murine HSCs were identified as described previously [31, 32]. Hypoxic cells were detected in BM by pimonidazole binding using a modified intracellular staining and flow cytometry method as described previously [33]. Please see details of these methods in the ESM (Methods 1.11).
Enumeration of monocytes and mobilisation from BM using G-CSF and AMD3100
Recombinant human G-CSF (125 μg/kg) was administered to control (n=6) and diabetic (n=6) mice every 12 h for a total of eight intraperitoneal injections [34], and blood was collected and analysed for Ly6Chi and CCR2hi monocytes. In addition, AMD3100 (5 mg/kg; Sigma, St Louis, MO, USA) was injected into the diabetic mice 3 h after the last G-CSF injection [34], and blood was also analysed for monocytes and endothelial progenitor cells.
Statistical analysis
Data are presented as mean±SEM. Student’s t test followed by Mann–Whitney or one-way ANOVA followed by Tukey’s post hoc test were used for statistical analysis using GraphPad Software (La Jolla, CA, USA). p<0.05 was considered significant.
Results
Long-standing type 1 diabetes results in deterioration of the BM microenvironment
LSK cells isolated from BM from 6-month-diabetic mice showed a 12% decrease, whereas after 12 months of diabetes a 55% decrease (p<0.05) in the total number of colonies was observed compared with controls (Fig. 1a, b). Colonies were able to incorporate DiI-labelled-acetylated LDL (DiI-Ac-LDL) (Fig. 1c); the cells gave rise to eEPCs and ECFCs (Fig. 1d) as well as expressing eNos (also known as Nos3) (Fig. 1e). eEPCs expressed Kdr, Cd68, Cd14 and Cd45, whereas ECFCs expressed Kdr, Cd31 (also known as Pecam) and Tie2 (also known as Tek). Compared with cells isolated from age-matched control mice, a significant reduction in the numbers of eEPCs was observed after 6 months of diabetes (p <0.05) (ESM Fig. 1a), while the total number of ECFCs showed a reduced trend, without reaching significance (ESM Fig. 1b). In addition, the total number of BMMNCs was reduced in 6-month-diabetic compared with age-matched control mice, although the numbers were comparable in 12-month-diabetic and age-matched control mice (ESM Table 2).
Fig. 1.

Effect of diabetes on the colony-forming ability of BM-derived LSK cells. (a) Total number of colonies formed by BM LSK cells isolated from mice with 6 and 12 months of diabetes was significantly lower than the cells from age-matched control mice. White bar, control; black bar, diabetic. *p<0.05 vs control, n=6 mice per group. (b) Cells demonstrate their ability to form colonies. (c) Cells take up DiI-Ac-LDL. (d) Cells give rise to ECFCs. (e) Cells express eNos
The BM microenvironment niches are composed of stromal/vascular/adipocyte cells and matrix which regulate HSC and progenitor cell behaviour. Adipocytes represent a key cell population which, when present in excess numbers, can negatively regulate haematopoiesis [35]. Less is known about the fat composition of type1 diabetic BM; therefore, we assessed fat content by non-invasive proton MRS in these mice. Unexpectedly, we observed a nearly 50% reduction in the BM fat ratio in 6-month-diabetic mice compared with control mice (Fig. 2), suggesting that the reduced numbers and function of LSK cells, eEPCs and ECFCs was not due to the changes in BM fat content.
Fig. 2.

Reduced fat content in the BM of diabetic mice. Representative single-voxel 1H MR spectra recorded non-invasively from within tibiae of (a) control and (b) diabetic mice. The taller peaks represent water, and the shorter peaks represent fat. (c) A significant reduction in marrow fat content was observed in the type 1 diabetic mice. *p< 0.05 vs control mice; n=10 mice for controls and n=14 mice for diabetic model
Diabetes results in a depletion of SP-HSCs
SP-HSCs were enumerated by low level binding and rapid efflux of Höechst 33342 (a cell-permeable DNA stain which is rapidly excluded by SP-HSCs because of ABC transporters) in 6- and 12-month-diabetic mice. Compared with controls (Fig. 3a), a 19% decrease in SP-HSCs was observed in the 6-month-diabetic mice (Fig. 3b), and a 93% decrease in 12-month-diabetic mice (Fig. 3c), showing a progressive depletion of this primitive stem cell population.
Fig. 3.
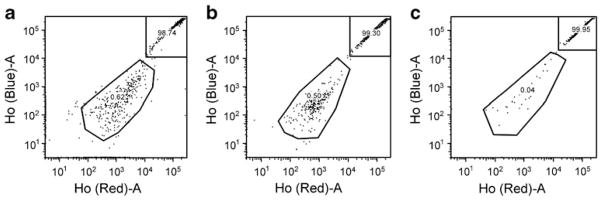
SP cells in BM are depleted in diabetic mice. SP-HSCs were detected using the ‘side population’ method of analysis in (a) control, (b) short-term-diabetic and (c) long-term-diabetic mice, the last of these showing reduced progenitor cells (11 month duration of type 1 diabetes) compared with control and short-term type 1 diabetes. n=3 mice per group; representative images of three independent experiments. Ho, Höechst
A subfraction of SP-HSCs demonstrate increased pimonidazole retention
To detect HSCs that were present in hypoxic niches, we administered the hypoxia probe pimonidazole (which in a hypoxic environment binds to –SH-containing molecules and accumulates in tissue) i.v. to diabetic and control mice before killing them. A pimonidazole-specific antibody was used to detect pimonidazole+ cells. In Fig. 4 (left panel), the majority of cells have higher fluorescence and are not shown on the scale. In all cases, the low Höechst regions encompass the HSCs. Region 1 (R1) represents mature haematopoietic cells (right panels), and region 2 (R2) represents the more primitive SP-HSCs (centre panels). As expected, SP-HSCs (R2) bound pimonidazole to a greater degree than the mature haematopoietic cells (Fig. 4). Cells in R1 and R2 from 4-month-diabetic mice showed only a small shift of pimonidazole staining compared with control mice. In contrast, cells from the 11-month-diabetic mice showed a prominent shift of pimonidazole staining compared with age-matched controls (R2 in Fig. 4), suggesting that more of the immature diabetic cells were hypoxic.
Fig. 4.

Low-oxygen microenvironment detection in diabetic and control mice BM. Low-oxygen microenvironments were detected using pimonidazole (PIM) uptake. The BMMNCs were stained with Höechst (Ho) 33342, and then the cells were washed to allow Höechst efflux. Cells from 4-month-diabetic (a–c), 4-month control (d–f), 11-month-diabetic (g–i) and 11-month control (j–l) mice are shown. The majority of cells show higher fluorescence and are off the scale (a, d, g and j). Region 1 (R1) (c, f, i and l) is composed of more mature cells, and region 2 (R2) (b, e, h and k) is primitive HSCs. As can be seen, the more differentiated cells essentially did not take up the pimonidazole. Representative images of three independent experiments; n=3 mice per group. MdCF, median channel fluorescence; FSC-H, forward scatter-height
Effect of diabetes on Nos, NADPH oxidase 2 (Nox2 [also known as Cybb]) and MMP expression
We measured all three Nos isoforms, nNos (also known as Nos1), eNos and iNos, in these cells. nNos was undetectable. eNos was undetectable in 6- and 12-month-diabetic mice and was significantly lower in 12-month control compared with 6-month control mice, suggesting an age-related decline in eNos (data not shown). BMMNCs isolated from the 12-month-diabetic mice showed a 31±3.3-fold increase in iNos compared with controls (p<0.05) (Fig. 5a), while iNos expression from 6-month-diabetic mice was comparable to controls.
Fig. 5.

Diabetes duration increases expression of genes associated with oxidative stress, stem cell mobilisation and level of inflammatory BM cytokines and growth factors. (a) Real-time PCR for iNos, Nox2, p22phox, p47phox and p67phox in BMMNCs isolated from mice with 12 months of established diabetes. All show significant increases vs age-matched control mice; *p<0.05 vs control mice, n=6 per group. (b) Measurement of selected cytokines, IL-1β, IL-3, IL-6, IL-10, TNF-α and M-CSF (*p<0.05 12-month control vs 12-month-diabetic), in BM supernatant fractions shows that duration of diabetes has a differential effect on cytokine expression. The key homing factor for endothelial progenitors, SDF-1, increases in the BM supernatant fraction in type1 diabetic mice (*p<0.05 12-month control vs 12-month-diabetic). n=4 mice per group
We reasoned that perhaps increased oxidative stress leading to peroxynitrite generation from iNos-generated NO may contribute to the stem cell dysfunction [36], therefore we measured expression of Nox, Nox2, Nox3, Nox4, p22phox (also known as Cyba), p47phox (also known as Ncf1) and p67phox (also known as Ncf2), key components that can contribute to ROS generation in BMMNCs. The only detected isoform in these cells was Nox2, which is consistent with a previous report [37]. Furthermore, no detectable difference in Nox2, p22phox, p47phox and p67phox expression was observed between 6-month-diabetic mice and controls (data not shown). However, by 12 months, a 2.0-fold, 1.3-fold, 2.6-fold and 13.0-fold increase in Nox2, p22phox, p47phox and p67phox was observed, respectively, in diabetic mice compared with controls (Fig. 5a).
BM cells express a wide array of adhesion molecules for attachment to the network of stromal cells. Proteolytic enzymes, such as MMPs, regulate progenitor release from these BM niches. The activity of MMP-9 in the BM supernatant fraction was measured, and age rather than diabetes (ESM Fig. 2) had the more profound effect.
Type 1 diabetes affects the levels of growth factors and cytokines in the BM microenvironment
The levels of stromal cell-derived factor (SDF)-1α, vascular endothelial growth factor (VEGF) and insulin-like growth factor binding protein-3 (IGFBP3), critical hypoxia-regulated factors that influence progenitor cell behaviour, were measured in BM supernatant fraction. SDF-1α levels showed a 1.96-fold increase after 12 months of diabetes compared with control mice (Fig. 5b). No change, however, was observed in VEGF levels among the groups (data not shown). IGFBP3, produced by BM stromal cells to support HSC proliferation, was comparable in controls and diabetic mice at 12 months (data not shown); however, Igfbp3 mRNA in the diabetic cells was 2.25-fold higher than in controls (p<0.05; data not shown), suggesting that in the diabetic BM supernatant fraction, IGFBP3 may be degraded [38]. There was no significant difference in IGF-1 and stem cell factor (data not shown) in diabetic compared with control mice.
We next compared the levels of IL-1β, IL-10, TNF-α, IL-6, IL-3, granulocyte-macrophage colony-stimulating factor (GM-CSF), G-CSF and M-CSF in BM supernatant fraction of the diabetic and control mice. Longer duration of diabetes resulted in increased IL-3, IL-10, IL-1β and TNF-α level compared with controls (Fig. 5b). In addition, GM-CSF was not detected, and G-CSF was not significantly altered (Fig. 5b). However, M-CSF, which promotes the generation of monocytes [24], was significantly increased in the diabetic supernatant fraction (5.86±0.68 pg/mg) compared with controls (2.52±0.31 pg/mg). This is consistent with our observations that more CFU-M and CFU-GM colonies were generated by BM cells isolated from diabetic mice (ESM Fig. 3).
CD45+/CCR2+/Ly6C+ positive monocytes are detected in the diabetic retina
We next asked whether there was an increase in extravasation of BM-derived cells in the diabetic retina, in particular the deleterious monocyte subset that is CCR2+. Using gfp chimeric mice made diabetic with STZ, we observed, as previously, that GFP+ cells enter the retina [39]. In Fig. 6, the inflammatory cells that infiltrated the retinas of the type 1 diabetic mice were examined. The CD45hi/CCR2low/neg/Ly6Clow/neg populations were CD11blow/neg, whereas CD45hi/CCR2+/Ly6Chi populations were CD11b+, consistent with an inflammatory monocytic phenotype. The inflammatory monocytes were expressed at approximately fivefold higher levels in diabetic compared with control mice. In addition, in both diabetic blood and kidney, CD45+/CCR2hi/Ly6Chi monocytes were increased compared with controls (ESM Fig. 4a, b).
Fig. 6.
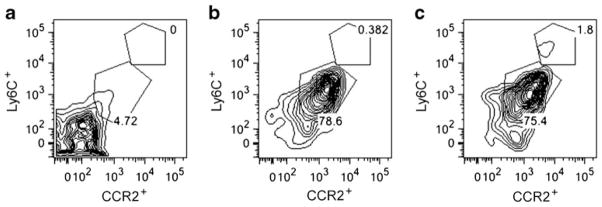
Diabetes increases inflammatory CD45+/CCR2+/Ly6C+ monocytes in retinas. The y-axis represents Ly6C, and the x-axis represents CCR2. (a) CD45− gate; (b, c) CD45+ gate. The control retina is shown in (b), and the diabetic retina in (c). The inflammatory infiltrate in STZ-induced diabetic retinas include a CD45hi/CCR2+/Ly6C+ population and a CD45hi/CCR2low/neg/Ly6C− population. The CD45hi/CCR2low/neg populations are CD11blow/neg, whereas CD45hi/CCR2+ populations are CD11b+ (consistent with an inflammatory monocytic phenotype). n=6 per group
We next evaluated mobilisation from the BM of monocytic cells and VEGFR2+/CD133+ cells, an EPC population. No significant difference in Ly6Chi/CCR2hi monocytes was found after G-CSF treatment in diabetic compared with control mice. However, in diabetic mice, injection of AMD3100 (a potent antagonist of the CXCR4 receptor) in addition to G-CSF resulted in a 78% increase in CCR2+/Ly6C+ monocytes in blood compared with injection of G-CSF alone (ESM Fig. 5). EPCs were also increased in the blood (about threefold compared with unstimulated control cells, n=3) and spleen of the mobilised diabetic mice (data not shown).
HSCs from type 1 diabetic gp130 knockout mice have increased ability to migrate coupled with increased generation of NO
The family of cytokines that bind GP130 includes IL-6, IL-11, IL-27, LIF, oncostatin M (OSM), ciliary neurotrophic factor and cardiotrophins and have been implicated in diabetes [40]. A threefold increase in OSM was observed in 12-month-diabetic mice compared with controls (Fig. 7). IL-27 was significantly higher after both 6 (2.4-fold change) and 12 months (2.6-fold change) of diabetes. LIF in the diabetic mice was threefold higher after 12 months compared with controls (Fig. 7). In contrast, IL-11, IL-6 and cardiotrophin-1 showed no difference between control and diabetic mice at either time point (data not shown). These results suggest that the BM supernatant fraction of type 1 diabetic mice shows increased production of selected ligands of GP130, specifically IL-27, LIF and OSM.
Fig. 7.
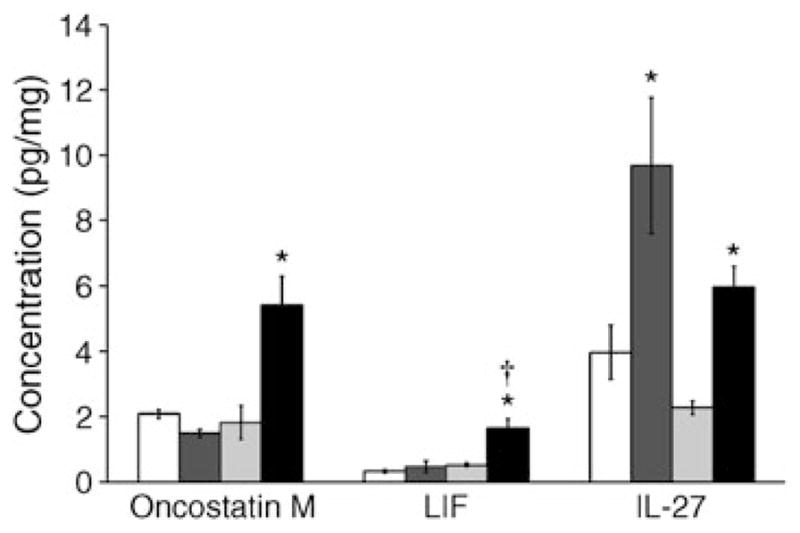
Type 1 diabetes results in increased levels of GP130 receptor ligands in the BM supernatant fraction. White bar, 6-month control; dark grey bar, 6-month diabetic; light grey bar, 12-month control; black bar, 12-month diabetic. The concentration of GP130 ligands, including OSM, IL-27 and LIF, was significantly increased after 12 months of type 1 diabetes compared with age-matched controls; *p<0.05 compared with controls; †p<0.05 compared with 6 months of diabetes. n=4 mice per group
Since IL-6 is increased in the serum of type 1 diabetic patients [41], we were surprised to find that IL-6 did not show a difference in the BM supernatant fraction between control and diabetic mice; however, the supernatant fraction represents the cumulative secretion of a vast number of different cell types. We therefore asked whether Il6 was increased in the diabetic HSCs. HSCs isolated from 6-month-diabetic mice showed a fivefold increase in Il6 (Fig. 8a) and a fourfold increase in Lif expression compared with controls (Fig. 8a). Moreover, pretreatment of the control HSCs with IL-6 (10 ng/ml) significantly reduced their migratory ability compared with untreated cells (p<0.05; Fig. 8b). Since IL-6, LIF, OSM and IL-27 were increased in diabetes and these factors mediate their effects through GP130, we next examined whether deletion of GP130 in BM cells would correct key aspects of diabetes-associated dysfunction such as migration and NO generation. As shown in Fig. 8c, d, HSCs isolated from diabetic gp130−/− mice showed a 1.8-fold increase in 4-amino-5-methyl-amino-2′,7′-difluorescein (DAF-FM) fluorescence in response to VEGF compared with wild-type diabetic mice. Moreover, HSCs isolated from diabetic gp130−/− mice showed a ninefold increase in migratory response to SDF-1α compared with diabetic wild-type mice (Fig. 8e). Thus, in the diabetic environment, GP130 signalling in the BM appears to exacerbate HSC dysfunction.
Fig. 8.

IL-6 reduces migratory ability of the HSCs, while GP130 deletion improves the function of diabetic HSCs. (a) Both Il6 and Lif expression were significantly greater in diabetic mice than age-matched controls; *p<0.05 vs control. (b) Pretreatment of HSCs with IL-6 (10 ng/ml) reduced their migratory ability towards SDF-1α; n=3; *p<0.05 vs untreated (c–j) Representative images of DAF-FM fluorescence showed that GP130 deletion had no effect on NO generation in response to VEGF challenge in HSCs of control mice. However, NO synthesis in type 1 diabetic HSCs was improved by loss of GP130 compared with age-matched diabetic HSCs with GP130 intact. (k) Cumulative quantitative measurements of NO release, expressed as percentage increase over time control. White bar, control; black bar, diabetic (*p<0.05 diabetic wild-type vs diabetic gp130−/−). (l) Increased NO bioavailability improved migratory capacity of the cells. Modified Boyden chamber assay showed an increase in migration of the diabetic gp130−/− HSCs in response to SDF-1α. White bar, control; black bar, diabetic *p<0.05 compared with wild-type (WT). n=6 mice per group. AFU, arbitrary fluorescence unit
Discussion
In this study, we identify several highly novel findings. True stem cells, the LTR-HSCs, appear to reside in the hypoxic niches of the BM where these cells may find protection from the harmful diabetic milieu. Key EPC populations found in the BM, responsible for systemic vascular repair, are decreased in long-term diabetes. This may be due to a shift in haematopoiesis towards generation of increased monocytes, specifically CCR2+ monocytes and away from the reparative EPCs. BM-derived monocytes can ‘home’ to the target tissues of diabetic complications, such as the retina and the kidney. An increase in detrimental monocytes along with reduced numbers and function of reparative EPCs may contribute to the pathogenesis of microvascular complications. Second, reducing GP130-mediated signalling can correct several aspects of diabetes-associated dysfunction in HSCs, such as migratory defects and NO generation, and may protect against the development of pathological changes.
Our studies also suggest that diabetes induces marked changes in HSCs within the BM compartment [42]. The low-oxygenated microenvironment maintains stem cell quiescence, with HSC differentiation occurring along the oxygen ascent towards the vasculature [43]. SP-HSCs that remain in the diabetic mouse are found only in the more hypoxic regions of the BM, which may represent a strategy to allow their survival and normal regulation. SP-HSCs have been shown to seek these niches for ‘protection’ and proper regulation by stromal cells. However, an alternative explanation is that, in diabetes, these regions of hypoxia simply increase as part of ‘generalised’ diabetes-induced ischaemia, as we observed an increase in the number of pimonidazole+ cells with the increase in duration of diabetes. One limitation of the study, however, is that we did not perform direct immunohistochemical localisation of these cells within the hypoxic BM niches.
Changes in fat/water ratio of the BM influence haematopoietic activity of progenitor populations [35]. In mice with type 1 diabetes for up to 6 months, using the technique of MRS, we observed a decrease in the fat peak. In contrast, studies by Botolin et al found increased adiposity [44, 45]. However, they measured the expression of adipocyte markers (peroxisome proliferator-activated receptor γ2, resistin and adipocyte fatty acid-binding protein) by RT-PCR and histology (trichrome staining to evaluate lipid-dense adipocytes) rather than the MRS approach we used. Furthermore, there were strain (BALB-C vs C57BL6) differences between the studies [45]. As type 1 diabetes is associated with decreased body, liver and peripheral adipose tissue weight compared with controls [46], our results suggest that the adipose content of the BM acts similarly to that of other sites. While our data suggest that there might be an influence of fat content on haematopoietic function, we are unable to draw any firm conclusions and this remains an important area of future research.
The loss of eNos mRNA in the diabetic BM progenitors probably contributes to their reparative dysfunction and reduced migratory function. Interestingly, the increased iNos expression, which may reflect the proinflammatory milieu, does not correct this defect, perhaps because of the increased oxidative stress which would lead to formation of peroxynitrite. Moreover, low levels of endothelial NO synthase contribute to low bioavailable NO. The increased oxidative stress, as observed by increased NADPH oxidase, further compromises bioavailable NO levels within these cells [36]. Our work is in agreement with that of Oikawa et al [42], which identified increased oxidative stress and activation of the NADPH oxidase system in the diabetic progenitors at early time points.
Our studies examined diabetes of greater duration, i.e. 48 weeks rather than 30 weeks, the longest duration of diabetes studied by Oikawa et al [42]. Interestingly, the age-related increase in SDF-1 and IGFBP3 levels in the BM would serve to trap progenitor cells within the BM compartment and potentially reduce their mobilisation into the circulation; this effect appeared to be independent of the presence of diabetes. While MMP, SDF-1 and IGFBP3 levels were more affected by the ageing process than the presence of diabetes, several diabetic-specific effects were also observed. In BM supernatant fraction from diabetic mice, we observed an increase in inflammatory cytokine levels for IL-1β, TNF-α and IL-3 and also IL-10. The increase in IL-10 may represent a physiological compensation by this anti-inflammatory factor. While IGF-1 was unchanged in our study, IGFBP3, which has profound effects on progenitor populations, increased with age, which we interpret as a beneficial ‘compensatory’ response much like the IL-10 response.
In addition, our results suggest that the reduced numbers of HSCs in diabetic BM may contribute to the pathogenesis of the vasodegenerative phase of diabetic retinopathy by the generation of fewer EPCs. As shown in Fig. 6, inflammatory monocytes can extravasate into the retina and contribute to retinal pathology, and we observed a similar increase in CCR2+ monocytes in the diabetic kidney. Previously, we showed a decrease in TH+ nerves in diabetic rats [39], and Maestroni et al [47] established that regulated production of monocytes was under an inhibitory noradrenergic tone, suggesting that loss of this noradrenergic influence, as is seen in rodents with diabetic peripheral neuropathy, would result in the generation of more monocytes [48], which is exactly what we observed in our type 1 diabetic model. We show that these mouse monocytes express CCR2+, which characterises them as reactive cells that are actively recruited into diabetic retina and kidney and can contribute to the observed pathology. Thus our studies strongly support the idea that type 1 diabetes-induced BM changes can drive vascular pathology and promote inflammation to promote diabetic complications.
In the BM supernatant fraction, we also examined the abundance of ligands that bind GP130 and found that OSM, IL-27 and LIF were increased at 6 months of diabetes compared with controls. Both LIF and IL-6 are involved in augmenting inflammatory responses [49] and were found to be highly expressed by the diabetic HSCs compared with control cells. Previously, it has been shown that mice that lacked GP130 in haematopoietic and endothelial cells developed BM dysfunction [27]. We found that GP130-deficient mice, when made diabetic with STZ, showed enhanced NO generation and migration compared with wild-type diabetic mice. These results support the notion that blocking GP130 is a viable strategy for correcting diabetes-associated progenitor dysfunction.
In summary, the present study demonstrates that significant changes occur in the BM of mice with 12 months duration of diabetes, with remarkable exhaustion of the most critical primitive HSC compartment. In addition to loss of these primitive cells, we observe a skew in haematopoiesis, with reduced numbers of reparative EPCs but increased numbers of inflammatory CCR2+ monocytes. These changes probably accelerate pathology by driving systemic and tissue inflammation in the absence of sufficient endothelial repair. Our studies also support the contention that a GP130 antagonist, by correcting diabetic EPC dysfunction, could serve to reduce diabetic microvascular complications.
Supplementary Material
Acknowledgments
The authors thank L.C. Shaw, University of Florida, for helping with image preparation and Y. Diao, University of Florida for technical help.
Funding This work was supported by the National Institutes of Health grants EY007739, EY012601, U01 HL087366 and DK090730 to M.B. Grant, 1R43HL093955 to S.H. Bartelmez and M.B. Grant, and EY00300 to T.S. Kern.
Abbreviations
- BM
Bone marrow
- BMMNC
Bone marrow mononuclear cell
- CFU
Colony forming units
- DAF-FM
4-Amino-5-methylamino-2′,7′-difluorescein
- DiI
1,1′-Dioctadecyl-3,3,3′,3′-tetramethyl-indocarbocyanine perchlorate
- ECFC
Endothelial colony-forming cell
- EPC
Endothelial progenitor cell
- eEPC
Early endothelial progenitor cell
- G-CSF
Granulocyte colony-stimulating factor
- GFP
Green fluorescent protein
- GM-CSF
Granulocyte-macrophage colony-stimulating factor
- GP
Glycoprotein
- IGFBP3
Insulin-like growth factor binding protein-3
- LIF
Leukaemia inhibitory factor
- LSK
Lineage− c-Kit+ Sca-1+
- LTR
Long term repopulating
- M-CSF
Monocyte/macrophage colony-stimulating factor
- MMP
Matrix metalloproteinase
- MRS
Magnetic resonance spectroscopy
- NO
Nitric oxide
- OSM
Oncostatin M
- ROS
Reactive oxygen species
- SDF-1
Stromal cell-derived factor-1
- SP
Side population
- STZ
Streptozotocin
- VEGF
Vascular endothelial growth factor
Footnotes
Contribution statement SH, JDA, SHB and MBG designed the studies and contributed equally to writing the manuscript. TSK provided material support and contributed to analysing and interpreting data and to writing the manuscript. YPRJ, VS, LS, JST, JD, SLC, NB and SC carried out experiments, analysed and interpreted data, and contributed to writing the manuscript. MSS designed experiments, interpreted data and contributed to writing the manuscript. DRS performed key experiments, analysed and interpreted data, and contributed to writing the manuscript. JDA, SHB and MBG take full financial responsibility for this article and confirm its originality. All authors gave final approval of the version to be published.
Electronic supplementary material The online version of this article (doi:10.1007/s00125-012-2781-0) contains peer-reviewed but unedited supplementary material, which is available to authorised users.
Duality of interest The authors declare that there is no duality of interest associated with this manuscript.
Contributor Information
S. Hazra, Pharmacology & Therapeutics, University of Florida, 1600 SW Archer Road, Academic Research Building, PO 100267, Gainesville, FL 32610-0267, USA
Y. P. R. Jarajapu, Pharmacology & Therapeutics, University of Florida, 1600 SW Archer Road, Academic Research Building, PO 100267, Gainesville, FL 32610-0267, USA
V. Stepps, BetaStem Therapeutics Inc, San Francisco, CA, USA
S. Caballero, Pharmacology & Therapeutics, University of Florida, 1600 SW Archer Road, Academic Research Building, PO 100267, Gainesville, FL 32610-0267, USA
J. S. Thinschmidt, Pharmacology & Therapeutics, University of Florida, 1600 SW Archer Road, Academic Research Building, PO 100267, Gainesville, FL 32610-0267, USA
L. Sautina, Division of Nephrology/Department of Medicine, University of Florida, Gainesville, FL, USA
N. Bengtsson, Pharmacology & Therapeutics, University of Florida, 1600 SW Archer Road, Academic Research Building, PO 100267, Gainesville, FL 32610-0267, USA
S. LiCalzi, Pharmacology & Therapeutics, University of Florida, 1600 SW Archer Road, Academic Research Building, PO 100267, Gainesville, FL 32610-0267, USA
J. Dominguez, Pharmacology & Therapeutics, University of Florida, 1600 SW Archer Road, Academic Research Building, PO 100267, Gainesville, FL 32610-0267, USA
T. S. Kern, Case Western Reserve University and Louis Stokes Veterans Administration Hospital, Cleveland, OH, USA
M. S. Segal, Division of Nephrology/Department of Medicine, University of Florida, Gainesville, FL, USA
J. D. Ash, University of Oklahoma, Oklahoma City, OK, USA
D. R. Saban, Schepens Eye Research Institute, Massachusetts Eye and Ear, Harvard Medical School, Boston, MA, USA
S. H. Bartelmez, BetaStem Therapeutics Inc, San Francisco, CA, USA
M. B. Grant, Email: grantma@ufl.edu, Pharmacology & Therapeutics, University of Florida, 1600 SW Archer Road, Academic Research Building, PO 100267, Gainesville, FL 32610-0267, USA
References
- 1.Fadini GP, Miorin M, Facco M, et al. Circulating endothelial progenitor cells are reduced in peripheral vascular complications of type 2 diabetes mellitus. J Am Coll Cardiol. 2005;45:1449–1457. doi: 10.1016/j.jacc.2004.11.067. [DOI] [PubMed] [Google Scholar]
- 2.Asahara T, Murohara T, Sullivan A, et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275:964–967. doi: 10.1126/science.275.5302.964. [DOI] [PubMed] [Google Scholar]
- 3.Orlic D, Kajstura J, Chimenti S, et al. Bone marrow cells regenerate infarcted myocardium. Nature. 2001;410:701–705. doi: 10.1038/35070587. [DOI] [PubMed] [Google Scholar]
- 4.Grant MB, May WS, Caballero S, et al. Adult hematopoietic stem cells provide functional hemangioblast activity during retinal neovascularization. Nat Med. 2002;8:607–612. doi: 10.1038/nm0602-607. [DOI] [PubMed] [Google Scholar]
- 5.Brunner S, Schernthaner GH, Satler M, et al. Correlation of different circulating endothelial progenitor cells to stages of diabetic retinopathy: first in vivo data. Invest Ophthalmol Vis Sci. 2009;50:392–398. doi: 10.1167/iovs.08-1748. [DOI] [PubMed] [Google Scholar]
- 6.Fadini GP, Sartore S, Baesso I, et al. Endothelial progenitor cells and the diabetic paradox. Diabetes Care. 2006;29:714–716. doi: 10.2337/diacare.29.03.06.dc05-1834. [DOI] [PubMed] [Google Scholar]
- 7.Fadini GP, Boscaro E, de Kreutzenberg S, et al. Time course and mechanisms of circulating progenitor cell reduction in the natural history of type 2 diabetes. Diabetes Care. 2010;33:1097–1102. doi: 10.2337/dc09-1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee IG, Chae SL, Kim JC. Involvement of circulating endothelial progenitor cells and vasculogenic factors in the pathogenesis of diabetic retinopathy. Eye. 2006;20:546–552. doi: 10.1038/sj.eye.6701920. [DOI] [PubMed] [Google Scholar]
- 9.Loomans CJ, van Haperen R, Duijs JM, et al. Differentiation of bone marrow-derived endothelial progenitor cells is shifted into a proinflammatory phenotype by hyperglycemia. Mol Med. 2009;15:152–159. doi: 10.2119/molmed.2009.00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fadini GP, Sartore S, Schiavon M, et al. Diabetes impairs progenitor cell mobilisation after hindlimb ischaemia-reperfusion injury in rats. Diabetologia. 2006;49:3075–3084. doi: 10.1007/s00125-006-0401-6. [DOI] [PubMed] [Google Scholar]
- 11.Caballero S, Sengupta N, Afzal A, et al. Ischemic vascular damage can be repaired by healthy, but not diabetic, endothelial progenitor cells. Diabetes. 2007;56:960–967. doi: 10.2337/db06-1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bhatwadekar AD, Guerin EP, Jarajapu YP, et al. Transient inhibition of transforming growth factor-beta1 in human diabetic CD34+ cells enhances vascular reparative functions. Diabetes. 2010;59:2010–2019. doi: 10.2337/db10-0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Orlandi A, Chavakis E, Seeger F, Tjwa M, Zeiher AM, Dimmeler S. Long-term diabetes impairs repopulation of hematopoietic progenitor cells and dysregulates the cytokine expression in the bone marrow microenvironment in mice. Basic Res Cardiol. 2010;105:703–712. doi: 10.1007/s00395-010-0109-0. [DOI] [PubMed] [Google Scholar]
- 14.Jarajapu YP, Caballero S, Verma A, et al. Blockade of NADPH oxidase restores vasoreparative function in diabetic CD34+ cells. Invest Ophthalmol Vis Sci. 2011;52:5093–5104. doi: 10.1167/iovs.10-70911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gallagher KA, Liu ZJ, Xiao M, et al. Diabetic impairments in NO-mediated endothelial progenitor cell mobilization and homing are reversed by hyperoxia and SDF-1 alpha. J Clin Invest. 2007;117:1249–1259. doi: 10.1172/JCI29710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Y, Ingram DA, Murphy MP, et al. Release of proinflammatory mediators and expression of proinflammatory adhesion molecules by endothelial progenitor cells. Am J Physiol Heart Circ Physiol. 2009;296:H1675–H1682. doi: 10.1152/ajpheart.00665.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Loomans CJ, de Koning EJ, Staal FJ, Rabelink TJ, Zonneveld AJ. Endothelial progenitor cell dysfunction in type 1 diabetes: another consequence of oxidative stress? Antioxid Redox Signal. 2005;7:1468–1475. doi: 10.1089/ars.2005.7.1468. [DOI] [PubMed] [Google Scholar]
- 18.Cotran RS, Pober JS. Cytokine-endothelial interactions in inflammation, immunity, and vascular injury. J Am Soc Nephrol. 1990;1:225–235. doi: 10.1681/ASN.V13225. [DOI] [PubMed] [Google Scholar]
- 19.Kern TS. Contributions of inflammatory processes to the development of the early stages of diabetic retinopathy. Exp Diabetes Res. 2007;2007:95103. doi: 10.1155/2007/95103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang M, Gan H, Shen Q, Tang W, Du X, Chen D. Proinflammatory CD14+CD16+ monocytes are associated with micro-inflammation in patients with type 2 diabetes mellitus and diabetic nephropathy uremia. Inflammation. 2012;35:388–396. doi: 10.1007/s10753-011-9374-9. [DOI] [PubMed] [Google Scholar]
- 21.Vozarova B, Weyer C, Hanson K, Tataranni PA, Bogardus C, Pratley RE. Circulating interleukin-6 in relation to adiposity, insulin action, and insulin secretion. Obes Res. 2001;9:414–417. doi: 10.1038/oby.2001.54. [DOI] [PubMed] [Google Scholar]
- 22.He T, Peterson TE, Katusic ZS. Paracrine mitogenic effect of human endothelial progenitor cells: role of interleukin-8. Am J Physiol Heart Circ Physiol. 2005;289:H968–H972. doi: 10.1152/ajpheart.01166.2004. [DOI] [PubMed] [Google Scholar]
- 23.Rusten LS, Smeland EB, Jacobsen FW, et al. Tumor necrosis factor-alpha inhibits stem cell factor-induced proliferation of human bone marrow progenitor cells in vitro. Role of p55 and p75 tumor necrosis factor receptors. J Clin Invest. 1994;94:165–172. doi: 10.1172/JCI117303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stanley ER, Guilbert LJ, Tushinski RJ, Bartelmez SH. CSF-1: a mononuclear phagocyte lineage-specific hemopoietic growth factor. J Cell Biochem. 1983;21:151–159. doi: 10.1002/jcb.240210206. [DOI] [PubMed] [Google Scholar]
- 25.Toledo-Corral CM, Banner LR. Early changes of LIFR and gp130 in sciatic nerve and muscle of diabetic mice. Acta Histochem. 2012;114:159–165. doi: 10.1016/j.acthis.2011.04.003. [DOI] [PubMed] [Google Scholar]
- 26.Feit-Leichman RA, Kinouchi R, Takeda M, et al. Vascular damage in a mouse model of diabetic retinopathy: relation to neuronal and glial changes. Invest Ophthalmol Vis Sci. 2005;46:4281–4287. doi: 10.1167/iovs.04-1361. [DOI] [PubMed] [Google Scholar]
- 27.Yao L, Yokota T, Xia L, Kincade PW, McEver RP. Bone marrow dysfunction in mice lacking the cytokine receptor gp130 in endothelial cells. Blood. 2005;106:4093–4101. doi: 10.1182/blood-2005-02-0671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Betz UA, Bloch W, van den Broek M, et al. Postnatally induced inactivation of gp130 in mice results in neurological, cardiac, hematopoietic, immunological, hepatic, and pulmonary defects. J Exp Med. 1998;188:1955–1965. doi: 10.1084/jem.188.10.1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kielczewski JL, Jarajapu YP, McFarland EL, et al. Insulin-like growth factor binding protein-3 mediates vascular repair by enhancing nitric oxide generation. Circ Res. 2009;105:897–905. doi: 10.1161/CIRCRESAHA.109.199059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grant MB, Caballero S, Tarnuzzer RW, et al. Matrix metalloproteinase expression in human retinal microvascular cells. Diabetes. 1998;47:1311–1317. doi: 10.2337/diab.47.8.1311. [DOI] [PubMed] [Google Scholar]
- 31.Goodell MA, Brose K, Paradis G, Conner AS, Mulligan RC. Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo. J Exp Med. 1996;183:1797–1806. doi: 10.1084/jem.183.4.1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sitnicka E, Ruscetti FW, Priestley GV, Wolf NS, Bartelmez SH. Transforming growth factor beta 1 directly and reversibly inhibits the initial cell divisions of long-term repopulating hematopoietic stem cells. Blood. 1996;88:82–88. [PubMed] [Google Scholar]
- 33.Olive PL, Luo CM, Banáth JP. Local hypoxia is produced at sites of intratumour injection. Br J Cancer. 2002;86:429–435. doi: 10.1038/sj.bjc.6600059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ferraro F, Lymperi S, Mendez-Ferrer S, et al. Diabetes impairs hematopoietic stem cell mobilization by altering niche function. Sci Transl Med. 2011;3:104ra101. doi: 10.1126/scitranslmed.3002191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Naveiras O, Nardi V, Wenzel PL, Hauschka PV, Fahey F, Daley GQ. Bone-marrow adipocytes as negative regulators of the haematopoietic microenvironment. Nature. 2009;460:259–263. doi: 10.1038/nature08099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 37.Schroder K, Kohnen A, Aicher A, et al. NADPH oxidase Nox2 is required for hypoxia-induced mobilization of endothelial progenitor cells. Circ Res. 2009;105:537–544. doi: 10.1161/CIRCRESAHA.109.205138. [DOI] [PubMed] [Google Scholar]
- 38.Bereket A, Lang CH, Blethen SL, Fan J, Frost RA, Wilson TA. Insulin-like growth factor binding protein-3 proteolysis in children with insulin-dependent diabetes mellitus: a possible role for insulin in the regulation of IGFBP-3 protease activity. J Clin Endocrinol Metab. 1995;80:2282–2288. doi: 10.1210/jcem.80.8.7543110. [DOI] [PubMed] [Google Scholar]
- 39.Busik JV, Tikhonenko M, Bhatwadekar A, et al. Diabetic retinopathy is associated with bone marrow neuropathy and a depressed peripheral clock. J Exp Med. 2009;206:2897–2906. doi: 10.1084/jem.20090889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.White UA, Stephens JM. The gp130 receptor cytokine family: regulators of adipocyte development and function. Curr Pharm Des. 2011;17:340–346. doi: 10.2174/138161211795164202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Targher G, Zenari L, Bertolini L, Muggeo M, Zoppini G. Elevated levels of interleukin-6 in young adults with type 1 diabetes without clinical evidence of microvascular and macrovascular complications. Diabetes Care. 2001;24:956–957. doi: 10.2337/diacare.24.5.956. [DOI] [PubMed] [Google Scholar]
- 42.Oikawa A, Siragusa M, Quaini F, et al. Diabetes mellitus induces bone marrow microangiopathy. Arterioscler Thromb Vasc Biol. 2010;30:498–508. doi: 10.1161/ATVBAHA.109.200154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jang YY, Sharkis SJ. A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low-oxygenic niche. Blood. 2007;110:3056–3063. doi: 10.1182/blood-2007-05-087759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Botolin S, Faugere MC, Malluche H, Orth M, Meyer R, McCabe LR. Increased bone adiposity and peroxisomal proliferator-activated receptor-gamma2 expression in type I diabetic mice. Endocrinology. 2005;146:3622–3631. doi: 10.1210/en.2004-1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Botolin S, McCabe LR. Bone loss and increased bone adiposity in spontaneous and pharmacologically induced diabetic mice. Endocrinology. 2007;148:198–205. doi: 10.1210/en.2006-1006. [DOI] [PubMed] [Google Scholar]
- 46.Motyl K, McCabe LR. Streptozotocin, type I diabetes severity and bone. Biol Proced Online. 2009;11:296–315. doi: 10.1007/s12575-009-9000-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maestroni GJ, Conti A, Pedrinis E. Effect of adrenergic agents on hematopoiesis after syngeneic bone marrow transplantation in mice. Blood. 1992;80:1178–1182. [PubMed] [Google Scholar]
- 48.Rameshwar P, Ganea D, Gascon P. In vitro stimulatory effect of substance P on hematopoiesis. Blood. 1993;81:391–398. [PubMed] [Google Scholar]
- 49.Ash J, McLeod DS, Lutty GA. Transgenic expression of leukemia inhibitory factor (LIF) blocks normal vascular development but not pathological neovascularization in the eye. Mol Vis. 2005;11:298–308. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.