

Significance
This study presents a unique approach (CopraRNA, for Comparative Prediction Algorithm for sRNA Targets) towards reliably predicting the targets of bacterial small regulatory RNAs (sRNAs). These molecules are important regulators of gene expression. Their detailed analysis thus far has been hampered by the lack of reliable algorithms to predict their mRNA targets. CopraRNA integrates phylogenetic information to predict sRNA targets at the genomic scale, reconstructs regulatory networks upon functional enrichment and network analysis, and predicts the sRNA domains for target recognition and interaction. Our results demonstrate that CopraRNA substantially improves the bioinformatic prediction of target genes and opens the field for the application to nonmodel bacteria.
Keywords: regulatory RNA, E. coli, RNA–RNA interaction
Abstract
Small RNAs (sRNAs) constitute a large and heterogeneous class of bacterial gene expression regulators. Much like eukaryotic microRNAs, these sRNAs typically target multiple mRNAs through short seed pairing, thereby acting as global posttranscriptional regulators. In some bacteria, evidence for hundreds to possibly more than 1,000 different sRNAs has been obtained by transcriptome sequencing. However, the experimental identification of possible targets and, therefore, their confirmation as functional regulators of gene expression has remained laborious. Here, we present a strategy that integrates phylogenetic information to predict sRNA targets at the genomic scale and reconstructs regulatory networks upon functional enrichment and network analysis (CopraRNA, for Comparative Prediction Algorithm for sRNA Targets). Furthermore, CopraRNA precisely predicts the sRNA domains for target recognition and interaction. When applied to several model sRNAs, CopraRNA revealed additional targets and functions for the sRNAs CyaR, FnrS, RybB, RyhB, SgrS, and Spot42. Moreover, the mRNAs gdhA, lrp, marA, nagZ, ptsI, sdhA, and yobF-cspC were suggested as regulatory hubs targeted by up to seven different sRNAs. The verification of many previously undetected targets by CopraRNA, even for extensively investigated sRNAs, demonstrates its advantages and shows that CopraRNA-based analyses can compete with experimental target prediction approaches. A Web interface allows high-confidence target prediction and efficient classification of bacterial sRNAs.
Small RNAs (sRNAs) are ubiquitous and important regulators of gene expression in bacteria. The most common and best investigated trans-acting sRNAs regulate their targets posttranscriptionally by RNA–RNA interactions, often depending on the RNA chaperone Hfq (1). Individual functions of model sRNAs have been discovered primarily through extensive experimental work and may be assigned to many different stress responses and signal transduction pathways, covering virtually all aspects of bacterial growth (1, 2) and virulence (3). One of the most intriguing conceptual advances has been the identification of sRNAs as posttranscriptional regulators that act globally within complex regulatory networks. Examples for such sRNAs are GcvB, which is a major regulator of amino acid metabolism and directly controls ∼1% of all Salmonella enterica mRNAs (4); MicA and RybB, which together constitute the repressor arm of the Sigma E response (5); and Spot42, a global regulator of catabolite repression (6). With the advent of high-throughput sequencing and comprehensive transcriptome analysis techniques, increasing numbers of new sRNAs have been detected in bacteria belonging to diverse taxa (7, 8). However, the experimental testing and verification of sRNA targets is costly, labor intensive, and may be challenging, even in model organisms. Moreover, for most environmentally and biotechnologically relevant microbes, experimental verification is hindered further by the lack of systems for their genetic manipulation.
The reliable computational prediction of sRNA targets promises a great reduction of required wet-laboratory analyses while enabling large-scale sRNA–mRNA network analyses in genetically intractable species. However, reliable in silico prediction of mRNA targets has been challenging because of the extreme heterogeneity of sRNAs in size, structure, and the typically short and imperfect sRNA–target complementarity (9). The existing tools for the genome-scale prediction of sRNA targets evaluate the strength of a particular sRNA–target interaction by either base pair complementarity (10) or thermodynamic models (11–13). The latter are built on the observed exponential correlation between repression strength and hybridization free energy (14), which can be corrected by an energy term that reflects the accessibility of the interaction sites (11, 12). However, despite continuous improvement of target prediction methods (15), even the most accurate methods integrating interaction site accessibility scoring and additional features, such as seed regions, produce many false positives and, thus, compromise the selection of putative targets for subsequent experimental investigation (16, 17).
Furthermore, the implementation of seed sequence conservation to improve sRNA target prediction has been difficult to achieve for bacterial systems because of the great flexibility of the interaction patterns (16). It is conceivable that the interaction is preserved while the actual interaction site is not. Therefore, to predict conserved interactions, it is necessary to combine evidence for interactions in different species without resorting to a consensus interaction-based approach.
Here, we introduce a computational approach that uses phylogenetic information from an extended model of sRNA–target evolution (CopraRNA, for Comparative Prediction Algorithm for sRNA Targets). CopraRNA depends solely on the conservation of target genes (i.e., conservation of target regulation) and does not require conservation of specific interaction sequences (SI Appendix, Figs. S1 and S2).
By introducing a generic approach combining predictions for homologous targets in distinct organisms, we reduced the hitherto existing high false positive rate (FPR) of single-organism target prediction. Using this strategy, CopraRNA matches microarray-based experimental sRNA target prediction with respect to the number of correctly identified direct targets (Fig. 1B and Table 1) and the characterization of physiological functions of these sRNAs. Thus, it constitutes a significant improvement of in silico sRNA target prediction and enables competitive and functional large-scale initial screening for sRNA targets without experimental effort and costs. Application of CopraRNA to previously characterized sRNAs proposed and partially verified additional targets and functions for the sRNAs cyclic AMP activated sRNA (CyaR), FNR regulated sRNA (FnrS), RybB, RyhB, sugar transport-related sRNA (SgrS), and Spot42. Also, it suggested the gdhA, lrp, marA, nagZ, ptsI, sdhA, and yobF-cspC mRNAs as hubs targeted by up to seven different sRNAs. A Web interface for CopraRNA has been set up under http://rna.informatik.uni-freiburg.de/CopraRNA/.
Fig. 1.

(A) Schematic overview of the CopraRNA pipeline. (B) Comparison of CopraRNA predictions with microarray results and other target prediction methods. Genome-wide target predictions for 18 sRNAs in E. coli and S. enterica with 101 experimentally verified targets from the literature. The plot shows the number of correctly predicted targets (true positive predictions, y axis) vs. the number of target predictions per sRNA (x axis) for our comparative method CopraRNA and the existing single-organism–based methods IntaRNA, TargetRNA, and RNApredator. The results, including the verifications from this study, are shown with solid lines, and the results based on the benchmark set only are demarcated with a dashed line. (Inset) total numbers of independently verified targets detected by either CopraRNA (46 targets) or microarray experiments (49 targets) for the sRNAs CyaR, FnrS, GcvB, MicF, RyhB, SgrS, and Spot42; 25 targets were identified by both methods. The numbers refer to our benchmark dataset (SI Appendix, Table S1) and to the table comparing CopraRNA with different microarray experiments (Table 1). Visualization of the predicted interaction domains in GcvB (C) and the predicted mRNA targets of GcvB (D). The density plots at the top give the relative frequency of a specific sRNA or mRNA nucleotide position in the predicted sRNA–target interactions. The plots combine all predictions with a P value ≤0.01 in all included homologs. Local maxima indicate distinct interaction domains and are marked with upright lines. The schematic alignment of homologous sRNAs and targets at the bottom show the predicted interaction domains. The aligned regions are displayed in gray, gaps in white, and predicted interaction regions in color (color differences are for contrast only). The locus tag and gene name (if available) of a representative cluster member are given on the right.
Table 1.
Comparison of CopraRNA predictions and published microarray studies
| CopraRNA |
Microarray |
|||||||
| sRNA | No. of candidates (P ≤ 0.01) | No. of candidates after postprocessing* | No. verified† | No. sig. diff. expr. genes‡ | No. verified† | Ref. | No. overlap§ verified/unverified | Overlap genes¶ |
| CyaR | 69 | 55 | 1 + 3‖ | 24 genes | 4 | 18 | 1/1 | fepA, ompX |
| 1 gene | 1 | 19 | 1/0 | ompX | ||||
| FnrS | 67 | 41 | 3 + 4‖ | 16 genes/11 operons | 6 + 1‖ | 20 | 3/0 | marA, sodB, yobA |
| 31 genes | 7 + 1‖ | 21 | 4/2 | adhP, marA, sfcA/maeA, sodB, ydhD/grxD, yobA | ||||
| GcvB | 60 | 34 | 14 | 54 genes | 16 | 4 | 10/3 | argT, aroP, brnQ, cycA, dppA, gdhA, gltI, lrp, oppA, serA, sstT, trpE, yifK |
| MicF | 50 | 30 | 4 | 5 genes | 4 | 22 | 2/0 | lrp, ompF |
| RyhB | 70 | 37 | 2 + 5‖ | 56 genes/18 operons | 3 + 1‖ | 23 | 3/3 | frdA, fumA, msrB, sdhA, sdhD, sodB |
| SgrS | 66 | 35 | 2 + 1‖ | 6 genes | 4 | 24 | 2/0 | ptsG, yigL |
| Spot42 | 85 | 48 | 4 + 3‖ | 16 genes | 7 | 6 | 3/0 | galk, gltA, xylF |
The candidates after postprocessing for these sRNAs are given in Table S5.
Top 15 targets + automatically and manually functionally enriched.
Verified targets after postprocessing regarding the benchmark list (SI Appendix, Table S1), published data, and this study.
Significantly differentially expressed genes with regard to the respective publications.
Genes detected by prediction and microarray (independently verified/unverified).
Independently verified targets are in boldface.
Verified in this study.
Results
Prediction Strategy.
CopraRNA begins with a genome-wide target prediction (12) for each considered organism, as summarized in Fig. 1A. The interaction energies are fitted to a general extreme value distribution and transformed into P values to normalize for organism-specific GC-content and dinucleotide frequency. These P values are combined for orthologous genes into a single P value per conserved interaction. Orthologous genes are determined based on the respective amino acid sequences (25); genes that are present in less than 50% of the investigated genomes are discarded. Two aspects require specific normalization. First, CopraRNA normalizes for the degree of overall dependency to account for the nonindependent P values that result from the general sequence conservation between related organisms. Second, the individual dependencies have to be calculated because, in most cases, the considered organisms will not be equidistant from each other. Thus, we additionally used species-specific weights that were calculated based on 16S rDNA-based phylogenetic trees. The combination of the P values used a modified z-transform method, which permits adjustment for dependency in the data and a weighting based on the phylogenetic relationship (26). We defined significance thresholds either on CopraRNA P values or on q-values (27); the latter provide correction for multiple testing by controlling the false discovery rate (FDR). Both methods have proven useful for the analysis of the benchmark dataset. The chosen P value threshold of 0.01 allows for the detection of approximately half of all verified benchmark targets (SI Appendix, Fig. S3A) and was applied for the functional enrichment and network analysis. The q-value gives a measure of how many false positive predictions are expected in the group of targets called significant. True positives are all experimentally verified targets (with regard to our benchmark dataset in SI Appendix, Table S1) within the positive predictions, whereas false positives are all positive predictions that are no real targets, i.e., in our case, those that have not been verified experimentally. Positive predictions (also called candidates below) are all targets that match the respective threshold criterion (e.g., a P value ≤0.01 or a given rank); they consist of true positive and false positive predictions (statistical terms are defined also in SI Appendix). A reliable bioinformatic prediction tool for sRNA targets should not predict more than ∼50% of false positive targets; therefore, we chose a q-value threshold of 0.5. The validity of this approach for CopraRNA was tested with the prediction for GcvB. We assume that GcvB, with its 22 verified targets, is so far the most thoroughly investigated sRNA (4). In the CopraRNA prediction of GcvB, 37 targets are predicted with a q-value ≤0.5. Of these, 35 have homologs in Escherichia coli or S. enterica, 11 of which have been verified. Fifteen of the 35 homologs are involved in amino acid metabolism or transport, i.e., they fit to the known biological function of GcvB. This corresponds to an FDR of 69% or 57%, respectively, with regard to currently known targets and is not very far from the statistical estimate of 50%. In general, the number of significant predictions with a q-value ≤0.5 is a rough approximation of the expected number of targets and the prediction quality of the tested sRNA. A detailed description of the CopraRNA procedure is provided in SI Appendix.
Benchmark with Experimentally Verified Targets.
To evaluate the accuracy of CopraRNA, we performed a benchmarking test on a set of 18 conserved enterobacterial sRNAs and their 101 experimentally verified mRNA targets (modified from ref. 16) using homologous sequences from three to eight organisms (SI Appendix, Fig. S4). Compared with predictions by the existing approaches IntaRNA (12), TargetRNA (10), and RNApredator (11) (Fig. 1B), CopraRNA showed a clear improvement in the sensitivity or true positive rate (sensitivity = 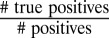 ) and positive predictive value (
) and positive predictive value ( ). Based on published data, CopraRNA’s top 1 target predictions were correct for 8 of 18 sRNAs (PPV: 44%), compared with 5 (PPV: 28%) for IntaRNA, 2 (PPV: 11%) for TargetRNA, and 1 (PPV: 6%) for RNApredator. When considering the top 5 and top 15 target predictions per sRNA, CopraRNA correctly detected 23 and 32, respectively, of all 101 targets (true positive rate: 23% and 32%, respectively), which constitutes a twofold increase in sensitivity compared with IntaRNA and a 2.9-fold and fourfold improvement compared with TargetRNA and RNApredator, respectively (SI Appendix, Table S2). In addition, our experimental verification (below) demonstrated that the existing lists of known targets are still incomplete, implying an underestimation of the true positive rate (Fig. 1B).
). Based on published data, CopraRNA’s top 1 target predictions were correct for 8 of 18 sRNAs (PPV: 44%), compared with 5 (PPV: 28%) for IntaRNA, 2 (PPV: 11%) for TargetRNA, and 1 (PPV: 6%) for RNApredator. When considering the top 5 and top 15 target predictions per sRNA, CopraRNA correctly detected 23 and 32, respectively, of all 101 targets (true positive rate: 23% and 32%, respectively), which constitutes a twofold increase in sensitivity compared with IntaRNA and a 2.9-fold and fourfold improvement compared with TargetRNA and RNApredator, respectively (SI Appendix, Table S2). In addition, our experimental verification (below) demonstrated that the existing lists of known targets are still incomplete, implying an underestimation of the true positive rate (Fig. 1B).
In many cases, the comparative approach resolved the problem of false negatives (i.e., verified targets missed in the prediction) in single-organism–based methods. Prominent examples are the GcvB targets lrp (4), oppA (4), and stm3903 (4); the RybB target ompN (28); and the Spot42 target gltA (6). The ranking of these targets improved from rank 95 to 3, rank 164 to 14, rank 1,297 to 40, rank 69 to 3, and rank 392 to 2, respectively (E. coli- or S. enterica-specific prediction vs. CopraRNA prediction). The benchmark dataset and the complete ranked list of all predictions are given in SI Appendix, Table S1 and Table S3.
Prediction of Interaction Domains.
In addition to the ranked list of predicted targets, CopraRNA provides comparative information on the putative interaction sites of the sRNA and its mRNA targets. These data are summarized in two density plots combining all predictions with a P value ≤0.01 for a specific sRNA (Fig. 1 C and D shows the GcvB example). Based on multiple sequence alignments, these plots visualize the frequency of single residues participating in the predicted sRNA–mRNA interactions. The plots are complemented by a series of schematic alignments for both sRNAs and mRNAs that highlight organism-specific predicted interactions. From these plots, the interaction domains of the sRNA can be inferred, as they provide the combined information of accessibility, complementarity, and phylogenetic conservation.
This visualization immediately highlights the two previously described interaction regions of GcvB (4) (Fig. 1C), the three different interaction regions of Spot42 (6), and the single 5′ located region of RybB (9) (SI Appendix, Fig. S5). In agreement with the published data for Spot42, gltA is targeted by the first single-stranded region (6) centered at position 6 in the multiple-sequence alignment (SI Appendix, Figs. S5 and S6). The newly identified targets sucC and gdhA base pair with the second and third interaction region of Spot42, respectively. For galK, all three regions are predicted to be involved in the interaction for four of the eight investigated organisms (SI Appendix, Fig. S5). As previously described (4), GcvB targets lrp and cycA via region “R2” of the sRNA (Fig. 1C), whereas most targets (e.g., dppA and oppA) interact with region “R1.” In the case of RprA, the full-length form appears to have two interaction domains, and only the distal site is retained after processing (29) (SI Appendix, Fig. S5), leading to a significant shift in the list of predicted targets. The mRNA plots are useful to obtain a rapid overview on the predicted interaction sites regarding their relative position and their phylogenetic conservation. The density plot also reveals the predominant interaction regions when using target sequences of the same length. For GcvB targets, there is a clear tendency toward the region near the start codon (Fig. 1D).
Functional Enrichment of Predicted Targets.
Many well-studied sRNAs control sets of functionally related genes [e.g., RyhB, nonessential iron-binding proteins (30), GcvB, amino acid biosynthesis genes (4)]. Therefore, we analyzed the top-ranked targets of all benchmark sRNAs for functional relationships based on automated functional enrichment using the database for annotation, visualization, and integrated discovery (DAVID) (31). A combination of CopraRNA and functional enrichment provided very clear results for several sRNAs and suggested their potential involvement in diverse cellular networks (Tables S4 and S5). The DAVID Web server clusters related terms and calculates a combined enrichment score. Table 2 shows representative terms for the most strongly enriched clusters of selected sRNAs. The accuracy of this approach is demonstrated exemplarily for GcvB: this sRNA has a broad set of 22 verified target mRNAs (4) and a clearly defined function as a regulator of amino acid metabolism and transport (4). GcvB has 60 positive predictions (P value ≤0.01, E. coli). Seven experimentally verified targets are in the top 10 list, which supports the prediction accuracy of our algorithm and represents a PPV of 70%. Among the 60 candidate targets, 19 were annotated with the term “cellular amino acid biosynthetic process” and were significantly enriched (enrichment score ∼6.65) over background (i.e., all genes included in the prediction output). In summary, 26 of the 60 predictions were grouped as amino acid related, including genes for 11 amino acid biosynthesis proteins, 9 amino acid transporters, and 4 peptide transporters. These results are complementary to the existing experimental findings and add several plausible candidates.
Table 2.
Results of the functional enrichment analysis using the DAVID Web server (31)
| sRNA | No. predicted | Enrichment score | Category | Term | No. |
| CyaR | 69 | 4.95 | UP_SEQ_FEATURE | Topological domain:Periplasmic | 26 |
| 3.45 | SP_PIR_KEYWORDS | Cell inner membrane | 32 | ||
| 2.15 | GOTERM_BP_FAT | GO:0005976∼polysaccharide metabolic process | 11 | ||
| FnrS | 67 | 2.43 | SP_PIR_KEYWORDS | Flavoprotein | 6 |
| 1.44 | GOTERM_MF_FAT | GO:0005506∼iron ion binding | 9 | ||
| 1.41 | GOTERM_MF_FAT | GO:0046872∼metal ion binding | 19 | ||
| GcvB | 60 | 6.65 | GOTERM_BP_FAT | GO:0008652∼cellular amino acid biosynthetic process | 19 |
| 4.12 | GOTERM_BP_FAT | GO:0006865∼amino acid transport | 9 | ||
| 2.78 | GOTERM_MF_FAT | GO:0015171∼amino acid transmembrane transporter activity | 5 | ||
| MicA | 46 | 1.97 | GOTERM_CC_FAT | GO:0009279∼cell outer membrane | 6 |
| 1.12 | GOTERM_BP_FAT | GO:0000271∼polysaccharide biosynthetic process | 6 | ||
| MicF | 50 | 2.36 | GOTERM_CC_FAT | GO:0044462∼external encapsulating structure part | 7 |
| 2.14 | GOTERM_CC_FAT | GO:0030312∼external encapsulating structure | 16 | ||
| 1.28 | SP_PIR_KEYWORDS | Lipoprotein | 5 | ||
| RyhB | 70 | 3.41 | GOTERM_MF_FAT | GO:0005506∼iron ion binding | 13 |
| 2.86 | GOTERM_MF_FAT | GO:0046872∼metal ion binding | 22 | ||
| 2.59 | GOTERM_MF_FAT | GO:0051536∼iron-sulfur cluster binding | 9 | ||
| SgrS | 66 | 1.62 | KEGG_PATHWAY | 02060: phosphotransferase system (PTS) | 5 |
| 1.36 | GOTERM_MF_FAT | GO:0046872∼metal ion binding | 17 | ||
| 1.35 | GOTERM_BP_FAT | GO:0051188∼cofactor biosynthetic process | 7 | ||
| Spot42 | 85 | 2.96 | GOTERM_BP_FAT | GO:0046356∼acetyl-CoA catabolic process | 7 |
| 2.53 | GOTERM_BP_FAT | GO:0006732∼coenzyme metabolic process | 12 | ||
| 1.83 | KEGG_PATHWAY | 00020:Citrate cycle, tricarboxylic acid cycle (TCA cycle) | 5 | ||
| FsrA | 54 | 4.77 | GOTERM_MF_FAT | GO:0051536∼iron-sulfur cluster binding | 8 |
| 3.81 | GOTERM_BP_FAT | GO:0022900∼electron transport chain | 6 | ||
| 3.69 | UP_SEQ_FEATURE | domain:4Fe-4S ferredoxin-type 2 | 4 | ||
| PrrF | 103 | 4.47 | GOTERM_MF_FAT | GO:0051536∼iron-sulfur cluster binding | 12 |
| 4.88 | GOTERM_MF_FAT | GO:0005506∼iron ion binding | 20 | ||
| 3.81 | SP_PIR_KEYWORDS | electron transport | 7 | ||
| SR1 | 50 | 1.88 | GOTERM_BP_FAT | GO:0030435∼sporulation resulting in formation of a cellular spore | 8 |
The top 3 significantly enriched terms (DAVID enrichment score ≥1.1) for 11 tested sRNAs are shown. For each sRNA, the number of predicted targets with a P value ≤0.01 (column 2), the score of the enriched functional cluster (column 3), the name and source of a representative term of this cluster (columns 4 and 5), and the number of unique genes in this cluster (column 6) are given. Individual gene members of the enriched terms are given in Table S5.
The known functions of GcvB were predicted almost completely by CopraRNA and the subsequent functional enrichment. The top 15 predictions and functionally enriched target candidates are shown in Fig. 2A.
Fig. 2.

Visualization of the functional enrichment analysis. All top 15 target predictions are shown plus predictions with a CopraRNA P value ≤0.01 that are functionally enriched (selected enriched terms). The edges connecting the sRNAs and targets are color coded according to the CopraRNA prediction P value, a darker color indicates a statistically more significant prediction. Previously experimentally verified targets from the literature [with regard to our benchmark list (SI Appendix, Table S1)] are marked with a black square, verifications from this study with a red square, and targets detected by microarrays with a blue square. Functionally enriched targets are color coded with respect to the enriched term. Results for (A) GcvB, (B) MicF, and (C) RyhB.
CopraRNA also returned the correct functional characterization for several other sRNAs. The predicted targets of MicA (Table 2 and SI Appendix, Fig. S7) and MicF were strongly enriched for outer membrane proteins, whereas the most strongly enriched cluster of RyhB targets consists of iron-binding proteins (Table 2 and Fig. 2 B and C).
Network Analysis of Predicted Targets.
Certain genes serve as regulatory hubs and are targeted by several sRNAs. For example, the mRNA encoding the alternative sigma factor RpoS is targeted directly by at least three sRNAs, the Arc-associated sRNA Z (ArcZ), DsrA, and the RpoS regulator RNA (RprA) (1), whereas the csgD mRNA is regulated by five different sRNAs, i.e., GcvB (32), the multicellular adhesive sRNA (McaS) (32, 33), the OmpR-regulated sRNA A/B (OmrA/B) (34), and RprA (35). Computational target prediction by CopraRNA allows the analysis of a high number of sRNAs, and the results can be combined to infer the gene regulatory network for a given organism. Indeed, our global network analysis based on the benchmark dataset predicted known and potential hotspots of sRNA-based regulation. In total, 15 mRNAs were predicted to be targeted by four or more sRNAs and ∼50 mRNAs by three or more sRNAs (Table S6). A striking example of an mRNA with multiple potential sRNA regulators encodes Lrp (leucine-responsive regulatory protein) and is predicted to be regulated by 7 of the 18 investigated sRNAs, including the previously identified regulators MicF (22, 36) and GcvB (4). The mRNA encoding the succinate dehydrogenase subunit SdhA has six predicted sRNA regulators, three of which were verified in this study (see below). We also detected multiple regulators of csgD and rpoS mRNAs. In addition to OmrA/B (34) and RprA (35), we predicted ChiX as a potential regulator of csgD. Another interesting example is the yobF-cspC dicistron with four potential regulators (CyaR, OmrA/B, and OxyS). From these, OxyS was previously shown to negatively regulate the yobF-cspC mRNA (10). The network obtained for 18 sRNAs and their previously verified and new targets is presented in Fig. 3A. In total, when using a P value threshold of 0.01, CopraRNA predicted 52 of the 101 benchmark targets. Furthermore, we verified 17 as yet unknown targets, uncovering connections between the regulatory networks of GcvB and Spot42, CyaR, RyhB and FnrS, and CyaR and SgrS. FnrS and RyhB share a dense overlapping regulon of at least four targets (Fig. 3A). Additionally, several operons were predicted to be influenced by multiple sRNAs: the sdhCDAB-sucABCD operon is targeted by five sRNAs at three different positions (Fig. 3B); Spot42 and RyhB each regulate two genes in the operon, sdhC (37) and sucC, as well as sdhD (37) and sdhA, respectively. In addition, the iscRUAB operon is regulated by both FnrS and RyhB (38) (Fig. 3C).
Fig. 3.

(A) Network of verified targets for the 18 sRNAs of the benchmark dataset. Visualization of the (B) sdhCDABsucABCD and (C) iscRSUAB operon with verified interaction sites; the promoters are annotated according to EcoCyc (52).
Experimental Verification of Predicted Targets.
Based on the benchmark results, we restricted the final set of target candidates for each sRNA to the top 15 predictions plus candidates that belong to the functional-enriched terms (Table S5). This approach provides a reasonable balance between sensitivity and specificity because it uses the high positive predictive value in the topmost predictions (SI Appendix, Fig. S3B) while allowing investigation of an extended target set. We selected 23 previously uncharacterized potential targets (SI Appendix, Table S7) for experimental testing using a GFP reporter system tailored to investigate posttranscriptional regulation (22). We verified 17 additional targets, which equals a success rate of ∼74%, and exemplarily proved the predicted interaction sites of yobF-CyaR, iscR-FnrS, nirB-RyhB, and gdhA-Spot42 through the introduction of compensatory mutations and for marA-FnrS, erpA-RyhB, marA-RyhB, and sucC-Spot42 by point mutations in their respective 5′UTRs (Fig. 4 A and B and SI Appendix, Fig. S8). Interestingly, the point mutations in the marA*1 construct resulted in an increased repression by wild-type RyhB, which indicates an improved RNA–RNA hybrid formation. Posttranscriptional repression of the remaining predicted targets was tested by flow cytometry (Fig. 4C) or Western blots (SI Appendix, Fig. S9). An overview of the constructs used and the respective mean fluorescence intensities is given in SI Appendix, Figs. S9 and S10. Most of the predicted interactions resemble the classic binding proximal to the translational start site. However, the binding sites for Spot42 in gdhA and icd align with positions +80 and +75 downstream from the start codon, deeply within the coding region. A direct inhibition of translation seems unlikely for these targets; rather, we assume a mechanism that reduces the half-life of the mRNAs, as shown for the ompD–MicC interaction in S. enterica (39, 40).
Fig. 4.

Verification of sRNA target candidates. Translational repression of 5′ UTR–gfp fusions when overexpressing the sRNA. The fold repression is the ratio of the GFP fluorescence of the respective translational 5′ UTR–GFP fusion in the presence of the control plasmid pJV300 and a plasmid for the overexpression of the respective sRNA, after subtraction of the background fluorescence. Compensatory point mutations in the UTR and sRNA are indicated with an asterisk. (A) Verification of the yobF–CyaR, nirB–RyhB, gdhA–Spot42, and iscR–FnrS interactions with compensatory point mutations. (B) Verification of the erpA–RyhB, marA–RyhB, marA–FnrS, and sucC–Spot42 interactions with point mutations in the 5′UTR. (C) Verification of the ptsI–CyaR, sdhA–CyaR, nagZ–RyhB, sdhA–RyhB, ptsI–SgrS, icd–Spot42, nagZ–FnrS, and sdhA–FnrS interactions without point mutations.
Performance of CopraRNA for sRNAs from Nonenterobacterial Species.
To evaluate the performance of CopraRNA for sRNAs that are not conserved in E. coli or S. enterica, we extended our benchmark dataset by five additional sRNAs from a wide range of bacterial families and phyla—the Fur-regulated sRNA A (FsrA) and SR1 (Firmicutes, Bacillaceae), LhrA (Firmicutes, Listeriaceae), the inhibitor of hctA translation (IhtA) (Chlamydiae), and PrrF (Proteobacteria, Pseudomonadaceae)—with a total of 17 experimentally verified targets (SI Appendix, Table S8). CopraRNA detects 11 of the 17 verified targets in the top 35 predictions, which resembles a true positive rate of ∼65% and a PPV of ∼6.3%. Again, this is at least ∼3.7 times better than the single-organism–specific methods (SI Appendix, Fig. S11). We also obtained intriguing functional enrichments for FsrA and PrrF (Table 2 and Table S5). The topmost enriched term for the predicted FsrA and PrrF targets is “GO:0051536∼iron-sulfur cluster binding” followed by other iron-related terms. This is in agreement with the known roles of these sRNAs in the iron stress response (30) and may hint at additional yet-unknown target genes of those sRNAs. The complete prediction dataset is given in Table S9.
Discussion
Comparison with Other Target Identification Strategies.
In this study, we present a comparative method for sRNA target identification in bacteria. The method is superior to existing bioinformatics tools (Fig. 1B) and works for a wide range of bacterial organisms. For seven tested benchmark sRNAs, CopraRNA can compete with microarray-based experiments for target detection (Table 1). CopraRNA is available as an easy-to-use Web interface (http://rna.informatik.uni-freiburg.de/CopraRNA/). True positive predictions are enriched by the downstream refinement of the prediction results through integration of existing data.
Using CopraRNA, we detected 17 as yet unknown targets for six sRNAs (Fig. 4 and SI Appendix, Fig S9). For the sRNAs FnrS, FsrA, GcvB, MicA, MicF, PrrF, RyhB, SgrS, and Spot42, bona fide physiological functions could be predicted accurately on our in silico results (Table 2). Compared with microarrays, CopraRNA has an advantage in that genetic modifications and time-consuming, expensive wet-laboratory experiments are not required for initial target screening. Additionally, CopraRNA is not biased by secondary effects, which might be picked up by experimental screening, and allows detection of targets not expressed under the tested conditions. Consequently, the predicted targets verify but also extend the existing microarray data.
However, CopraRNA also comes with certain limitations. The primary limitation of bioinformatic target prediction methods is that most predictions correspond to false positive predictions. The comparative approach of CopraRNA reduces this problem to the extent that further experimental analysis becomes much more reasonable than with existing tools, but it does not solve this problem completely. In our benchmark assay, half of the 101 known targets are detected with a P value threshold of 0.01 (SI Appendix, Fig S3A). At this threshold, an average of 65 targets is predicted for each sRNA and the FPR is ∼95% (SI Appendix, Fig S3B). Thus, a reasonable sensitivity of 50% comes with a low specificity of 5%. In fact, this is a strong improvement, as the other tools tested reach a maximum sensitivity of 25% (IntaRNA) at 65 predictions per sRNA, and e.g., IntaRNA needs 226 predictions per sRNA to reach a sensitivity of 50%. Nevertheless, a low specificity challenges investigators to follow up on the predictions. For that reason, we do not stick to the P value threshold strictly, but focus on the top 15 list and on the predictions (P ≤ 0.01) suggested by further postprocessing steps. These steps may include automatic and manual functional enrichment (Fig. 2), network analysis (Fig. 3), overlaps with transcription factor regulons (Fig. 5 and SI Appendix, Fig S13), or correlation patterns coming from microarray data (41, 42). This combined strategy was very successful in retaining sensitivity while enhancing specificity. We demonstrated this by the experimental verification of 73% of the selected 23 predicted targets that were not characterized previously. These results also show that the FPR is at least slightly overestimated because of previously unknown targets (SI Appendix, Fig S3B; compare dashed and solid blue lines). Another challenge is a prediction without a meaningful postprocessing result, caused, e.g., by the lack of additional data or lower prediction quality. For these cases, we control the FDR statistically by calculating a q-value. The average q-value at prediction rank 65 is ∼0.54 and therefore judged by the current benchmark data, rather too optimistic. Nevertheless, the q-value distribution is valuable to roughly estimate the general prediction quality for a given sRNA. For example, we could not predict known targets for ArcZ. This less informative prediction is accompanied correctly by a rapidly growing q-value and only 10 predictions with q ≤ 0.5. On the other side, the good prediction for GcvB has 38 predictions with q ≤ 0.5, and as described above, the q-value fits well to the benchmark dataset. CopraRNA generally requires the conservation of an sRNA and also a substantial level of target conservation in the selected species. Therefore, single-organism–specific targets are likely to be missed, as are interactions that generally are not predictable by the underlying IntaRNA algorithm (e.g., double-kissing hairpin complexes). For example, the metE–FnrS interaction [verified in E. coli (20)] seems to be conserved or detectable only in three of the eight included species (SI Appendix, Fig. S12). This results in a high combined P value of 0.54 and a rank of 1,969 in the combined prediction and shows the importance of carefully selecting species. A small evolutionary distance favors sensitivity, and a large distance favors specificity. The downstream functional enrichment analysis relies on the availability of the organism in the DAVID database (31), and the results depend on the annotation quality of the genome of interest. Of note, CopraRNA is a target prediction tool for sRNAs that are expected to act in trans; it is not suitable for the differentiation of a trans-acting RNA from other types of transcripts. However, the functional enrichment analysis, the conservation plots, and the q-value distribution provided by CopraRNA might provide a hint as to whether a given conserved RNA is a functional trans-acting sRNA.
Fig. 5.

Partial regulatory network around FNR, ArcA, and FnrS. The figure shows verified FnrS targets, as well as predicted targets (CopraRNA P value ≤0.01) regulated by FNR or ArcA. For the transcription factors, only selected targets are displayed.
Additional Targets and Functions of Previously Characterized sRNAs.
The inspection of the benchmark dataset revealed additional targets and functions, even for sRNAs extensively characterized in the past. For the cAMP receptor protein (CRP)-regulated sRNA CyaR (18, 19), we detected as yet unidentified targets in primary metabolism (sdhA) and the phosphotransferase system (ptsI), constituting previously unreported links of the CyaR regulon to carbon metabolism. Furthermore, with regard to the yobF-cspC operon, we found a potential explanation for the indirect negative effect of CyaR on the rpoS mRNA, which was detected in a screen with 26 sRNAs (43). The yobF gene is organized together with cspC in a dicistronic operon, and the RNA chaperone CspC is a posttranscriptional stabilizer of the rpoS message (44).
FnrS is involved in gene regulation after the shift from aerobic to anaerobic conditions, and its expression is activated by the transcription factors FNR and ArcA (20, 21). The combination of existing information (45) with our predictions and verifications for FnrS results in a remarkable complex regulatory network (Fig. 5): (i) FnrS transduces the signal to several non-FNR and -ArcA targets. These include the target nagZ and the two transcription factor mRNAs iscR and marA. (ii) The prediction also revealed several target candidates, which are controlled simultaneously by FNR and ArcA, which would establish multi-output feed-forward loops. Although the transcription factor MarA is not directly regulated by FNR or ArcA, four genes that are activated by MarA (acnA, fumC, sodA, zwf) are repressed by ArcA and/or FNR. These four genes are involved in the resistance to superoxide (46) and provide a reasonable explanation for the repression of marA by FnrS at anaerobic conditions. The repression of the transcription factor IscR may be part of the observed O2-dependent expression of the iscR regulon (47).
FnrS shares three targets with RyhB. Both sRNAs regulate the mRNA encoding MarA, which is involved in the response to antimicrobial compounds and oxidative stress (46), and of the mRNA for the β-N-acetylglucosaminidase NagZ, which permits resistance to β-lactams in Pseudomonas aeruginosa (48). Interestingly, both MarA and NagZ are not obviously involved in iron homeostasis. For the iron stress-induced sRNA RyhB, we predicted mRNAs for 13 iron-containing proteins as targets and verified the posttranscriptional regulation of erpA, the mRNA of an A-type carrier (ATC) protein involved in iron–sulfur cluster biogenesis (49), and of nirB, which codes for a subunit of nitrite reductase.
Regarding the dual-function RNA SgrS, we predicted interactions with mRNAs of additional components of the phosphotransferase system (chhB, cmtB and fruA) and verified the posttranscriptional regulation of ptsI (Fig. 4), which codes for the non–sugar-specific enzyme I component of the PTS. Furthermore, we detected the recently described positive regulated sugar phosphatase mRNA yigL (50) as a direct target.
We also predicted and verified targets for the CRP-repressed Spot42 sRNA which is involved in catabolite repression and controls a range of genes in central and secondary metabolism and sugar transport (6). Our predictions show a large, 18-gene overlap with the CRP regulon and point to an even broader regulatory role for Spot42 in primary metabolism involving the citrate cycle and acetyl-CoA–dependent processes (Table 2, Tables S4 and S5, and SI Appendix, Fig. S13). Our successful experimental validation of the targets gdhA, icd, and sucC proves the accuracy of our predictions.
In sum, CopraRNA allows for an efficient screening of large numbers of sRNAs and has proven superior compared with existing methods. Using this tool, we obtained compelling evidence that sRNAs are global regulators of large sets of mRNAs, comparable to protein transcription factors and eukaryotic microRNAs. We also show that it is a common concept that mRNAs are targeted by multiple sRNAs and correctly predicted the regulatory hubs csgD and rpoS. Furthermore, we proposed and partially verified gdhA, lrp, marA, nagZ, ptsI, sdhA, and yobF-cspC as hubs targeted by up to seven different sRNAs. Finally, we present examples for complex posttranscriptional events at the operon level, including multiple targeting by the same, as well as different, sRNAs.
Methods
Experimental Methods.
Bacterial strains and growth.
Cells were grown in Luria–Bertani (LB) broth or on LB plates at 37 °C. Antibiotics (where appropriate) were applied at the following concentrations: 100 mg⋅mL−1 ampicillin and 25 mg⋅mL−1 chloramphenicol.
Plasmid construction.
The plasmids for the overexpression of FnrS and CyaR and those for the translational superfolder–GFP fusions were constructed as described previously (22).
Oligonucleotides and plasmids.
Oligonucleotides and plasmids are listed in SI Appendix, Tables S10 and S11.
Fluorescence measurements.
Overnight cultures were used to inoculate (1:100) fresh cultures, and cultivation was continued to OD600 = 2.0. Culture samples equivalent to 1 OD were harvested by centrifugation and resuspended in PBS. Aliquots of 100 µL were transferred to a 96-well microtiter plate, and relative GFP levels were measured in a Victor3 fluorimeter (Perkin-Elmer). A wild-type strain was measured in parallel to subtract autofluorescence levels. All samples were measured in biological triplicates. This method was used to analyze the RyhB–nirB and the CyaR–yobF interactions.
Flow cytometry-based fluorescence measurements.
Single bacterial colonies were inoculated in 200 µL LB medium in 96-well microtiter plates containing ampicillin and chloramphenicol and grown at 37 °C, 100 rpm for 12–15 h. Cells were diluted 1/5 in LB and fixed with formaldehyde (Roti-Histofix 10%; Carl Roth GmbH) to an final concentration of 1% (wt/vol) and measured directly on an Accuri C6 flow cytometer (BD Biosciences). The mean fluorescence of 50,000 events was averaged for 6–12 independent biological replicates. The fold repression was calculated as the ratio of the mean GFP fluorescence of the respective translational UTR–GFP fusion in the presence of the control plasmid pJV300 and a plasmid for the overexpression of the respective sRNA, after subtraction of the background fluorescence. Background fluorescence was measured with the control plasmids pXG-0 and pJV300 (22):
 |
The respective mean fluorescences after subtraction of the background fluorescence are shown in SI Appendix, Fig. S9. Western blots were performed as described in ref. 9.
Theoretical Methods.
Benchmark analysis.
For the benchmark analysis, we conducted whole-genome target predictions for E. coli (NC_000913) and S. enterica (NC_003197, NC_003277) based on the sequences 200 nt upstream and 100 nt downstream of the annotated start codons as the input (the first nucleotide of the start codon corresponds to position 201). The Web server of RNApredator used the whole gene for target prediction. Otherwise, all the tools were used with the given standard parameters. The P value threshold of TargetRNA was set to 0.99 to obtain the top 100 predictions. The benchmark dataset included 18 sRNAs and a total of 101 previously published targets (SI Appendix, Table S1). Some targets were verified in both E. coli and S. enterica; the total number of verified sRNA–target pairs is 113, but we used only the nonredundant dataset. We included only targets for which a direct posttranscriptional regulation by an sRNA was verified experimentally. Targets detected only by RT-PCR, microarrays, or Northern blots and not verified further were excluded.
Functional enrichment.
Functional enrichments (functional annotation clustering) were performed on the DAVID Web server (31) for all benchmark sRNA predictions. For each sRNA, the target candidates (P ≤ 0.01) were tested against all the genes on the list as background. Obvious artifacts, i.e., predicted interactions with the complementary strand of the genomic coding region of the respective sRNA, were excluded. Enrichments were performed for E. coli. The standard parameters were changed to a “Similarity Threshold” of 0.85 and an “Initial Group Membership” and “Final Group Membership” of 2. Our threshold for a functional-enriched term was a DAVID enrichment score of ≥1.1. Networks were visualized using Cytoscape (51).
CopraRNA algorithm.
To reduce the number of false positive hits in the interaction predictions, we searched for interactions that are conserved in various species. However, for several reasons, it is conceivable that the interaction is preserved whereas the actual interaction site is not. To be able to still predict conserved interactions, it is necessary to combine the evidence for interactions in the different species without resorting to a consensus-based approach. In addition to the Web server version, a stand-alone version of CopraRNA is available (www.bioinf.uni-freiburg.de/Software/). A more detailed description of CopraRNA, with a focus on the calculation of P values, may be found in SI Appendix.
Supplementary Material
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft Focus Program “Sensory and Regulatory RNAs in Prokaryotes” (SPP1258); Bundesministerium für Bildung und Forschung (BMBF) Grant 0316165 (to W.R.H, R.B., and J.V.); and the Excellence Initiative of the German Federal and State Governments (EXC 294 to R.B.). K.P. was supported by a postdoctoral fellowship from the Human Frontiers in Science Program.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1303248110/-/DCSupplemental.
References
- 1.Storz G, Vogel J, Wassarman KM. Regulation by small RNAs in bacteria: Expanding frontiers. Mol Cell. 2011;43(6):880–891. doi: 10.1016/j.molcel.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gottesman S, Storz G. Bacterial small RNA regulators: Versatile roles and rapidly evolving variations. Cold Spring Harb Perspect Biol. 2011;3(12):a003798. doi: 10.1101/cshperspect.a003798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Papenfort K, Vogel J. Regulatory RNA in bacterial pathogens. Cell Host Microbe. 2010;8(1):116–127. doi: 10.1016/j.chom.2010.06.008. [DOI] [PubMed] [Google Scholar]
- 4.Sharma CM, et al. Pervasive post-transcriptional control of genes involved in amino acid metabolism by the Hfq-dependent GcvB small RNA. Mol Microbiol. 2011;81(5):1144–1165. doi: 10.1111/j.1365-2958.2011.07751.x. [DOI] [PubMed] [Google Scholar]
- 5.Gogol EB, Rhodius VA, Papenfort K, Vogel J, Gross CA. Small RNAs endow a transcriptional activator with essential repressor functions for single-tier control of a global stress regulon. Proc Natl Acad Sci USA. 2011;108(31):12875–12880. doi: 10.1073/pnas.1109379108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beisel CL, Storz G. The base-pairing RNA spot 42 participates in a multioutput feedforward loop to help enact catabolite repression in Escherichia coli. Mol Cell. 2011;41(3):286–297. doi: 10.1016/j.molcel.2010.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sharma CM, et al. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature. 2010;464(7286):250–255. doi: 10.1038/nature08756. [DOI] [PubMed] [Google Scholar]
- 8.Mitschke J, et al. An experimentally anchored map of transcriptional start sites in the model cyanobacterium Synechocystis sp. PCC6803. Proc Natl Acad Sci USA. 2011;108(5):2124–2129. doi: 10.1073/pnas.1015154108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Papenfort K, Bouvier M, Mika F, Sharma CM, Vogel J. Evidence for an autonomous 5′ target recognition domain in an Hfq-associated small RNA. Proc Natl Acad Sci USA. 2010;107(47):20435–20440. doi: 10.1073/pnas.1009784107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tjaden B, et al. Target prediction for small, noncoding RNAs in bacteria. Nucleic Acids Res. 2006;34(9):2791–2802. doi: 10.1093/nar/gkl356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eggenhofer F, Tafer H, Stadler PF, Hofacker IL. RNApredator: Fast accessibility-based prediction of sRNA targets. Nucleic Acids Res. 2011;39(Web Server issue):W149–W154. doi: 10.1093/nar/gkr467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Busch A, Richter AS, Backofen R. IntaRNA: Efficient prediction of bacterial sRNA targets incorporating target site accessibility and seed regions. Bioinformatics. 2008;24(24):2849–2856. doi: 10.1093/bioinformatics/btn544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rehmsmeier M, Steffen P, Höchsmann M, Giegerich R. Fast and effective prediction of microRNA/target duplexes. RNA. 2004;10(10):1507–1517. doi: 10.1261/rna.5248604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hao Y, et al. Quantifying the sequence-function relation in gene silencing by bacterial small RNAs. Proc Natl Acad Sci USA. 2011;108(30):12473–12478. doi: 10.1073/pnas.1100432108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Backofen R, Hess WR. Computational prediction of sRNAs and their targets in bacteria. RNA Biol. 2010;7(1):33–42. doi: 10.4161/rna.7.1.10655. [DOI] [PubMed] [Google Scholar]
- 16.Richter AS, Backofen R. Accessibility and conservation: General features of bacterial small RNA-mRNA interactions? RNA Biol. 2012;9(7):954–965. doi: 10.4161/rna.20294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beisel CL, Updegrove TB, Janson BJ, Storz G. Multiple factors dictate target selection by Hfq-binding small RNAs. EMBO J. 2012;31(8):1961–1974. doi: 10.1038/emboj.2012.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Lay N, Gottesman S. The Crp-activated small noncoding regulatory RNA CyaR (RyeE) links nutritional status to group behavior. J Bacteriol. 2009;191(2):461–476. doi: 10.1128/JB.01157-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Papenfort K, et al. Systematic deletion of Salmonella small RNA genes identifies CyaR, a conserved CRP-dependent riboregulator of OmpX synthesis. Mol Microbiol. 2008;68(4):890–906. doi: 10.1111/j.1365-2958.2008.06189.x. [DOI] [PubMed] [Google Scholar]
- 20.Boysen A, Møller-Jensen J, Kallipolitis B, Valentin-Hansen P, Overgaard M. Translational regulation of gene expression by an anaerobically induced small non-coding RNA in Escherichia coli. J Biol Chem. 2010;285(14):10690–10702. doi: 10.1074/jbc.M109.089755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Durand S, Storz G. Reprogramming of anaerobic metabolism by the FnrS small RNA. Mol Microbiol. 2010;75(5):1215–1231. doi: 10.1111/j.1365-2958.2010.07044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Corcoran CP, et al. Superfolder GFP reporters validate diverse new mRNA targets of the classic porin regulator, MicF RNA. Mol Microbiol. 2012;84(3):428–445. doi: 10.1111/j.1365-2958.2012.08031.x. [DOI] [PubMed] [Google Scholar]
- 23.Massé E, Vanderpool CK, Gottesman S. Effect of RyhB small RNA on global iron use in Escherichia coli. J Bacteriol. 2005;187(20):6962–6971. doi: 10.1128/JB.187.20.6962-6971.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Papenfort K, Podkaminski D, Hinton JCD, Vogel J. The ancestral SgrS RNA discriminates horizontally acquired Salmonella mRNAs through a single G-U wobble pair. Proc Natl Acad Sci USA. 2012;109(13):E757–E764. doi: 10.1073/pnas.1119414109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Uchiyama I. MBGD: A platform for microbial comparative genomics based on the automated construction of orthologous groups. Nucleic Acids Res. 2007;35(Database issue):D343–D346. doi: 10.1093/nar/gkl978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hartung J. A note on combining dependent tests of significance. Biom J. 1999;41:849–855. [Google Scholar]
- 27.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci USA. 2003;100(16):9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bouvier M, Sharma CM, Mika F, Nierhaus KH, Vogel J. Small RNA binding to 5′ mRNA coding region inhibits translational initiation. Mol Cell. 2008;32(6):827–837. doi: 10.1016/j.molcel.2008.10.027. [DOI] [PubMed] [Google Scholar]
- 29.Argaman L, et al. Novel small RNA-encoding genes in the intergenic regions of Escherichia coli. Curr Biol. 2001;11(12):941–950. doi: 10.1016/s0960-9822(01)00270-6. [DOI] [PubMed] [Google Scholar]
- 30.Salvail H, Massé E. Regulating iron storage and metabolism with RNA: An overview of posttranscriptional controls of intracellular iron homeostasis. Wiley Interdiscip Rev RNA. 2012;3(1):26–36. doi: 10.1002/wrna.102. [DOI] [PubMed] [Google Scholar]
- 31.Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 32.Jørgensen MG, et al. Small regulatory RNAs control the multi-cellular adhesive lifestyle of Escherichia coli. Mol Microbiol. 2012;84(1):36–50. doi: 10.1111/j.1365-2958.2012.07976.x. [DOI] [PubMed] [Google Scholar]
- 33.Thomason MK, Fontaine F, De Lay N, Storz G. A small RNA that regulates motility and biofilm formation in response to changes in nutrient availability in Escherichia coli. Mol Microbiol. 2012;84(1):17–35. doi: 10.1111/j.1365-2958.2012.07965.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Holmqvist E, et al. Two antisense RNAs target the transcriptional regulator CsgD to inhibit curli synthesis. EMBO J. 2010;29(11):1840–1850. doi: 10.1038/emboj.2010.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mika F, et al. Targeting of csgD by the small regulatory RNA RprA links stationary phase, biofilm formation and cell envelope stress in Escherichia coli. Mol Microbiol. 2012;84(1):51–65. doi: 10.1111/j.1365-2958.2012.08002.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holmqvist E, Unoson C, Reimegård J, Wagner EGH. A mixed double negative feedback loop between the sRNA MicF and the global regulator Lrp. Mol Microbiol. 2012;84(3):414–427. doi: 10.1111/j.1365-2958.2012.07994.x. [DOI] [PubMed] [Google Scholar]
- 37.Desnoyers G, Massé E. Noncanonical repression of translation initiation through small RNA recruitment of the RNA chaperone Hfq. Genes Dev. 2012;26(7):726–739. doi: 10.1101/gad.182493.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Desnoyers G, Morissette A, Prévost K, Massé E. Small RNA-induced differential degradation of the polycistronic mRNA iscRSUA. EMBO J. 2009;28(11):1551–1561. doi: 10.1038/emboj.2009.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pfeiffer V, Papenfort K, Lucchini S, Hinton JCD, Vogel J. Coding sequence targeting by MicC RNA reveals bacterial mRNA silencing downstream of translational initiation. Nat Struct Mol Biol. 2009;16(8):840–846. doi: 10.1038/nsmb.1631. [DOI] [PubMed] [Google Scholar]
- 40.Bandyra KJ, et al. The seed region of a small RNA drives the controlled destruction of the target mRNA by the endoribonuclease RNase E. Mol Cell. 2012;47(6):943–953. doi: 10.1016/j.molcel.2012.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hernández-Prieto MA, et al. Iron deprivation in Synechocystis: Inference of pathways, non-coding RNAs, and regulatory elements from comprehensive expression profiling. G3 (Bethesda) 2012;2(12):1475–1495. doi: 10.1534/g3.112.003863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Modi SR, Camacho DM, Kohanski MA, Walker GC, Collins JJ. Functional characterization of bacterial sRNAs using a network biology approach. Proc Natl Acad Sci USA. 2011;108(37):15522–15527. doi: 10.1073/pnas.1104318108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mandin P, Gottesman S. Integrating anaerobic/aerobic sensing and the general stress response through the ArcZ small RNA. EMBO J. 2010;29(18):3094–3107. doi: 10.1038/emboj.2010.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cohen-Or I, Shenhar Y, Biran D, Ron EZ. CspC regulates rpoS transcript levels and complements hfq deletions. Res Microbiol. 2010;161(8):694–700. doi: 10.1016/j.resmic.2010.06.009. [DOI] [PubMed] [Google Scholar]
- 45.Gama-Castro S, et al. RegulonDB version 7.0: Transcriptional regulation of Escherichia coli K-12 integrated within genetic sensory response units (Gensor Units) Nucleic Acids Res. 2011;39(Database issue):D98–D105. doi: 10.1093/nar/gkq1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martin RG, Rosner JL. Promoter discrimination at class I MarA regulon promoters mediated by glutamic acid 89 of the MarA transcriptional activator of Escherichia coli. J Bacteriol. 2011;193(2):506–515. doi: 10.1128/JB.00360-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Giel JL, Rodionov D, Liu M, Blattner FR, Kiley PJ. IscR-dependent gene expression links iron-sulphur cluster assembly to the control of O2-regulated genes in Escherichia coli. Mol Microbiol. 2006;60(4):1058–1075. doi: 10.1111/j.1365-2958.2006.05160.x. [DOI] [PubMed] [Google Scholar]
- 48.Zamorano L, et al. NagZ inactivation prevents and reverts β-lactam resistance, driven by AmpD and PBP 4 mutations, in Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2010;54(9):3557–3563. doi: 10.1128/AAC.00385-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pinske C, Sawers RG. A-type carrier protein ErpA is essential for formation of an active formate-nitrate respiratory pathway in Escherichia coli K-12. J Bacteriol. 2012;194(2):346–353. doi: 10.1128/JB.06024-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Papenfort K, Sun Y, Miyakoshi M, Vanderpool CK, Vogel J. Small RNA-mediated activation of sugar phosphatase mRNA regulates glucose homeostasis. Cell. 2013;153(2):426–437. doi: 10.1016/j.cell.2013.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cline MS, et al. Integration of biological networks and gene expression data using Cytoscape. Nat Protoc. 2007;2(10):2366–2382. doi: 10.1038/nprot.2007.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Keseler IM, et al. EcoCyc: Fusing model organism databases with systems biology. Nucleic Acids Res. 2013;41(Database issue):D605–D612. doi: 10.1093/nar/gks1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.