

Abstract
Objective
Genetic alterations may contribute to chronic pancreatitis (CP) in Chinese young patients. This study was designed to investigate mutations of cationic trypsinogen (PRSS1), pancreatic secretory trypsin inhibitor or serine protease inhibitor Kazal type 1 (SPINK1), cystic fibrosis transmembrane conductance regulator (CFTR), chymotrypsin C (CTRC) and CLDN2 genes and the copy number variations (CNVs) of PRSS1 and asses associations with the development of idiopathic CP (ICP) in Chinese children.
Design
Retrospective.
Setting
A single center.
Participants
75 ICP Chinese children (40 boys and 35 girls).
Primary and secondary outcome measures
Mutations of PRSS1, SPINK1, CFTR, CTRC and CLDN2 genes and CNVs.
Results
7 patients had heterozygous mutations in PRSS1, that is, N29I (n=1), R122H or R122C (n=6). The CNVs of PRSS1 in five patients had abnormal copies (1 copy (n=4), five copies (n=1)). 43 patients had IVS3+2T>C (rs148954387) (10 homozygous and 33 heterozygous) in SPINK1. None of the PRSS1 mutation patients carried a SPINK1 mutation. Frequency of PRSS1 and SPINK1 mutations was 9.3% and 57.3%, respectively, with an overall frequency of 66.6% (50/75). In addition, one patient had a novel deletion of CFTR (GCTTCCTA from c.500 to c.508 leading to the shortened polypeptide molecule via a stop codon). Another patient had a novel missense in CLDN2 exon 2 (c.592A>C mutation). Clinically, patients with SPINK1 mutations had a higher rate of pancreatic duct stones, pancreatic pseudocyst and pancreatic calcification than those without SPINK1 mutations (p<0.05).
Conclusions
SPINK1 mutations were more commonly associated with Chinese children with ICP. SPINK1 IVS3+2T>C mutation may play an important role in the pathogenesis of Chinese paediatric ICP. However, further study is needed to confirm and to investigate the role of these genes in the development of Chinese ICP.
Keywords: Gastroenterology, Genetics
Article summary.
Article focus
Genetic alterations may contribute to chronic pancreatitis in Chinese young patients.
China is different from the Western countries with regard to ethnic and cultural backgrounds, socioeconomic status, climatic conditions and dietary habits.
Key messages
SPINK1 IVS3+2T>C mutation was more commonly associated with Chinese children with idiopathic chronic pancreatitis (ICP).
Paediatric patients with SPINK1 IVS3+2T>C mutation had a higher rate of pancreatic duct stones, pancreatic pseudocyst and pancreatic calcification than those without.
SPINK1 IVS3+2T>C mutation may play an important role in the pathogenesis of Chinese paediatric ICP, which may show some insights into a novel target for therapy.
Strengths and limitations of this study
To the best of our knowledge, this is the first study to determine the spectrum and frequency of PRSS1, SPINK1, CFTR, CTRC and CLDN2 gene mutations and PRSS1 copy number variations in unrelated children with chronic pancreatitis in China.
Further study is needed to confirm and to investigate the role of these genes in the development of Chinese ICP.
Introduction
Chronic pancreatitis (CP) is an inflammatory disease characterised by irreversible destruction of the pancreatic normal structure and function and is associated with persistent abdominal pain or steatorrhoea. In adults, alcohol abuse is an important cause of CP, while other factors, such as anatomical changes, metabolic disease, trauma and heredity, may also cause or associate with CP. However, up to 10–25% of patients with CP have no clear risk factors and are classified as having idiopathic chronic pancreatitis (ICP).1 In children, it is estimated that ICP accounts for approximately 40–60% of all children with chronic pancreatitis in Western countries, but the reported rate was as high as 73.8% in China.2 The pathogenesis of ICP, especially in children, remains poorly understood. Because conventional risk factors such as alcohol abuse are uncommon in children, it has been reported that environmental risk factors may play a limited role in the pathogenesis of ICP in children; thus, patients with ICP at these ages are thought to be suitable for studies of genetic defects.3
During the last two decades, genetic factors have been identified in patients with CP and these factors are believed to play an important role in the pathogenesis of CP.4 For example, previous studies reported identification of mutations in genes encoding cationic trypsinogen (UniGene name: protease serine 1: PRSS1) (OMIM 276000),5 cystic fibrosis transmembrane conductance regulator (CFTR) (OMIM 602421),6 the pancreatic secretory trypsin inhibitor or serine protease inhibitor Kazal type 1 (PSTI or SPINK1) (OMIM 167790)3 and chymotrypsin C (CTRC) (OMIM 601405),7 all of which reveal that hereditary pancreatitis is a more common form of CP than was once believed.1 Recently, a newly detected candidate gene known as CLDN2 has been shown to be associated with sporadic and alcohol-related CP.8 Thus, genetic study may reveal a genetic basis for significant percentage of patients with so-called ‘idiopathic’ CP. It becomes acceptable that development of CP requires a combination of genetic predisposition and environmental, structural or toxic insult.9 Therefore, identification of genetic and environmental risk factors could offer the potential tool for risk assessment, early diagnosis and earlier intervention of CP.10
Many studies have discovered that mutations plus the copy number variations (CNVs) in PRSS1 (trypsinogen), SPINK1 (serine protease inhibitor Kazal-type 1), CFTR (cystic fibrosis transmembrane conductance regulator) and CTRC (chymotrypsin C, also known as caldecrin) were causally linked to the pathogenesis of ICP,11–13 while patients with PRSS1 or SPINK1 mutations may be at a higher risk of developing pancreatic cancer.14 CFTR variants were associated with idiopathic and alcoholic CP. Furthermore, CLDN2 was shown to be strongly associated with CP, suggesting that it probably acts as a disease modifier to accelerate the development and progression of CP through a non-trypsin-dependent process since CLDN2 is a highly regulated tight junction protein to form ion and water channels between endothelial cells.8 However, as the largest populated country in the world, the incidence of CP in China, including paediatric CP, has risen rapidly,2 15 but there have been only a few studies reporting that genetic factors contributed to the pathogenesis of CP, but none of these studies focused on ICP in children.13 16 In addition, China is different from the Western countries with regard to ethnic and cultural backgrounds, socioeconomic status, climatic conditions and dietary habits. In this pilot study, we identified mutations in PRSS1, SPINK1, CFTR, CTRC and CLDN2 genes and CNVs of PRSS1 to determine the spectrum and frequency of the mutations and CNVs in unrelated children with ICP in the mainland of China. The data from this study will provide information on the genetic basis of paediatric ICP in China.
Patients and methods
Study population and diagnosis criteria
We recruited 75 patients with ICP under 18 years from Changhai Hospital between January 1997 and December 2008. There was no history of tobacco smoking or alcohol consumption in these patients. All patients originated from the Han ethnicity in the mainland of China. The diagnose criteria of CP and ICP was defined as a condition characterised by typical history (abdominal pain, diabetes mellitus and/or steatorrhoea) and the presence of any one of the following findings: (1) ductal changes on endoscopic retrograde cholangiopancreatography (ERCP); (2) pancreatic calcification on imaging examination; or (3) histological evidence of CP.17 18 Furthermore, affected individuals were classified as having ICP if precipitating risk factors (such as alcohol abuse, trauma, previous medication, infection, metabolic disorders and/or a positive family history) were absent.19 This study was approved by the Ethics Committee of Changhai Hospital, Shanghai, and a written informed consent was given by their parents or legal guardians according to the ethical guidelines of the Declaration of Helsinki.
DNA isolation
Peripheral blood samples were collected from each patient in an ethylene diamine tetra acetic acid (EDTA)-anticoagulated tube and frozen at −20°C for subsequent DNA extraction. Genomic DNA was isolated from 500 μL samples using the Lab-aid 800 automatic nucleic acid extraction machine following a standard protocol and quantified using a NanoDrop machine (Thermo, Wilmington, USA).
PCR analyses of gene mutations
DNA samples from patients were subjected to PCR analyses of gene mutations. Specifically, primers flanking the targeted regions of PRSS1, SPINK1, CFTR, CTRC and CLDN2 genes were designed and synthesised according to the nucleotide sequence published by NCBI as shown in online supplementary table S1. The data on mutation analyses were confirmed by DNA sequence and repeated using PCR. PCR was performed in the GeneAmp 9700 System (Applied Biosystems, Foster City, California, USA) using a 15 μL reaction mixture containing 7.5 μL 2×Taq Mix (Vivantis, USA), 5.7 μL ddH2O, 1.5 μL DNA templates (10 ng/μL) and 0.15 μL of each primer (20 mM). PCR amplification was set at an initial 6 min denaturation at 94°C, followed by 35 cycles of 30 s at 94°C, 30 s at 58°C (PRSS1, SPINK1) or 56°C (CFTR, CTRC, CLDN2), 45 s at 72°C and a final extension at 72°C for 7 min. PCR products were then incubated with 0.1 U shrimp alkaline phosphatase at 37°C for 1 h, followed by heat inactivation at 85°C for 20 min and then sequenced using an ABI Prism BigDye Terminator Cycle Sequencing Kit, V.3.1 on an ABI Prism 3730 sequencer (Applied Biosystems). Data passed a duplicate quality-control test using four samples and showed 100% concordance.
Vector NT1, Chromas and Bioedit software were applied to analyse the results.
Detection of gene CNVs
A predesigned and validated CNV assay kit to assess PRSS1 CNVs was obtained from AB Life Technologies (Hs03184214_cn) (see details in online supplementary table S2). RT-PCR was performed using 2×Taqman Genotyping Master Mix 5 μL (Vivantis), ddH2O 0.5 μL, DNA templates (10 ng/μL) 4 μL, 0.25 μL Taqman Copy Number Assay (Vivantis) and 0.25 Taqman Copy Number Assay (Vivantis) in a total volume of 10 μL. Cycle conditions were as follows: initial denaturation for 10 min at 95°C, followed by 40 cycles of (15 s at 95°C, and 60 s at 60°C) in a 7900 RT-PCR thermal cycler (Applied Biosystems). The assays were performed in triplicate and repeated at least once for each sample. PRSS1 gene copy number was calculated compared to the proportion to RNAseP reference assay (AB Life Technologies cat#4403326). Data were analysed using Copy Caller Software (V.1.0 from AB Life Technologies).
Statistical analyses
Continuous data were reported as mean±SD analysed using a Student's t test and/or U-test. Fisher's exact test or χ2 test was used to analyse the categorical data. The gene mutations or CNVs were associated with age at diagnosis, pancreatic calcification, changes in pancreatic duct (stenosis or dilation), pancreatic calcification and pancreatic pseudocyst for clinical significance of these mutations.19
The age at onset was divided into two subgroups according to the mean value. A p value of less than 0.05 was considered statistically significant and all reported p values were two-sided.
Results
Baseline clinical characteristics of study participants
A total of 75 unrelated children were included in this study, that is, 40 boys and 35 girls and age at first onset was 11.91±3.79 years (ranged between 3 and 18 years). Clinically, 81.3% (61/75) of the patients showed acute pancreatitis, 13 patients began with pure abdominal pain, 3 patients with weight loss, 1 patient with diarrhoea and 1 patient with high blood glucose.
Imaging examinations, including CT, MRCP or ERCP, detected pancreatic duct stones in 45 patients, changes in pancreatic duct (stenosis or dilation) in 57 patients, pancreatic calcification in 15 patients, pancreatic pseudocyst in 15 patients and pancreatic divisum in four patients (table 1). Laboratory tests showed that six patients had increased levels of CA199, a biomarker for pancreatic cancer, six patients had high blood cholesterol, two had increased blood glucose and two patients had low blood calcium levels.
Table 1.
Characteristics of the ICP study participants
| Age, median (range), years | 11.91 (3–18) |
|---|---|
| Sex, n (%) | |
| Female | 35 |
| Male | 40 |
| Clinical symptoms, n (%) | |
| Acute pancreatitis | 61 |
| Pure abdominal pain | 13 |
| Weight loss | 3 |
| Diarrhoea | 1 |
| High-blood glucose | 1 |
| Other | 0 |
| Imaging examination (CT, MRCP or ERCP) | |
| Pancreatic duct stenosis or dilation | 57 |
| Pancreatic duct stones | 45 |
| Pancreatic pseudocyst | 15 |
| Pancreatic calcification | 15 |
| Other | 4* |
| Laboratory tests | |
| Increased CA199 | 6 |
| Increased blood cholesterol | 6 |
| Increased blood glucose | 2 |
| Decreased blood calcium | 2 |
*Pancreatic Divisum.
ERCP, endoscopic retrograde cholangiopancreatography; ICP, idiopathic chronic pancreatitis.
Analyses of PRSS1, SPINK1, CFTR and CTRC gene mutations in these patients
We then analysed mutations of PRSS1, SPINK1, CFTR and CTRC gene in these 75 patients and found three types of heterozygous PRSS1 mutations in seven patients, including N29I (n=1) in exon 2, and R122H (n=6) with c.365G>A (n=5) and c.364C> T (n=1) in exon 3(see online supplementary figure S1). A single gene mutation of SPINK1 occurred in 43 patients, including 10 homozygous and 33 heterozygous SPINK1 mutation (IVS3+2T>C). However, there was no single patient with PRSS1 mutation who had a SPINK1 mutation, making 9.3% (7/75) and 57.3% (43/75) of, PRSS1 and SPINK1 mutation rates, respectively, and an overall rate of 66.6% (50/75) of patients who at least had one PRSS1 or SPINK1 mutation (table 2).
Table 2.
PRSS1, SPINK1, CFTR and CTRC gene mutations in the patients with ICP
| Gene mutations | Region | Functional class | Positive, n (%) |
|---|---|---|---|
| PRSS1 | |||
| A16V | Exon 2 | Missense | 0 |
| N29I | Exon 2 | Missense | 1 (1.3) |
| E79K | Exon 3 | Missense | 0 |
| R116C | Exon 3 | Missense | 0 |
| A121T | Exon 3 | Missense | 0 |
| R122H or R122C | Exon 3 | Missense | 6 (8.0)* |
| SPINK1 | |||
| N34S | Exon 3 | Missense | 0 |
| IVS3+2T>C | IVS 3 | Splicing | 43 (57.3)† |
| CFTR | |||
| R117H | Exon 4 | Missense | 0‡ |
| F508del | Exon 11 | Del | 0 |
| c.2562T>G | Exon 15 | Nonsense | 51 (68.0)§ |
| c.4389G>A | Exon 27 | Nonsense | 1 (1.3) |
| CTRC | |||
| c.143A>G | Exon 3 | Missense | 0 |
| c.180C>T | Exon 3 | Nonsense | 0 |
| c.217G>A | Exon 3 | Missense | 0 |
| c.703G>A | Exon 7 | Missense | 0 |
| c.760C>T | Exon 7 | Missense | 0 |
| p.K247_R254del | Exon 7 | Del | 0 |
| CLDN2 | |||
| c.22G>A | Exon 1 | Nonsense | 2 |
| c.327A>T | Exon 2 | Nonsense | 1 |
| c.592A>C | Exon 2 | Missense | 1 |
| c.768T>C | Exon 2 | Nonsense | 22 |
*One was c.364 C>T and other five were c.365 G>A.
†Thirty-three were heterozygous and 10 were homozygous.
‡One patient has the deletions of GCTTCCTA from c.500 to c.508.
§Thirty-seven were heterozygous and 14 were homozygous.
Furthermore, one patient had CFTR gene deletions of GCTTCCTA sequences between c.500 and c.508 at exon 4, leading to an early stop codon (figure 1). CFTR gene C.2562 T>G polymorphism was also detected in 51 patients, 14 of whom were homozygous. Heterozygous CFTR gene c.4389G>A mutation was identified in another patient. Moreover, there were four types of TG-repeats and poly-T tract including (TG)10-T7(n=2), (TG)11-T7(n=55), (TG)12-T5(n=5) and (TG)12-T7(n=13) in the junction of intron 8 and exon 9 and (TG)11-T7 of CFTR gene found in 73.3% of these patients (see online supplementary table S3). In addition, CFTR gene V allele was slightly more frequent than the M allele at codon 470 (59.3% and 40.7%, respectively). The dominant genotype was M/V followed by V/V and M/M (see online supplementary table 4).
Figure 1.

A Novel cystic fibrosis transmembrane conductance regulator gene deletion detected using DNA sequence of PCR products.
However, we screened six types of CTRC mutations, including c.143A>G, c.217G>A, c.180C>T in exon 3, c.703G>A, c.760C>T and p.K247_R254del in exon 7 in these 75 patients, but did not find any mutations. In addition, we screened both exons of CLDN2 and found four types of heterozygous mutations in a total of 26 patients (table 2), that is, c.22G>A at exon 1, c.327A>T, c.592A>C and c.768T>C at exon 2. C.592A>C is a missense mutation, while the other three types are nonsense. However, none of these patients had more than one type of CLDN2 mutation.
Analyses of PRSS1 gene CNVs in these patients
PRSS1 gene copy numbers were normal in most patients. Specifically, PRSS1 gene copy number in 70 patients had two copies detected using the probe Hs03184214_cn, whereas four patients had only one copy and another patient had five copies (figure 2).
Figure 2.
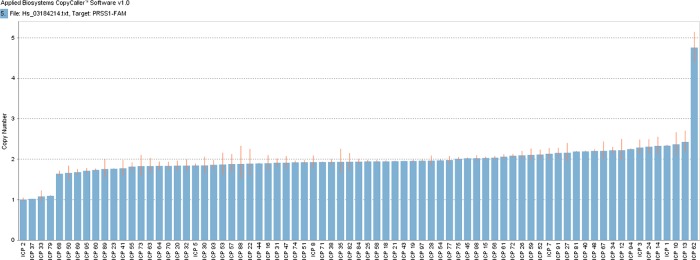
Copy number variations (Hs03184214_cn) of PRSS1 gene in 75 Chinese patients with ICP.
Association of mutations with clinicopathological data
We associated these genetic alterations with clinicopathological data from the 75 patients. Our data showed that three mutations of IVS3+2T>C in SPINK1 gene, M470V and c.2562 T>G in CFTR gene had relatively higher frequencies than other genetic alterations and were associated with clinicopathological data. Briefly, the rates of pancreatic duct stones, pancreatic pseudocyst and pancreatic calcification were higher in patients with a SPINK1 gene IVS3+2T>C mutation than that of patients without IVS3+2T>C (69.8% vs, 46.9% p=0.045; 11.6% vs 31.25% p=0.036; 27.9% vs 9.4% p=0.047, respectively). The rate of pancreatic pseudocyst was lower in patients with the SPINK1 gene IVS3+2T>C mutation than that of patients without IVS3+2T>C (11.6% vs 31.25% p=0.036; table 3). However, there was no statistical significance between age at diagnosis of patients with and without IVS3+2T>C mutation. M470V and c.2562 T>G were not significantly associated with these clinical characteristics.
Table 3.
Association of gene mutation (IVS3+2T>C, M470V and c.2562T>G) with clinicopathological data from 75 patients
| Mutations | Age at diagnosis |
Pancreatic duct stenosis or dilation |
Pancreatic duct stones |
Pancreatic pseudocyst |
Pancreatic calcification |
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| <12 | ≥12 | χ2 | p Value | Yes | No | χ2 | p Value | Yes | No | χ2 | p Value | Yes | No | χ2 | p Value | Yes | No | χ2 | p Value | |
| IVS3+2T>C | ||||||||||||||||||||
| Yes | 17 | 26 | 0.815 | 0.367 | 34 | 9 | 0.521 | 0.471 | 30 | 13 | 4.006 | 0.045 | 5 | 38 | 4.415 | 0.036 | 12 | 31 | 3.938 | 0.047 |
| No | 16 | 16 | 23 | 9 | 15 | 17 | 10 | 22 | 3 | 29 | ||||||||||
| M470V | ||||||||||||||||||||
| MM | 18 | 23 | 0.199 | 0.905 | 32 | 9 | 0.305 | 0.859 | 25 | 16 | 0.773 | 0.679 | 9 | 32 | 1.566 | 0.457 | 11 | 30 | 2.664 | 0.264 |
| MV | 5 | 5 | 7 | 3 | 7 | 3 | 3 | 7 | 1 | 9 | ||||||||||
| VV | 10 | 14 | 18 | 6 | 13 | 11 | 3 | 21 | 3 | 21 | ||||||||||
| c.2562T>G | ||||||||||||||||||||
| Yes | 23 | 28 | 0.078 | 0.780 | 39 | 12 | 0.019 | 0.899 | 32 | 19 | 0.500 | 0.479 | 12 | 39 | 1.241 | 0.265 | 12 | 39 | 1.241 | 0.265 |
| No | 10 | 14 | 18 | 6 | 13 | 11 | 3 | 21 | 3 | 21 | ||||||||||
Discussion
In the current study, we revealed PRSS1, SPINK1, CFTR, CTRC and CLDN2 gene mutations in patients with ICP, especially PRSS1 and SPINK1 gene mutations, occurred in almost 70% of Chinese children with ICP. To the best of our knowledge, this is the first study to determine the spectrum and frequency of PRSS1, SPINK1, CFTR, CTRC and CLDN2 gene mutations and PRSS1 CNVs in unrelated CP children in China. The data indicate that genetic changes occurring in Chinese patients with ICP could be associated with ICP development. Further study will investigate how these gene alterations contribute to ICP development.
Our current data on the frequency of PRSS1 mutations were similar to those (9.3% vs 9–23%) of a previous study on children with CP or ICP,20 however, our data on the frequency of SPINK1 mutations (57.3%) appeared higher than those (19–40%) reported in the previous study.3 Moreover, Witt et al20 first showed PRSS1 gene mutations (3/30 cases at 3×A16 V) in German paediatric CP patients. Thereafter, they showed that in a study of 96 unrelated CP children, PRSS1 gene mutations (5×A16 V, 1×N29I and 5×R122H) occurred in 11 (11.5%) patients.3 In a review of 164 unrelated children with CP, the frequency was reported to be 9.1% (n=15, 8×A16 V, 5×R122H and 2×N29I).21 PRSS1 gene mutations were detected in two (12.5%, 1×R122H and 1×A16 V) of 16 patients classified as having early-onset ICP in a Swiss study22 and in 11 (23.1%) of 52 children with CP (6×R122H, 4×R122C, 1×N29I) in a Polish study.23 In our current study, PRSS1 mutations were found in 9.3% of 75 Chinese paediatric patients with ICP, which included N29I and R122H mutations. Heterozygous mutations of the PRSS1 gene commonly occurred in CP patients in the Western populations20 23 and was the only form of mutation in our patients, indicating that the main spectrum and frequency of PRSS1 gene mutations in Chinese children with ICP are similar to those reported in paediatric patients in Western countries.20–23 The PRSS1 mutations seem to be one of the predisposing factors for ICP, irrespective of race, although the third most common PRSS1 mutation (ie, A16 V) is not found in our study. However, these spectrum and frequency are different from most previous Asian studies, in which PRSS1 mutations were at a low frequency or even absent.13 24 25 In a previous study on 129 Chinese patients with ICP (34 early-onset and 95 late-onset using a cut-off age of 35 years), Chang et al13 showed PRSS1 mutations in 6 (4.6%) patients; in 2 (5.9%) patient with early onset (1×R116C and 1×C139S) and 4 (4.2%) patients with late onset (1×L104P, 1×R116C, 1×T137M and 1×C139S). These mutations are all considered relatively uncommon mutations in Western countries.26 The potential reason for this discrepancy may be due to the sampling bias, that is, Chang's study included both children and adults, whereas our current study only included children.
One of the most significant findings in our current study was the high frequency of SPINK1 IVS3+2T>C mutation, which was first reported by Kume et al They showed a SPINK1 IVS3+2T>C mutation in 13–16% of unrelated Japanese patients with ICP.27 However, two additional studies from Western countries reported IVS 3+2T>C mutation only in one (1.0%) of 96 and in 3 (2.7%) of 112 paediatric patients with ICP.3 12 The IVS 3+2T>C mutation has been found in three (1.7%) of 172 German patients with CP, but was thought to be a rare polymorphism and not a mutation.28 However, a recent Chinese study on 129 patients with ICP revealed an IVS 3+2T>C mutation in 8.5% of patients and the mutation was predominantly responsible for early-onset ICP (29.4% in early onset vs 1.1% in late onset).13 Alternatively, Pfutzer et al12 showed a frequency of N34S mutation in SPINK1 in 40.4% (23/57) of American children with ICP, whereas Truninger et al11 reported a frequency of the mutation at 43% (6/14) German patients with early-onset ICP. In patients with tropical calcific pancreatitis, which is an idiopathic, juvenile, non-alcoholic form of CP widely prevalent in several tropical countries, such as India, N34S mutation can reach 46%. In the current study, IVS 3+2T>C mutations were found in 57.3% (43/75) of unrelated Chinese children with ICP, but we did not find any N34S mutations in the current study. These data suggest that the spectrum and frequency of SPINK1 mutations vary geographically among different populations; IVS 3+2T>C mutations are more common in Chinese children with ICP, whereas N34S mutations are more frequent in Western populations. The underlying role and molecular mechanisms of the IVS3+2T>C mutation in CP development are being explored. For example, this mutation was in complete linkage disequilibrium with −215G>A mutation, which might alter the efficiency of the SPINK1 gene transcription. IVS3+2T>C mutation affects the splicing donor site that is highly conserved in eukaryotes.3 IVS3+2T>C mutations can cause skipping of the whole of exon 3, where the trypsin binding site is located, leading to the loss of the trypsin binding site,27 altered expression of SPINK1 protein in CP patients with the IVS3+2T>C mutation, affecting the protease/antiprotease balance within the pancreas. However, further studies are necessary to elucidate the underlying molecular mechanisms.
In addition, the second significant finding of our current study is CFTR gene polymorphisms, such as M470V (n=51), c.2562T>G (n=51), TG repeats and poly T tract in Chinese children with ICP. We found that 68% (51/75) patients had both c.2562 T>G and M470V polymorphisms, including heterozygous and homozygous alleles and one patient with heterozygous c.4389G>A mutation. Both c.2562T>G and c.4389G>A mutations are nonsense, while an obstructive tubulopathy of the pancreas due to the CFTR dysfunction is thought to play a primary role in CP development, although the exact pathogenic process of pancreatitis associated with CFTR mutations is still under investigation. The function of CFTR in the pancreas is to dilute and alkalinise the protein-rich acinar secretions, so that the formation of protein plugs that lead to pancreatic injury may be prevented. A M470V polymorphism on exon 10 affects the intrinsic chloride activity, and thereby affects CFTR protein function. The TG repeats and poly-T tract can influence CFTR at transcription levels because these intronic variants could lead to reductions in protein synthesis and expression, or altered splicing to compromise the intracellular transport and/or activity. Huang et al29 conducted the first comprehensive study on the functional polymorphisms of CFTR in Chinese healthy subjects and found that T7 was the most common haplotype (93.6%) and (TG)11 and (TG)12 were the dominant haplotypes in the junction of intron 8 and exon 9. Our current data also validated (TG)11-T7 as the most common type. The poly-T, TG-repeats and M470V distributions were similar to those studies on other East Asians.30 31 In addition, a diverse range of CFTR loss-of-function variants have been reported to be associated with ICP and alcoholic CP, whereas their functional effects remain to be defined. Recently, Whitcomb et al reported that the coinheritance of CFTR R75Q and SPINK1 variants is significantly higher in patients with ICP than in controls (8.75% vs 0.38%). Using patch-clamp techniques, they also found that the CFTR genotype caused a selective defect in biocarbonate conductance.32 Another study from Australia showed that symptomatic pancreatitis occurs in 20% of pancreatic sufficient cystic fibrosis patients. To evaluate genotype–phenotype interactions, they developed the pancreatic insufficiency prevalence (PIP) score to determine severity in a large number of CFTR mutations and found that specific CFTR genotypes were associated with pancreatitis, that is, patients carrying genotypes with mild phenotypic effects may have a greater risk of developing pancreatitis than patients carrying genotypes with moderate–severe phenotypic consequences at any given time.33 In addition, common CFTR haplotypes seem to modulate susceptibility to CP.
Although multiple rare CTRC gene mutations have been associated with CP in European and Asian populations, our current study did not find any CTRC mutations in Chinese children with ICP, indicating that CTRC mutation varies geographically or ethnically. According to the biochemical activities and the functional properties of CTRC variants, Zhou and Sahin-Tóth hypothesised three mutually non-exclusive models to demonstrate the possible role of CTRC variants in predisposing to CP: (1) Impaired trypsinogen and/or trypsin degradation; (2) Impaired activation of A-type carboxylpeptidases; and (3) Induction of endoplasmic reticulum stress.34 We infer that CTRC might play a limited role, if any, in the pathogenesis of CP in China.
However, until 2012, genetic variation in CLDN2 has not been previously associated with disease in humans. A two-stage (discovery and replication) genome-wide association study (GWAS) showed that CLDN2 genotype confers the greatest risk for CP, and its alleles via interacting with alcohol consumption, can amplify the risk. These data could partially explain the higher frequency of alcohol-related pancreatitis in men than women while the real causal relationship between CP and CLDN2 has been ambiguously defined. Our current data are the first study on CLDN2 SNPs in Chinese patients ICP to report four novel SNPs. Only one of these is a missense mutation known as c.592A>C in CDS of CLDN2 gene, making the amino acid change from Met to Leu. This patient was a girl who was diagnosed as ICP after several episodes of acute pancreatitis since she was 13 years old. All of these clinical characteristics showed no deviances from other patients. Claudin-2 encoded by CLDN2 is normally expressed at low levels in the tight junction between cells of the pancreatic ducts and in pancreatic islets. But when stressed, acinar cells can also express claudin-2, proved by porcine models of acute pancreatitis.35 Besides, the CLDN2 promoter includes a nuclear factor (NF)-κB-binding site,36 and CLDN2 expression is enhanced in other cells under conditions associated with injury and stress.
In an attempt to associate these mutations with clinical parameters from these patients with ICP, we did not observe any association between the gene mutations and an earlier age of CP diagnosis, which is contrary to a previous study.37 However, we showed that patients with IVS3+2T>C mutation were more likely to have pancreatic duct stones or pancreatic calcification than those without such a mutation. It has been well known that patients with pancreatic calcification are more severe than those without pancreatic calcification. Therefore, our observations, along with previous findings27 suggest that a IVS3+2T>C mutation in SPINK1 predisposes to more severity of CP. Moreover, patients with CP have a markedly increased risk in developing pancreatic cancer compared to the general population14 15 and PRSS1 or SPINK1 mutation may be a predictor for pancreatic cancer development in patients with CP. The IVS3+2T>C mutation was present in 0.6% of the sporadic pancreatic patients with cancer. Thus, CP patients with PRSS1 or SPINK1 mutation should avoid any risk factors, including alcohol and tobacco, be monitored for any signs or symptoms (pain, weight loss, jaundice and/or abdominal mass) or with serum markers and imaging examination for pancreatic cancer.
CNVs often occur in human cancers and the compositions of CNVs may contain deletion, amplification, deletion plus amplification, multiple alleles and complicated locus. Lafrate and Sebat38 were the first to report CNVs in human genome in 2004. To date, many studies proved that both CNVs and SNP can affect gene expressions. However, the effect of PRSS1 gene CNVs on ICP has not been fully studied. In our study, four patients with one copy were found not to be complicated with any mutations screened above. Therefore, reduced CNV can also be detected in patients with ICP, contradicting the hypothesis that reduced CNV may be a protect factor, which needs further studies.
Supplementary Material
Acknowledgments
The authors would like to thank Medjaden Bioscience Limited, Hong Kong, China, for assistance in preparation of this manuscript.
Footnotes
Contributors: ZL and Z-SL conceived the project. X-TS, WW, X-L W and D-Z Z completed the DNA isolation, PCR analyses of gene mutations and detection of gene copy number variations. CS, TX, L-HH, X-WL, B Y, M-YL, FJ and JG collected the peripheral blood samples and clinical data. L-MB and YL completed the statistical analyses.
Funding: This work was supported by grants from the National Natural Science Foundation of China (No. 81270541, to ZL; No. 81170332, to WW), Shanghai Rising-Star Program (No. 13QA1404600, to ZL), Outstanding Young Scholars Fund of Second Military Medical University (to ZL), and Disciplinary Joint Research Projects of Changhai Hospital (No. CH125510312, to ZL), the China postdoctoral science foundation funded project (2012M510110 to D-Z Z).
Competing interests: None.
Patient consent: Obtained.
Ethics approval: The Ethics Committee of Changhai Hospital, Shanghai.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data sharing statement: No additional data are available.
References
- 1.Keller J, Layer P. Idiopathic chronic pancreatitis. Best Pract Res Clin Gastroenterol 2008;22:105–13 [DOI] [PubMed] [Google Scholar]
- 2.Wang W, Liao Z, Li ZS, et al. Chronic pancreatitis in Chinese children: etiology, clinical presentation and imaging diagnosis. J Gastroenterol Hepatol 2009;24:1862–8 [DOI] [PubMed] [Google Scholar]
- 3.Witt H, Luck W, Hennies HC, et al. Mutations in the gene encoding the serine protease inhibitor, Kazal type 1 are associated with chronic pancreatitis. Nat Genet 2000;25:213–16 [DOI] [PubMed] [Google Scholar]
- 4.Chen JM, Férec C. Genetics and pathogenesis of chronic pancreatitis: the 2012 update. Clin Res Hepatol Gastroenterol 2012;36:334–40 [DOI] [PubMed] [Google Scholar]
- 5.Whitcomb DC, Gorry MC, Peston RA, et al. Hereditary pancreatitis is caused by a mutations in the cationic trypsinogen. Nat Genet 1996;14:141–5 [DOI] [PubMed] [Google Scholar]
- 6.Sharer N, Schwarz M, Malone G, et al. Mutations of the cystic fibrosis gene in patients with chronic pancreatitis. N Engl J Med 1998;339:645–52 [DOI] [PubMed] [Google Scholar]
- 7.Rosendahl J, Witt H, Szmola R, et al. Chymotrypsin C (CTRC) mutations that diminish activity or secretion are associated with chronic pancreatitis. Nat Genet 2008;40:78–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Whitcomb DC, LaRusch J, Krasinskas AM, et al. Common genetic variants in the CLDN2 and PRSS1-PRSS2 loci alter risk for alcohol-related and sporadic pancreatitis. Nat Genet 2012;44:1349–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Witt H, Apte MV, Keim V, et al. Chronic pancreatitis: challenges and advances in pathogenesis, genetics, diagnosis, and therapy. Gastroenterology 2007;132:1557–73 [DOI] [PubMed] [Google Scholar]
- 10.Treiber M, Schlag C, Schmid RM. Genetics of pancreatitis: a guide for clinicians. Curr Gastroenterol Rep 2008;10:122–7 [DOI] [PubMed] [Google Scholar]
- 11.Truninger K, Witt H, Kock J, et al. Mutations of the serine protease inhibitor, Kazal type 1 gene, in patients with idiopathic chronic pancreatitis. Am J Gastroenterol 2002;97:1133–7 [DOI] [PubMed] [Google Scholar]
- 12.Pfutzer RH, Barmada MM, Brunskil AP, et al. SPINK1/PSTI polymorphisms act as disease modifiers in familial and idiopathic chronic pancreatitis. Gastroenterology 2000;119:615–23 [DOI] [PubMed] [Google Scholar]
- 13.Chang YT, Wei SC, L PC, et al. Association and differential role of PRSS1 and SPINK1 mutations in early-onset and late-onset idiopathic chronic pancreatitis in Chinese subjects. Gut 2009;58:885. [DOI] [PubMed] [Google Scholar]
- 14.Rebours V, Boutron-Ruault MC, Schnee M, et al. The natural history of hereditary pancreatitis: a national series. Gut 2009;58:97–103 [DOI] [PubMed] [Google Scholar]
- 15.Wang LW, Li ZS, Li SD, et al. Prevalence and clinical features of chronic pancreatitis in China: a retrospective multicenter analysis over 10 years. Pancreas 2009;38:248–54 [DOI] [PubMed] [Google Scholar]
- 16.Chang MC, Chang YT, Wei SC, et al. Association of novel chymotrypsin C gene variations and haplotypes in patients with chronic pancreatitis in Chinese in Taiwan. Pancreatology 2009;9:287–92 [DOI] [PubMed] [Google Scholar]
- 17.Tandon RK, Sato N, Garg PK, Consensus Study Group Chronic pancreatitis: Asia-Pacific consensus report. J Gastroenterol Hepatol 2002;17:508–18 [DOI] [PubMed] [Google Scholar]
- 18.Axon AT, Classen M, Cotton PB. Pancreatography in chronic pancreatitis: International definitions. Gut 1984;25:1107–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Witt H, Sahin-Tóth M, Landt O, et al. A degradation-sensitive anionic trypsinogen (PRSS2) variant protects against chronic pancreatitis. Nat Genet 2006;38:668–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Witt H, Luck W, Becker M. A signal peptide cleavage site mutations in the cationic trysinogen is strongly associated with chronic pancreatitis. Gastroenterology 1999;117:7–10 [DOI] [PubMed] [Google Scholar]
- 21.Witt H. Gene mutations in children with chronic pancreatitis. Pancreatology 2001;1:432–8 [DOI] [PubMed] [Google Scholar]
- 22.Truninger K, Kock J, Wirth HP, et al. Trypsinogen gene mutations in patients with chronic or recurrent acute pancreatitis. Pancreas 2001;22:18–23 [DOI] [PubMed] [Google Scholar]
- 23.Sobczynska-Tomaszewska A, Bak D, Oralewska B, et al. Analysis of CFTR, SPINK1, PRSS1 and AAT mutations in children with acute or chronic pancreatitis. J Pediatr Gastroenterol Nutr 2006;43:299–306 [DOI] [PubMed] [Google Scholar]
- 24.Nishimori I, Kamakura M, Fujikawa-Adachi K, et al. Mutations in exons 2 and 3 of the cationic trypsinogen gene in Japanese families with hereditary pancreatitis. Gut 1999;44:259–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oh HC, Kim MH, Choi KS, et al. Analysis of PRSS1 and SPINK1 mutations in Korean patients with idiopathic and familial pancreatitis. Pancreas 2009;38:180–3 [DOI] [PubMed] [Google Scholar]
- 26.Teich N, Rosendahl J, Toth M, et al. Mutations of human cationic trypsinogen (PRSS1) and chronic pancreatitis. Hum Mutat 2006;27:721–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kume K, Masamune A, Kikuta K, et al. [−215G>A; IVS3+2T>C] Mutation in the SPINK1 gene causes exon 3 skipping and loss of the trypsin binding site. Gut 2006;55:1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kalinin VN, Kaifi JT, Schwarzenbach H, et al. Association of rare SPINK1 gene mutation with another base substitution in chronic pancreatitis patients. World J Gastroenterol 2006;12:5352–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang Q, Ding W, Wei MX. Comparative analysis of common CFTR polymorphisms poly-T, TG-repeats and M470V in a healthy Chinese population. World J Gastroenterol 2008;14:1925–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nam MH, Hijikata M, Tuan le A, et al. Variations of the CFTR gene in the Hanoi-Vietnamese. Am J Med Genet A 2005;136:249–53 [DOI] [PubMed] [Google Scholar]
- 31.Fujiki K, Ishiguro H, Ko SB, et al. Genetic evidence for CFTR dysfunction in Japanese: background for chronic pancreatitis. J Med Genet 2004;41:e55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schneider A, Larusch J, Sun X, et al. Combined bicarbonate conductance-impairing variants in CFTR and SPINK1 variants are associated with chronic pancreatitis in patients without cystic fibrosis. Gastroenterology 2011;140:162–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ooi CY, Peter R. Cystic fibrosis transmembrane conductance regulator (CFTR) gene mutations in pancreatitis. J Cyst Fibros 2012;11:355–62 [DOI] [PubMed] [Google Scholar]
- 34.Zhou J, Sahin-Tóth M. Chymotrypsin C mutations in chronic pancreatitis. J Gastroenterol Hepatol 2011;26:1238–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee JH, Kim KS, Kim TJ, et al. Immunohistochemical analysis of claudin expression in pancreatic cystic tumors. Oncol Rep 2011;25:971–8 [DOI] [PubMed] [Google Scholar]
- 36.Sakaguchi T, Gu X, Golden HM, et al. Cloning of the human claudin-2 5’-flanking region revealed a TATA-less promoter with conserved binding sites in mouse and human for caudal-related homeodomain proteins and hepatocyte nuclear factor-1alpha. J Biol Chem 2002;277:21361–70 [DOI] [PubMed] [Google Scholar]
- 37.Howes N, Lerch MM, Greenhalf W, et al. Clinical and genetic characteristics of hereditary pancreatitis in Europe. Clin Gastroenterol Hepatol 2004;2:252–61 [DOI] [PubMed] [Google Scholar]
- 38.Sebat J, Lakshmi B, Troge J, et al. Large-scale copy number polymorphism in the human genome. Science 2004;305:525–8 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.