

Significance
We describe a genomic disorder that causes obesity, intellectual disability, and seizures. Children with this syndrome carry an unbalanced chromosome translocation that results in the duplication of over 100 genes, including G protein β3 (GNB3). Although GNB3 polymorphisms have been associated with obesity, hypertension, and diabetes, the mechanism of GNB3 pathogenesis is unknown. We created a transgenic mouse model that carries a duplication of GNB3, weighs significantly more than wild-type mice, and has excess abdominal fat. GNB3 is highly expressed in the brain and may be important for signaling related to satiety and/or metabolism.
Abstract
Obesity is a highly heritable condition and a risk factor for other diseases, including type 2 diabetes, cardiovascular disease, hypertension, and cancer. Recently, genomic copy number variation (CNV) has been implicated in cases of early onset obesity that may be comorbid with intellectual disability. Here, we describe a recurrent CNV that causes a syndrome associated with intellectual disability, seizures, macrocephaly, and obesity. This unbalanced chromosome translocation leads to duplication of over 100 genes on chromosome 12, including the obesity candidate gene G protein β3 (GNB3). We generated a transgenic mouse model that carries an extra copy of GNB3, weighs significantly more than its wild-type littermates, and has excess intraabdominal fat accumulation. GNB3 is highly expressed in the brain, consistent with G-protein signaling involved in satiety and/or metabolism. These functional data connect GNB3 duplication and overexpression to elevated body mass index and provide evidence for a genetic syndrome caused by a recurrent CNV.
Body weight exhibits a heritability of ∼70% (1–3), with both rare and common alleles contributing to obesity. Few cases of obesity are explained by mutations in single genes, with most confined to the well-known leptin/melanocortin pathway (4, 5). Genome-wide association studies (GWASs) have identified alleles that confer a small risk for elevated body mass index (BMI) and other measures of obesity, and some common variants involve candidate genes that are highly expressed in the central nervous system (6–9). However, the functional mechanism by which most GWAS loci contribute to obesity is unknown.
Recently, large deletions and duplications that represent copy number variation (CNV) have been linked to early onset obesity in children (10–14). Rare CNVs have proven to be highly penetrant in familial and spontaneous cases of obesity that are sometimes concomitant with intellectual disability (ID) and/or other neurodevelopmental disorders. Rare but recurrent CNVs are particularly useful to pinpoint critical regions or candidate genes, because their shared genotype can be correlated with a common phenotype. For example, recurrent deletions of 16p11.2 are enriched in multiple obesity cohorts vs. controls (10–12).
Mouse models have been instrumental in unraveling the genetic mechanisms and signaling pathways involved in obesity, which are critical for the development of therapeutics. Notably, ob/ob mutant mice that lack leptin are morbidly obese and exhibit other features, including hyperphagia, infertility, decreased immune function, and energy and body temperature dysfunction (15, 16). In humans, congenital leptin deficiency causes early onset hyperphagia, hypogonadism, impaired immunity, and severe obesity (17). For both ob/ob mice and leptin-deficient humans, leptin replacement ameliorates many of these symptoms (18–20). Mouse models of other metabolic disorders have also proven insightful in teasing apart the physiological effects of genes involved in obesity (21–24).
Here, we report a syndrome caused by a recurrent chromosomal translocation associated with obesity as well as ID, seizures, macrocephaly, and eczema. Moreover, we have recapitulated the obesity phenotype in a transgenic mouse model and identified the gene responsible for elevated weight gain. This CNV duplication causes a highly penetrant form of early onset obesity.
Results
Genomic Disorder Is Caused by a Recurrent Unbalanced Chromosome Translocation.
We recruited seven unrelated patients with a previous diagnosis of an unbalanced translocation between chromosomes 8 and 12 and fine-mapped the breakpoints. All seven subjects share the same genomic rearrangement, resulting in a 7.0-Mb loss of 8p and an 8.5-Mb gain of 12p (Fig. S1). There are 23 and 107 RefSeq genes within the 8p deletion and 12p duplication, respectively. In each unbalanced translocation der(8)t(8;12)(p23.1;p13.31), which for brevity, we refer to as der(8)t(8;12), the breakpoints are in segmental duplications shared between chromosome bands 8p23.1 and 12p13.31, spanning ∼280 kb (Figs. S2 and S3). Recurrent translocations between paralogous segments of the genome are consistent with rearrangement by nonallelic homologous recombination (NAHR) (25–30).
Children with the der(8)t(8;12) exhibit developmental delays, ID, seizures, macrocephaly, eczema, and mild dysmorphic features (Fig. 1, Table 1, SI Materials and Methods, and Table S1). Notably, six of seven children with the unbalanced translocation are obese, with BMI values above the 95th percentile (Fig. 2 and Table 1). Typically, the weight gain occurs in the first few years of life and continues into adulthood, even when on restricted diets. Subjects 1–4, 6, and 7 have a clinical diagnosis of obesity based on height and weight measurements. To rule out overgrowth, which is characterized as height 2–3 SDs greater than the mean for age and sex (31, 32), we analyzed the heights of children with the der(8)t(8;12) and found that none met the clinical criteria (Table S1).
Fig. 1.

Children with the der(8)t(8;12) exhibit features including a thin upper lip, short nose, periorbital fullness, malar hypoplasia, narrow palpebral fissures, high forehead, and prominent chin. Facial features of subject 1 at (A) 22 mo, (B) 5 y, and (C) 13 y; subject 2 at (D) 4 y, (E) 11 y, and (F) 15 y; subject 3 at (G) 4 y and (H) 11 y; (I) subject 4 at 9 y; (J) subject 6 at 4 y; and (K) subject 7 at 5 y are shown. (L) Pedigrees of 7 families. Filled symbols indicate affected individuals with the unbalanced translocation.
Table 1.
Clinical features of subjects
| Subject | Age | GNB3 | Height (cm; %) | Weight (kg; %) | BMI (%) | HC (cm; %) | DD | S | H | DF | G | PC | O | E | SP | DP |
| 1 | 29 y, 0 mo | CCT | 167.6 | 85.3 | 30.4 | 57.2 | + | + | + | + | + | + | + | + | + | + |
| 2 | 15 y, 6 mo | CCC | 174 (50) | 90.2 (>95) | 29.8 (>95) | 61.5 (>98) | + | + | + | + | − | N | + | + | + | + |
| 3 | 11 y, 8 mo | CCC | 137.5 (10–25) | 49.8 (90–95) | 26.3 (>95) | 57 (>98) | + | + | + | + | + | + | + | + | + | + |
| 4 | 8 y, 0 mo | TTT | 135 (90) | 42 (>95) | 23.0 (>95) | 55 (>98) | + | + | + | + | − | N | + | + | − | − |
| 5 | 6 y, 11 mo | CCC | 114.3 (10–25) | 22.2 (50) | 17 (50–75) | 55.9 (>98) | + | − | + | + | − | + | − | + | + | + |
| 6 | 4 y, 10 mo | CTT | 115 (90–95) | 27.3 (>95) | 20.6 (>95) | 52 (90–98) | + | + | − | + | + | + | N | − | + | − |
| 7 | 4 y, 8 mo | CCT | 115.5 (>95) | 26 (>95) | 19.5 (>95) | 52 (90–98) | + | − | − | + | + | + | + | + | + | − |
| 7/7 | 5/7 | 5/7 | 7/7 | 4/7 | 5/5 | 5/6 | 6/7 | 6/7 | 4/7 |
Listed are age at assessment, GNB3 genotype (Fig. S9 and Table S2), height, weight, BMI, head circumference (HC), and percentiles (%). Subject 1 is an adult; her measurements do not fall within childhood growth percentiles. Subjects exhibit features (+) including developmental delay (DD), seizure (S), hypotonia (H), dysmorphic features (DF), abnormal gait (G), poor coordination (PC), ocular problems (O), eczema (E), social personality (SP), and dental/palate abnormalities (DP). Features not formally evaluated (N) or not present (−) are indicated. Detailed clinical information is located in SI Materials and Methods.
Fig. 2.

BMI for (A) male and (B) female subjects. The 5th, 50th, and 95th BMI percentile curves for boys and girls from the Centers for Disease Control and Prevention are plotted. BMI values were calculated using the reported height and weight with a childhood BMI calculator (47).
Obesity Candidate Gene G protein β3 Is Duplicated.
Among 107 genes duplicated in children with the unbalanced translocation, one gene of particular note is G protein β3 (GNB3). Polymorphisms in GNB3 are associated with hypertension, diabetes, and obesity (33). The most studied polymorphism is a cytosine-to-thymine change (c.825C > T, commonly known as C825T) in exon 10 of GNB3 that is synonymous at the amino acid level (TCC to TCT Serine). However, the T haplotype is linked to a shorter in-frame splice variant, GNB3-s, which lacks part of exon 9 (34). Although total mRNA expression and protein levels are not significantly altered by the C825T polymorphism (34, 35), cell culture assays have shown that cells derived from CT heterozygotes or TT homozygotes have increased activation of G proteins compared with CC homozygotes (34). We reasoned that an extra copy of GNB3 because of the der(8)t(8;12) could, like the C825T polymorphism, alter G protein signaling and confer increased risk of obesity.
Mouse Model of GNB3 Duplication Recapitulates Obesity Phenotype.
To test this hypothesis, we developed BAC transgenic mice that carry an extra copy of human GNB3. The BAC insert sequence is 184 kb and contains 14 genes from 12p13.31, including the T variant of GNB3. We confirmed the BAC insertion by FISH and array comparative genome hybridization (Fig. S4) and bred founders with wild-type animals to generate offspring. Starting at weaning, we weighed 30 transgenic (GNB3/+) and 22 wild-type (+/+) littermates derived from a single founder (Line 5) (Fig. 3). Taqman quantitative PCR assays revealed two copies of the BAC in GNB3/+ mice (Fig. S5). We used a linear mixed model to analyze the longitudinal effects of the presence of the BAC transgene on mouse weight. The weight of the mice is significantly associated with transgene genotype (P = 0.00199). On average, male and female GNB3/+ mice are 4.1–7.5% and 6.0–14.3% heavier, respectively, than their +/+ littermates from age 7 wk (Fig. 3). We found similar weight differences between +/+ and GNB3/+ animals from two other transgenic founder lines (Lines 3 and 4) (Fig. S6), ruling out insertional effects of the transgene. Furthermore, we dissected and weighed gonadal white adipose tissue (WAT) in GNB3/+ and +/+ littermates. WAT percent (WAT/body weight) of GNB3/+ mice is significantly greater than +/+ mice in females (P = 0.0122) and males (P < 0.0001; Student t test) (Fig. 4A).
Fig. 3.
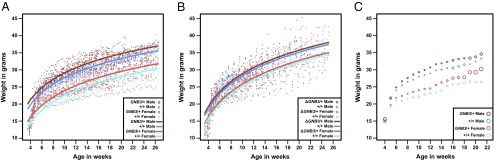
Plots of mouse weight by age; (A) 52 (14 GNB3/+ male, 13 +/+ male, 16 GNB3/+ female, and 9 +/+ female mice) and (B) 91 (25 ΔGNB3/+ male, 31 +/+ male, 20 ΔGNB3/+ female, and 15 +/+ female mice) mice were weighed three times a week beginning at weaning. Each dot represents the weight of a single mouse at a given age. Lines indicate predicted mouse weight by age based on presence/absence of the transgene and mouse sex. (C) Weight gain of GNB3/+ and +/+ animals. Circles depict average weekly values of mouse weight ± SEM.
Fig. 4.

(A) Gonadal WAT was extracted from 20-wk-old mice (19 GNB3/+ male, 12 +/+ male, 14 GNB3/+ female, and 11 +/+ female mice) and weighed to calculate WAT percent. GNB3/+ WAT percent is significantly greater than +/+ WAT percent in *females (P = 0.0122) and **males (P < 0.0001; Student t test). (B) GNB3 is highly expressed in GNB3/+ brains. Mean Ct values from qRT-PCR of human GNB3, mouse Gnb3, human MLF2, and mouse Mlf2 transcripts in whole brains from eight 8-wk-old male mice are plotted. Data from GNB3/+ and +/+ littermates are shown as solid bars; data from ΔGNB3/+ and +/+ littermates are shown as hatched bars. Lower Ct values correspond to greater gene expression; the absence of Ct data indicates no detection of transcript. All qRT-PCR experiments were performed in triplicate with an internal mouse β-actin (Actb) control. SEM for 40 Actb triplicates was ±0.05 Ct; maximum SEM for other transcripts was ±0.31 Ct.
To measure transgenic GNB3 and endogenous Gnb3 expression, we performed quantitative RT-PCR (qRT-PCR). Using RNA from brains of GNB3/+ and +/+ littermates, we detect human GNB3 expression from GNB3/+ but not +/+ mice. In addition, human GNB3 expression is significantly greater than endogenous Gnb3, as measured by cycle threshold (Ct) values from multiple primer pairs (Fig. 4B). These qRT-PCR experiments measure total GNB3 and Gnb3 transcript levels rather than the relative contributions of full-length transcripts and splice variants; however, these data are consistent with overexpression of GNB3 from the human transgene in GNB3/+ mice. Because of the similarity between the β-subunits of G proteins, we cannot distinguish GNB3 from related proteins by Western blot. Using a GNB-common antibody, we detected high levels of protein in mouse brain and adipose tissues (Figs. S7 and S8). However, we cannot specifically measure GNB3 abundance in +/+ vs. GNB3/+ animals, because the antibody is predicted to detect multiple human and mouse β-subunits.
To confirm that GNB3 is responsible for the obesity phenotype in transgenic animals, we replaced GNB3 with a neomycin resistance gene to recombineer a construct that retains the other 13 genes in the BAC but not GNB3 (ΔGNB3). We generated ΔGNB3 transgenic mice and compared the weights of ΔGNB3/+ and +/+ littermates. ΔGNB3/+ mice have three copies of the BAC transgene (Fig. S5). The weight of the mice is not significantly associated with the transgenic ΔGNB3/+ genotype (P = 0.104) (Fig. 3B). To rule out the unlikely possibility that the entire ΔGNB3 construct was silenced, we also measured expression of MLF2, another human gene on the BAC. We performed qRT-PCR to amplify GNB3, Gnb3, MLF2, and Mlf2 transcripts in brains from ΔGNB3/+ and +/+ littermates (Fig. 4B). MLF2 is expressed at similar levels in the brains of ΔGNB3/+ and GNB3/+ animals, consistent with normal expression from the ΔGNB3 BAC. These data support our hypothesis that duplication of GNB3 is responsible for the obesity phenotype.
Translocation Formation and Incidence.
The unbalanced translocation between chromosomes 8 and 12 is rare; it may be inherited from a parent with the balanced translocation, or it may be a new (de novo) event in the proband. We analyzed the inheritance of the der(8)t(8;12) by FISH and found that four rearrangements were de novo and three rearrangements were inherited from a mother who carries the balanced translocation (Fig. 1). To determine the parental origin of the four de novo rearrangements, we performed genome-wide SNP genotyping using the Affymetrix 6.0 platform. All four de novo translocations were derived from the maternal chromosomes 8 and 12, consistent with a maternal bias in this NAHR-mediated translocation.
Copies of the segmental duplications that mediate the translocation between chromosomes 8 and 12 are also present on the short arm of chromosome 4. A recurrent translocation between chromosomes 4 and 8, also mediated by NAHR, is a well-known genomic rearrangement that, when inherited in an unbalanced fashion, causes Wolf–Hirschhorn syndrome [der(4)t(4;8)] (25, 36) or a milder ID syndrome [der(8)t(4;8)] (37). Familial studies have shown that the translocation between chromosomes 4 and 8 always originates maternally (25). Because of the homology between segmental duplications on chromosomes 4, 8, and 12, recurrent translocations between chromosomes 4 and 12 are also possible. There is one report of a der(4)t(4;12) unbalanced translocation in a child with ID, macrosomy, macrocephaly, dysmorphic features, and epilepsy (38).
Given the incidence of the der(8)t(8;12), we would expect to find children with the reciprocal unbalanced translocation resulting in 8p trisomy and 12p monosomy. However, the unbalanced der(12)t(8;12) is not reported in large studies of pathogenic CNV, including the Unique Rare Chromosome Disorder Support Group database, the Database of Chromosomal Imbalance and Phenotype in Humans using Ensembl Resources, the International Standards for Cytogenomic Arrays Consortium, or Signature Genomic Laboratories (39–41). Individuals with the der(12)t(8;12) are likely missing from CNV studies due to embryonic lethality caused by heterozygous loss of many genes on 12p. Trisomy for portions of 12p has been reported previously in children with ID and epilepsy (42). However, children with large deletions of 12p are absent from the literature, consistent with a chromosome rearrangement incompatible with life.
Discussion
Although the der(8)t(8;12) unbalanced translocation has been previously reported in three children with ID (27, 42, 43), our finding connects this genomic rearrangement with ID, seizures, macrocephaly, eczema, and obesity in multiple individuals and suggests that, like other recurrent CNVs (44), the der(8)t(8;12) translocation constitutes a unique genomic syndrome. As is the case for the recurrent translocation between chromosomes 4 and 8, all of the de novo der(8)t(8;12) translocations are maternally derived. Because NAHR occurs during meiosis (45, 46), this maternal bias in translocation formation could suggest a higher risk for aberrant interchromosomal recombination during female meioses. Additional studies of der(8)t(8;12) families are necessary to investigate this maternal bias in a larger cohort.
The der(8)t(8;12) unbalanced translocation is one of several newly discovered CNVs associated with obesity. Collectively, such CNVs contribute to the missing heritability of obesity that is not explained by rare single-gene mutations or more common variants identified by GWASs. Polymorphisms in GNB3 have been recognized as a risk factor for obesity and related disorders; however, this study connects GNB3 gene duplication to a highly penetrant form of obesity. Furthermore, our mouse model of GNB3 duplication recapitulates the obesity phenotype and reveals overexpression of GNB3 in the brains of transgenic animals. In both transgenic mice and humans with the der(8)t(8;12), GNB3 duplication leads to moderate elevation in body weight. Control experiments with ΔGNB3 mice confirm that GNB3 is largely responsible for the weight gain phenotype; however, we cannot rule out additional subtle effects from other genes within the CNV. It is possible that smaller duplications, including GNB3 and/or variation in GNB3 expression, are also associated with childhood obesity. However, small duplications of GNB3 have not been described in databases of clinically recognized CNVs (39–41). The absence of small GNB3 duplications is not surprising, because most children referred for clinical cytogenetic testing are referred for neurodevelopmental disorders rather than isolated obesity.
We have linked the obesity phenotype to 1 of ∼130 genes deleted or duplicated as part of the der(8)t(8;12) unbalanced translocation, but it is important to point out that there are many genes on 8p and/or 12p that could be responsible for other syndromic features, including ID, seizures, macrocephaly, eczema, and dysmorphic facies. Nevertheless, our studies connect GNB3 gene dosage to obesity and provide a model of the functional significance of GNB3 in BMI. Rare CNVs that include G protein-coupled receptor genes are enriched in cases of early onset obesity, and pathway analysis has revealed gene networks involving GNB3 (14). As in our study, these CNV data suggest that G proteins play an important role in the genetics of obesity. We anticipate that human and mouse studies of GNB3 duplication and risk alleles will lead to a better understanding of cell signaling, physiology, and behaviors involved in childhood obesity. Additional investigation into GNB3-related pathways may point to novel therapeutics for the growing obesity epidemic.
Materials and Methods
Human Subjects.
This study was approved by the Emory University Institutional Review Board. We recruited subjects from the Unique Rare Chromosome Disorder Support Group (www.rarechromo.org) and clinical cytogenetics laboratories. Samples, photos, and clinical information were obtained with informed consent per our human subjects protocol. We received peripheral blood and/or DNA samples from subjects and their parents. Samples from families 1–5 were EBV-transformed to create lymphoblastoid cell lines using standard methods.
Statistical Analysis.
To analyze the longitudinal effect of the BAC transgenes on mouse weight, we used the R package nlme to model weight (grams) as a linear function of presence/absence of the transgene, sex, age [log(days)], litter, and a mouse-specific random effect to account for the repeated observations of each mouse. The P value corresponding to presence/absence of the transgene was used to assess significance of the association between the transgene and mouse weight in the presence of the other variables.
Experimental details for array comparative genome hybridization, genotyping, and mouse studies are described in SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank the families for participating in this study. Helen Zhang and the Emory Transgenic Mouse Core generated transgenic animals, and the Emory Integrated Genomics Core performed qRT-PCR and BAC copy number experiments. We also thank Cheryl Strauss for editorial assistance. This project was supported by National Institutes of Mental Health Grant 5R01MH92902 (to M.K.R.) and the Emory Discovery Fund.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
3The Unique Rare Chromosome Disorder Support Group authors are Beverly A. Searle and Sarah L. Wynn.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1305999110/-/DCSupplemental.
References
- 1.Feinleib M, et al. The NHLBI twin study of cardiovascular disease risk factors: Methodology and summary of results. Am J Epidemiol. 1977;106(4):284–285. doi: 10.1093/oxfordjournals.aje.a112464. [DOI] [PubMed] [Google Scholar]
- 2.Stunkard AJ, Harris JR, Pedersen NL, McClearn GE. The body-mass index of twins who have been reared apart. N Engl J Med. 1990;322(21):1483–1487. doi: 10.1056/NEJM199005243222102. [DOI] [PubMed] [Google Scholar]
- 3.Moll PP, Burns TL, Lauer RM. The genetic and environmental sources of body mass index variability: The Muscatine Ponderosity Family Study. Am J Hum Genet. 1991;49(6):1243–1255. [PMC free article] [PubMed] [Google Scholar]
- 4.Blakemore AI, Froguel P. Investigation of Mendelian forms of obesity holds out the prospect of personalized medicine. Ann N Y Acad Sci. 2010;1214:180–189. doi: 10.1111/j.1749-6632.2010.05880.x. [DOI] [PubMed] [Google Scholar]
- 5.Choquet H, Meyre D. Molecular basis of obesity: Current status and future prospects. Curr Genomics. 2011;12(3):154–168. doi: 10.2174/138920211795677921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thorleifsson G, et al. Genome-wide association yields new sequence variants at seven loci that associate with measures of obesity. Nat Genet. 2009;41(1):18–24. doi: 10.1038/ng.274. [DOI] [PubMed] [Google Scholar]
- 7.Willer CJ, et al. Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat Genet. 2009;41(1):25–34. doi: 10.1038/ng.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Speliotes EK, et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet. 2010;42(11):937–948. doi: 10.1038/ng.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bradfield JP, et al. A genome-wide association meta-analysis identifies new childhood obesity loci. Nat Genet. 2012;44(5):526–531. doi: 10.1038/ng.2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bachmann-Gagescu R, et al. Recurrent 200-kb deletions of 16p11.2 that include the SH2B1 gene are associated with developmental delay and obesity. Genet Med. 2010;12(10):641–647. doi: 10.1097/GIM.0b013e3181ef4286. [DOI] [PubMed] [Google Scholar]
- 11.Bochukova EG, et al. Large, rare chromosomal deletions associated with severe early-onset obesity. Nature. 2010;463(7281):666–670. doi: 10.1038/nature08689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Walters RG, et al. A new highly penetrant form of obesity due to deletions on chromosome 16p11.2. Nature. 2010;463(7281):671–675. doi: 10.1038/nature08727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Glessner JT, et al. A genome-wide study reveals copy number variants exclusive to childhood obesity cases. Am J Hum Genet. 2010;87(5):661–666. doi: 10.1016/j.ajhg.2010.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wheeler E, et al. Genome-wide SNP and CNV analysis identifies common and low-frequency variants associated with severe early-onset obesity. Nat Genet. 2013;45(5):513–517. doi: 10.1038/ng.2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Coleman DL. Obese and diabetes: Two mutant genes causing diabetes-obesity syndromes in mice. Diabetologia. 1978;14(3):141–148. doi: 10.1007/BF00429772. [DOI] [PubMed] [Google Scholar]
- 16.Friedman JM, Halaas JL. Leptin and the regulation of body weight in mammals. Nature. 1998;395(6704):763–770. doi: 10.1038/27376. [DOI] [PubMed] [Google Scholar]
- 17.Montague CT, et al. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature. 1997;387(6636):903–908. doi: 10.1038/43185. [DOI] [PubMed] [Google Scholar]
- 18.Farooqi IS, et al. Effects of recombinant leptin therapy in a child with congenital leptin deficiency. N Engl J Med. 1999;341(12):879–884. doi: 10.1056/NEJM199909163411204. [DOI] [PubMed] [Google Scholar]
- 19.Paz-Filho G, Wong ML, Licinio J. Ten years of leptin replacement therapy. Obes Rev. 2011;12(5):e315–e323. doi: 10.1111/j.1467-789X.2010.00840.x. [DOI] [PubMed] [Google Scholar]
- 20.Coppari R, Bjørbæk C. Leptin revisited: Its mechanism of action and potential for treating diabetes. Nat Rev Drug Discov. 2012;11(9):692–708. doi: 10.1038/nrd3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ren D, Li M, Duan C, Rui L. Identification of SH2-B as a key regulator of leptin sensitivity, energy balance, and body weight in mice. Cell Metab. 2005;2(2):95–104. doi: 10.1016/j.cmet.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 22.Burns B, et al. Rai1 haploinsufficiency causes reduced Bdnf expression resulting in hyperphagia, obesity and altered fat distribution in mice and humans with no evidence of metabolic syndrome. Hum Mol Genet. 2010;19(20):4026–4042. doi: 10.1093/hmg/ddq317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lacaria M, et al. A duplication CNV that conveys traits reciprocal to metabolic syndrome and protects against diet-induced obesity in mice and men. PLoS Genet. 2012;8(5):e1002713. doi: 10.1371/journal.pgen.1002713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yeung F, et al. Nontelomeric role for Rap1 in regulating metabolism and protecting against obesity. Cell Rep. 2013;3(6):1847–1856. doi: 10.1016/j.celrep.2013.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Giglio S, et al. Heterozygous submicroscopic inversions involving olfactory receptor-gene clusters mediate the recurrent t(4;8)(p16;p23) translocation. Am J Hum Genet. 2002;71(2):276–285. doi: 10.1086/341610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.South ST, et al. Co-occurrence of 4p16.3 deletions with both paternal and maternal duplications of 11p15: Modification of the Wolf-Hirschhorn syndrome phenotype by genetic alterations predicted to result in either a Beckwith-Wiedemann or Russell-Silver phenotype. Am J Med Genet A. 2008;146A(20):2691–2697. doi: 10.1002/ajmg.a.32516. [DOI] [PubMed] [Google Scholar]
- 27.Ou Z, et al. Observation and prediction of recurrent human translocations mediated by NAHR between nonhomologous chromosomes. Genome Res. 2011;21(1):33–46. doi: 10.1101/gr.111609.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luo Y, et al. Diverse mutational mechanisms cause pathogenic subtelomeric rearrangements. Hum Mol Genet. 2011;20(19):3769–3778. doi: 10.1093/hmg/ddr293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hermetz KE, Surti U, Cody JD, Rudd MK. A recurrent translocation is mediated by homologous recombination between HERV-H elements. Mol Cytogenet. 2012;5(1):6. doi: 10.1186/1755-8166-5-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Robberecht C, Voet T, Esteki MZ, Nowakowska BA, Vermeesch JR. Nonallelic homologous recombination between retrotransposable elements is a driver of de novo unbalanced translocations. Genome Res. 2013;23(3):411–418. doi: 10.1101/gr.145631.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Visser R, Kant SG, Wit JM, Breuning MH. Overgrowth syndromes:from classical to new. Pediatr Endocrinol Rev. 2009;6(3):375–394. [PubMed] [Google Scholar]
- 32.Verge CF, Mowat D. Overgrowth. Arch Dis Child. 2010;95(6):458–463. doi: 10.1136/adc.2009.157693. [DOI] [PubMed] [Google Scholar]
- 33.Klenke S, Kussmann M, Siffert W. The GNB3 C825T polymorphism as a pharmacogenetic marker in the treatment of hypertension, obesity, and depression. Pharmacogenet Genomics. 2011;21(9):594–606. doi: 10.1097/FPC.0b013e3283491153. [DOI] [PubMed] [Google Scholar]
- 34.Siffert W, et al. Association of a human G-protein beta3 subunit variant with hypertension. Nat Genet. 1998;18(1):45–48. doi: 10.1038/ng0198-45. [DOI] [PubMed] [Google Scholar]
- 35.Sun A, Ge J, Siffert W, Frey UH. Quantification of allele-specific G-protein beta3 subunit mRNA transcripts in different human cells and tissues by Pyrosequencing. Eur J Hum Genet. 2005;13(3):361–369. doi: 10.1038/sj.ejhg.5201334. [DOI] [PubMed] [Google Scholar]
- 36.Wheeler PG, Weaver DD, Palmer CG. Familial translocation resulting in Wolf-Hirschhorn syndrome in two related unbalanced individuals: Clinical evaluation of a 39-year-old man with Wolf-Hirschhorn syndrome. Am J Med Genet. 1995;55(4):462–465. doi: 10.1002/ajmg.1320550414. [DOI] [PubMed] [Google Scholar]
- 37.Tranebjaerg L, Petersen A, Hove K, Rehder H, Mikkelsen M. Clinical and cytogenetic studies in a large (4;8) translocation family with pre- and postnatal Wolf syndrome. Ann Genet. 1984;27(4):224–229. [PubMed] [Google Scholar]
- 38.Benussi DG, et al. Trisomy 12p and monosomy 4p: Phenotype-genotype correlation. Genet Test Mol Biomarkers. 2009;13(2):199–204. doi: 10.1089/gtmb.2008.0109. [DOI] [PubMed] [Google Scholar]
- 39.Firth HV, et al. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am J Hum Genet. 2009;84(4):524–533. doi: 10.1016/j.ajhg.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cooper GM, et al. A copy number variation morbidity map of developmental delay. Nat Genet. 2011;43(9):838–846. doi: 10.1038/ng.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kaminsky EB, et al. An evidence-based approach to establish the functional and clinical significance of copy number variants in intellectual and developmental disabilities. Genet Med. 2011;13(9):777–784. doi: 10.1097/GIM.0b013e31822c79f9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Segel R, et al. The natural history of trisomy 12p. Am J Med Genet A. 2006;140(7):695–703. doi: 10.1002/ajmg.a.31143. [DOI] [PubMed] [Google Scholar]
- 43.Margari L, et al. Molecular cytogenetic characterization and genotype/phenotype analysis in a patient with a de novo 8p23.2p23.3 deletion/12p13.31p13.33 duplication. Am J Med Genet A. 2012;158A(7):1713–1718. doi: 10.1002/ajmg.a.35400. [DOI] [PubMed] [Google Scholar]
- 44.Mefford HC, Eichler EE. Duplication hotspots, rare genomic disorders, and common disease. Curr Opin Genet Dev. 2009;19(3):196–204. doi: 10.1016/j.gde.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lupski JR. Hotspots of homologous recombination in the human genome: Not all homologous sequences are equal. Genome Biol. 2004;5(10):242. doi: 10.1186/gb-2004-5-10-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Turner DJ, et al. Germline rates of de novo meiotic deletions and duplications causing several genomic disorders. Nat Genet. 2008;40(1):90–95. doi: 10.1038/ng.2007.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Flegal KM, Wei R, Ogden C. Weight-for-stature compared with body mass index-for-age growth charts for the United States from the Centers for Disease Control and Prevention. Am J Clin Nutr. 2002;75(4):761–766. doi: 10.1093/ajcn/75.4.761. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.