

Abstract
Lumen formation is a critical event in biological tube formation, yet its molecular mechanisms remain poorly understood. Specifically, how lumen expansion is coordinated with other processes of tubulogenesis is not well known, and the role of membrane transporters in tubulogenesis during development has not been adequately addressed. Here we identify a solute carrier 26 (Slc26) family protein as an essential regulator of tubulogenesis using the notochord of the invertebrate chordate Ciona intestinalis as a model. Ci-Slc26aα is indispensable for lumen formation and expansion, but not for apical/luminal membrane formation and lumen connection. Ci-Slc26aα acts as an anion transporter, mediating the electrogenic exchange of sulfate or oxalate for chloride or bicarbonate and electroneutral chloride:bicarbonate exchange. Mutant rescue assays show that this transport activity is essential for Ci-Slc26aα’s in vivo function. Our work reveals the consequences and relationships of several key processes in lumen formation, and establishes an in vivo assay for studying the molecular basis of the transport properties of SLC26 family transporters and their related diseases.
Biological tubular systems serve essential functions in embryogenesis, organogenesis, and adult physiology. The development of biological tubes is an interesting paradigm not only for biologists, but also for physicians, given that many devastating diseases are caused by failure of proper tubulogenesis or malfunction of tubular structures (1). A biological tube, in its simplest configuration, is composed of an epithelium enclosing a lumen. The tubular epithelium can either arise from a preexisting epithelium by wrapping or budding, or develop de novo from nonepithelial cells. In the latter case, nonepithelial cells undergo a mesenchymal-to-epithelial transition and become polarized, with a portion of the cell membrane, the apical domain, coming into direct contact with the future lumen (2). Exocytosis at the apical/luminal domain contributes to both apical membrane biogenesis and initial lumen formation.
Recently, several transmembrane proteins, including transporters, pumps, and channels localized in the developing tubular epithelia, have been implicated in lumen expansion and tube size control (3). Among these, claudin and Na+K+-ATPase regulate lumen size in the development of the zebrafish gut (4) and the Drosophila trachea (5, 6) through their junctional activity. Interestingly, at least for Na+K+-ATPase, its pumping activity is dispensable (5). A bona fide apical transporter has not yet been identified and implicated in lumen expansion.
Transepithelial ion transport is critically important for luminal fluid secretion and absorption in many tubular structures. The solute carrier 26 (SLC26) sulfate transporters, so-called because the first member, SLC26A1, was identified as a liver SO42− transporter (7), compose a family of multidomain transmembrane anion exchangers capable of transporting a wide range of monovalent and divalent anions (8). Most SLC26 family members are found in tubular systems of various internal organs, where they are involved in important physiological processes. Mutations in several members of this family are implicated in genetic diseases, including SLC26A2 in chondrodysplasias, SLC26A3 in chloride-losing diarrhea, and SLC26A4 in Pendred syndrome and nonsyndromic deafness (9). Nonetheless, the roles of Slc26 family proteins in development remain largely unknown.
An slc26 gene has been shown to be expressed in the developing notochord of the simple chordate Ciona intestinalis. The notochord in a Ciona embryo undergoes a series of morphogenetic processes to construct a tube, which serves as a hydroskeleton in the larva to support swimming (10). The notochord initially is a solid cord consisting of 40 cells organized in tandem. During tubulogenesis, individual cells undergo a mesenchymal-to-epithelial transition through which apical/luminal domains emerge at the opposite ends of each cell. Subsequently, extracellular lumens appear and expand between neighboring cells. At a later stage, cells initiate a bidirectional crawling movement, which results in conversion of the cells to an endothelial-like morphology, along with lumen coalescence (11). A recent investigation demonstrated that the formation of dorsal longitudinal anastomotic vessels in vertebrates involves a de novo establishment of lumens at both sides of the endothelial cells and the connection of these separate lumens via cell shape changes (12). This is remarkably similar to what occurs in the Ciona notochord. We previously showed that an Slc26 protein is specifically localized at the apical/luminal domain of the Ciona notochord (11), suggesting a role in tubulogenesis.
Here we used morpholino (MO) knockdown and rescue experiments to reveal a requirement for Ci-Slc26aα in notochord tube formation. We found that Ci-Slc26aα is essential for lumen expansion, but not for apical/luminal membrane specification and biogenesis or for lumen connection. Ci-Slc26aα behaves as an osmotically neutral anion antiporter, mediating the electrogenic exchange of SO42− or C2O42− for Cl− or HCO3− and electroneutral Cl−:HCO3− exchange in vitro, and this transport activity is critical for its in vivo function in lumen expansion.
Results and Discussion
Ci-Slc26aα Is an Slc26 Family Anion Transporter.
Ci-slc26aα was originally identified as a target of Brachyury in the developing notochord (clone 309h) (13), with a protein sequence similar to that of the SLC26 family sulfate transporters. An antibody raised against the C-terminal portion (D658-N762) of 309h (Ci-SLC26-2) showed a specific localization in the apical/luminal domains at lumen formation stages (11). We isolated and sequenced the full-length cDNA from C. intestinalis in Norway. Ci-slc26aα encodes 805 amino acid residues and has a 91.7% identity with 309h, with the divergence resulting from alternative splicing and polymorphisms. According to the Simple Modular Architecture Research Tool (SMART) (14), Ci-Slc26aα is a transmembrane protein with 12 intramembrane segments. Pfam analysis (15) detects a domain structure common to all SLC26 family members, with a GLY motif at the N terminus, a sulfate transporter domain in the transmembrane region, and a sulfate transporter and anti-sigma factor antagonist (STAS) domain in the C terminus (Fig. 1A). The best match to Ci-Slc26aα in vertebrates is SLC26A5, an SLC26 family member capable of exchanging SO42− for Cl− in nonmammalian vertebrates (16); in mammals, it also serves as a motor protein in the inner ear (17). Reciprocally, Ci-Slc26aα is also the best match to the vertebrate SLC26A5 in the Ciona genome.
Fig. 1.

Ci-Slc26aα is an SLC26 family protein. (A) Domain structure of Ci-Slc26aα. (B) Neighbor-joining tree of 134 sulfate transporters. Bootstrap values are based on 100 replicates, and only those >90 are shown on the branches. (C) Electrogenic anion transport mediated by Ci-Slc26aα in CHO cells. Transport currents occurred in the presence of either sulfate or oxalate (10 mM, intracellular), but not when solely monovalent anions were present (10 mM intracellular and 100 mM extracellular Cl−). (D) Transport current reversal potential shifted strongly to more negative voltages as the extracellular Cl− concentration decreased, indicating coupled antiport of Cl− against the divalent anion. (E) Inhibition of sulfate transport current by 30 mM salicylate. Intracellular and extracellular Cl− concentrations were both 10 mM; intracellular and extracellular sulfate concentrations were 10 and 1 mM, respectively. (F) Ratiometric measurement of Cl−/HCO3− exchange by Ci-Slc26aα. CHO cells expressing Ci-Slc26aα (red) or lacking expression (black) were loaded with the ratiometric pH dye BCECF and superfused with HCO3−. On removal of Cl−, a rise in intracellular pH indicates Cl−/HCO3− antiport driven by the inward Cl− gradient. (G) Initial rates of pH change were 0.013 ± 0.002 s−1 (n = 12 cells) for Ci-Slc26aα cells and and 0.002 ± 0.002 s−1 (n = 9) control cells. Ci, Ciona intestinalis; Mt, Molgula tectiformis; Sp, Strongylocentrotus purpuratus; Bf, Branchiostoma floridae; Dm, Drosophila melanogaster; Rl, Rhizobium leguminosarum.
To explore the homology of Ci-Slc26aα, we constructed a phylogenetic tree with 134 known or predicted sulfate transporters from various organisms. The majority of SLC26 members cluster into monophyletic groups A1–A9, whereas Ci-Slc26aα locates in a sister lineage, which together with A1–A9 forms the sister group to A11 and plant sulfate transporters (Fig. 1B). Although our results demonstrate that Ci-Slc26aα is clearly an SLC26 family member, we found no support for Ci-Slc26aα as a homolog of any SLC26 subgroup, including SLC26A5. The placement of Ci-Slc26aα near the root of the SLC26A1–9 cluster, along with the relatively small number of Ciona slc26 genes (5) in the genome, suggest that Ci-Slc26aα may have ancestral functions that have diversified in the vertebrate SLC26 subgroups.
To experimentally examine the transport properties of Ci-Slc26aα, we expressed Ci-Slc26aα in CHO cells and performed patch-clamp recordings and fluorometric intracellular pH measurements. Ci-Slc26aα generated robust transport currents in the presence of sulfate or oxalate (Fig. 1C), indicating electrogenic transport of both divalent substrates. Transport occurred at millimolar concentrations of the divalent substrates with a half-maximal rate at 2.5 mM extracellular SO42− (Fig. S1 A and B). Transport rates and reversal potential were strongly dependent on Cl− concentrations. Reduction of extracellular Cl− shifted the current reversal to more negative potentials (Fig. 1D), indicating electrogenic exchange of the divalent anion against Cl− (16). Transport was blocked by salicylate (Fig. 1E), a known inhibitor of transport by vertebrate SLC26A5 homologs (16). The reversal potential of the transport current was consistent with the stoichiometric 1:1 exchange of the divalent substrate versus Cl−, as shown previously for vertebrate homologs (16). In addition, HCO3− could replace Cl− as the monovalent substrate anion (Fig. S1C).
Given that SLC26 transporters function via an alternating access mechanism (16, 18), Ci-Slc26aα most likely also can act as an electroneutral Cl−:HCO3− exchanger. Consistent with this idea, in the presence of extracellular HCO3−, removal of extracellular Cl− induced robust intracellular alkalinization (Fig. 1 F and G), indicating an influx of HCO3− in exchange for Cl−. The addition of HCO3− did not induce measurable electric currents, demonstrating that Ci-Slc26aα functions in an electroneutral 1 Cl−:1 HCO3− exchange mode. However, Ci-Slc26aα exhibited only negligible uncoupled transport activity, in contrast to mammalian SLC26A5 and SLC26A6, which also can mediate uncoupled anion currents of NO3− and SCN− (19, 20). Based on the domain organization, phylogeny, and transport characteristics, we conclude that Ci-Slc26aα is a Ciona SLC26 family transporter. We designate this protein Ci-Slc26aα to reflect the lack of basis for assigning it to a vertebrate SLC26 subgroup.
Expression of Ci-Slc26aα Correlates with a Function in Lumen Formation.
We confirmed and extended the known expression data for Ci-slc26aα using our full-length clone and in situ hybridization. Ci-slc26aα first appears in the notochord at neurula stage, where it persists to late tailbud stage (Fig. 2A and Fig. S2) (13). At early tailbud stage, Ci-Slc26aα expression also begins in the sensory vesicle. The notochord expression was correlated with antibody staining showing that Ci-Slc26aα localizes specifically at the apical/luminal domain of individual notochord cells throughout the lumen formation process (Fig. 2B) (11). Ci-Slc26aα-mCherry driven by the notochord-specific Brachyury enhancer recapitulated both the temporary and spatial localization of the endogenous protein (Fig. 2 E–H). This occurred despite the earlier transcriptional activity of the Brachyury element, suggesting that the apical/luminal localization of Ci-Slc26aα in notochord cells is regulated at the protein level.
Fig. 2.

Expression and localization of Ci-Slc26aα. (A) Ci-slc26aα is expressed in the sensory vesicle and the notochord at early tailbud stage. (B) Diagram of the notochord at mid-tailbud stage during lumen formation. (C) Diagram of the sensory vesicle. (D) Ci-Slc26aα localizes to the apical side of the sensory vesicle epithelium and plasma membrane of the otolith. Ci-Slc26aα-mCherry localizes to the apical/luminal domain of the notochord cells during lumen expansion (E), during cell migration and lumen connection (F and G), and after tube formation (H). n, notochord; sv, sensory vesicle.
The Ciona sensory vesicle consists of the conspicuously pigmented ocellus and otolith (Fig. 2C), which serve as the larva’s photoreceptor and gravity sensing organ (21), analogous to the vertebrate eye and a part of the inner ear, respectively. Both organs protrude into the sensory vesicle lumen, which is enclosed by the sensory vesicle epithelium (Fig. 3F). Ci-Slc26aα is specifically localized at the luminal side of the epithelium and the plasma membrane of the otolith cell (Fig. 2D). Together, these results indicate that Ci-Slc26aα is present at the apical/luminal domain of lumen-forming cells. The presence of Ci-Slc26aα in the sensory vesicle epithelium and otolith is similar to the expression of mammalian SLC26A4 (also known as pendrin, after Pendred syndrome), in the epithelia of the vestibular labyrinth and the endolymphatic sac (22), which are responsible for sensing spatial orientation.
Fig. 3.
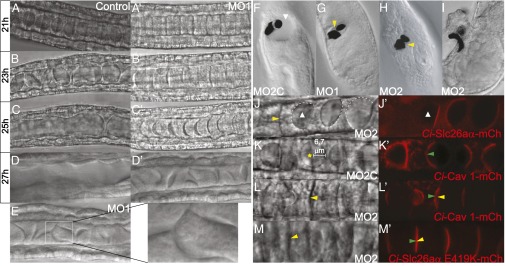
Ci-Slc26aα is essential for lumen expansion, but not for apical/luminal membrane specification and biogenesis or lumen connection. (A–D′) Notochord in control (A–D) and Ci-Slc26aαMO1–injected embryos (A′–D′). (E) Narrow tube in Ci-Slc26aαMO1–injected notochord. (F–I) Sensory vesicle in control embryo (F; Ci-Slc26aαMO2C–injected) and embryos injected with Ci-Slc26aαMO1 (G) or Ci-Slc26aαMO2 (H and I). (J and J′) Ci-Slc26aα expression rescued the lumen formation defect. (K and K′) Ci-Caveolin 1-mCherry was localized at the apical/luminal domain in control MO-injected embryos. (L–M′) Apical/luminal membrane, marked by Ci-Cav-1-mCherry (L and L′) or Ci-Slc26aα E419K-mCherry (M and M′), developed in Ci-Slc26aαMO2–injected notochord cells. The white dashed line in J outlines the apical/luminal membranes of cells next to Ci-Slc26aα-mCherry–expressing cells, the white arrowhead indicates the lumen, the yellow arrowhead indicates the absence of lumen, and the green arrowhead indicates the presence of apical/luminal membrane. mCh, mCherry.
Ci-Slc26aα Is Essential in Lumen Expansion, but Not in Other Processes of Lumen Formation.
We used MO knockdown to study the function of Ci-Slc26aα. Notochord development preceding lumen formation was not affected in embryos injected with Ci-Slc26aαMO1, an MO designed to block the initiation of Ci-Slc26aα translation (Fig. 3A′). After the cell elongation stage, when extracellular lumen pockets appeared and expanded in the notochord of control embryos (Fig. 3 A–C), the extracellular space between adjacent notochord cells in Ci-Slc26aαMO1–injected embryos developed only narrow pockets of lumens that slowly acquired a crescent shape (Fig. 3 A′–C′). These significantly smaller lumens were eventually tilted as a result of the crawling movement of the notochord cells (Fig. 3D′), and some of them were connected to one another to form narrow and short tubes locally (Fig. 3E). This finding suggests that full expansion of the lumens is not required for cell movement and lumen connection. Of interest, notochord cells in ascidians that do not construct a tubular notochord also execute extensive cell movement (10), supporting the notion that lumen formation is not a prerequisite for subsequent cell shape changes. In addition, lumen coalescence by endothelial cell rearrangement during zebrafish vasculogenesis does not appear to depend on significant lumen expansion (12, 23). The Ci-Slc26aαMO1 lumen formation defect was also obtained using a splice MO, Ci-Slc26aαMO2 (Fig. S3 and Fig. 3J, yellow arrowhead), which reduced the normal transcript by ∼80% (Fig. S4), but was not observed in embryos injected with a control MO, Ci-Slc26aαMO2C (Fig. 3K), which has the reverse sequence of Ci-Slc26aαMO2. In addition, reintroduction of Ci-Slc26aα-mCherry to MO knockdown embryos by electroporation (which results in mosaic expression of the construct) fully rescued the lumen formation defect around the Ci-Slc26aα–positive cells (Fig. 3 J and J′).
Ci-Slc26aα knockdown also caused malformation of the sensory vesicles. Injecting moderate amounts (20 fmol) of Ci-Slc26aαMO1 or Ci-Slc26aαMO2 significantly reduced the sensory vesicle luminal space (Fig. 3 G and H). Higher amounts (50 fmol) sometimes resulted in a displacement and exclusion of the pigmented organs in the head (Fig. 3I and Movies S1 and S2). These results suggest that Ci-Slc26aα plays a similar role in lumen formation in the sensory vesicle as it does in the notochord. Mammalian SLC26A4 is also involved in ion homeostasis of the inner ear fluids. Patients with Pendred syndrome, caused by mutations in SLC26A4, have an enlarged vestibular aqueduct resulting from disruption of the balance between fluid secretion in the vestibular labyrinth and fluid absorption in the endolymphatic sac (22).
In our rescue experiment, cells that did not express any detectable Ci-Slc26aα-mCherry but were adjacent to cells that did express the construct also developed a large luminal membrane (outlined with dashed lines in Fig. 3J). This finding suggests that Ci-Slc26aα is not involved in apical/luminal membrane specification and biogenesis. To confirm this result and to identify the precise role of Ci-Slc26aα, we examined apical/luminal domain formation using an mCherry fusion of Ci-Caveolin 1 (Cav 1), an integral membrane protein that is enriched at the apical/luminal domain of notochord cells (Fig. 3 K and K′; embryo injected with the control MO, Ci-Slc26aαMO2C). In Ci-Slc26aαMO2 knockdown embryos, a significant area of the lateral surface of notochord cells became positive for Ci-Cav 1-mCherry (Fig. 3 L and L′), even when the lumen was not visible (compared with the control shown in Fig. 3 K and K′). In addition, a Ci-Slc26aα E419K mutant, which is not able to rescue the MO phenotype (Fig. 3M), was recruited to a large area of the notochord cell interfaces (Fig. 3M′).
The foregoing results suggest that disruption of Ci-Slc26aα specifically affects lumen expansion, but not apical/luminal domain differentiation and expansion. The uncoupling of lumen expansion from luminal membrane biogenesis underscores the importance of a mechanism independent of exocytosis in lumen formation during tubulogenesis. The extent of the contribution of this mechanism can be theorized by calculating the difference between the total volume of the lumen (Vlumen, considered here as spherical, with a radius of R) and the lumen volume contributed by the sum of the secretory vesicles (Vvesicles, each with an average radius of r), considering that all luminal membrane is supplied by vesicle secretion (Alumen = Avesicles, where A is surface area). The number of vesicles required to form Alumen is 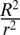 , and the difference, expressed as
, and the difference, expressed as  , is
, is 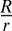 . Given that the mature extracellular lumen is several orders of magnitude larger than individual vesicles, the contribution to the lumen by secretory vesicles becomes almost negligible, despite the fact that the vesicles supply the entire luminal membrane. Secretory vesicles in notochord cells during lumen formation are located near the apical/luminal membrane and have an average r of 0.05 μm (Fig. S5). To create a typical lumen (indicated by a star in Fig. 3K) with an R of 6.7 μm, 17,956 secretory vesicles, each supplying 0.03 μm2 of membrane, are needed to generate a surface area of 564.10 μm2 (Alumen). However, these vesicles can contribute to only 9.40 μm3 of the lumen volume (Vvesicles), which is 134-fold short of the actual lumen volume of 1,259.83 μm3 (Vlumen). Our experimental results showing that significant apical/luminal membrane is generated without any visible lumen in MO knockdown embryos validate this model (Fig. 3 M and M′).
. Given that the mature extracellular lumen is several orders of magnitude larger than individual vesicles, the contribution to the lumen by secretory vesicles becomes almost negligible, despite the fact that the vesicles supply the entire luminal membrane. Secretory vesicles in notochord cells during lumen formation are located near the apical/luminal membrane and have an average r of 0.05 μm (Fig. S5). To create a typical lumen (indicated by a star in Fig. 3K) with an R of 6.7 μm, 17,956 secretory vesicles, each supplying 0.03 μm2 of membrane, are needed to generate a surface area of 564.10 μm2 (Alumen). However, these vesicles can contribute to only 9.40 μm3 of the lumen volume (Vvesicles), which is 134-fold short of the actual lumen volume of 1,259.83 μm3 (Vlumen). Our experimental results showing that significant apical/luminal membrane is generated without any visible lumen in MO knockdown embryos validate this model (Fig. 3 M and M′).
Transport Activity of Ci-Slc26aα Is Essential for Its in Vivo Function.
To test whether the transport activity of Ci-Slc26aα is essential for its function in lumen expansion, we sought to correlate the ability of Ci-Slc26aα mutants to rescue the MO knockdown phenotype in vivo, with their ability to transport in vitro. We found that none of the truncation mutants (Fig. S6A), which lacked the GLY motif, a part of the sulfate transporter domain and the STAS domain, or the STAS domain, were able to target to the plasma membrane (Fig. S6B). We then generated mutants with single amino acid substitutions in regions that are highly conserved with human SLC26 proteins (Fig. S7A). Mutations of homologous residues have been reported in patients with SLC26 disease (Table S1), so we reasoned that these mutations were likely to elicit a phenotype. We found that all of the point mutants localized at the apical domain at the onset of lumen formation and did not cause abnormality in the notochord when expressed in normal embryos (Fig. S7B). However, when expressed in Ci-Slc26aα MO2-injected embryos, Ci-Slc26aα G245V, which is analogous to the G209V mutation in SLC26A4 from patients with Pendred syndrome and nonsyndromic hearing loss (24, 25), failed to rescue the lumen formation defect (Fig. 4B). Ci-Slc26aα L485P, which corresponds to the L483P mutation in SLC26A2 from patients with achondrogenesis type 1B (26), also failed to rescue the lumen formation defect (Fig. 4D). In contrast, the Ci-Slc26aα Y591C mutant, which corresponds to Y556C and Y556H in human SLC26A4 from patients with Pendred syndrome (25, 27, 28), was able to rescue lumen formation (Fig. 4E). Ci-Slc26aα R444C, analogous to R409P also from patients with Pendred syndrome (29, 30), partially rescued the defective lumen formation phenotype (Fig. 4C).
Fig. 4.

In vivo function and in vitro transport activity of Ci-Slc26aα mutants. (A–E) Mosaic expression (red channel) of WT (A) and Ci-Slc26aα point mutants (B–E) in Ci-Slc26aαMO2–injected embryos. Green channel, fluorescein-tagged Ci-Slc26aαMO2C; bright channel, transilluminated images. The white arrowhead indicates the lumen, and the yellow arrowhead indicates a substantial reduction of lumen. (F) Representative whole-cell transport currents of WT and Ci-Slc26aα point mutants expressed in CHO cells. Divalent substrates (10 mM) were applied to the extracellular side, yielding electrical outward currents for functional transporters. Intracellular and extracellular Cl− concentrations were 10 mM and 100 mM, respectively. Mutant R444C also shows transport in the absence of divalent anions and has an altered current–voltage relationship indicative of uncoupled Cl− permeation as an additional transport mode (Fig. S7 D and E). (G) Summary of data obtained from experiments in F. Bars indicate currents on application of the substrate measured at 0 mV membrane potential and normalized to the cell surface (mean ± SEM).
We next tested the transport activity of these mutants in vitro. All mutants localized to the plasma membrane when expressed in CHO cells (Fig. S7C). Whole-cell patch-clamp recordings showed that Y591C exhibited robust transport currents, R444C maintained a substantial current, L485P retained only miniscule transport activity, and G245V was completely dysfunctional (Fig. 4 F and G and Fig. S7 D and E). These results correlate closely with the ability of the mutants to rescue the lumen formation defect in vivo, indicating that the transport activity of Ci-Slc26aα is critical for tube development (Table S1).
Our study adds an Slc26 family protein to the collection of membrane proteins that are specifically involved in tubulogenesis. Ci-Slc26aα is an apical transporter, exerting a role in tubulogenesis through its anion transport activity. The failure of lumen expansion after disruption of Ci-Slc26aα reveals a nonredundant role for this transporter in vivo. The immediate consequence of its action in the context of lumen development remains poorly understood. Extensive luminal expansion can be accomplished by a cross-membrane osmotic gradient that drives fluid efflux, or by the action of high molecular weight gel-forming glycoproteins that expand in the luminal space on hydration. Ci-Slc26aα–mediated sulfate transport might be required to provide the substrate for the sulfation of luminal glycoaminoglycans; however, because sulfation occurs in intracellular compartments, such a role would imply sulfate influx from the extracellular fluid. This mechanism appears unlikely to be responsible for the early phase of lumen formation, given that Ci-Slc26aα is located specifically at the apical membrane surrounding the forming lumen, where the nascent extracellular fluid lacks a sulfate source. Consequently, we suggest that Ci-Slc26aα is involved initially in providing an osmotic drive for water influx to inflate the notochord lumen.
Ci-Slc26aα is an osmotically neutral anion exchanger, excluding the possibility that it alone can establish a concentration gradient to mobilize water accumulation in the lumen. The complete picture is likely more complex, with other transporters and channels present and collaborating with Ci-Slc26aα to regulate luminal osmolarity. Of note, it has been reported that the iodide efflux activity of SLC26A4, a known osmotically neutral anion exchanger (31), is significantly reduced in the G209V mutant isolated from patients with Pendred syndrome or nonsyndromic hearing loss associated with an enlarged vestibular aqueduct (25). The fact that Ci-Slc26aα G245V, which no longer has any transport activity, fails to support lumen expansion suggests that Ciona notochord tubulogenesis can serve as an in vivo model for exploring the complex molecular pathogenesis of these diseases. Once significant lumen has formed, Ci-Slc26aα may then shuttle sulfate into the cytoplasm, providing the substrate for the sulfation of luminal glycoaminoglycans that will facilitate further lumen expansion. It has been reported that the proteoglycans synthesized by chondrocytes from a patient with achondrogenesis type 1B homozygous for an L483P substitution in SLC26A2 were severly undersulfated (26). Ci-Slc26aα L485P, which is unable to transport sulfate, fails to support lumen expansion, validating the importance of the structural element at and around this residue in maintaining the protein function in vivo.
Materials and Methods
Animals and Embryos.
Adult C. intestinalis were collected from Grimstadfjorden or purchased from Station Biologique de Roscoff. Animals were kept in the Sars Center ascidian facility for 1–3 wk before experiments. Eggs and sperm dissected from different animals were mixed for fertilization. Embryos were dechorionated and cultured at 16 °C to stages of interest.
In Situ Hybridization.
In situ hybridization was carried out as described by Wada et al. (32) with the following modifications. Embryos were fixed for 2 h at room temperature using 4% PFA in a PBS solution containing 0.1% Tween-20 (PBS-T), 100 mM MOPS buffer, and 0.5 M NaCl. Before hybridization, the embryos were digested for 30 min with 12 μg/mL protease K in PBS-T at 37 °C.
Immunohistochemistry.
Anti–Ci-Slc26aα and staining procedures were performed as described previously (11).
Clone and Constructs.
Ci-slc26aα was amplified from Norwegian C. intestinalis cDNA using the primer pair CAGAAAAAATGGCCGAAGAAAATTCGATTC and CATTTGCGTCGTTTCGGC. The PCR product was cloned into PCR8/GW/TOPO (Invitrogen) and then subcloned into a destination vector, B3-eBra-bpfog-B5::B1-ccdB/CmR-B2-mCherry, modified from Minos-B3-eBra-bpfog-B5::B1-ccdB/CmR-B2-mCherry (11) in which the Minos terminal repeats were removed and the orientation of ColE1 was reversed, to obtain Ci-Slc26aα-mCherry by the Gateway recombination method. For electrophysiology, Ci-Slc26aα was amplified with CAGGGCTAGCATGGCCGAAGAAAATTCGA and GCGTGGATCCCATTTGCGTCGTTTCGG and cloned into pEGFP-N1 by ligation at NheI and BamHI to create pEGFP-Ci-Slc26aα. Ci-caveolin 1, corresponding to gene model KYOTOGRAIL2005.177.18.1, was amplified with primers CAGAAAAAATGGACAATGTGGAACTTGATTC and GTCTCTCCTCAGTGACATCACA, cloned into PCR8/GW/TOPO, and recombined into Minos-B3-eBra-bpfog-B5::B1-ccdB/CmR-B2-mCherry to obtain Ci-Cav 1-mCherry.
Phylogeny.
The protein sequences of 134 sulfate transporters in 44 organisms were collected from GenBank, aligned using ClustalX, and trimmed. Paup4.0b (Sinauer Associates) was used to build the neighbor-joining phylogeny tree, using maximum likelihood optimality criterion and the JTT amino acid substitution model. Bootstrap values were calculated based on 100 replicates.
Electroporation.
Electroporation was performed as described previously (33) with some modifications. Here, 100 μL of dechorionated fertilized eggs were mixed with 50 μg of plasmid DNA and electroporated in 4-mm cuvettes with a Gene Pulser Xcell System (Bio-Rad), using a time constant protocol (50 V, 15 ms).
MO Microinjection.
Three fluorescein-tagged MOs—Ci-Slc26aαMO1 (CGTGAATCGAATTTTCTTCTGCCAT), Ci-Slc26aαMO2 (AACTGGTGCTTACATGTATAAGGCC), and Ci-Slc26aαMO2C (CCGGAATATGTACATTCGTGGTCAA)—were synthesized by Gene Tools. MOs were dissolved in H2O to make a 1 mM solution. Microinjection was carried out as described by Christiaen et al. (34). Successfully injected embryos were selected based on the presence of fluorescence for further study.
Mutagenesis.
The Quikchange Site-Directed Mutagenesis Kit (Stratagene) was used to generate Ci-Slc26aα point mutants from Ci-Slc26aα-mCherry and pEGFP-Ci-Slc26aα.
Rescue Experiment.
Eggs successfully injected with MOs were fertilized and then electroporated with the Ci-Slc26aα-mCherry construct. The embryos were then washed and cultured at 16 °C.
Anion Transport.
Ci-Slc26aα and mutant-containing pEGFP plasmids were transfected into CHO cells using JetPEI transfection reagent (Polyplus). Patch-clamp recordings of electrogenic transport currents were performed as described previously (16, 35). In brief, cells with unequivocal and comparable membrane fluorescence were selected at 24–48 h after transfection. Whole-cell patch-clamp recordings were carried out at room temperature (20–22 °C) with EPC10 amplifiers (Heka) controlled by Patchmaster software (Heka). Electrogenic anion transport was measured as the ionic transport current in the response to command voltage ramps (300 ms; ∼0.8 V/s).
Patch electrodes were filled with one of the following intracellular solutions: 160 mM CsCl; 10 mM CsCl and 150 mM K-aspartate; 10 mM CsCl, 130 mM K-aspartate, and 10 mM Cs2SO4; or 10 mM CsCl, 130 mM K-aspartate, and 10 mM Cs2-oxalate. All intracellular solutions contained 1 mM Hepes and 1 mM K2EGTA and were adjusted to pH 7.3 with KOH. When divalent transport substrates were applied through the patch pipette, extracellular solutions were 100 mM NaCl, 10 mM Hepes, 2 mM Mg-gluconate, and 60 mM Na-gluconate. For variation of extracellular Cl− concentration, Cl− was replaced by equimolar gluconate. The solution for extracellular application of divalent anions was10 mM NaCl, 10 mM Na2SO4 or Na-oxalate, 140 mM Na-gluconate, 10 mM Hepes, and 2 mM Mg-gluconate, adjusted to pH 7.4 with NaOH. Na-salicylate (10 mM) was substituted for Na-gluconate.
Electroneutral monovalent transport was measured with the ratiometric pH-sensitive dye 2′,7′-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein (BCECF) (36). Cells were loaded with acetoxymethyl ester derivative of BCECF (BCECF-AM) (Invitrogen), excited alternately at 430 nm and 500 nm with an Oligochrome wavelength switching light source (TILL Photonics). Fluorescence emission from individual cells was recorded at 535 ± 15 nm with a photodiode-based photometry setup (TILL Photonics). pH was measured as the ratio of fluorescence intensities measured at the two excitation wavelengths and calibrated with the nigericin/K+ method (37). Cells were superfused with extracellular solution containing 24 mM NaHCO3 and 120 mM NaCl (pH 7.4; bubbled with 5% CO2), and Cl−:HCO3− exchange was induced by the removal of chloride (substitution by equimolar gluconate).
Microscopy.
Nomarski images were obtained using a Nikon Eclipse E800 microscope equipped with a 40× objective (NA 1.00) and a SPOT RTKE CCD camera (Diagnostic Instruments). Confocal images were obtained with a Leica TCS SP5 confocal laser-scanning microscope equipped with a 40× oil-immersion objective (NA 1.25). If necessary, embryos were sedated using 0.015% MS222 (A5040; Sigma-Aldrich). Image analysis and processing were performed with Leica TCS SP5 systems LAS AF software packages and ImageJ.
Transmission Electron Microscopy.
Tailbud-stage embryos were fixed in 4% paraformaldehyde and postfixed in 1% osmium tetroxide. The samples were embedded in Durcupan resin (Fluka). Then 60-nm sections were counterstained with uranyl acetate and lead citrate and observed under a JEOL JEM 1230 transmission electron microscope.
Supplementary Material
Acknowledgments
We thank Drs. Gemma Richards and Dmitry Gorbunov for useful discussions and a critical reading of the manuscript, and Drs. Daniela Schreiber and Lucas Leclère for technical expertise. This work was supported by the Norwegian Research Council (Grants 133335/V40 and 183302/S10, to D.J.), the Deutsche Forschungsgemeinschaft (Grant OL 240/4, to D.O.), and a research grant from the University Medical Center Giessen and Marburg (to D.O.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. C.S. is a guest editor invited by the Editorial Board.
Data deposition: The Ci-Slc26aα sequence reported in this paper has been deposited in the GenBank database (accession no. KF494348).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1220884110/-/DCSupplemental.
References
- 1.Lubarsky B, Krasnow MA. Tube morphogenesis: Making and shaping biological tubes. Cell. 2003;112(1):19–28. doi: 10.1016/s0092-8674(02)01283-7. [DOI] [PubMed] [Google Scholar]
- 2.Datta A, Bryant DM, Mostov KE. Molecular regulation of lumen morphogenesis. Curr Biol. 2011;21(3):R126–R136. doi: 10.1016/j.cub.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bryant DM, Mostov KE. From cells to organs: Building polarized tissue. Nat Rev Mol Cell Biol. 2008;9(11):887–901. doi: 10.1038/nrm2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bagnat M, Cheung ID, Mostov KE, Stainier DY. Genetic control of single lumen formation in the zebrafish gut. Nat Cell Biol. 2007;9(8):954–960. doi: 10.1038/ncb1621. [DOI] [PubMed] [Google Scholar]
- 5.Paul SM, Palladino MJ, Beitel GJ. A pump-independent function of the Na,K-ATPase is required for epithelial junction function and tracheal tube-size control. Development. 2007;134(1):147–155. doi: 10.1242/dev.02710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu VM, Schulte J, Hirschi A, Tepass U, Beitel GJ. Sinuous is a Drosophila claudin required for septate junction organization and epithelial tube size control. J Cell Biol. 2004;164(2):313–323. doi: 10.1083/jcb.200309134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bissig M, Hagenbuch B, Stieger B, Koller T, Meier PJ. Functional expression cloning of the canalicular sulfate transport system of rat hepatocytes. J Biol Chem. 1994;269(4):3017–3021. [PubMed] [Google Scholar]
- 8.Mount DB, Romero MF. The SLC26 gene family of multifunctional anion exchangers. Pflugers Arch. 2004;447(5):710–721. doi: 10.1007/s00424-003-1090-3. [DOI] [PubMed] [Google Scholar]
- 9.Dawson PA, Markovich D. Pathogenetics of the human SLC26 transporters. Curr Med Chem. 2005;12(4):385–396. doi: 10.2174/0929867053363144. [DOI] [PubMed] [Google Scholar]
- 10.Jiang D, Smith WC. Ascidian notochord morphogenesis. Dev Dyn. 2007;236(7):1748–1757. doi: 10.1002/dvdy.21184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dong B, et al. Tube formation by complex cellular processes in Ciona intestinalis notochord. Dev Biol. 2009;330(2):237–249. doi: 10.1016/j.ydbio.2009.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Herwig L, et al. Distinct cellular mechanisms of blood vessel fusion in the zebrafish embryo. Curr Biol. 2011;21(22):1942–1948. doi: 10.1016/j.cub.2011.10.016. [DOI] [PubMed] [Google Scholar]
- 13.Hotta K, Takahashi H, Erives A, Levine M, Satoh N. Temporal expression patterns of 39 Brachyury-downstream genes associated with notochord formation in the Ciona intestinalis embryo. Dev Growth Differ. 1999;41(6):657–664. doi: 10.1046/j.1440-169x.1999.00467.x. [DOI] [PubMed] [Google Scholar]
- 14.Schultz J, Milpetz F, Bork P, Ponting CP. SMART, a simple modular architecture research tool: Identification of signaling domains. Proc Natl Acad Sci USA. 1998;95(11):5857–5864. doi: 10.1073/pnas.95.11.5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dorwart MR, Shcheynikov N, Yang D, Muallem S. The solute carrier 26 family of proteins in epithelial ion transport. Physiology (Bethesda) 2008;23:104–114. doi: 10.1152/physiol.00037.2007. [DOI] [PubMed] [Google Scholar]
- 16.Schaechinger TJ, Oliver D. Nonmammalian orthologs of prestin (SLC26A5) are electrogenic divalent/chloride anion exchangers. Proc Natl Acad Sci USA. 2007;104(18):7693–7698. doi: 10.1073/pnas.0608583104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zheng J, et al. Prestin is the motor protein of cochlear outer hair cells. Nature. 2000;405(6783):149–155. doi: 10.1038/35012009. [DOI] [PubMed] [Google Scholar]
- 18.Muallem D, Ashmore J. An anion antiporter model of prestin, the outer hair cell motor protein. Biophys J. 2006;90(11):4035–4045. doi: 10.1529/biophysj.105.073254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schänzler M, Fahlke C. Anion transport by the cochlear motor protein prestin. J Physiol. 2012;590(Pt 2):259–272. doi: 10.1113/jphysiol.2011.209577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ohana E, Shcheynikov N, Yang D, So I, Muallem S. Determinants of coupled transport and uncoupled current by the electrogenic SLC26 transporters. J Gen Physiol. 2011;137(2):239–251. doi: 10.1085/jgp.201010531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meinertzhagen IA, Lemaire P, Okamura Y. The neurobiology of the ascidian tadpole larva: Recent developments in an ancient chordate. Annu Rev Neurosci. 2004;27:453–485. doi: 10.1146/annurev.neuro.27.070203.144255. [DOI] [PubMed] [Google Scholar]
- 22.Wangemann P. The role of pendrin in the development of the murine inner ear. Cell Physiol Biochem. 2011;28(3):527–534. doi: 10.1159/000335113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blum Y, et al. Complex cell rearrangements during intersegmental vessel sprouting and vessel fusion in the zebrafish embryo. Dev Biol. 2008;316(2):312–322. doi: 10.1016/j.ydbio.2008.01.038. [DOI] [PubMed] [Google Scholar]
- 24.Campbell C, et al. Pendred syndrome, DFNB4, and PDS/SLC26A4 identification of eight novel mutations and possible genotype–phenotype correlations. Hum Mutat. 2001;17(5):403–411. doi: 10.1002/humu.1116. [DOI] [PubMed] [Google Scholar]
- 25.Taylor JP, Metcalfe RA, Watson PF, Weetman AP, Trembath RC. Mutations of the PDS gene, encoding pendrin, are associated with protein mislocalization and loss of iodide efflux: Implications for thyroid dysfunction in Pendred syndrome. J Clin Endocrinol Metab. 2002;87(4):1778–1784. doi: 10.1210/jcem.87.4.8435. [DOI] [PubMed] [Google Scholar]
- 26.Rossi A, Bonaventure J, Delezoide AL, Cetta G, Superti-Furga A. Undersulfation of proteoglycans synthesized by chondrocytes from a patient with achondrogenesis type 1B homozygous for an L483P substitution in the diastrophic dysplasia sulfate transporter. J Biol Chem. 1996;271(31):18456–18464. doi: 10.1074/jbc.271.31.18456. [DOI] [PubMed] [Google Scholar]
- 27.Coyle B, et al. Molecular analysis of the PDS gene in Pendred syndrome. Hum Mol Genet. 1998;7(7):1105–1112. doi: 10.1093/hmg/7.7.1105. [DOI] [PubMed] [Google Scholar]
- 28.Van Hauwe P, et al. Two frequent missense mutations in Pendred syndrome. Hum Mol Genet. 1998;7(7):1099–1104. doi: 10.1093/hmg/7.7.1099. [DOI] [PubMed] [Google Scholar]
- 29.Fugazzola L, et al. Differential diagnosis between Pendred and pseudo-Pendred syndromes: Clinical, radiologic, and molecular studies. Pediatr Res. 2002;51(4):479–484. doi: 10.1203/00006450-200204000-00013. [DOI] [PubMed] [Google Scholar]
- 30.Park HJ, et al. Origins and frequencies of SLC26A4 (PDS) mutations in east and south Asians: Global implications for the epidemiology of deafness. J Med Genet. 2003;40(4):242–248. doi: 10.1136/jmg.40.4.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ohana E, Yang D, Shcheynikov N, Muallem S. Diverse transport modes by the solute carrier 26 family of anion transporters. J Physiol. 2009;587(Pt 10):2179–2185. doi: 10.1113/jphysiol.2008.164863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wada S, Katsuyama Y, Yasugi S, Saiga H. Spatially and temporally regulated expression of the LIM class homeobox gene Hrlim suggests multiple distinct functions in development of the ascidian, Halocynthia roretzi. Mech Dev. 1995;51(1):115–126. doi: 10.1016/0925-4773(95)00359-9. [DOI] [PubMed] [Google Scholar]
- 33.Christiaen L, Wagner E, Shi W, Levine M. Electroporation of transgenic DNAs in the sea squirt Ciona. Cold Spring Harb Protoc. 2009 doi: 10.1101/pdb.prot5345. doi:10.1101/pdb.prot5345. [DOI] [PubMed] [Google Scholar]
- 34.Christiaen L, Wagner E, Shi W, Levine M. Microinjection of morpholino oligos and RNAs in sea squirt (Ciona) embryos. Cold Spring Harb Protoc. 2009 doi: 10.1101/pdb.prot5347. doi:10.1101/pdb.prot5347. [DOI] [PubMed] [Google Scholar]
- 35.Schaechinger TJ, et al. A synthetic prestin reveals protein domains and molecular operation of outer hair cell piezoelectricity. EMBO J. 2011;30(14):2793–2804. doi: 10.1038/emboj.2011.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rink TJ, Tsien RY, Pozzan T. Cytoplasmic pH and free Mg2+ in lymphocytes. J Cell Biol. 1982;95(1):189–196. doi: 10.1083/jcb.95.1.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Negulescu PA, Machen TE. Intracellular ion activities and membrane transport in parietal cells measured with fluorescent dyes. Methods Enzymol. 1990;192:38–81. doi: 10.1016/0076-6879(90)92062-i. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.