

Abstract
Adaptation under fasting conditions is critical for survival in animals. Sirtuin 1 (SIRT1), a protein deacetylase, plays an essential role in adaptive metabolic and endocrine responses under fasting conditions by modifying the acetylation status of various proteins. Fasting induces growth hormone (GH) resistance in the liver, leading to decreased serum insulin-like growth factor-I (IGF-I) levels as an endocrine adaptation for malnutrition; however, the underlying mechanisms of this action remain to be fully elucidated. Here we report that in vivo knockdown of SIRT1 in the liver restored the fasting-induced decrease in serum IGF-I levels and enhanced the GH-dependent increase in IGF-I levels, indicating that SIRT1 negatively regulates GH-dependent IGF-I production in the liver. In vitro analysis using hepatocytes demonstrated that SIRT1 suppresses GH-dependent IGF-I expression, accompanied by decreased tyrosine phosphorylation on signal transducer and activator of transcription (STAT) 5. GST pull-down assays revealed that SIRT1 interacts directly with STAT5. When the lysine residues adjacent to the SH2 domain of STAT5 were mutated, STAT5 acetylation decreased concomitant with a decrease in its transcriptional activity. Knockdown of SIRT1 enhanced the acetylation and GH-induced tyrosine phosphorylation of STAT5, as well as the GH-induced interaction of the GH receptor with STAT5. These data indicate that SIRT1 negatively regulates GH-induced STAT5 phosphorylation and IGF-I production via deacetylation of STAT5 in the liver. In addition, our findings explain the underlying mechanisms of GH resistance under fasting conditions, which is a known element of endocrine adaptation during fasting.
Adaptation under fasting conditions is critical for survival in animals and involves various metabolic and endocrine changes (1). In particular, the endocrine system plays an essential role in this physiological adaptation (2). Various endocrine changes generally direct energy utilization toward survival functions and away from growth and reproduction. A representative endocrine adaptation occurs in the growth hormone (GH)–insulin-like growth factor-I (IGF-I) axis. Starvation and malnutrition lower circulating IGF-I levels despite elevated GH secretion, indicating the presence of GH resistance (3). Indeed, GH administration in fasted rats fails to increase the concentrations of circulating IGF-I (4). Exogenous GH given to GH-deficient fasted human subjects causes only a twofold increase in serum IGF-I concentrations, compared with a 10-fold increase in normally fed subjects (5).
GH binds to the GH receptor (GHR) and causes GHR to activate Janus kinase 2 (JAK2). JAK2 then phosphorylates GHR and recruits signal transducer and activator of transcription (STAT) 5 through an interaction between the STAT5 SH2 domain and the phosphorylated tyrosine (Tyr) in GHR (6). As a result, JAK2 is able to phosphorylate STAT5, leading to its dimerization and translocation to the nucleus, where it binds to the regulatory elements of target genes, including IGF-I (7). The reduction in serum IGF-I level is caused mainly by a decreased IGF-I mRNA level in the liver, where most circulating IGF-I is produced (8, 9).
Previous results may explain the development of GH resistance in the liver under fasting conditions. Insulin regulates GHR expression in the liver, and a low insulin concentration in the portal vein during fasting is associated with decreased expression of GHR in hepatocytes (10, 11). In another study, fibroblast growth factor 21 (FGF-21), a hormone induced by fasting, was found to cause GH resistance by decreasing the active form of STAT5, resulting in decreased growth to conserve energy during starvation (12). These data suggest that GH resistance under fasting is caused by several mechanisms; however, whether these mechanisms can fully explain the GH resistance status, especially in a physiological setting, remains to be seen.
Sirtuin 1 (SIRT1) is a class III histone deacetylase of the sirtuin family that is uniquely dependent on NAD for catalysis. The abundance and activity of this nutrient-sensing deacetylase increase with caloric restriction (CR) to help preserve euglycemia and promote efficient energy utilization (13). For example, although SIRT1 deacetylates transducer of regulated CREB protein 2 (TORC2) and suppresses TORC2-mediated gluconeogenesis by its degradation (14), it is thought to mainly increase gluconeogenesis mediated by the deacetylation and activation of peroxisome proliferator-activated receptor gamma coactivator 1 (PGC-1) (15) and forkhead box protein O1 (FOXO1) (16). In addition, SIRT1 deacetylates STAT3, resulting in the stimulation of gluconeogenesis through the inhibition of STAT3 activity (17).
In this study, we examined the involvement of SIRT1 in the regulation of GH-dependent IGF-I production in the liver, and explored the mechanisms underlying this involvement.
Results
SIRT1 Modulates IGF-I Production in the Liver Under Fasting Conditions.
To explore the involvement of SIRT1 in the regulation of IGF-I under fasting conditions, we knocked down the SIRT1 protein in mice using an antisense oligonucleotide in the liver (Fig. S1 A and B). Serum IGF-I levels decreased significantly after these mice were subjected to 48-h fasting,; however, knockdown of SIRT1 in the liver partially restored this decrease (Fig. 1A). mRNA expression levels of IGF-I in the liver under fasting conditions were also restored by SIRT1 knockdown (Fig. 1B). We also examined representative target genes of GH in the liver. The expression levels of igfbp3 and socs2 were up-regulated after SIRT1 knockdown (Fig. 1C), suggesting that SIRT1 modulates GH action in the liver.
Fig. 1.

SIRT1 regulates GH-dependent IGF-I production in vivo. (A) Knockdown of SIRT1 in the liver restored the decrement of serum IGF-I levels under 48-h fasting. (B and C) Samples obtained after 48-h fasting. (B) Expression of IGF-I in the liver was increased by knockdown of SIRT1. (C) Expression of igfbp3 and SOCS2, regulated by GH, was increased by knockdown of SIRT1. (D) Knockdown of SIRT1 in the liver enhanced a GH-dependent increase in serum IGF-I levels in hypophysectomized mice under 48-h fasting. *P < 0.05; **P < 0.01. N.S., not significant.
We then investigated the effect of SIRT1 knockdown on the induction of IGF-I by GH stimulation under fasting conditions using hypophysectomized mice, considered an appropriate model in which to observe the GH-induced increase in serum IGF-I levels in vivo (18) (Fig. S1 C–E). In the control mice, serum IGF-I levels were increased after GH stimulation (Fig. 1D), as described previously (18). Intriguingly, knockdown of SIRT1 significantly enhanced the GH-induced elevation of serum IGF-I levels (Fig. 1D). These data suggest that SIRT1 negatively regulates GH-induced IGF-I production in the liver.
SIRT1 Negatively Regulates GH-Induced IGF-I mRNA Production in Hepatocytes.
To explore the involvement of SIRT1 in GH-dependent IGF-I production in hepatocytes, we next examined the effects of activation or inhibition of SIRT1 on GH-induced IGF-I mRNA production in HepG2 cells (a human hepatocellular carcinoma cell line) and rat primary hepatocytes. Overexpression of WT SIRT1 suppressed GH-induced IGF-I mRNA production, whereas dominant-negative (DN) SIRT1 (H363Y, a catalytically inactive mutant) enhanced IGF-I mRNA production in HepG2 cells, indicating that SIRT1 negatively regulates GH-dependent IGF-I production in HepG2 cells (Fig. 2A).
Fig. 2.

Involvement of SIRT1 in GH-induced IGF-I mRNA production and Tyr phosphorylation of STAT5. The effect of the changes in SIRT1 activity on GH-dependent IGF-I mRNA production was evaluated in human hepatocellular carcinoma cell line HepG2 and primary rat hepatocytes. (A) Overexpression of WT SIRT1 or DN SIRT1 (H363Y mutant) in HepG2 cells. Cells were stimulated with GH (500 ng/mL) for 6 h. IGF-1 mRNA expression levels were measured using quantitative real-time PCR and normalized to those of β-actin. Data are mean ± SEM of six samples per group, expressed relative to the control group. *P < 0.05; **P < 0.01. (B) Knockdown of SIRT1 by siRNA in primary rat hepatocytes. Cells were stimulated with GH (500 ng/mL) for 24 h. (C–I) GH-stimulated Tyr phosphorylation of STAT5 was analyzed by immunoblotting. HepG2 cells and primary rat hepatocytes were stimulated with GH for 15 min, after which the lysate was subjected to immunoblot analysis. (C and D) Pretreatment of SIRT1 activator (10 mM NAD for 30 min) and overexpression of DN SIRT1 in GHR-expressed HepG2 cells. (E and F) Pretreatment of NAD or SIRT1 inhibitor with 10 mM nicotinamide for 6 h. (G–I) Knockdown of SIRT1 by siRNA in primary rat hepatocytes. Phospho-STAT5 signals were normalized to total STAT5. Data are mean ± SEM. n = 4 per group. *P < 0.05; **P < 0.01 versus the control group at the same time point. N.S., not significant.
We also investigated the role of endogenous SIRT1 in rat primary hepatocytes. Consistent with the foregoing results, knockdown of SIRT1 enhanced GH-induced IGF-I mRNA production in rat primary hepatocytes (Fig. 2B). Treatment with SIRT1 stimulators (NAD and resveratrol) (Fig. S2A) or inhibitors (sirtinol and nicotinamide) (Fig. S2B) in HepG2 cells and rat primary hepatocytes (Fig. S2C), and knockdown of SIRT1 in HepG2 cells (Fig. S2D), showed compatible results. These results clearly indicate that SIRT1 negatively regulates GH-induced IGF-I mRNA production in hepatocytes.
SIRT1 Negatively Regulates GH-Induced Tyr Phosphorylation on STAT5.
To clarify the underlying mechanisms of SIRT1 regulation, we analyzed GH-induced Tyr phosphorylation on STAT5 in hepatocytes. Exogenous NAD treatment has been shown to activate SIRT1 by increasing the intracellular NAD/NADH ratio in vitro (19). We found that treatment with NAD inhibited GH-dependent STAT5 phosphorylation in HepG2 cells (Fig. 2 C and D). Importantly, overexpression of dominant negative SIRT1 restored this decrease in STAT5 phosphorylation, indicating that the effect of NAD treatment depends on the endogenous SIRT1 activity.
Whereas treatment with NAD inhibited GH-dependent STAT5 phosphorylation, treatment with the SIRT1 inhibitors nicotinamide (Fig. 2 E and F) and sirtinol (Fig. S3 A and B) enhanced STAT5 phosphorylation in rat primary hepatocytes in a dose-dependent manner. Treatment with resveratrol suppressed GH-dependent STAT5 phosphorylation (Fig. S3C), overexpression of DN SIRT1 enhanced it, and WT SIRT1 inhibited it (Fig. S3D). In addition, knockdown of SIRT1 in rat primary hepatocytes (Fig. 2 G and H) led to significant enhancement in GH-induced Tyr phosphorylation on STAT5 (Fig. 2 H and I). These results indicate that SIRT1 negatively regulates GH-induced Tyr phosphorylation on STAT5.
Fasting-Induced GH Resistance Is Restored by Nicotinamide Treatment.
We next investigated whether GH resistance under fasting conditions could be restored by treatment with nicotinamide, a SIRT1 inhibitor, in vivo. The protein level and activity of SIRT1 protein reportedly increase under fasting conditions in the liver (15, 20). In our mice under fasting conditions, SIRT1 protein levels were increased (Fig. 3 A and B) and the association between SIRT1 and STAT5 was enhanced (Fig. 3A). Simultaneously, acetylation of lysine (Lys) on STAT5 decreased (Fig. 3 A and C), and GH-induced Tyr phosphorylation was inhibited (Fig. 3 A and D) compared with mice under fed conditions. However, when the SIRT1 inhibitor nicotinamide was administered to mice under fasting conditions, although the interaction between SIRT1 and STAT5 remained unchanged (Fig. 3A), Lys acetylation of STAT5 was restored to the levels measured under fed conditions (Fig. 3 A and C), and STAT5 Tyr phosphorylation in response to GH stimulation was restored as well (Fig. 3 A and D). In contrast to STAT5, fasting did not affect Lys acetylation levels in GHR and JAK2 (Fig. S4). Tyr phosphorylation levels of JAK2 (Fig. 3 A and E) and ERK (Fig. 3 A and F) were unchanged under fasting conditions and nicotinamide administration, suggesting that SIRT1 specifically affects the Lys acetylation and Tyr phosphorylation status of STAT5 in GHR signaling.
Fig. 3.
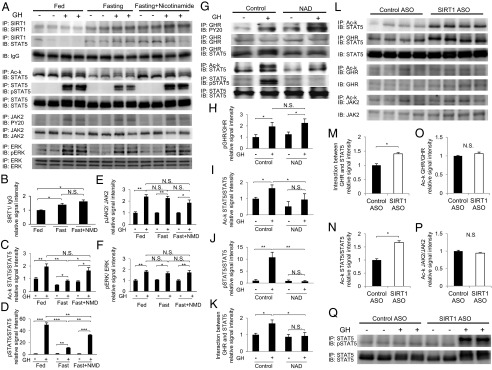
SIRT1 regulates acetylation and GH-induced Tyr phosphorylation of STAT5 in vivo. (A) The SIRT1 inhibitor nicotinamide reversed the blunted response of GH signaling under fasting conditions. Male C57BL/6J mice were treated with nicotinamide during fasting (150 mg/kg, i.p.). After 24 h, GH (3 mg/kg, i.p.) was administered; 15 min later, mice were killed, and liver protein was extracted and analyzed by immunoblotting. (B–F) Quantitative analysis was performed by scanning densitometry. The levels of acetylated and phosphorylated protein were normalized to those of total protein. (G) Treatment with NAD decreased Lys acetylation of STAT5 and inhibited GH-induced Tyr phosphorylation of STAT5 under fed conditions. Male C57BL/6J mice were treated with NAD (5 mg/kg, i.p.). After 15 min, GH (3 mg/kg, i.p.) was administered for an additional 15 min, after which the mice were killed and liver protein was extracted and analyzed by immunoblotting. (H–K) Quantitative analysis was performed by scanning densitometry. (L–Q) Knockdown of SIRT1 in mice increased the interaction between STAT5 and GHR (M), concomitant with increased Lys acetylation of STAT5 (N). There were no changes in Lys acetylation of GHR (O) or JAK2 (P). Knockdown of SIRT1 in hypophysectomized mice markedly enhanced the blunted response of Tyr phosphorylation in STAT5 by GH under fasting conditions (Q). After 48-h fasting, GH (3 mg/kg, i.p.) was administered for 15 min, after which the mice were killed and liver protein was extracted and analyzed by immunoblotting (Fig. S1 A and B). Quantitative analysis was performed by scanning densitometry. Data are mean ± SEM. n = 4 per group. *P < 0.05; **P < 0.01; ***P < 0.005. N.S., not significant.
NAD Treatment Inhibits GH-Induced Interaction of GHR and STAT5, as well as Tyr Phosphorylation of STAT5, Under Fed Conditions.
We next investigated the effect of NAD administration on the activation of SIRT1 in the liver under fed conditions in vivo. Although Tyr phosphorylation of GHR by GH stimulation was not affected by NAD treatment (Fig. 3 G and H), Lys acetylation of STAT5 was significantly decreased (Fig. 3 G and I), corresponding to a reduction in GH-induced Tyr phosphorylation on STAT5 (Fig. 3 G and J). Furthermore, the GH-induced interaction between GHR and STAT5 was significantly decreased by NAD treatment (Fig. 3 G and K). These data demonstrate that treatment with NAD resembles the effect of fasting in terms of changes in GH signaling. These data also reveal that the Lys acetylation status of STAT5 and the interaction of GHR and STAT5 are closely associated with GH-induced Tyr phosphorylation on STAT5 in vivo. Because NAD treatment may activate not only SIRT1, but also other NAD-dependent enzymes, we further investigated the specificity of SIRT1 in vivo.
SIRT1 Negatively Regulates STAT5 Activation by GH in Vivo Under Fasting Conditions.
We examined the effects of SIRT1 knockdown on the activation of STAT5 in vivo (Fig. S1B). Knockdown of SIRT1 increased the interaction between GHR and STAT5 (Fig. 3 L and M), concomitant with increased Lys acetylation on STAT5 (Fig. 3 L and N). In contrast, Lys acetylation on GHR and JAK2 was unchanged by knockdown of SIRT1 (Fig. 3 L, O, and P). Tyr phosphorylation on STAT5 normally induced by GH was not detected in hypophysectomized SIRT1 knockdown mice, demonstrating a blunted response to GH under fasting conditions, as described previously (21); however, GH-induced Tyr phosphorylation of STAT5 was markedly enhanced when SIRT1 was knocked down (Fig. 3Q).
SIRT1 Interacts Directly with STAT5 and Deacetylates Lys Residues on STAT5.
To investigate the direct interaction of SIRT1 and STAT5, we performed GST pull-down assays in vitro. As shown in Fig. 4A, the GST-SIRT1 fusion protein and the in vitro-translated FLAG-STAT5B protein exhibited direct interaction. To determine the region in STAT5B responsible for binding SIRT1, we generated a set of 3 V5-tagged STAT5B deletion mutants (T1, T2, and T3) that were designed to test each domain in STAT5B (Fig. 4B). Immunoprecipitation analysis showed that the N-terminal domain, coiled-coil domain, and DNA-binding domain of STAT5B were necessary for the interaction between STAT5B and SIRT1 (Fig. 4B).
Fig. 4.

Protein–protein interaction between SIRT1 and STAT5B. (A) GST-SIRT1 fusion protein interacted with FLAG-tagged STAT5B. (Upper) Coomassie brilliant blue staining of GST fusion proteins. (Lower) Immunoblot of STAT5B. (B) SIRT1 interacts with the N-terminal, coiled-coil, and DNA-binding domains of STAT5B. Truncated mutants of V5-tagged STAT5B and FLAG-tagged SIRT1 cDNA were transfected and analyzed for interactions by immunoprecipitation in HEK293 cells. ND, N-terminal domain; CCD, coiled-coil domain; DNA BD, DNA-binding domain; LD, linker domain; SH2D, Src homology 2 domain; TAD, transactivation domain. (C) SIRT1 interacts with and deacetylates STAT5B. WT or DN SIRT1 (H363Y) was expressed, and the interaction with STAT5B, as well as the acetylation status of STAT5B, were analyzed by immunoprecipitation.
We then explored whether SIRT1 deacetylates STAT5B in vitro. Although the overexpression of WT SIRT1 decreased the acetylation of Lys in STAT5B, overexpression of DN SIRT1 did not affect the level of acetylated Lys in STAT5B irrespective of the interaction of SIRT1 and STAT5B (Fig. 4C), demonstrating that the deacetylation enzymatic activity of SIRT1 is not necessary for this interaction. These results indicate that SIRT1 deacetylates STAT5B by direct interaction.
GH-Dependent Tyr Phosphorylation Relies on the Acetylation of Lys Residues Adjacent to the SH2 Domain of STAT5B.
It was recently reported that activation of STAT3 requires the acetylation of its Lys residues (22). Furthermore, prolactin-induced Tyr phosphorylation of STAT5 requires STAT5 acetylation. In particular, the acetylation of Lys residues (681K, 694K, 701K, and 705K) adjacent to Tyr-699 are important for STAT5 activation by prolactin (23). To investigate the significance of the STAT5B Lys residues neighboring Y699 and the SH2 domain for the activation of STAT5B, we generated Lys mutants of STAT5B and analyzed their functional properties in vitro (Fig. 5A). Interestingly, Lys acetylation in WT STAT5B increased in parallel with Tyr phosphorylation after GH treatment (Fig. 5B). In contrast, the Lys mutants showed significantly decreased Lys acetylation compared with WT (Fig. 5 B and C). Furthermore, GH-induced Tyr phosphorylation in STAT5B was substantially impaired concomitant with the decrease in Lys acetylation (Fig. 5 B and C). In particular, the K681R mutant displayed significantly reduced Tyr phosphorylation, suggesting that this Lys residue is critical for STAT5B activation by GH.
Fig. 5.

Lys residues adjacent to the SH2 domain in STAT5B are critical for GH-dependent STAT5B Tyr phosphorylation and transcriptional activity. (A) Locations of Lys residues that were converted to arginine residue by site-directed mutagenesis in STAT5B. (B and C) HEK293 cells transfected with GHR cDNA and WT or Lys mutant (K681R, K694R, K701R, or K705R) STAT5 cDNAs were stimulated with 500 ng/mL GH for 30 min, after which the lysates were analyzed by immunoblotting. The values of acetylated and phosphorylated STAT5B signals were normalized to those of total STAT5. (D) HEK293 cells were transfected with GHR cDNA, WT, or Lys mutants of STAT5B cDNAs and a pLHRE-luciferase reporter, followed by treatment with 500 ng/mL GH for 24 h before measurement of luciferase activity. Data are mean ± SEM. n = 4 per group. *P < 0.05; **P < 0.01; ***P < 0.005. N.S., not significant. (E and F) The mechanisms through which SIRT1 regulates STAT5 activation by GH. (E) In the fed condition, the SH2 domain of STAT5 recognizes and binds to Tyr-phosphorylated GHR, causing JAK2 to phosphorylate and activate STAT5. (F) In the fasting condition, SIRT1 is activated and interacts with STAT5, thereby deacetylating Lys residues adjacent to the SH2 domain of STAT5. This results in an impaired ability to bind Tyr-phosphorylated GHR, which inhibits activation of STAT5.
We further examined the effects of mutating these Lys residues on transcriptional activity using a STAT5B-responsive lactogenic hormone-responsive element (pLHRE) reporter gene. As expected, the Lys mutants of STAT5B showed impaired transcriptional activity that corresponded with the levels of Lys acetylation and Tyr phosphorylation (Fig. 5D). These results clearly demonstrate that Lys acetylation is critical for the activation of STAT5B by GH stimulation.
Discussion
In this study, we have shown that SIRT1 negatively regulates GH-induced Tyr phosphorylation on STAT5 in hepatocytes in vitro and in the liver in vivo. In both cases, this inhibition resulted in reduced GH-induced IGF-I production, indicating the GH resistance status. GH resistance caused by malnutrition or fasting is considered an adaptive response that blunts growth and conserves energy for survival. Under fasting conditions, the reduced serum IGF-I level impairs growth and decreases insulin sensitivity, whereas elevated levels of GH mobilize free fatty acids from adipose tissue and play an important role in preventing hypoglycemia by inducing insulin resistance (24, 25). Sirtuins have emerged as important sensors of energy status and modulators for adaptation in mammals (26). In particular, SIRT1 is a regulator of gluconeogenesis and lipolysis during fasting. In the context of preventing hypoglycemia during fasting, it is reasonable that activated SIRT1 negatively regulates GH-dependent IGF-I production in the liver as an adaptive mechanism.
Several animal models are available for analyzing the role of SIRT1 in the liver. Because SIRT1 activity is tightly regulated by energy intake and this regulation is a relatively acute survival response to malnutrition, we chose this model using knockdown rather than liver-specific KO mice, which is a chronic SIRT1-deficient model. Indeed, knockdown models have been used to demonstrate that SIRT1 is involved in the regulation of gluconeogenesis (17) and lipid metabolic response in the liver (27). On the other hand, using liver-specific KO mice, Chen et al. (28) found that SIRT1 might not be functional in the liver on CR. Although the precise mechanism responsible for this discrepancy is unknown, it may be related to the difference in the condition of fasting or CR, or to some compensatory mechanisms for the chronic absence of SIRT1 in the liver.
A previous study suggested that inhibition of GH-induced IGF-I production during fasting is caused by decreased insulin concentration in the portal vein, which reduces GHR expression (29) and translocation (11) in the liver. We examined the effects of SIRT1 on GHR expression, but found no effect of SIRT1 level on GHR mRNA level (Fig. S5). Another study reported that fasting induced a reduction in phosphorylated GHR level with no change in GHR protein level (21), suggesting that GH resistance occurs as a result of the GHR status; however, in this study, SIRT1 had no effect on GHR protein levels and the phosphorylation of GHR by GH, but had a strong direct affect on STAT5 activity, suggesting that STAT5 is a primary regulatory target under fasting conditions. It was recently reported that FGF21, an adaptive hormone for starvation, inhibits STAT5 signaling and blunts growth, leading to GH resistance (12). Given that FGF21 activates SIRT1 by increasing the NAD/NADH ratio (30), FGF21 also may inhibit STAT5 mediated by SIRT1 activation. Another report demonstrated that SIRT1 activators contribute to the suppression of T-cell proliferation by down-regulating STAT5A/B expression and suppressing pSTAT5A/B signaling in response to IL-2 (31). Taken together, these data indicate that although GH signaling is modulated at various levels under fasting conditions, SIRT1 specifically regulates STAT5 activity, and this underlying mechanism plays an important role in the physiological adaptive response.
GST pull-down assays and experiments using WT and DN SIRT1 demonstrated that SIRT1 interacts directly with STAT5 and deacetylates its Lys residues. Our data also show that STAT5 acetylation is associated with a GH-induced interaction between STAT5 and GHR and is necessary for the transcriptional activity of STAT5. In contrast to STAT5, the activity of SIRT1 did not affect the acetylation or GH-induced Tyr phosphorylation of GHR and JAK2. These data suggest the following model of SIRT1 involvement in GH signaling. Under fasting conditions, activated SIRT1 interacts with and deacetylates STAT5. The deacetylized STAT5 has a reduced ability to interact with phosphorylated Tyr residues in GHR and thereby decrease its phosphorylation and effect on transcriptional activity (Fig. 5 E and F), indicating that SIRT1 negatively regulates the GH-dependent recruitment of STAT5 to GHR through the deacetylation of STAT5. Lys mutant analysis of STAT5B has demonstrated that K681 is the most critical mutant for the acetylation and Tyr phosphorylation of STAT5B. This mutant resides in the hydrophilic region neighboring the SH2 domain that is essential for the interaction with GHR (23). Interestingly, STAT3, which stimulates gluconeogenesis in the liver, is known to be regulated by SIRT1 via the deacetylation of several critical Lys residues in STAT3 (17). Together with our data, the Lys acetylation status of STAT proteins modulated by SIRT1 may integrate energy status and modulate hormone and cytokine signaling for this adaptation.
SIRT1 modifies the GH–IGF-I axis through various mechanisms. Brain-specific SIRT1 deletion in mice impairs GH secretion owing to alterations in hypothalamic function (32), suggesting that SIRT1 in the hypothalamus regulates GH secretion. As our findings demonstrate, SIRT1 also regulates IGF-I production in the liver, thereby reducing the serum concentration of IGF-I. Moreover, IGF-binding protein-1 levels in the liver are significantly increased in SIRT1 KO mice, reducing the bioavailability of IGF-I (33). Given that both increased GH secretion and decreased serum IGF-I levels are necessary for adaptation under fasting conditions, SIRT1 in the brain and liver may cooperatively regulate the GH–IGF-I axis in these conditions. On the other hand, SIRT1 directly deacetylates and activates insulin receptor substrate (IRS)-2 in H4IIE cells (34), and SIRT1 inhibition reduces IGF-I/IRS-2/Ras/ERK1/2 signaling in neurons (35). These results suggest that SIRT1 positively regulates IGF-I action. Overall, it appears that SIRT1 modulates the action of IGF-I in a tissue- and context-dependent manner.
More interestingly, both the GH–IGF-I axis and SIRT1 are essentially involved in regulation of the lifespan. In rodents, SIRT1 is required for lifespan extension by CR (35), whereas reduced GH/IGF-I signaling is associated with an extended lifespan in various species (36). Although several mechanisms have been proposed to explain the role of SIRT1 in lifespan regulation, the crosstalk between SIRT1 and the GH–IGF-I axis in the liver may contribute to the CR-mediated extension of the lifespan.
In conclusion, this study provides evidence that SIRT1 negatively regulates GH-induced IGF-I production via deacetylation of STAT5 in the liver, leading to GH resistance under fasting conditions. Furthermore, our results imply a pathway through which SIRT1 and GH/IGF-I signaling jointly regulate various metabolic processes in the adaptation to starvation and regulation of the lifespan.
Materials and Methods
Cell culture, primary hepatocyte isolation, and plasmid and siRNA transfection are described in SI Materials and Methods. SIRT1 knockdown in vivo was performed as described previously (17). Protocols for antibodies, immunoprecipitation, immunoblot analysis, GST pull-down experiments, luciferase reporter gene assay, quantitative real-time PCR, measurement of serum IGF-I levels by ELISA, and animal experiments are described in SI Materials and Methods.
Statistical Analysis.
All data are reported as mean ± SEM. Statistical analyses were performed using Student’s t test for comparison of two groups and one-way ANOVA with Scheffé’s F test for multiple group comparisons. A P value < 0.05 was considered statistically significant. Representative results from at least three independent experiments are shown unless stated otherwise.
Supplementary Material
Acknowledgments
We thank Drs. K. Chihara, Y. Okimura, H. Kaji, and A. E. Handayaningshi for their support and valuable suggestions and C. Ogata and K. Imura for their excellent technical assistance. This work was supported in part by Grants-in-Aid for Scientific Research (B) 18390276 and 20390262 and Grant-in-Aid for Scientific Research on Innovative Areas 20200079 from the Ministry of Education, Culture, Sports, Science, and Technology of Japan; by the Health and Labor Science Research Grants program; and by the Foundation for Growth Science.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. R.d.C. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1220606110/-/DCSupplemental.
References
- 1.Cahill GF., Jr Starvation in man. N Engl J Med. 1970;282(12):668–675. doi: 10.1056/NEJM197003192821209. [DOI] [PubMed] [Google Scholar]
- 2.Lawson EA, Klibanski A. Endocrine abnormalities in anorexia nervosa. Nat Clin Pract Endocrinol Metab. 2008;4(7):407–414. doi: 10.1038/ncpendmet0872. [DOI] [PubMed] [Google Scholar]
- 3.Thissen JP, Pucilowska JB, Underwood LE. Differential regulation of insulin-like growth factor I (IGF-I) and IGF-binding protein-1 messenger ribonucleic acids by amino acid availability and growth hormone in rat hepatocyte primary culture. Endocrinology. 1994;134(3):1570–1576. doi: 10.1210/endo.134.3.7509741. [DOI] [PubMed] [Google Scholar]
- 4.Thissen JP, Ketelslegers JM, Underwood LE. Nutritional regulation of the insulin-like growth factors. Endocr Rev. 1994;15(1):80–101. doi: 10.1210/edrv-15-1-80. [DOI] [PubMed] [Google Scholar]
- 5.Merimee TJ, Zapf J, Froesch ER. Insulin-like growth factors in the fed and fasted states. J Clin Endocrinol Metab. 1982;55(5):999–1002. doi: 10.1210/jcem-55-5-999. [DOI] [PubMed] [Google Scholar]
- 6.Uyttendaele I, et al. Mammalian protein–protein interaction trap (MAPPIT) analysis of STAT5, CIS, and SOCS2 interactions with the growth hormone receptor. Mol Endocrinol. 2007;21(11):2821–2831. doi: 10.1210/me.2006-0541. [DOI] [PubMed] [Google Scholar]
- 7.Rotwein P. Mapping the growth hormone–Stat5b–IGF-I transcriptional circuit. Trends Endocrinol Metab. 2012;23(4):186–193. doi: 10.1016/j.tem.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bornfeldt KE, Arnqvist HJ, Enberg B, Mathews LS, Norstedt G. Regulation of insulin-like growth factor-I and growth hormone receptor gene expression by diabetes and nutritional state in rat tissues. J Endocrinol. 1989;122(3):651–656. doi: 10.1677/joe.0.1220651. [DOI] [PubMed] [Google Scholar]
- 9.Yakar S, et al. Normal growth and development in the absence of hepatic insulin-like growth factor I. Proc Natl Acad Sci USA. 1999;96(13):7324–7329. doi: 10.1073/pnas.96.13.7324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baxter RC, Bryson JM, Turtle JR. Somatogenic receptors of rat liver: Regulation by insulin. Endocrinology. 1980;107(4):1176–1181. doi: 10.1210/endo-107-4-1176. [DOI] [PubMed] [Google Scholar]
- 11.Leung KC, Doyle N, Ballesteros M, Waters MJ, Ho KK. Insulin regulation of human hepatic growth hormone receptors: Divergent effects on biosynthesis and surface translocation. J Clin Endocrinol Metab. 2000;85(12):4712–4720. doi: 10.1210/jcem.85.12.7017. [DOI] [PubMed] [Google Scholar]
- 12.Inagaki T, et al. Inhibition of growth hormone signaling by the fasting-induced hormone FGF21. Cell Metab. 2008;8(1):77–83. doi: 10.1016/j.cmet.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gillum MP, Erion DM, Shulman GI. Sirtuin-1 regulation of mammalian metabolism. Trends Mol Med. 2010;17(1):8–13. doi: 10.1016/j.molmed.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu Y, et al. A fasting inducible switch modulates gluconeogenesis via activator/coactivator exchange. Nature. 2008;456(7219):269–273. doi: 10.1038/nature07349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rodgers JT, et al. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434(7029):113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 16.Frescas D, Valenti L, Accili D. Nuclear trapping of the forkhead transcription factor FoxO1 via Sirt-dependent deacetylation promotes expression of glucogenetic genes. J Biol Chem. 2005;280(21):20589–20595. doi: 10.1074/jbc.M412357200. [DOI] [PubMed] [Google Scholar]
- 17.Nie Y, et al. STAT3 inhibition of gluconeogenesis is downregulated by SirT1. Nat Cell Biol. 2009;11(4):492–500. doi: 10.1038/ncb1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sondergaard M, Dagnaes-Hansen F, Flyvbjerg A, Jensen TG. Normalization of growth in hypophysectomized mice using hydrodynamic transfer of the human growth hormone gene. Am J Physiol Endocrinol Metab. 2003;285(2):E427–E432. doi: 10.1152/ajpendo.00573.2002. [DOI] [PubMed] [Google Scholar]
- 19.Zhuo L, et al. NAD blocks high glucose-induced mesangial hypertrophy via activation of the sirtuins-AMPK-mTOR pathway. Cell Physiol Biochem. 2011;27(6):681–690. doi: 10.1159/000330077. [DOI] [PubMed] [Google Scholar]
- 20.Nemoto S, Fergusson MM, Finkel T. Nutrient availability regulates SIRT1 through a forkhead-dependent pathway. Science. 2004;306(5704):2105–2108. doi: 10.1126/science.1101731. [DOI] [PubMed] [Google Scholar]
- 21.Beauloye V, et al. Impairment of liver GH receptor signaling by fasting. Endocrinology. 2002;143(3):792–800. doi: 10.1210/endo.143.3.8692. [DOI] [PubMed] [Google Scholar]
- 22.Yuan ZL, Guan YJ, Chatterjee D, Chin YE. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science. 2005;307(5707):269–273. doi: 10.1126/science.1105166. [DOI] [PubMed] [Google Scholar]
- 23.Ma L, et al. Acetylation modulates prolactin receptor dimerization. Proc Natl Acad Sci USA. 2010;107(45):19314–19319. doi: 10.1073/pnas.1010253107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao TJ, et al. Ghrelin O-acyltransferase (GOAT) is essential for growth hormone-mediated survival of calorie-restricted mice. Proc Natl Acad Sci USA. 2010;107(16):7467–7472. doi: 10.1073/pnas.1002271107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nass RM, Gaylinn BD, Rogol AD, Thorner MO. Ghrelin and growth hormone: Story in reverse. Proc Natl Acad Sci USA. 2010;107(19):8501–8502. doi: 10.1073/pnas.1002941107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chalkiadaki A, Guarente L. Sirtuins mediate mammalian metabolic responses to nutrient availability. Nat Rev Endocrinol. 2012;8(5):287–296. doi: 10.1038/nrendo.2011.225. [DOI] [PubMed] [Google Scholar]
- 27.Rodgers JT, Puigserver P. Fasting-dependent glucose and lipid metabolic response through hepatic sirtuin 1. Proc Natl Acad Sci USA. 2007;104(31):12861–12866. doi: 10.1073/pnas.0702509104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen D, et al. Tissue-specific regulation of SIRT1 by calorie restriction. Genes Dev. 2008;22(13):1753–1757. doi: 10.1101/gad.1650608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Straus DS, Takemoto CD. Effect of fasting on insulin-like growth factor-I (IGF-I) and growth hormone receptor mRNA levels and IGF-I gene transcription in rat liver. Mol Endocrinol. 1990;4(1):91–100. doi: 10.1210/mend-4-1-91. [DOI] [PubMed] [Google Scholar]
- 30.Chau MD, Gao J, Yang Q, Wu Z, Gromada J. Fibroblast growth factor 21 regulates energy metabolism by activating the AMPK-SIRT1-PGC-1alpha pathway. Proc Natl Acad Sci USA. 2010;107(28):12553–12558. doi: 10.1073/pnas.1006962107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gardner PJ, et al. SIRT1 activation protects against autoimmune T cell-driven retinal disease in mice via inhibition of IL-2/Stat5 signaling. J Autoimmun. 2013;42:117–129. doi: 10.1016/j.jaut.2013.01.011. [DOI] [PubMed] [Google Scholar]
- 32.Cohen DE, Supinski AM, Bonkowski MS, Donmez G, Guarente LP. Neuronal SIRT1 regulates endocrine and behavioral responses to calorie restriction. Genes Dev. 2009;23(24):2812–2817. doi: 10.1101/gad.1839209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li H, et al. SirT1 modulates the estrogen–insulin-like growth factor-1 signaling for postnatal development of mammary gland in mice. Breast Cancer Res. 2007;9(1):R1. doi: 10.1186/bcr1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang J. The direct involvement of SirT1 in insulin-induced insulin receptor substrate-2 tyrosine phosphorylation. J Biol Chem. 2007;282(47):34356–34364. doi: 10.1074/jbc.M706644200. [DOI] [PubMed] [Google Scholar]
- 35.Li Y, Xu W, McBurney MW, Longo VD. SirT1 inhibition reduces IGF-I/IRS-2/Ras/ERK1/2 signaling and protects neurons. Cell Metab. 2008;8(1):38–48. doi: 10.1016/j.cmet.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Berryman DE, Christiansen JS, Johannsson G, Thorner MO, Kopchick JJ. Role of the GH/IGF-1 axis in lifespan and healthspan: Lessons from animal models. Growth Horm IGF Res. 2008;18(6):455–471. doi: 10.1016/j.ghir.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.