

Significance
Nitric oxide from endothelial cells is important for regulating cardiovascular function. Recent data suggest that red blood cells are a source for nitric oxide. We examined the function of red blood cell nitric oxide and the regulation of its formation by the enzyme arginase. We show that red blood cells contain arginase that inhibits nitric oxide export. The function was tested in a heart model subjected to ischemia. The recovery of heart function after ischemia was improved following inhibition of red blood cell arginase. This effect depended on the specific protein producing nitric oxide. The results demonstrate an important function by red blood cells in regulating heart function during ischemia via nitric oxide production under tight control by arginase.
Keywords: ischemia, reperfusion, infarction, erythrocyte
Abstract
The theory that red blood cells (RBCs) generate and release nitric oxide (NO)-like bioactivity has gained considerable interest. However, it remains unclear whether it can be produced by endothelial NO synthase (eNOS), which is present in RBCs, and whether NO can escape scavenging by hemoglobin. The aim of this study was to test the hypothesis that arginase reciprocally controls NO formation in RBCs by competition with eNOS for their common substrate arginine and that RBC-derived NO is functionally active following arginase blockade. We show that rodent and human RBCs contain functional arginase 1 and that pharmacological inhibition of arginase increases export of eNOS-derived nitrogen oxides from RBCs under basal conditions. The functional importance was tested in an ex vivo model of myocardial ischemia-reperfusion injury. Inhibitors of arginase significantly improved postischemic functional recovery in rat hearts if administered in whole blood or with RBCs in plasma. By contrast, arginase inhibition did not improve postischemic recovery when administered with buffer solution or plasma alone. The protective effect of arginase inhibition was lost in the presence of a NOS inhibitor. Moreover, hearts from eNOS−/− mice were protected when the arginase inhibitor was given with blood from wild-type donors. In contrast, when hearts from wild-type mice were given blood from eNOS−/− mice, the arginase inhibitor failed to protect against ischemia-reperfusion. These results strongly support the notion that RBCs contain functional eNOS and release NO-like bioactivity. This process is under tight control by arginase 1 and is of functional importance during ischemia-reperfusion.
Nitric oxide (NO) is a biological messenger that is a key regulator of cardiovascular function by inducing vasodilation, inhibition of platelet aggregation, and leukocyte adhesion (1). Reduced bioavailability of endothelium-derived NO is closely associated with development of several cardiovascular diseases including atherosclerosis, ischemia-reperfusion injury, and hypertension. The vascular effects of NO have traditionally been considered to be mediated by endothelium-derived NO after formation by the constitutively expressed endothelial NO synthase (eNOS). An alternative source of NO is nitrite that can be converted to NO in cardiac tissue during ischemia or hypoxia (2–4). In 1996, Stamler and colleagues suggested a role for red blood cells (RBCs) in exporting NO bioactivity and regulating blood flow (5). In this model, RBCs contain NO in the form of S-nitrosylated hemoglobin, which is in equilibrium with small nitrosothiols that are exported preferentially under deoxygenated conditions (5, 6). RBCs thereby provide NO-based vasodilatory activity through S-nitrosothiols when deoxygenated. It was also suggested that the source of RBC NO is eNOS (5). However, it was assumed that eNOS was exclusively vascular in origin, and mechanisms regulating RBC formation and export of NO bioactivity have been a matter of significant debate over the years (7). Another mechanism for NO generation by RBCs has been proposed in which deoxygenated hemoglobin converts inorganic nitrite to NO followed by export of NO bioactivity (8). More recently, RBCs were shown to contain eNOS protein (9–12), but it remains controversial whether RBC eNOS is functional and whether significant amounts of NO is formed and exported as a result of eNOS activity partly because hemoglobin in the RBC effectively scavenges NO and, thereby, limits export and functional effect of NO (13, 14).
Human RBCs also express the enzyme arginase (15). Endothelial cell arginase has emerged as an important regulator of NO production by competing with eNOS for their common substrate l-arginine (16, 17). Thus, increased arginase activity induced by reactive oxygen species, proinflammatory cytokines, and hypoxia may limit the pool of l-arginine available for NO production in endothelial cells (16). Accordingly, inhibition of arginase increases endothelium-derived NO formation and improves endothelium-dependent vasodilatation in animal models and patients with coronary artery disease (17–19). Furthermore, inhibition of arginase reduces myocardial infarct size in both a rat and pig model of coronary artery ligation and reperfusion in vivo (20, 21). The effect of arginase inhibition was blocked by a NOS inhibitor and an NO scavenger, demonstrating that it was mediated via enhanced NO formation due to a shift in the metabolism of arginine from arginase to NOS.
The role of arginase in RBCs is unknown. We hypothesized that the high levels of arginase expressed under basal physiological conditions in RBCs regulates NO production. The present study was therefore designed to investigate the regulatory role of arginase on release of NO bioactivity from RBCs and the functional effect of this release by using an isolated heart model of ischemia-reperfusion known to be responsive to enhanced bioavailability of NO.
Results
Functionally Active Arginase 1 Is Expressed in Rodent and Human RBCs.
Earlier studies have detected arginase 1 expression in human RBCs, but the general notion is that this enzyme is lacking in rodents (15). Using Western blot analysis, we demonstrate arginase 1 protein in RBCs from rats, mice, and humans (Fig. 1A). In addition, arginase activity was detected in rat RBCs, and this activity was effectively inhibited by the arginase inhibitor Nω-hydroxy-nor-l-arginine (nor-NOHA; Fig. 1B). In contrast, arginase 2 could not be detected in RBCs of the species tested but was present in renal tissue (Fig. 1A) as predicted from earlier studies (22).
Fig. 1.

Arginase expression and activity in RBCs. (A) Western blot of arginase 1 expression in human, rat, and mouse RBCs and rat liver (positive control) and arginase 2 expression in RBCs and rat kidney (positive control). (B) Arginase activity in rat RBCs (n = 3) following incubation with vehicle or the arginase inhibitor nor-NOHA. Data are shown as mean ± SEM. Significant differences between treatments are shown; *P < 0.05 (t test).
Arginase Inhibition Increases Nitrate and Nitrite Release from RBCs.
To study RBC nitrogen oxide export, we incubated fresh washed RBCs from rodents and humans as well as human blood with different concentrations of nor-NOHA, and the extracellular accumulation of nitrate and nitrite was measured. Nor-NOHA caused a dose-dependent increase in nitrate as well as nitrite both in the presence and absence of l-arginine supplementation (Fig. 2). The increase in nitrate induced by nor-NOHA was prevented by Nω-nitro-l-arginine methyl ester (l-NAME) (Fig. 2C). To further study whether the nitrate accumulation was due to eNOS activity, we compared RBC nitrogen oxide export from eNOS−/− and wild-type mice. nor-NOHA induced an increase in nitrate in the RBC medium from wild-type mice but not from eNOS−/− mice (Fig. 2D).
Fig. 2.
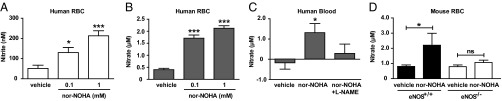
Export of nitrogen oxides (nitrite and nitrate) from human RBCs (n = 6; A and B) following incubation with vehicle or nor-NOHA in the presence of l-arginine. Nitrate in human blood (n = 9; C) incubated with vehicle, nor-NOHA, or nor-NOHA + l-NAME in the absence of l-arginine. Nitrate export from RBCs of wild-type and eNOS−/− mice (n = 5; D) following incubation with vehicle or nor-NOHA with l-arginine. Data are shown as mean ± SEM. Significant differences from vehicle treatments are shown; *P < 0.05, **P < 0.01, ***P < 0.001 (one-way ANOVA).
The Cardioprotective Effect of Arginase Inhibition Depends on RBCs.
An ex vivo myocardial ischemia-reperfusion injury model was used to test the functional effects of NO bioactivity exported from RBCs. Previous studies have shown that increase in NO bioavailability is highly protective during ischemia reperfusion (23–25). The first set of experiments was performed by using vehicle and nor-NOHA (0.1–1 mM) in Krebs–Henseleit (KH) buffer. Administration of nor-NOHA in buffer at the onset of ischemia did not affect recovery of left ventricular developed pressure (LVDP) or left ventricular end-diastolic pressure (LVEDP) during reperfusion in comparison with vehicle (Fig. 3).
Fig. 3.

Effect of vehicle (n = 9) and nor-NOHA (1 mM; n = 7) on recovery of LVDP and LVEDP. Isolated and Langendorff-perfused rat hearts were subjected to 25 min of global ischemia followed by 60 min of reperfusion. Vehicle or nor-NOHA was administered in KH buffer at the onset of ischemia. LVDP is expressed as the percentage recovery from preischemia and LVEDP in absolute pressure. Data are shown as mean ± SEM. There was no significant difference between the treatments (two-way ANOVA).
In contrast, when nor-NOHA (1 mM) was administered in whole blood, significant improvements in the postischemic recovery of LVDP, the positive first derivative of LV pressure (dP/dt), and coronary flow were observed (Fig. 4). Furthermore, LVEDP during reperfusion was significantly lower in hearts given nor-NOHA in blood in comparison with those given vehicle in blood (Fig. 4). Comparable improvement in improved postischemic LV systolic and diastolic function could be reproduced with another structurally unrelated arginase inhibitor, 2(S)-amino-6-boronohexanoic acid (ABH; 1 mM), administered in blood (Fig. 5).
Fig. 4.

Effect of vehicle (n = 10) and nor-NOHA (1 mM; n = 16) on recovery of LVDP, LVEDP, dP/dt, and coronary flow (CF) of rat hearts during reperfusion following 25 min of global ischemia. Vehicle and nor-NOHA was administered in blood at the onset of ischemia. LVDP, dP/dt, and CF are expressed as percentage recovery from preischemia and LVEDP in absolute pressure. Data are shown as mean ± SEM. Significant differences between treatments are shown (two-way ANOVA).
Fig. 5.

Effect of vehicle (n = 5) and ABH (1 mM; n = 6) on recovery of LVDP and LVEDP. Vehicle or ABH was administered in blood at the onset of ischemia. LVDP is expressed as percentage recovery from preischemia and LVEDP in absolute pressure. Data are shown as mean ± SEM. Significant differences between treatments are shown (two-way ANOVA).
To clarify the role of RBCs in the cardiac protection induced by arginase inhibition, buffy coat was removed, leaving RBCs and plasma as solvent for nor-NOHA. In the presence of RBC and plasma, nor-NOHA markedly enhanced LVDP and reduced LVEDP following ischemia (Fig. 6A). By contrast, when nor-NOHA was dissolved in plasma alone (without RBCs), it failed to improve postischemic myocardial function in comparison with vehicle (Fig. 6B). Taken together, these observations clearly illustrate that the cardioprotective effect of arginase inhibition depends on the presence of RBCs.
Fig. 6.

Effect of vehicle and nor-NOHA (1 mM) on recovery of LVDP and LVEDP in rat hearts during reperfusion following 25 min of global ischemia. Vehicle and nor-NOHA were administered in RBC + plasma (n = 8; A) or plasma only (n = 7; B) at the onset of ischemia. LVDP is expressed as percentage recovery from preischemia and LVEDP in absolute pressure. Data are shown as mean ± SEM. Significant differences between treatments are shown (two-way ANOVA).
Cardioprotective Effect of Arginase Inhibition Depends on eNOS.
Next, the involvement of NOS in the cardioprotective effect of nor-NOHA in blood was investigated by administration of l-NAME with blood. l-NAME alone did not affect postischemic functional recovery per se, but it abolished the improvement in LVDP and LVEDP induced by nor-NOHA (Fig. 7). To investigate whether the cardioprotective effect of arginase inhibition involved the eNOS isoform, experiments were performed in eNOS+/+ and eNOS−/− mice. Nor-NOHA (1 mM) administered in blood from eNOS+/+ mice improved postischemic functional recovery in eNOS+/+ hearts (Fig. 8A). Also, when nor-NOHA in eNOS+/+ blood was given to hearts from eNOS−/− mice, a significant improvement in LV function during recovery was observed (Fig. 8B). By contrast, nor-NOHA in eNOS−/− blood failed to affect postischemic LV systolic or diastolic function in eNOS+/+ hearts in comparison with vehicle (Fig. 8C). These experiments demonstrate the pivotal role of RBC eNOS in cardioprotection afforded by arginase inhibition.
Fig. 7.

Effect of the nitric oxide synthase inhibitor l-NAME on cardioprotection induced by arginase inhibition in rat hearts. Recovery of LVDP and LVEDP during reperfusion is shown. l-NAME (0.1 mM) or vehicle was added in blood 5 min before nor-NOHA (1 mM) or vehicle and administered at the onset of ischemia. Data are shown as mean ± SEM (n = 6–16). Significance denotes difference between the group given nor-NOHA + vehicle and the other groups (two-way ANOVA).
Fig. 8.

Pivotal role of eNOS in blood for cardioprotective effect of arginase inhibition using mouse hearts. Isolated eNOS+/+ and eNOS−/− mouse hearts were subjected to 45 min of global ischemia and 60 min of reperfusion in a Langendorff system. Vehicle or nor-NOHA (1 mM) was given in blood from eNOS+/+ and eNOS−/− at the onset of ischemia. Functional recoveries of LVDP and LVEDP during reperfusion are shown. (A) Nor-NOHA (n = 7) or vehicle (n = 5) in eNOS+/+ blood was administrated to eNOS+/+ hearts. (B) nor-NOHA (n = 6) or vehicle (n = 6) in eNOS+/+ blood was administrated to eNOS−/− hearts. (C) nor-NOHA (n = 6) or vehicle (n = 8) in eNOS−/− blood was administrated to eNOS+/+ hearts. Data are shown as mean ± SEM. Significant differences between treatments are shown (two-way ANOVA).
Discussion
Here we show that RBCs are capable of generating and exporting NO bioactivity that exerts functional effect in a target tissue. The NO bioactivity is generated by a RBC eNOS whose activity is under strict control by arginase 1 that is expressed in both human and rodent RBCs. Inhibition of arginase activity in RBCs induces release of nitrogen oxides. Moreover, arginase inhibition protects from myocardial ischemia-reperfusion injury via a mechanism that depends on RBC-derived eNOS activity. These observations provide evidence for an important mechanism by which arginase 1 exerts a tonic inhibitory effect on NO production by eNOS in RBCs and demonstrate functional eNOS activity and NO export from RBCs.
Previous studies have demonstrated that RBCs contain NO and chemically related species, but the capability of these cells to export NO has been debated and the issue has remained controversial. Stamler and coworkers (5) originally suggested that RBCs can export NO bioactivity that contributes to the regulation of blood flow. Furthermore, RBCs may carry eNOS protein, but it has remained controversial whether the enzyme is functionally active and whether RBC-derived NO can escape the scavenging by hemoglobin (10, 11). Because it is known that arginase is able to regulate NO production in endothelial cells by competition with eNOS for their common substrate, we tested the hypothesis that arginase regulates NO production in RBCs. We show that both rat and mouse RBCs express a functional arginase 1 protein and that inhibition of arginase results in export of nitrogen oxides and NO bioactivity from RBCs via an eNOS-dependent effect. Because RBCs were found to express only arginase 1, it can be concluded that this isoform is responsible for the functional effect of arginase in the present study. The tight control of eNOS by arginase may explain previous difficulties in demonstrating functional eNOS activity and NO export from RBCs.
The functional relevance of the arginase-regulated NO production in RBCs was evaluated in a model of myocardial ischemia-reperfusion. It is well documented that NO plays a key role in protection from ischemia-reperfusion injury (23–25). Our data clearly show that RBCs were required for the protection induced by nor-NOHA because it was lost when administered in plasma or buffer. The cardioprotective effect of arginase inhibition in blood was blocked by l-NAME, demonstrating dependence on NOS activity. The crucial role of RBC eNOS was revealed in experiments using blood from eNOS-deficient mice given to wild-type recipient hearts. In these experiments, the cardioprotective effects of nor-NOHA were again abolished. In contrast, hearts from eNOS-deficient mice were protected if nor-NOHA was given with blood from wild-type mice. This latter example also illustrates that eNOS in cardiac tissue and blood vessels is not required for the protection. Taken together, these series of experiments provide strong functional evidence for eNOS-derived NO export from RBCs mediating protection from ischemia-reperfusion injury. Furthermore, this effect of RBC eNOS is tightly regulated by arginase.
In the present study, we did not attempt to address the exact mechanism for RBC nitrogen oxide generation, handling, and export. Thus, we cannot tell whether eNOS directly releases NO from the RBC or whether intermediates such as S-nitrosothiols (5) or nitrite (8) are formed inside the RBC and are transported between the RBC and the target tissue. Previous studies have demonstrated that S-nitrosothiols derived from RBCs induce coronary dilatation (26), which may suggest a role of S-nitrosothiols. Nitrite may also be involved because it has been shown to be reduced to NO in the ischemic myocardium and to protect against ischemia-reperfusion injury (2, 4). Moreover, nitrite may form NO and S-nitrosothiols inside the RBC (8, 27, 28). Whether S-nitrosothiols, nitrite, or free NO are involved in eNOS-derived cardioprotective effects of RBCs needs to be clarified in future studies. At this stage, our data demonstrate cardioprotective NO bioactivity from RBC eNOS, which is under strict control by arginase. The suggestion that NO, derived from a RBC eNOS, is able to exert important biological effects in adjacent cells is in line with recent observations. Thus, NO released from RBCs suppresses platelet activation and aggregation (11) and induces dilatation of mesenteric arteries (12). Recently, it was further suggested that NO derived from RBC eNOS is involved in blood pressure regulation in vivo (29). These observations clearly suggest that the increased NO production in RBCs may translate into tissue protective effects. Of additional importance is the fact that NO increases the deformability of RBCs, which is crucial for the adequate passage of RBCs through the microvasculature (11). A remaining key question will be to further elucidate the importance of arginase as a regulator of RBC NO formation and export under normal physiological conditions. It is tempting to speculate that mechanisms exist in vivo for rapid dynamic control of arginase activity, NO export, and control of vascular function. It would also be of importance to evaluate the involvement of RBC arginase in regulation and control of NO export in cardiovascular disease including hypertension, atherosclerosis, and diabetes. Recent data demonstrate that arginase inhibition improves endothelial function in patients with coronary artery disease and diabetes (19). However, it is unknown whether this vasculoprotective effect involves RBC arginase. The present findings are also of interest in the context of the observed association between blood transfusion and increased risk of myocardial infarction and mortality following myocardial infarction (30). It is tempting to speculate that reduced RBC NO bioactivity and increased arginase activity in stored RBCs may contribute to this observation (31–33).
Certain limitations with the present study deserve commenting. At this stage, we cannot entirely rule out the possibility that RBC eNOS controls the release of another factor unrelated to NO that, in turn, mediates the cardioprotective effects. However, our earlier in vivo studies demonstrating that the cardioprotective effects of arginase inhibition were abolished not only by a NOS inhibitor but also by coadministration of an NO scavenger (21) argues against such effect. It should also be emphasized that these data were obtained in an ex vivo model and any extrapolation to the in vivo situation should be done with some caution.
In conclusion, the present study demonstrates an essential role of arginase 1 in control of eNOS function in RBCs. Inhibition of arginase unravels an important functional effect of RBC-derived NO that mediates protection against myocardial ischemia-reperfusion injury. These observations provide evidence for an important regulatory role of RBCs in control of vascular NO bioactivity with physiological and pathophysiological consequences in ischemia-reperfusion.
Materials and Methods
The study was approved by the regional ethical committee for animal experiments in Stockholm and conforms to the Guide for Care and Use of Laboratory Animals published by the National Institutes of Health (Publication 85–23, revised 1996). Wistar rats (10 wk old) were purchased from Charles River, and eNOS−/− mice and their wild-type controls (C57/6BL 12 wk old) were purchased from Jackson Laboratory.
Heart Isolation and Perfusion.
Hearts were isolated from rats or mice and perfused in a Langendorff system as described (34, 35). Briefly, the animals were anesthetized (i.p. in mice, i.m. in rats) with fentanyl (1 mg⋅kg−1) + fluanisone (Hypnorm; Janssen Pharmaceutica; 50 mg⋅kg−1) and midazolam (Dormicum; Hoffman-La Roche; 25 mg⋅kg−1). Heparin (250 IU) was injected i.p. in mice and i.v. in rats. The hearts were excised and placed in ice-cold buffer. The ascending aorta was cannulated, and retrogradely perfused with gassed (5% CO2 in O2) KH buffer (118.5 mM NaCl, 25.0 mM NaHCO3, 4.7 mM KCl, 1.2 mM KH2PO4, 1.2 mM MgSO4, 11.1 mM glucose, and 2.4 mM CaCl2) at a constant pressure (55 mmHg and 75 mmHg for mouse and rat hearts, respectively) at 37 °C. A balloon connected to a pressure transducer was inserted into the left ventricle through the left atrium for recording of isovolumetric LVDP, and its positive first derivative dP/dt and LVEDP. The balloon was adjusted to give a baseline LVEDP of 6 mmHg.
Experimental Protocol.
After start of perfusion, all hearts were allowed to stabilize for at least 25 min, after which baseline parameters were registered. Global ischemia was induced by clamping the inflow tubing. The duration of global ischemia was 25 min for rat hearts and 45 min for mouse hearts. Reperfusion was initiated by releasing the clamp and was maintained for 60 min. At the start of ischemia, vehicle (KH buffer) or the arginase inhibitors nor-NOHA (0.1–1 mM; Bachem) or ABH (1 mM; kind gift from Corridor Pharmaceuticals) was injected into the coronary circulation via a sidearm in the perfusion system. The arginase inhibitors were initially dissolved in 0.9% NaCl and stored frozen until used in the experiments. In separate experiments, vehicle and nor-NOHA were diluted in KH buffer, whole blood, or RBCs + plasma before being administered to the hearts. ABH was administered in blood only. The NOS inhibitor l-NAME (0.1 mM; Sigma Aldrich) was diluted in blood 5 min before addition of nor-NOHA or KH buffer. All solutions were incubated at 37 °C for 25 min and were administered in volumes of 3 mL to rat hearts and 0.4 mL to mouse hearts.
Blood Sampling and Isolation of Blood Components.
Whole blood was collected from the thoracic cavity after removal of the heart used in the experiment. In experiments using eNOS+/+ and eNOS −/− mice, one heart from each genotype was used in parallel experiments by using blood from the opposite genotype. Blood components were separated by immediate centrifugation at 2,000 × g for 10 min at +4 °C. RBCs + plasma was obtained by removing the buffy coat including some plasma and top part of RBC layer after which the remaining RBCs and plasma were gently mixed. Plasma was obtained by collection after centrifugation. RBCs were obtained by discarding the buffy coat and plasma. Successful removal of white blood cells (>99%) and platelets (≥98%) was verified by differential cell counting. RBCs were either used immediately or stored frozen at −80 °C for determination of arginase protein expression and activity.
In separate experiments, RBCs were isolated from healthy volunteers (men age 45 ± 4 y) and anesthetized mice. Heparinized blood (20 mL of human, 1 mL of mouse) were centrifuged at 2,000 × g (human) or 500 × g (mouse) and +4 °C for 5 min. The RBCs were washed three times with 2 volumes of nitrite/nitrate-free KH buffer containing 3 mM l-arginine. After the washing step, the same volume of l-arginine-containing KH buffer was added to the RBCs and gently mixed and incubated with nor-NOHA (0.1–1 mM) at 37 °C for 30 min. The samples were then centrifuged at 2,000 × g and 4 °C for 10 min and the supernatants were stored at -80 °C until analyzed.
Additional blood from healthy volunteers (men age 40 ± 3 y) was diluted (1:5) in nitrite/nitrate-free PBS. The diluted blood from each donor was treated with PBS (vehicle), nor-NOHA (3 mM), or l-NAME (3 mM) + nor-NOHA. The treated blood samples were harvested at 0 (baseline) and 30 min. The samples were centrifuged at 800 × g and 4 °C for 30 min, and the supernatants were stored at −80 °C until analyzed. The changes in nitrate concentrations from baseline to the 30-min time point were determined.
Western Blot.
RBCs from mice and rats were lysed by using RIPA lysis buffer (Amresco) containing protease inhibitors (Roche). The protocol of Western blot was described (20, 36). Briefly, proteins were separated by 12% SDS/PAGE, transferred to nitrocellulose membrane, and blocked in 5% nonfat dried milk for 2 h at room temperature. Membranes were incubated overnight at 4 °C by using rabbit anti-arginase 1 (1:2,000; HPA003595, Atlas, Sigma). Membranes were then washed in Tris-buffered saline with 0.1% Tween 20 and incubated with IRDye 800-conjugated goat anti-rabbit IgG (1:15,000; LI-COR Biosciences), and imunoreactive bands were visualized by using infrared fluorescence (IR-Odyssey scanner; LI-COR Biosciences).
Arginase Activity.
Rat RBCs were lysed by using fresh 1 mM EDTA, Trion X-100 (0.1%, MERCK, Darmstadt, Gemany) and protease inhibitors (Roche) in PBS. Arginase activity was determined as described in detail (20, 37). Each sample was incubated at 37 °C for 1 h with either l-arginine (50 mM Tris⋅HCl at pH 9.7) + vehicle, l-arginine + ABH (0.1 mM; Enzo Clinical Labs) or with l-arginine + nor-NOHA (1 mM). The concentration of the end product urea was determined by using spectrophotometry. The inhibition induced by arginase inhibitor was calculated as the difference between the urea production from vehicle-treated and inhibitor-treated samples.
Nitrate Determination.
Nitrate and nitrite were measured with a dedicated HPLCy (HPLC) system (ENO-20; EiCom) as described (38).
Calculations and Statistics.
LVDP and dP/dt during reperfusion are expressed in percentage of the preischemic values. LVEDP is expressed in absolute values. The differences in functional cardiac parameters were analyzed by two-way ANOVA. Differences in nitrate/nitrite production were analyzed by one-way ANOVA. Differences in arginase activity were analyzed by Student’s t test. All statistical analysis was calculated by using GraphPad Prism (V5.01).
Acknowledgments
We thank Pellina Jonsson, Marita Wallin, and Carina Nihlén for technical assistance and Prof. Malte Kelm and Miriam Cortese-Krott (University of Düsseldorf) for helpful technical advice on how to measure NOx release from human RBCs. This work was supported by Swedish Research Council Grant 10857 and grants from the Swedish Heart and Lung Foundation, the European Foundation for the Study of Diabetes, the Stockholm County Council (ALF), Karolinska Institutet/Stockholm County Council Strategic Cardiovascular Programme, the King Gustav V and Queen Victoria Foundation, the Torsten Söderbergs Foundation, and the Novo Nordisk Foundation.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
References
- 1.Moncada S, Higgs A. The L-arginine-nitric oxide pathway. N Engl J Med. 1993;329(27):2002–2012. doi: 10.1056/NEJM199312303292706. [DOI] [PubMed] [Google Scholar]
- 2.Duranski MR, et al. Cytoprotective effects of nitrite during in vivo ischemia-reperfusion of the heart and liver. J Clin Invest. 2005;115(5):1232–1240. doi: 10.1172/JCI22493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lundberg JO, Weitzberg E. NO-synthase independent NO generation in mammals. Biochem Biophys Res Commun. 2010;396(1):39–45. doi: 10.1016/j.bbrc.2010.02.136. [DOI] [PubMed] [Google Scholar]
- 4.Webb A, et al. Reduction of nitrite to nitric oxide during ischemia protects against myocardial ischemia-reperfusion damage. Proc Natl Acad Sci USA. 2004;101(37):13683–13688. doi: 10.1073/pnas.0402927101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jia L, Bonaventura C, Bonaventura J, Stamler JS. S-nitrosohaemoglobin: A dynamic activity of blood involved in vascular control. Nature. 1996;380(6571):221–226. doi: 10.1038/380221a0. [DOI] [PubMed] [Google Scholar]
- 6.Doctor A, et al. Hemoglobin conformation couples erythrocyte S-nitrosothiol content to O2 gradients. Proc Natl Acad Sci USA. 2005;102(16):5709–5714. doi: 10.1073/pnas.0407490102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gladwin MT, Lancaster JR, Jr, Freeman BA, Schechter AN. Nitric oxide’s reactions with hemoglobin: A view through the SNO-storm. Nat Med. 2003;9(5):496–500. doi: 10.1038/nm0503-496. [DOI] [PubMed] [Google Scholar]
- 8.Cosby K, et al. Nitrite reduction to nitric oxide by deoxyhemoglobin vasodilates the human circulation. Nat Med. 2003;9(12):1498–1505. doi: 10.1038/nm954. [DOI] [PubMed] [Google Scholar]
- 9.Cortese-Krott MM, et al. Human red blood cells at work: Identification and visualization of erythrocytic eNOS activity in health and disease. Blood. 2012;120(20):4229–4237. doi: 10.1182/blood-2012-07-442277. [DOI] [PubMed] [Google Scholar]
- 10.Kang ES, et al. Normal circulating adult human red blood cells contain inactive NOS proteins. J Lab Clin Med. 2000;135(6):444–451. doi: 10.1067/mlc.2000.106805. [DOI] [PubMed] [Google Scholar]
- 11.Kleinbongard P, et al. Red blood cells express a functional endothelial nitric oxide synthase. Blood. 2006;107(7):2943–2951. doi: 10.1182/blood-2005-10-3992. [DOI] [PubMed] [Google Scholar]
- 12. Ulker P, Gunduz F, Meiselman HJ, Baskurt OK (October 17, 2012) Nitric oxide generated by red blood cells following exposure to shear stress dilates isolated small mesenteric arteries under hypoxic conditions. Clin Hemorheol Microcirc, 10.3233/CH-2012-1618. [DOI] [PubMed]
- 13.Hobbs AJ, Gladwin MT, Patel RP, Williams DL, Butler AR. Haemoglobin: NO transporter, NO inactivator or NOne of the above? Trends Pharmacol Sci. 2002;23(9):406–411. doi: 10.1016/s0165-6147(02)02067-9. [DOI] [PubMed] [Google Scholar]
- 14.Joshi MS, et al. Nitric oxide is consumed, rather than conserved, by reaction with oxyhemoglobin under physiological conditions. Proc Natl Acad Sci USA. 2002;99(16):10341–10346. doi: 10.1073/pnas.152149699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spector EB, Rice SC, Kern RM, Hendrickson R, Cederbaum SD. Comparison of arginase activity in red blood cells of lower mammals, primates, and man: Evolution to high activity in primates. Am J Hum Genet. 1985;37(6):1138–1145. [PMC free article] [PubMed] [Google Scholar]
- 16.Durante W, Johnson FK, Johnson RA. Arginase: A critical regulator of nitric oxide synthesis and vascular function. Clin Exp Pharmacol Physiol. 2007;34(9):906–911. doi: 10.1111/j.1440-1681.2007.04638.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pernow J, Jung C. Arginase as a potential target in the treatment of cardiovascular disease: Reversal of arginine steal? Cardiovasc Res. 2013;98(3):334–343. doi: 10.1093/cvr/cvt036. [DOI] [PubMed] [Google Scholar]
- 18.Ryoo S, et al. Endothelial arginase II: A novel target for the treatment of atherosclerosis. Circ Res. 2008;102(8):923–932. doi: 10.1161/CIRCRESAHA.107.169573. [DOI] [PubMed] [Google Scholar]
- 19.Shemyakin A, et al. Arginase inhibition improves endothelial function in patients with coronary artery disease and type 2 diabetes mellitus. Circulation. 2012;126(25):2943–2950. doi: 10.1161/CIRCULATIONAHA.112.140335. [DOI] [PubMed] [Google Scholar]
- 20.Gonon AT, et al. Local arginase inhibition during early reperfusion mediates cardioprotection via increased nitric oxide production. PLoS ONE. 2012;7(7):e42038. doi: 10.1371/journal.pone.0042038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jung C, Gonon AT, Sjöquist PO, Lundberg JO, Pernow J. Arginase inhibition mediates cardioprotection during ischaemia-reperfusion. Cardiovasc Res. 2010;85(1):147–154. doi: 10.1093/cvr/cvp303. [DOI] [PubMed] [Google Scholar]
- 22.Levillain O. Expression and function of arginine-producing and consuming-enzymes in the kidney. Amino Acids. 2012;42(4):1237–1252. doi: 10.1007/s00726-011-0897-z. [DOI] [PubMed] [Google Scholar]
- 23.Jones SP, Bolli R. The ubiquitous role of nitric oxide in cardioprotection. J Mol Cell Cardiol. 2006;40(1):16–23. doi: 10.1016/j.yjmcc.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 24.Vinten-Johansen J, et al. Nitric oxide and the vascular endothelium in myocardial ischemia-reperfusion injury. Ann N Y Acad Sci. 1999;874:354–370. doi: 10.1111/j.1749-6632.1999.tb09251.x. [DOI] [PubMed] [Google Scholar]
- 25.Heusch G, Boengler K, Schulz R. Cardioprotection: Nitric oxide, protein kinases, and mitochondria. Circulation. 2008;118(19):1915–1919. doi: 10.1161/CIRCULATIONAHA.108.805242. [DOI] [PubMed] [Google Scholar]
- 26.Reynolds JD, et al. S-nitrosohemoglobin deficiency: A mechanism for loss of physiological activity in banked blood. Proc Natl Acad Sci USA. 2007;104(43):17058–17062. doi: 10.1073/pnas.0707958104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Angelo M, Singel DJ, Stamler JS. An S-nitrosothiol (SNO) synthase function of hemoglobin that utilizes nitrite as a substrate. Proc Natl Acad Sci USA. 2006;103(22):8366–8371. doi: 10.1073/pnas.0600942103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Basu S, et al. Catalytic generation of N2O3 by the concerted nitrite reductase and anhydrase activity of hemoglobin. Nat Chem Biol. 2007;3(12):785–794. doi: 10.1038/nchembio.2007.46. [DOI] [PubMed] [Google Scholar]
- 29.Wood KC, et al. Circulating blood endothelial nitric oxide synthase contributes to the regulation of systemic blood pressure and nitrite homeostasis. Arterioscler Thromb Vasc Biol. 2013;33(8):1861–1871. doi: 10.1161/ATVBAHA.112.301068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chatterjee S, Wetterslev J, Sharma A, Lichstein E, Mukherjee D. Association of blood transfusion with increased mortality in myocardial infarction: A meta-analysis and diversity-adjusted study sequential analysis. JAMA Intern Med. 2013;173(2):132–139. doi: 10.1001/2013.jamainternmed.1001. [DOI] [PubMed] [Google Scholar]
- 31.Bennett-Guerrero E, et al. Evolution of adverse changes in stored RBCs. Proc Natl Acad Sci USA. 2007;104(43):17063–17068. doi: 10.1073/pnas.0708160104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mollinedo F, et al. (May 29, 2013) Arginase as a new concern in blood transfusion. Blood Transfus, 10.2450/2013.0237-12. [DOI] [PMC free article] [PubMed]
- 33.Sherwood MW, Rao SV. Acute coronary syndromes: Blood transfusion in patients with acute MI and anaemia. Nat Rev Cardiol. 2013;10(4):186–187. doi: 10.1038/nrcardio.2013.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang JN, et al. Sex differences in mouse heart rate and body temperature and in their regulation by adenosine A1 receptors. Acta Physiol (Oxf) 2007;190(1):63–75. doi: 10.1111/j.1365-201X.2007.01690.x. [DOI] [PubMed] [Google Scholar]
- 35.Tähepõld P, Vaage J, Starkopf J, Valen G. Hyperoxia elicits myocardial protection through a nuclear factor kappaB-dependent mechanism in the rat heart. J Thorac Cardiovasc Surg. 2003;125(3):650–660. doi: 10.1067/mtc.2003.36. [DOI] [PubMed] [Google Scholar]
- 36.Gonon AT, et al. Adiponectin protects against myocardial ischaemia-reperfusion injury via AMP-activated protein kinase, Akt, and nitric oxide. Cardiovasc Res. 2008;78(1):116–122. doi: 10.1093/cvr/cvn017. [DOI] [PubMed] [Google Scholar]
- 37.Berkowitz DE, et al. Arginase reciprocally regulates nitric oxide synthase activity and contributes to endothelial dysfunction in aging blood vessels. Circulation. 2003;108(16):2000–2006. doi: 10.1161/01.CIR.0000092948.04444.C7. [DOI] [PubMed] [Google Scholar]
- 38.Jansson EA, et al. A mammalian functional nitrate reductase that regulates nitrite and nitric oxide homeostasis. Nat Chem Biol. 2008;4(7):411–417. doi: 10.1038/nchembio.92. [DOI] [PubMed] [Google Scholar]