

Abstract
This article reviews the antitumor and anti-HIV activities of naturally occurring triterpenoids, including the lupane, ursane, oleanane, lanostane, dammarane, and miscellaneous scaffolds. Structure–activity relationships of selected natural compounds and their synthetic derivatives are also discussed.
1 Introduction
Natural products are an excellent reservoir of biologically active compounds. For centuries, extracts from natural products have been a main source of folk medicines, and even today, many cultures still employ them directly for medicinal purposes. Among the classes of identified natural products, triterpenoids, one of the largest families, have been studied intensively for their diverse structures and variety of biological activities. As a continued study of naturally occurring drug candidates, this review describes the research progress over the last three years (2006–2008) on triterpenoids possessing cytotoxic or anti-HIV activity, with focus on the occurrence, biological activities, and structure–activity relationships of selected compounds and their synthetic derivatives.
2 Potential antitumor effects of triterpenoids
2.1 The lupane group
Betulinic acid (1) is a naturally occurring pentacyclic triterpene belonging to the lupane family. It is present in many terrestrial plant species, e.g., Ancistrocladus heyneanus, Diospyros leucomelas, Syzygium formosanum, Tetracera boliviana, Tryphyllum peltatum, and Ziziphus vulgaris.1–3 Compound 1 can also be obtained in quantity from the bark of the London plane tree, Platanus acerifolia.4,5 Table 1 lists other plant sources of 1 reported since 2006.
Table 1.
Sources of betulinic acid (1)
| Part used | Plant origin |
|---|---|
| Roots | Ainsliaea acerifolia26 |
| Helicteres angustifolia27 | |
| Mimosa pudica28 | |
| Root bark | Harungana madagascariensis29 |
| Ziziphus cambodiana30 | |
| Bark | Alnus hirsuta31 |
| Platanus orientalis32 | |
| Stems | Hydnocarpus hainanensis33 |
| Stem bark | Barringtonia racemosa34 |
| Dichapetalum gelonioides35 | |
| Diospyros crassiflora36 | |
| Diospyros decandra37 | |
| Fagara tessmannii38 | |
| Psorospermum glaberrimum39 | |
| Terminalia superba40 | |
| Heartwood | Diospyros paniculata41 |
| Aerial parts | Gouania ulmifolia42 |
| Lippia dulcis43 | |
| Ocimum basilicum44 | |
| Perovskia abrotanoides45 | |
| Scoparia dulcis46 | |
| Stems and leaves | Jasminum lanceolarium47 |
| Leaves | Lycopus lucidus48 |
| Salacia chinensis49 | |
| Ugni molinae50 | |
| Leaves and twigs | Clematoclethra actinidioides51 |
| Cratoxylum arborescens52 | |
| Hulls | Prunus dulcis12 |
| Whole plant | Cunila spicata53 |
| Dracocephalum forrestii54 | |
| D. komarovi55 | |
| Hyptis fasciculata53 |
Trumbull and co-workers6 identified the inhibitory activities of compound 1 against tumor promotion in 1976. Later, its action was identified as a melanoma-specific cytotoxicity mediated by the induction of apoptosis.7 The cytotoxicity as well as mechanism of action of 1 have been reviewed.8–11 Although it was initially identified as a melanoma-specific cytotoxic agent, in more recent studies, 1 showed antiproliferative activity against MCF-7 cells (GI50: 0.27 µM),12 as well as neuroblastoma (SKNAS), rhabdomyosarcoma-medulloblastoma (TE671), breast carcinoma (T47D), lung carcinoma (A549), colon adenocarcinoma (HT-29), multiple myeloma (RPMI8226), cervical carcinoma (HPCC), and glioblastoma multiforme (HPGBM) cell lines (IC50: 2.4–4.5 µM).13 It also showed broad-spectrum cytotoxicity towards lung, colorectal, breast, prostate, and cervical cancer cell lines,14 as well as drug-resistant colon adenocarcinoma cell lines (SNU-C5/WT, SNU-C5/5FU-R, and SNU-C5/OXT-R).15

Compound 1 induces apoptosis11,16 through the mitochondrial pathway.15,17,18 A typical decrease in bcl-2 and cyclin D1 gene expression and increase in bax gene expression was observed in several cancer cell lines treated with 1,13,19 which agreed with prior literature.18 Subsequent studies on its anticancer mechanism revealed that 1 is a potent activator of the chymotrypsin-like activity of the proteasome.20 In addition, 1 decreases expression of vascular endothelial growth factor (VEGF) and the antiapoptotic protein survivin in prostate cancer cells (LNCaP) by activating selective proteasome-dependent degradation of the transcription factor’s specificity proteins.21 Compound 1 also inhibits NF-κB expression in androgen-refractory human prostate cancer cells (PC-3), which exhibit high constitutive NF-κB expression.19
Furthermore, although 1 reportedly is an inhibitor of human topoisomerases (topo) I and IIα,22,23 it does not show synergistic effects with other topo-I inhibitors. In contrast, 1 inhibited the formation of topo-I DNA cleavable complexes (apoptotic mediatora) induced by camptothecin, staurosporine, and etoposide in prostate cancer cells.24 This study also indicated that the tumor death mediated by 1 can be counteracted by the mitogen-activated protein kinase 1 (MAPK1) inhibitor U0126 in melanoma cells.25
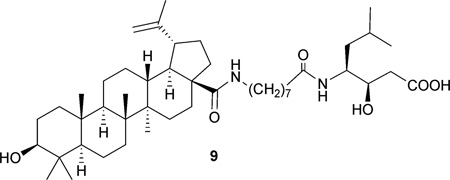
Structure–activity relationship (SAR) studies of 1 have led to the preparation of numerous C-3 hydroxyl, C-20 double bond, C-28 carboxylic acid, and miscellaneous modified analogues. In most cases, C-3 modification led to altered cytotoxic potency and selectivity, while reduction of the C-20–C-29 double bond and replacement of the C-28 carboxyl group with ester and amide side chains generally gave derivatives inactive against cancer cell lines.9 Recent approaches revealed similar results. Compound 2, the C-3 epimer of 1, demonstrated decreased cytotoxicity when compared with 1.56 Betulinic C-3 esters 3 and 4 exhibited better activity against KB cells.57 3-O-Glycosidated derivatives 5 and 6 were 8- to 12-fold more potent than 1 (IC50: 2.6–3.9 µM) against cancer cells and showed better selectivity compared with healthy cells (IC50: 31 µM).58 The C-28 monosaccharide derivatives (7) showed slightly increased cytotoxicity, whereas trisaccharide analogues (8) were inactive.59 In anti-HIV studies, adding a C-28 side chain was a successful structure modification strategy; however, the resulting potent anti-HIV lead 9 (IC9564) lost the ability to activate chymotrypsin-proteasomal activity compared with 1.20
2-Cyano-3,12-dioxooleana-1,9(11)-dien-28-oic acid (CDDO) is a promising anticancer synthetic oleanolic acid analogue (see Section 2.2), and this same CDDO modification concept (incorporating a 2-substituted-1-en-3-one A-ring in the triterpenoid skeleton) was also applied to 1. The resulting A-ring-modified 2-substituted-1-en-3-one derivatives (10–14) showed enhanced cytotoxic or cytostatic activity against pancreatic (Panc-28), colon (SW480), leukemia (U937), and myeloma (RPMI 8226) cancer cell lines.60,61 Compared with 10–12, analogues 13 and 14, which have a methylcarboxy rather than cyano group at C-2, showed increased potency in an apoptosis assay.61 Expanding the ring system by incorporating an indole or pyrazine ring at C-2 and C-3 afforded heterocyclic analogues (15–19), which were more potent than the parent compound, 1, against pancreatic (MIAPaCa2), ovarian (PA-1), and other (SW620, A549, HT-29, K-562, K-562-tax, and -b2–4) cancer cell lines.62,63 Non-typical synthetic approaches to C-30 and E-ring modifications led to the oxo-lupane analogues 20–23, which also showed enhanced cytotoxicity against CEM cells (IC50: 5–7 µM).64


Compound 1 has also been evaluated for additional biological activities. It reduces the activity of cholesterol acyltransferase (ACAT-1 and ACAT-2)48 and the formation of triglyceride (TG) by inhibiting diacylglycerol acyltransferase in human HepG2 cells.31 When studied for cardiovascular activity, 1 demonstrated up-regulation of eNOS and down-regulation of NADPHoxidase in human endothelial cells.65 The proinflammatory effect of 1 was proved by blocking the interaction of C1q with antibodies in the classical pathway.66 Moreover, 1 displayed anti-inflammatory activity through the suppression of both NO and PGE2 generation by macrophages, with IC50 values of 0.7 and 0.6 µM, respectively.67 Compound 1 is also a competitive inhibitor of the protease (3CL) of SARS-CoV, a highly virulent coronavirus, with a Ki value of 8.2 µM.68 In other experiments, 1 and various C-3 modified analogues showed antimicrobial activity, with inhibitory effects toward Mycobacterium tuberculosis (MIC: 10–109 µM),30,69 Trichophyton soudanense (MIC: 54.8 µM), and T. mentagrophytes (MIC: 27.4 µM).70 Compound 1 also inhibited the invasion of Porphyromomas falciparum into erythrocytes, with IC50 values of 13.8 µM,71 and blocked the binding effects of heat-labile entero-toxin (LT) of E. coli in intestinal epithelial cells, resulting in the suppression of LT-induced diarrhea.72
Lupeol (24), a very common lupane-triterpene widely distributed in the plant kingdom, occurs across a multitude of taxonomically diverse genera and geographical areas. In a survey of the Chemical Abstracts database, more than 100 literature references during the period 2006 to 2008 reported the occurrence of lupeol. A recently published review described the chemopreventive prospects, molecular target, and mechanism of action of 24.73 In an earlier investigation of apoptotic induction and cell arrest in various cancer cells, further molecular-level phenomena involving MAPK, PI3K/Akt, death receptor, and NF-κB signaling pathways were also revealed.73 The following in vitro and in vivo studies showed similar results; 24 inhibited the growth of LNCaP cells (IC50: 75 µM) and significantly reduced testosterone-induced prostate changes in mice by a factor of 1.4.74 Compound 24 arrested the cell cycle at the G(1)-S phase in PC-3 cells75 or the G2/M phase in 451Lu cells and 451Lu xenografts in nude mice.76 In addition, 24 caused induction of capase-3, and elevation of the Bcl-2/Bax ratio in LNCaP, PC-3 and 451Lu cells were observed, which are characteristics of mitochondria-mediated apoptosis.74–76 Interestingly, pre-treatment or post-treatment with 24 at a dose of 200 mg/mouse showed 56% and 43% preventive effects, respectively, against DMBA-induced DNA breakage.77

Betulin (25), a C-28 hydroxylated lupane commonly isolated together with 1, generally showed lower activity in cytotoxic assays than 1.12,59,62,78,79 However, exceptions were found against A-549, B16-F1, DLD-1, and WS1 cell lines.58 In an SAR study of glycoside derivatives, glycosidation at C-3 or C-28 of 25 gave 26 and 27, which showed slightly enhanced cytotoxic activity.59 In other literature reports, derivatives 28 and 29 with C-3 or C-28 monosaccharide units, respectively, lost cytotoxicity.58 In contrast, the 3-O-glycosylated derivative of lupeol (30) showed 7–12-fold improved cytotoxicity.58
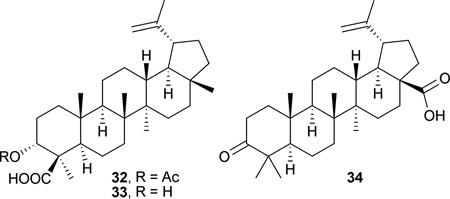
28-Hydroxy-3-oxo-lup-20(29)-en-30-al (31), obtained from the stems of Microtropis fokienensis and Perrottetia arisanensis, showed cytotoxic effects toward Hep3B, MCF7, Ca9–22, and HL60 cell lines (IC50: 1.4–4.7 µM).56 The acetyl ester 32 isolated from Boswellia carteri was cytotoxic against IMR-32 (IC50: 4.1 µM) and SK-N-SH (IC50: 4.7 µM) cell lines, while its free acid analogue 33 was less potent (IC50: >30.9 µM).80 Betulonic acid (34) from Betula platyphylla showed cytotoxic effects against KB (IC50: 2.3 µM) and KB-C2 (IC50: 3.8 µM) cell lines.81
2.2 The ursane and oleanane groups
The gum resin of genus Boswellia (olibanum or frankincense) has been used traditionally in Asia and Africa as a folk medicine for the treatment of arthritis and inflammatory disorders.82 The main active components of the resin are boswellic acids (BAs, 35–40), which are pentacyclic triterpene carboxylic acids isolated from various Boswellia species (Burseraceae), including B. carterii, B. frereana, B. papyrifera, B. sacra, and B. serrata.83,84 Chemically, BAs can differ in the positions of the two methyl groups on ring-E; β-BAs (35–38) have 19,20-dimethyl substitution, while α-BAs (39–40) have 20,20-dimethyl substitution, classified as the ursane and oleanane groups of triterpenes, respectively.

The relative potency of BAs, proposed mechanisms of action, and molecular targets, including non-redox-type 5-lipoxygenase (5-LOX), platelet 12-lipoxygenase (p12-LO), human leukocytes elastase (HLE), cytochrome P450 enzymes (CYP 2C8/2C9/3A4), topoisomerases I and IIa, and NF-κB kinase (IKK α/b), have been reviewed.83,85 An inhibitory effect on cycloxygenase-1 (COX-1) by 38 was also identified.86 These targets are indicative of the anti-inflammatory (5-LOX, p12-LO, HLE, and COX-1) and anticancer (topo I/IIa and IKK α/β) activities of the BAs.
Fig. 1 summarizes the signal transduction scheme of 38-induced apoptosis. Although the effector caspases and other markers have been identified, the primary molecular targets as well as the precise role of mitochondria are still unclear. For details, see the review by Poeckel et al.83
Fig. 1.

Simplified apoptotic signaling scheme and modulation by AKBA (from ref. 83): (+) positive or upregulatory effects of AKBA; (−) negative or inhibitory effects. IKK is the only target for which a direct inhibition by AKBA has been postulated. n.e.: no effect.
Compound 38, which is commonly known as AKBA (3-O-acetyl-11-keto-β-boswellic acid), has been the most well explored BA. A recent discovery revealed that 38 showed moderate to low toxicity against human skin-derived normal cell lines.87 However, 38 induced apoptosis in PC-3 and LNCaP cells through a death receptor (DR-5)-mediated pathway, which is a signal transduction cascade involving the activation of capase-8 and capase-3 in apoptosis.88 The inhibitory effect of 38 was significant in comparison with those of indomethacin and cyclophosphamide against bFGF-induced angiogenesis using an in vivo matrigel plug assay.89 Two C-3 acetylated BA derivatives, 36 and 40, showed almost comparable activity with cisplatin against neuroblastoma (NB-39 and SK-N-SH) cell lines.80 Compound 40 triggered apoptosis by activation of caspase-3 and induction of DNA fragmentation in PC-3 cells and PC-3 xenotransplants in chick chorioallantoic membrane.90 Compound 36 showed similar inhibitory effects to those of β-carotene, reported as 380 and 397 molar ratios, respectively, to 32 pM TPA used as inhibitor, in the TPA-mediated EBV-EA activation assay.80 In several anti-inflammatory studies, treatment with BAs from B. serrata diminished chemical- or stress-induced edema, arthritis, and gastric ulcer, with moderate to low activity.91,92 Compound 38 demonstrated a synergistic effect in combination with indomethacin on kainic acid-induced excitotoxicity and oxidative damage in vitro,87,93 and with glucosamine for the treatment of murine edema and arthritis.94
In anti-inflammatory and other assays, β-BAs generally show greater potency than α-BAs. In contrast, 3-O-acetyl-α-boswellic acid (AαBA, 40) was the most potent analogue (IC50: 1–3 µM) compared with β-derivatives against purified topo I.83 The cytotoxic SAR of BAs has seldom been addressed, but in recent studies, 40 showed higher cytotoxic activity than 39, indicating the importance of a C-3 acetyl group for optimal potency of α-BAs.80 Furthermore, 4-amino β-BA analogues (41–43) displayed enhanced cytotoxicity compared with the corresponding β-BAs possessing a 4-carboxylic acid, while C-3 β-epimers of these amino analogues (44) did not show improved activity.95 A CDDO-like (see Section 2.2) analogue (45) showed enhanced activity against A-549, A-431, HL-60, MCF-7, T47D, and HT1080 cell lines, with sub-micromolar EC50 values (EC50: 0.10–0.59 µM).96

Ursolic acid (3β-hydroxy-urs-12-en-28-oic acid (46) is a prevalent pentacyclic triterpenoid carboxylic acid. It has been found in various plants in both aglycone and glycoside forms, and traditional uses of plants containing 46 in folk medicine are abundant. Modern studies have shown that 46 possesses many biological activities, such as anti-oxidative, anti-inflammatory, antitumor, and hepato-protective activity. The diverse inflammatory effects of 46 were reviewed by Ikeda et al. in 2008.97 This review also summarized the inhibitory activity of 46 on cancer cells, which may be due to suppression of inhibitors of the NF-κB pathway and p65 phosphorylation, thereby causing down-regulation of the expression of downstream oncogenes. Compound 46 may also reduce skin tumor formation by inhibiting the binding of carcinogen to epidermal DNA or cell membrane. Furthermore, 46 induces cell differentiation and apoptosis in certain cancer cell lines. For details, see the review by Ikeda et al.97
Current work has provided more evidence on the bioactivity of 46. Inhibitory effects were observed on both the PI3K-Akt and MAPK P44/42 pathways, which are associated with cell apoptosis in endometrial cancer cell lines (SNG-II and HEC108).98 Compound 46 exhibited chemopreventive effects during the cancer initiation phase of an in vivo inhibitory assay of aberrant crypt foci (ACF), which are putative precursors of colon cancer, and increased neutral sphingomyelinase (N-SMase) activity.99 Compound 46 also inhibited endogenous reverse transcriptase (RT), an enzyme involved in the control of cell proliferation and differentiation, in melanoma (A375) and anaplastic carcinoma (ARO) cell lines. Down-regulation of the expression of two cancer-related genes, c-myc and cyclin-D1, in A375 and/or ARO cells was stimulated by 46.100 In contrast, 46 reduced the amount of L1210 cell growth inhibition induced by doxorubicin. The result might be due to scavenging of ROS involved in the apoptotic processes induced by doxorubicin.101 A similar inhibitory pattern was observed on H2O2-induced DNA breakage in HL-60 cell lines treated with 46.102

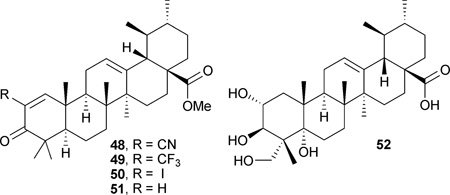
In other SAR studies, compounds with an acetyl moiety on the C-3 hydroxy group (47) showed greater potency than those with a free hydroxyl.103 In addition, the CDDO modification strategy was applied. The C-2 cyano or trifluoromethyl derivatives of 1-en-3-one-ursolic acid (48 and 49) showed better activity than C-2 iodo- and non-substituted analogues (50 and 51) in anti-proliferation assays using KU7, 253JB-V, Panc-1, and Panc-28 cancer cell lines (IC50: 0.17–1.13 µM).104
The bioactivities of other naturally occurring ursane-triterpenes have also been reported. Topical application of asiatic acid (52) resulted in a significant reduction in skin tumor formation induced by TPA, and TPA-induced [3H]thymidine incorporation was lessened. Concurrently, 52 inhibited the TPA-induced generation of NO and expression of iNOS and COX-2, which are important factors in tumor promotion.105 In addition, 52 induced apoptosis in PPC-1 and U-87MG cancer cells. The apoptotic phenomena included early activation of caspase-2, −3, and −8 in PPC-1 cells111 and caspase-9 and −3 in U-87MG cells.112 Intracellular release of Ca2+ was observed in both cell lines, and U-87MG cell death was alleviated by BAPTA/AM, an intracellular Ca2+ inhibitor. These results implied that the cell death is attributable to Ca2+-mediated necrotic apoptosis.106
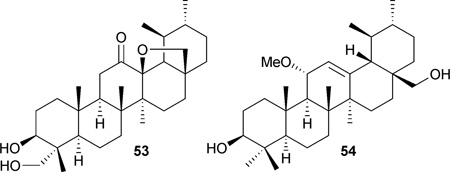
Two new ursane-type triterpenes, microfokienoxane C (53) and 3β,28-dihydroxy-11α-methoxyurs-12-ene (54), were isolated from the leaves of Microtropis fokienensis. Compound 53 was cytotoxic against HepG2 and Hep3B cancer cell lines, with IC50 values of 8.1 and 9.5 µM, respectively. Compound 54 showed activity against the HepG2 cell line, with an IC50 value of 9.2 µM.107
Glycyrrhizin (55) and its aglycone, glycyrrhetic acid (56, GA), are abundant constituents of licorice (most familiarly the species Glycyrrhiza glabra). The aglycone 56 exists as 18α- and 18b-isomers, and 18b-GA was more effective than 18α-GA in inhibiting the mutagenicity induced by various mutagens.108 A recent review on the pharmacological effects of Glycyrrhiza sp. described the broad-spectrum of bioactivities of 55 and 56, including anti-inflammatory, antiviral, hepatoprotective, antitumor, and immunomodulatory.109 For details about prior studies on antitumor activities of 55 and 56, see the reviews by Wang and Nixon108 and Asl et al.109

Although several studies have found that GA demonstrates cytotoxic or apoptotic activity, most of the results have shown only moderate or low potency. Thus, investigation has focused more on the preparation of active derivatives and chemosensitizing activity. Two 56 analogues, 57 and 58, which possess a C-3 alkoxyimino group and a C-29 carboxylic acid or methyl ester, showed slightly stronger antiproliferative and apoptotic activities than 56. The GI50 value improved from 63 µM to 19 µM against HL-60 cells. However, the C-29 free acid (58) is important, because the methyl ester analogue (57) was less potent in the apoptosis assay.110 The application of 2-cyano-1-ene-3-one (CDDO) modification on GA was successful, as methyl 2-cyano-3,11-dioxo-18β-olean-1,12-dien-30-oate (59, β-CDODA-Me) was active against HL-60 cells at a sub-micromolar concentration (EC50: 0.93 µM).96 Another study reported that 59 and the 2-trifluoromethyl derivative (60) were more cytotoxic against KU7 (IC50: 0.38–1.59 µM), Panc-1 (IC50: 0.82–1.22 µM), Panc-28 (IC50: 1.14–1.80 µM), and 253JB-V cells (IC50: 0.25–0.67 µM) compared to 56. The relative potencies of 59 and 60 were dependent on cell line.111

A mechanism study on 59 revealed that its potent antitumor activity may be due to differential induction of two tumor-suppressor genes, Krüppel-like factor-4 (KLF-4) and caveolin-1, in colon cancer cells.112 Subsequent pro-apoptotic responses were also induced through activation of kinases ATF3, NAG-1, and p21 or through kinase-independent inhibition of AR and PSA, resulting in the growth inhibition of LNCaP cells.113 Compound 56 showed chemosensitizing effects with various clinical oncology drugs; it enhanced the sensitivity of vinblastine against KB-C2 cells and doxorubicin against KB/MRP cells. In addition, 56 partly reversed multidrug resistance in P-gp-expressing cells (KB-C2) or multidrug resistance protein 1 (MRP1)-expressing cells (KB/MRP), by increasing the intracellular accumulation of the antitumor drugs.114 In an interesting study, chemical conjugates of 56 and paclitaxel were synthesized. However, although the conjugated compounds (61) showed better activity than 56, they were still less potent than paclitaxel itself.115 Based on these findings, it is likely that glycyrrhetic acid analogues elicit antitumor activity through multiple pathways.
Oleanolic acid (62, OA), a well known triterpene occurring in numerous varieties of plants, possesses many biological properties, including anti-inflammatory, trypanocidal, anti-HIV, and cytotoxic activities.116 In 1988, OA was reported to suppress TPA-induced tumor promotion.117 Synthetic A-ring modified OA analogues with improved cytotoxicity were pursued, and 2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid (63, CDDO) as well as its C-28 methyl ester, CDDO-Me (64, RTA402), were designed and synthesized by Honda et al. in 1998. Both of these synthetic derivatives strongly inhibited the production of NO induced by IFN-γ in mouse macrophages (IC50: 0.4 nM).118 Further derivatization of CDDO to yield imidazolides (65, CDDO-Im), amides (66 and 67), or a dinitrile (68, Di-CDDO), significantly increased the anti-NO activity. Among these derivatives, CDDO-Im (65), was 100 times more potent than CDDO, with an IC50 value of 1 pM.119 In addition to the strong anti-inflammatory activity, CDDO derivatives were reported to exhibit other bioactivities, including cytoprotection, cancer cell growth inhibition, and apoptosis induction.120 Preliminary studies confirmed that OAs have the potential to become an important new treatment for multiple forms of cancer, and both 63 and 64 are currently in phase I clinical trials for the treatment of metastatic or unresectable solid tumors or lymphoma. The pharmacological effects induced by OA-related derivatives were summarized in very recent reviews by Liby et al.,120 Sogno et al.121 and Sultana et al.116

Xanifolia-Y (69) was isolated from Xanthoceras sorbifolia, and several hydrolyzed derivatives (70–72) were prepared. Glycoside 69, which has angeloyl groups on C-21 and C-22, was more cytotoxic (IC50: 2.63 µM) to OVCAR3 cells than either the non-esterified glucoside or the di-esterified aglycone (70 and 71, IC50: >120 µM and 14.2 µM, respectively). Thus, the ester moieties on C-21 and C-22 are required for the cytotoxicity, while hydrolysis of the trisaccharide slightly lowered activity.122
Maslinic acid (73), an oleanane triterpene identified from Crataegus oxyacantha in 1953,123 was recently also isolated from apple peel along with the derivatives 74 and 75. All three compounds were evaluated for antitumor activity, and 74 together with 75 showed minor antiproliferative effects against HepG2 (EC50: 17.9–20.6 µM), MCF-7 (EC50: 20.9–29.2 µM), and Caco-2 (EC50: 8.9–14.2 µM), while 73 showed an effect only on the Caco-2 cell line (EC50: 15.4 µM).124 Compound 73 was also isolated quantitatively from olive fruit, but showed only a weak anti-proliferative effect against the HT-29 cell line (EC50: 101.2 µM) without any necrotic effects.125 25-Hydroxy-3-oxoolean-12-en-28-oic acid (76, amooranin) was isolated from the stem bark of Amoora rohituka. It was cytotoxic against the SW620 cell line (IC50: 6.2 µM) and delayed tumor growth rate in SW620 xenografts in nude mice. Concurrently, a microarray study of 76 showed that it caused down-regulation of VEGF and JUN genes, which are associated with angiogenesis and antiproliferation of cancer cells, respectively.126 Capilliposide B (77), an epoxy-oleanane saponin isolated from Lysimachia capillipes, was cytotoxic against human A-2780 cells, with an IC50 value of 0.08 µM.127 In a semi-synthetic approach,128,129 analogues were prepared from 77 and screened for cytotoxicity. However, only the 3-O-β-d-glucopyranoside (78) and 3-O-β-d-galactopyranoside (79) derivatives showed enhanced antitumor activity (IC50: 30–40 µm).128 Two D:C-friedooleanane-type compounds (multiflorane-type), 80 and 81, isolated from the stems of Lagenaria siceraria, showed significant cytotoxicity toward the human SKHEP-1 hepatocellular cancer cell line with IC50 values of 8.0 and 4.6 µm, respectively.130

2.3 The lanostane group
Ganoderic acid D (82) isolated from Ganoderma lucidum was assessed for anti-proliferation activity and showed an IC50 value of 17.3 µm against human cervical carcinoma cells (HeLa). Furthermore, the 82-treated HeLa cells were arrested at the G2/ M phase of the cell cycle with apoptosis. A simultaneous proteomic study led to the identification of 21 genes regulated by 82, while the 14-3-3 protein-related genes may play an important role for 82-induced cytotoxicity.131 Ganoderiol F (83) derived from Ganoderma amboinense, was found to inhibit proliferation of cancer cell lines, including chronic myelogenous leukemia (K562, IC50: 4 µm) and hepatocellular carcinoma (HepG2 and Huh7, IC50: 17 µm and 8.5 µm, respectively). Activation of MAPK/ EKR and up-regulation of CDK inhibitor p16 were found in early stages of treatment with 83 and were presumed to cause cell-cycle arrest at the G1 phase and trigger premature senescence of HepG2 cells.132 Dehydrotrametenolic acid (84), a lanostane triterpene isolated from the sclerotium of Poria cocos, selectively inhibited H-ras-transformed J82 cells and induced apoptosis through the caspase-3 pathway. Cells treated with 84 were arrested at the G2/M phase and accumulated at the sub-G1 phase, with a high GI50 value of 40 µm. Compound 84 also regulated the expression of H-Ras, Akt, and ERK, which are the downstream proteins of H-Ras signaling pathways.133 In addition, a subsequent study of the same fungus yielded 12 lanostane triterpenes (85–96), which showed inhibitory effects in the EBV-EA activation assay in Raji cells. Compounds 90 and 96 also showed inhibitory effects in DMBA- and TPA-mediated in vivo murine skin tests.134


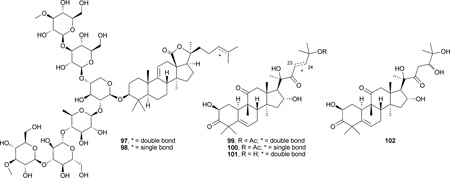
Impatienside A (97) and bivittoside D (98), two lanostane-type triterpenes containing C-3 hexasaccharide moieties, were isolated from the sea cucumber Holothuria impatiens. The two glycosides exhibited in vitro cytotoxicity equal or slightly better than those of the clinical antitumor drug etoposide (VP-16) against seven different human tumor cells, including colon cancer (HCT-116 and HT-29), lung cancer (A549), hepatoma (HepG2), prostate cancer (DU145), breast cancer (MCF-7), and nasopharyngeal cancer (KB), with IC50 values of 0.25–1.9 µm.135
Cucurbitane-type triterpenes, isomers of lanostanes, constitute a group of diverse substances that are known for their bitterness and toxicity. They were isolated initially from Cucurbitaceae plants and were later found in other plant families. Besides cytotoxicity and anticancer activity, cucurbitacins also exhibit other wide ranging in vitro or in vivo pharmacological effects, such as purgative, anti-inflammatory, and anti-fertility. However, these compounds have had only very limited usage due to their non-specific toxicity. The distribution, structure characterization, and bioactivities were reviewed by Chen et al. in 2005.136 More recent studies showed that cucurbitacin B (99) possessed weak inhibitory effects on laryngeal squamous cell carcinoma (Hep-2, IC50: 39.06 µm), which may be due to the inhibition of STAT-3, a transcription activator in cell growth.137 23,24-Dihydrocucurbitacin B (100), isolated from roots of Trichosanthes kirilowii, inhibited the proliferation of human breast cancer cells (Bcap37, IC50: 1.5 µm) and arrested the cell cycle at the G2/M phase. The antiproliferation activity was reduced by the caspase-family inhibitor Z-VAD-FMK, suggesting a mitochondria-dependent pathway.138 Cucurbitacins D (101) and J (102) elicited anti-proliferative effects on both hepatocellular carcinoma BEL-7402 cells (IC50: 1.41 and 1.37 µm, respectively) and malignant melanoma SK-MEL-28 cells (IC50: 1.22 and 1.28 µm, respectively).27

2.4 The dammarane–euphane group
Ginsenosides are a series of compounds comprised of more than 60 triterpenes and related glycosides isolated from the leaves, stems, berries, and roots of different Panax species. Recently, ginsenosides from natural resources were evaluated for their antitumor activity. However, compared to other triterpenes, most ginsenosides showed low to no cytotoxicity (IC50: >20 µm) against various cancer cell lines.139–144 The anti-angiogenic properties of 20(S)-protopanaxadiol (103) and 20(S)-protopanaxatriol (104) were evaluated in an in vitro angiogenesis assay using HUVECs, in which the compounds demonstrated strong anti-proliferative activity (EC50: 2.16–6.64 µm).142 Furthermore, 20(S)-25-methoxyprotopanaxadiol (105) induced apoptosis and cell cycle arrest in the G1 phase, inhibited proliferation of T98G, HPAC, A-549, H1299 and PC-3 cell lines (IC50: 5.0, 5.8, 5.7, 4.9, and 5.8 µm, respectively), and was 5- to 15-fold more potent than 104.145,146 In addition, three semi-synthetic derivatives with a C-20 sugar moiety (106–108) showed significant cytotoxicity against MCF, SK-MEL-2, and B16 cancer cell lines147 (see Table 2).
Table 2.
Cytotoxicity of 106–108 against MCF, SK-MEL-2, and B16 cancer cell lines.145
| Cell line | IC50 (µm) |
||
|---|---|---|---|
| 106 | 107 | 108 | |
| MCF-7 | 0.62 | 2.69 | 1.86 |
| SK-MEL-2 | 1.82 | 2.19 | 0.19 |
| B16 | 7.62 | 6.66 | 0.37 |
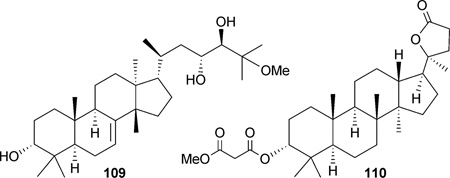
25-Methoxyhispidol A (109), a tirucallane triterpene isolated from the fruit of Poncirus trifoliata, demonstrated a slight antiproliferative effect (IC50: 21.0 µm) against the SKHEP-1 cell line, which was postulated to occur through arrest in the G0/G1 cell cycle and induction of apoptosis.148 The dammarane derivative 110 was isolated from the flower spikes of Betula platyphylla.81,149 Compound 110 inhibited the efflux of rhoda-mine 123 KB-C2 cells, implicating MDR-reversing effects by modulating P-gp.149
Five dichapetalin-type triterpenoids, dichapetalins A (111), I (112), J (113), K (114), and L (115), were isolated from the stem bark of Dichapetalum gelonioides. Compounds 111–113 showed selective inhibitory activity (IC50: 0.3–0.8 µm) against the SW626 human ovarian cancer cell line, while 114 and 115 showed broader cytotoxicity against Lu1, LNCaP, SW626, MCF-7, and HUVEC cell lines (IC50: 1.6–14.2 µm).35 Acutissimatriterpenes A, B, and E (116–118) were isolated from the aerial parts of Phyllanthus acutissima.150 Compounds 116 and 117 exhibited cytotoxicity only against P-388 tumor cell line, while compound 118 showed cytotoxicity against P-388, MCF-7, and Lu-a tumor cell lines (IC50: 0.007–7.100 µm). Most impressively, 118 demonstrated remarkable cytotoxicity against P-388 (IC50: 0.007 µm), and was more potent than ellipticine, the positive control (IC50: 0.813 µm).150 However due to the complex structures and the limited extent of the study, SAR conclusions could not be readily established for dichapetalin-type triterpenoids.
2.5 Other triterpenoids
Quinonoid triterpenes
Celastrol (119), a quinone methide triterpenoid, was isolated recently from Celastrus orbiculatus,151 Cheiloclinium cognatum,152 and Maytenus chuchuhuasca.153 It was already known that celastrol exhibits anti-inflammatory,154,155 antioxidant,155 neuro-protective,156,157 and anti-infective properties, and its strong cytotoxic effect was reported as early as 1981.158 Very recent studies showed that 119 was cytotoxic against Panc-1 pancreatic cancer cells (IC50: 3 µm) in vitro, active against Panc-1 xenografts in vivo in mice, and inhibited tumor metastasis in the RIP1-Tag2 transgenic mouse model of pancreatic islet carcinoma.159

Previous investigation on the strong antitumor effect of 119 prompted a series of studies on its molecular target. It enhanced the cytotoxic effects of TNF, paclitaxel, and doxorubicin, as well as the TNF-κB pathway and NF-κB-regulated gene products.160 Further pharmacological study showed that 119 inhibited the activation of NF-κB by blocking the phosphorylation and degradation of IkB kinase in the upstream NF-κB signal transduction cascade.151 Both 119 and gedunin (136) were found to inhibit HSP90 function by acting outside the ATP-binding pocket, which is similar to the action of the existing drugs cisplatin and novobiocin.159,161,162 A concurrent study revealed that 119 suppressed the activation of aryl hydrocarbon receptor-dependent gene expression in oral leukoplakia (MSK-Leuk1) cells, which implicates a chemopreventive effect.163 Investigation of its anti-angiogenesis effect revealed that 119 suppressed angiogenesis by acting on the VEGF tyrosine kinase receptors, VEGFR-1 and VEGFR-2, but not VEGF itself.164 Compound 119 also showed both in vitro and in vivo chymotrypsin-like proteasomal inhibitory activity toward purified proteasome (20S and 26S proteasome, IC50: 2.5–5 µm) in PC-3 or LNCaP cells, and inhibited PC-3 xenografted tumors in nude mice.165
Subsequent SAR studies on semi-synthetic analogues of 119 concluded that the quinone methide moiety is important for cytotoxic activity. Dihydrocelastrol (120) and 6-oxo-pristimerol (121), derivatives with an aromatized quinone methide moiety, were inactive in both cytotoxicity166 and anti-tubulin polymerization assays.153 The C-29 free acid present in 119 is not required for its ability to induce apoptosis; C-29 ester or amide derivatives (122) showed an equal or lower potency in inhibiting the growth of human and mouse melanoma tumor cells.166 Analogues that lack C-29, such as tingenone (123) and 22β-hydroxytingenone (124), showed lower activity than 119 in in vitro tubulin polymerization inhibition and cytotoxicity assays against RPMI8226 cell lines.153

Tetranortriterpenes (limonoids)
Triterpenoids belonging to the limonoid family occur as glucosides and aglycones in fruits of Rutaceae and Meliaceae plant families. They show putative antitumor properties, and have been sufficiently reviewed regarding their distribution,167 biosynthetic pathway,168,169 and bioactivity.170,171 The latest study on the antiproliferative effects of three limonoids, limonin (125), nomilin (126) and obacunone (127), confirmed that, as either aglycones (125–127) or glycosides (128–130), these compounds exert versatile action against neuroblastoma cancer cells (SH-SY5Y). They induced apoptosis, arrested the cancer cell cycle at the G1 phase, and showed an aneuploidic effect, which elevated the aneuploid numbers in treated cancer cells compared with untreated ones. Both aglycones and glucosides were toxic to the cells, but aglycones were less effective in causing rapid cell death.172

3α,7α-Dideacetylkhivorin (131) was isolated from Khaya senegalensis, and subsequent cytotoxic screening showed that it was active against SiHa (IC50: 0.11 µM), MCF-7 (IC50: 0.14 µM), and Caco-2 (IC50: 0.07 µM) cancer cell lines.173 Nimbolide (132), a limonoid extracted from the flowers of Azadirachta indica (neem tree), exhibited potent cytotoxic activity against U937 (IC50: 1.24 µM), HL-60 (IC50: 1.12 µM), and THP1 (IC50: 1.42 µM) cells and antiproliferation activity against B16 (IC50: 1.74 µM) cell lines. In a follow-up study using the U937 cell line, 132 increased the number of aneuploid cells and the appearance of annexin V positive cells, implicating the occurrence of cell arrest and apoptosis, correspondingly.174 Two nimbolide derivatives, 133 and 134, were synthesized from 132, but showed weak activity in the brine shrimp lethality bioassay, indicating the importance of the lactone ring.175

Gedunin (135), a limonoid with antimalarial, insecticidal, and antitumor activities, was recently isolated from Xylocarpus granatum176 and Cedrela sinensis.177 It exerted weak antitumor activity against the CaCo-2 cell line, with an IC50 value of 16.83 µM.176 However, 135 and its derivatives 136–140 showed moderate cytotoxicity against P-388 murine leukemia cells, with IC50 values ranging from 6.0–17.1 µM. In the same study, 141– 143 exhibited very weak or no activity (IC50: >116 µM), suggesting that an oxygenated functionality at the 11α-position is critical.177 Further modification at C-7 on 135 gave two active analogues with C-7 carbamate (144) and ketone (145) groups. These compounds demonstrated antiproliferative activity comparable to 135, and acted by modulating heat shock protein (Hsp90) in MCF-7 and SkBr3 cells.178 Methyl angolensate (146), a natural tetranortriterpenoid derived from the root callus of Soymida febrifuga,179 possessed low cytotoxic activity (IC50: 100.0 µM), but did induce apoptosis by triggering the intrinsic pathway.180

Quassinoids
Quassinoids are bitter constituents and secondary metabolites exclusively of Simaroubaceous origin. They are regarded biogenetically as degraded triterpenoids. Quassinoids have diverse bioactivities including antifeedant, insecticidal, herbicidal, antiparasitic, antimalarial, and antitumor effects.181
As early as the 1970s, reports ascribed a wide range of antitumor activity to bruceantin, a quassinoid triterpene, which elicited major interest in quassinoids as promising lead drugs. However, the complex chemical structures of quassinoids have limited their anticancer potential. For more details, see the review by Vieira et al.181
6α-Tigloyloxychaparrinone (147) was isolated from Ailanthus integrifolia in 1978,182 and its antitumor activities have been reported.183,184 More recent studies led to the isolation of 147 from A. altissima. It showed inhibitory effects on hypoxia-induced VEGF, erythropoietin expression, and the activation of HIF-1α, a transcription factor protein that promotes tumor cell adaptation and survival under hypoxic conditions.185 Brusatol (148) showed cytotoxic activity against P-388 murine cancer cells, with an IC50 value of 0.012 µM. The A-ring modified compounds 149 and 150 showed reduced activity, indicating that the C-2 enolic oxygen is essential for activity. The 11-ketone analogue 151 showed even weaker activity, indicating that a β-hydroxy group at C-11 is also important.186

Triterpenes from marine sources
Numerous sodwanone triterpenes have been purified from Axinella species. Among them, sodwanone V (152) inhibited activation of HIF-1 in T47D (IC50: 15 µM) and PC-3 (IC50: 15 µM) cells. It also showed weak cytotoxicity against MDA-MB-231 breast tumor cells (IC50: 23 µM). 10,11-Dihydrosodwanone B (153) showed moderate cytotoxicity against T47D cells (IC50: 22 µM). Four derivatives, 154–157, inhibited hypoxia-induced HIF-1 activation in T47D cells (IC50: 20–25 µM).187

Two sipholane-type triterpenes, sipholenol A (158) and sipholenone A (159), were isolated from the marine sponge Callyspongia siphonella. They showed cytotoxic activity against the MCF-7 breast cancer cell line, with IC50 values of 1.2 and 0.9 µM, respectively. At a dose of 0.026 µM per pellet, 159 also showed anti-angiogenic activity in the chorioallantoic membrane (CAM) assay.188 In a chemosensitization study, 158 potentiated the cytotoxicity of the antitumor drugs colchicine, vinblastine, and paclitaxel against MDR nasopharyngeal cancer cell lines (KB-C2 and KB-V1), by inhibiting P-gp(ABCB1)-mediated drug efflux.189

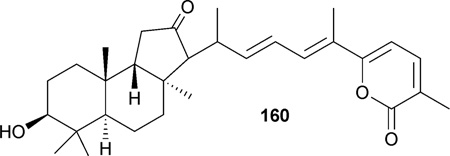
Jaspolide B (160), a novel isomalabaricane-type triterpene isolated from marine sponges, Jaspis spp,190 exhibited cytotoxicity toward hepatoma (Bel-7402, IC50: 29.1 µM; HepG2, IC50: 29.5 µM) and human promyeloleukemic (HL-60, IC50: 0.78 µM) cell lines.191,192 Subsequent studies showed 160 induces apoptosis and cell cycle arrest in HL-60 at the G2/M phase191 and in Bel-7402 at the G1 phase.192
3 Anti-HIV effects of triterpenoids
3.1 The lupane group
In 1994, the authors’ Natural Products Research Laboratories first discovered that betulinic acid (1, BA) showed an inhibitory effect against HIV-1IIIB replication, with an EC50 value of 1.4 µM.193 Since then, betulinic acid has been used as a pre-eminent molecular scaffold in the development of triterpene analogues as anti-HIV drugs. Systematic structural modifications were carried out on the C-3 hydroxyl, A-ring, C-19 isopropenyl group, and C-28 carboxyl moiety, which led to the discovery of many potent anti-HIV agents (Table 3).194,195
Table 3.
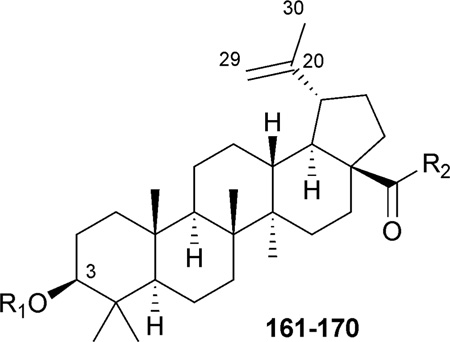
Structures and anti-HIV activity of C-3- and/or C-28-modified betulinic acid (1) derivatives 161–170a
 | |||
|---|---|---|---|
| No. | R1 | R2 | IC50 (µM) |
| 161 (Bevirimat) | DSB | OH | 0.00035196 |
| 0.0013197 | |||
| 0.011205 | |||
| 162 | 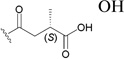 |
OH | 0.0087197 |
| 163 | 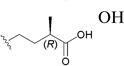 |
OH | 0.12197 |
| 9 | H | 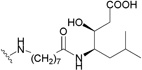 |
0.053202 |
| 164 | H | 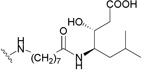 |
0.05–2.00199 |
| 165 | H | -NH-(CH2)7-NHCOCH3 | 0.047202 |
| 166 | DSB | -NH-(CH2)7-NHCOCH3 | 0.0026202 |
| 167 | DSB | 0.007205 | |
| 168 | DSB | 0.006205 | |
| 169 | DSB | 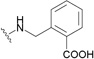 |
0.006206 |
| 170 | DSB | 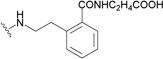 |
0.003206 |
DSB: 3′,3′-dimethylsuccinyl.
The C-3 hydroxy group can be readily acylated with a wide variety of anhydrides and acid chlorides. Such modification resulted ultimately in the identification of bevirimat (3-O-3′,3′-dimethylsuccinyl BA, 161), which displayed a significant anti-HIV EC50 value of 0.35 nM.196 The SAR study also showed that an ester functionality at C-3, an appropriate length of the C-3 side chain, a terminal carboxylic acid moiety, and dimethyl substitution at the C-3′ position are critical for the enhanced antiviral potency of 161. Further SAR demonstrated that, within the 3′-dimethyl moiety, the 3′S-methyl contributed more towards the activity, because 3′S-monomethylsuccinyl-substituted BA (3′S-MSB, 162) and 161 showed similar antiviral potency (EC50: 8.7 and 1.3 nM, respectively), while 3′R-MSB (163) showed much reduced activity (EC50: 0.12 µM).197
Mechanism of action studies revealed that 161 retained its nanomolar antiviral potency against HIV-1 isolates resistant to reverse transcriptase (RT) and protease (PR) inhibitors. To define the target of 161 (bevirimat), scientists from Panacos Pharmaceuticals Inc. and the National Institutes of Health characterized the virus produced by cells treated with the compound.198 The studies showed that 161 inhibited the processing of the viral Gag polyprotein at a specific step, the conversion of the capsid precursor p25 to mature capsid (CA) p24, in a dose-dependent manner.198 The impaired cleavage of p25 to p24 led to the production of morphologically defective, non-infectious, immature viral particles (Fig. 2). Therefore, 161 was designated as amaturation inhibitor (MI),a new class in the antiretroviral family. It is currently in Phase IIb clinical trials and will enter Phase III clinical trials in 2009.
Fig. 2.

Bevirimat (161) targets Gag at the CA-SP1 cleavage site to interfere with the processing of p25 into mature p24, resulting in the production of morphologically defective, non-infectious viral particles. (Panacos, XVII international HIV drug resistance workshop, Spain, 2008).
Conversely, C-28 ω-aminoalkanoic acid derivatives of 1 were discovered to function by blocking the viral entry into the host cells.199,200 Incremental chain lengthening significantly influenced the anti-HIV potency of the derivatives, while the introduction of a second aminoalkanoic acid at the end of the C-28 side chain could further modulate the antiviral potency.195 This research led to the discovery of RPR103611 (164),199 a statine derivative, which inhibits the infectivity of several HIV-1 strains with EC50 values in the range of 0.05–2 µM, and 9 (IC9564),201 a stereoisomer of 164, which showed equipotency in the antiviral testing. A43-D (165) was designed with a slightly simplified C-28 substituent.202 It also showed similar anti-HIV activity compared to 9 and 164.
A mechanism of action study discovered that C-28-modified BA analogues function at a post-binding step necessary for virus entry to the host cell. Detailed study found that 9 induced a nonproductive gp120 conformation that is not able to trigger a conformational rearrangement in gp41 for membrane fusion.203 Evidence also indicated that 9 can target the V3 loop of gp120, a domain involved in chemokine receptor binding.204 The exact binding target of triterpene derivatives is still under investigation.
Modification of the C-19 isopropenyl group showed little effect on the anti-HIV activity. Neither saturation of the 20(29) double bond nor allylic substitution at C-30 through an ether or thioether linkage influenced the anti-HIV activity of the 1 derivatives significantly.205 Further research demonstrated that the C-30 position serves as a good place to incorporate water-solubilizing moieties and to increase the hydrophilicity of this compound class.205
Interestingly, as indicated, the anti-HIV-1 targets of triterpene analogues can vary depending on the side chain modification positions. C-28-modified analogues of 1 are potent HIV entry inhibitors, while C-3-modified derivatives function by blocking virus maturation. A combination of these two modifications led to the identification of A12-2 (166), a bifunctional HIV inhibitor that interferes with both viral entry and maturation.202 Compounds 167 and 168, in which a secondary amine forms the C-28 amide bond, exhibited significantly increased metabolic stability in pooled human liver microsomes.205 Compounds 166–168 showed very potent anti-HIV activity, which was slightly better than that of 161. Gerrish et al. synthesized another series of 3,28-disubstituted 1-analogues.206 These compounds contained a dimethylsuccinyl ester side chain at C-3 and variously substituted benzyl and phenethyl amides at C-28. Several compounds in this series showed potent anti-HIV activity, and 169 and 170 showed the best pharmacokinetic/pharmacodynamic (PK/PD) properties.206 Generally, substitutions at the ortho-position of the benzyl or phenyl ring provided the best activity. The 3,28-dimodified derivatives may serve as attractive promising leads to develop a next generation of 1-derived HIV-1 inhibitors as clinical trials candidates.
2β-Carboxyl-3β-hydroxyl-norlupA(1)-20(29)-en-28-oic acid (171), a lupane-like triterpene with a rearranged five-membered A-ring, was isolated from the thorns of Gleditsia sinensis. It showed significant anti-HIV activity, with an EC50 value of <0.13 µM.207 The fact that 171 differs from 1 only in the A-ring configuration suggests that A-ring functionalities are important pharmacophores for the antiviral activity of the lupane-type triterpenes.
A new 2,3-seco-lupane triterpene, 16β-hydroxy-2,3-seco-lup-20(29)-ene-2,3-dioic acid (172), was obtained during a search for antiviral agents in the stems of Stauntonia obovatifoliola.208 Compound 172 exhibited good anti-HIV-1 protease activity (EC50: 17.8 µM), compared with the control 62 (EC50: 54.38 µM), a known HIV-1 protease inhibitor.208 This result suggested that a seco A-ring with dual polar moieties may increase the antiviral protease activity of triterpenes. In a concurrent study, lupane-, 30-noroleanane-, and oleanane-type triterpenes showed similar anti-HIV protease activity during testing. When analyzing the structures more carefully, it was discovered that the 16β-OH group is important to the enhanced antiviral potency of lupane triterpenes, as seen with compounds 172–175.

3.2 The ursane and oleanane groups
The oleanane-type triterpene maslinic acid (73) inhibits the HIV-1 protease. In order to further explore the antiviral activity, several different α- and ω-amino acids (single, dipeptide, or tripeptide) were coupled at the C-28 position of 73 by using both solution- and solid-phase synthetic methods. However, only compounds 176 [with 11-aminoundecanoic acid (11AUA) at C-28] and 177 (with a C-28 conjugate dipeptide of 11AUA and l-valine) showed approximately 50% inhibition activity against virus replication at 10 µM. These derivatives showed no cytotoxicity, although 73 itself displayed both cytotoxic and antiviral activities.209
Prior research on glycyrrhetic acid (56) and its synthetic derivatives mainly led to negative results.210,211 The modifications focused on substitutions of the C-3 hydroxy and C-30 carboxylic acid groups. Because the rigid triterpene scaffold determines the relative position of the functional side chains, the skeleton could be very important to the anti-HIV and other bioactivities. Therefore, the distance from the hydroxy group to the carboxylic moiety and the dihedral angles of the D/E rings were predicted using modeling software. The results provided a possible explanation for why analogues of 56 showed fewer anti-HIV effects than analogues of betulinic (1) or moronic acid (181).210,211

A recent paper reported the synthesis and anti-HIV activity of a group of analogues of α-d-glucosamine-conjugated glycyrrhetic acid (56).212 The glucosamine was linked to the 3β-hydroxy group through a spacer containing phthalic, maleic, or succinic acid residues. Although such modifications slightly increased the anti-HIV activity of 56, the antiviral potency of these derivatives remained only moderate. The most potent analogue 3-O-[3-(N-2-deoxy-α-d-glucopyranos-2-yl)-carbamoyl]-phthaloyl-11-deoxy-glycyrrhetic acid (178) had an anti-HIV EC50 value of 2.0 µM.212

The anti-HIV and other biological activities of oleanolic acid (62, OA) and its derivatives were reviewed by Sultana et al. in 2007.116 Basically, OA was identified as an anti-HIV principle, because it inhibited HIV-1 replication with an EC50 value of 3.7 µM and a therapeutic index (TI) of 12.8.213 Modification of OA at the C-3 position led to the discovery of 3-O-3′,3′-dimethylsuccinyl-OA (179), which had significant antiviral EC50 (0.86 nM) and TI (22 400) values.213 However, due to differences in the screening system, the reported activity of 62 may vary in different publications.211 The mechanism of action of 179 is similar to that of 161, which functions as a maturation inhibitor and blocks the cleavage of p25 to p24, resulting in the production of morphologically defective, non-infectious HIV particles. The SAR of 3-O-acyl OAs with different succinyl or glutaryl substitution showed similar trends compared with 3-O-acyl BA analogues, suggesting that the C-3 side chain is a very important pharmacophore, while the triterpene backbone may serve as a docker allowing the side chains to interact with the targets.

Previously, the optimal C-3 and C-28 side chains were identified in bi-functional betulinic acid (BA) analogues. Therefore, to extend the research focus on the replacement of triterpenoid docker, some 3,28-disubstituted OA derivatives were also prepared.210 Compound 180, an OA derivative with 1,7-dia-minoheptane linked at the C-28 position and 3′,3′-dimethyl-succinyl linked at the C-3 position, showed anti-HIV activity, with an EC50 value of 0.95 µM.210 This activity is lower than the identically substituted BA analogue, 166 (EC50: 0.0026 µM), indicating that BA is a better-fit docker than OA.
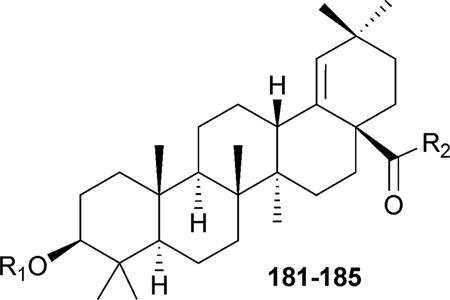
Moronic acid (181, MA) was isolated from Brazilian propolis, a herbal medicine used worldwide. It exhibited significant anti-HIV activity with an EC50 value of 0.2 µM and TI of 186.214 In 2006, the authors’ group reported the discovery of a series of MA derivatives that exhibited remarkable anti-HIV potency. Compounds 182–184, which contain C-3 3′,3′-dimethylsuccinyl substitution and different C-28 amide side chains, exhibited potent anti-HIV activity in H9 lymphocytes with EC50 values of 0.017, 0.0156, and 0.007 µM, respectively (Table 4).211 These activity results are similar to those of 161 (EC50: 0.007 µM), a drug candidate that is currently in phase IIb anti-AIDS clinical trials. Another 3,28-disubstituted MA derivative (185) was synthesized and had an antiviral EC50 value of 0.055 µM (Table 4).210 Mechanism of action study revealed that 3,28-disubstituted betulinic acid or moronic acid analogues exhibit both anti-fusion and anti-maturation activity. Thus, they were designated as bifunctional triterpenes.208,213 From the SAR, it was discovered that a longer C-28 side chain (as in 184 and 185) is helpful for enhanced antiviral potency.
Table 4.
Structures and anti-HIV activity of moronic acid (181) and its C-3- and C-28- modified derivatives 182–185

Another oleanane-type triterpene, papyriogenin A (186), was isolated from the flowers, pith, leaves, and fruit of Tetrapanax papyriferus. It showed antiviral activity, with an EC50 value of less than 1.7 µM in HIV-1IIIB-infected H9 lymphocytes.215
C-3-modified ursolic acid (UA),211,216 as well as 3- and 28-modified UA analogues (187–189),210,211 were synthesized and evaluated for anti-HIV activity (Table 5). The C-3-(3′,3′-dimethylsuccinyl)- and C-3-(3′,3′-dimethylglutaryl)-substituted derivatives of UA (188 and 189) exhibited anti-HIV replication, with EC50 values of 0.08 and 0.68 µM, respectively.211 Compound 187, which was evaluated with a different assay system, also displayed anti-HIV activity, with an EC50 value of 0.23 µM.210 When the activities of triterpenes with the same side chain but different scaffolds were compared, it was discovered that, among the synthesized compounds, the most effective molecular scaffold for anti-HIV activity remained betulinic acid (166, EC50 value: 0.0026 mM), followed by moronic, ursolic, and oleanolic acids.210
Table 5.
Structures and anti-HIV activity of C-3- and C-28-modified ursolic acid derivatives (187–190)
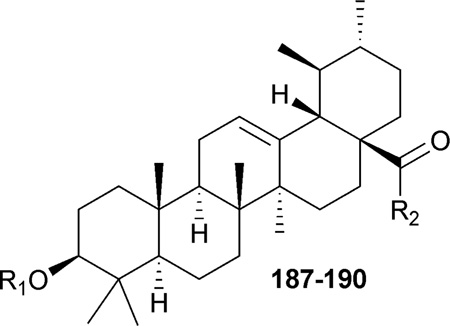
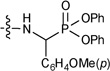
In addition, some a-aminophosphonates were conjugated to the C-28 of UA.217 However, all of the resulting analogues showed zero-to-weak anti-HIV entry activity. The best compound (190) inhibited viral entry by only 20% at a concentration of 0.1 µM.217 This result further proved the conclusion that a longer C-28 side chain is very important for triterpene-derived HIV entry inhibitors, with a carbon chain length of 7–10 being optimal.
3.3 The lanostane group
Plants of the genus Schisandra are known to contain dibenzo-cycloctadiene lignans, as well as lanostane and cycloartane triterpenes. In 2003, Sun’s group began a phytochemical study of several species of the Schisandra genus collected in the Yunnan province of China, which led to the discovery of a series of norcycloartane triterpenoids. During 2006–2008, Sun’s group also identified several nortriterpenoids and bisnortriterpenoids from various species of Schisandra that possessed weak to moderate anti-HIV-1 activity.218–220
Rubriflordilactones A (191) and B (192) were isolated from the leaves and stems of Schisandra rubriflora.221 They are highly unsaturated rearranged bisnortriterpenoids that contain a biosynthetically modified aromatic D-ring. The anti-HIV activity of both compounds was tested in HIV-1IIIB-infected C8166 cell lines, and their cytotoxicity was evaluated in C8166 and MT-4 cells. Compound 191 showed only weak antiviral potency; however, 192 presented moderate anti-HIV activity, with an EC50 of 21.1 µM and TI of 12.39.221

Rubriflorins A–C (193–195), three highly oxygenated nortriterpenoids, were also isolated from the leaves and stems Schisandra rubriflora.222 They feature an opened A-ring. Compounds 193 and 194 exhibited moderate anti-HIV-1 activity, with EC50 values of 17.9 and 29.2 µM, respectively, while 195 showed weak HIV-1 inhibition activity, with an EC50 value of 143.1 µM.222 Rubriflorins D–J (196–202) were found in the same plant shortly after the discovery of 193–195.223 They also contain an opened A-ring and showed moderate to weak anti-HIV activity, with EC50 values ranging from 26.4 to 167.0 µM.223

Sphenadilactones A (203) and B (204) were isolated from the leaves and stems of Schisandra sphenanthera.224 Their structural elucidation was accomplished by extensive NMR and single-crystal X-ray crystallography. However, an erratum was later published to correct the stereochemistry of Me-27, which should have an α- rather than β-orientation.225 Compound 203 exhibited very weak anti-HIV-1 activity, with an EC50 of 237.8 µM in HIV-1IIIB-infected C8166 cells.224 No obvious cytotoxicity was observed when both compounds were tested against three human tumor cell lines, K562, A549, and HT-29. A minor nortriterpenoid, sphenadilactone C (205), was also isolated in a subsequent study of Schisandra sphenanthera.226 It is characterized by the presence of a partial enol structure, and is the first example of an acetamide-bearing highly oxygenated nortriterpenoid from a cycloartane. Compound 205 also exhibited very weak antiviral activity against HIV-1IIIB, with an EC50 of 47.8 mM.226

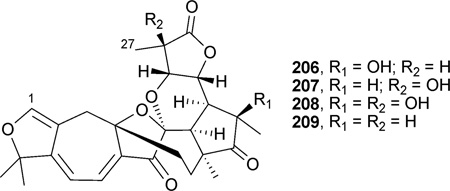
Re-investigation of Schisandra sphenanthera resulted in the isolation of sphenalactones A–D (206–209), which have an unusual 27-carbon skeleton derived from cycloartane, due to the loss of C-2, C-3, and CH3-28.227 This is the first class in the Schisandra species and the first case in the cycloartane triterpenoid family in which C-2 and C-3 are degraded in the molecule. Compounds 208–209 demonstrated weak anti-HIV-1 replication activity, with EC50 values of 185.1, 153.7, 105.4, and 76.2 µM, respectively.227
Schintrilactones A (210) and B (211), two new nortriterpenoids with modified five-membered D-ring and δ-lactone E-ring, were isolated from Schisandra chinensis. Both compounds have a unique carbon skeleton, with an unstable stereogenic center at C-20. The two diastereomers slowly interconvert through an enol intermediate. Compounds 210 and 211 demonstrated anti-HIV-1 activity, with EC50 values of 33.9 and 68.2 µM, respectively, compared with AZT as positive control (EC50: 8.46 µM).228 Nortriterpenoids schindilactones D–G (212–215) and bisnortriterpenoids wuweizidilactones G (216) and H (217) were also isolated from the stems and leaves of Schisandra chinensis. However, they demonstrated only weak antiviral activity, with EC50 values ranging from 32 to 181 µM.229 Wilsonianadilactones A–C (218–220) were isolated from the stems and leaves of Schisandra wilsoniana, a climbing plant distributed in the Yunnan province of China. All three compounds showed weak anti-HIV-1 activity, with EC50 values of 38.3, 99.1, and 118.6 µM in an in vitro antiviral assay.230

Lancifodilactone H (221), a trinorcycloartane triterpenoid with a biosynthetically modified seven-membered lactone ring, and lancifoic acid A (222), a new A ring-secocycloartane triterpenoid, were isolated from the stems and leaves of Schisandra lancifolia. They exhibited moderate anti-HIV activity, with EC50 values of 37.4 and 33.2 µM, respectively.231
The genus Kadsura belongs to the family Schisandraceae. In an investigation of Kadsura heteroclita, a new nortriterpenoid, long-ipedlactone J (223), was isolated. When tested in HIV-1IIIB infected C8166 cells, 223 showed a moderate anti-HIV EC50 of 7.1 µM.232
Four new lanostane triterpenes, colossolactones V, VI, VII,and VIII (224–227), were isolated from the fruiting bodies of the Vietnamese mushroom Ganoderma colossum.233 With seco A- and E-rings, 224 and 225 differ structurally from previous isolated colossolactones, which have a seven-membered lactone A-ring and six-membered lactone E-ring. Compound 226 has a lactone E-ring, but seco A-ring, while compound 227 has both A- and E-lactone rings. These three new triterpenoids, together with three previously isolated compounds, collosolactone E (228), schisanlactone A (229), and colossolactone G (230), were tested against HIV-1 protease. Compounds 224, 226, 228, and 229 showed anti-protease activity, with EC50 values of 14.6, 24.7, 15.3, and 10.8 µM, respectively, while 225, 227, and 230 were inactive.233
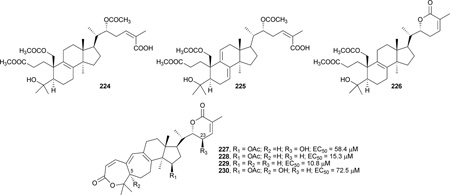
The presence of a hydroxy group at C-23 (R3) or C-5 (R2) in the tested compounds reduced the activity (i.e., 227 vs. 228 and 230 vs. 228), suggesting that the hydrophobicity of the triterpene skeleton is important to the anti-HIV-1 protease inhibition activity. For the A-ring seco compounds, 224 (with a single double bond at C-8–C-9) exhibited higher activity than the related 225 with two double bonds at C-7–C-8 and C-9–C-11. The unsaturation pattern would alter the three-dimensional structures and, consequently, the spatial arrangement of pharmacophores in the compounds, which have a significant influence on activity. On the other hand, the presence of the seco or lactone A- and/or E-ring did not significantly influence the anti-HIV protease activity of the analogues.
Two unusual octanorcucurbitacins, endecaphyllacins A (231) and B (232), were isolated from the tubers of Hemsleya endecaphylla.234 These two compounds, together with three known cucurbitane analogues including 99, 100, and cucurbitacin I (233), were evaluated for their ability to prevent the cytopathic effects of HIV-1 in C8166 cells. Although compounds 231 and 232 showed only moderate anti-HIV activity (EC50: 88.27 and 33.70 µM), 99, 100, and 233 showed significant potency (EC50: 0.16, 0.23, and 1.36 µM, respectively) in this assay.234 The high activity of 99, 100, and 233 may be due to the presence of the C-17 functional side chain. However, because 99 is the first reported common cucurbitane analogue with potent antiviral activity, repeated evaluation may be necessary to confirm the compound’s potency. Kuguacins A–E (234–238) were isolated from roots of Momordica charantia.235 They also belong to the cucurbitacin family. Compounds 236 and 238 displayed moderate anti-HIV-1 activity (EC50: 19.7 and 59.6 µM; TI: 23.68 and 7.81, respectively). Although compounds 234 and 235 showed moderate antiviral activity with EC50 values of 22.8 and 24.9 µM, they were also cytotoxic, resulting in lower TI values of 7.01 and 3.09, respectively. The cucurbitacin analogues hemslecins A and B (239 and 240) were discovered as antibacterial drugs from Hemsleya jinfushanensis some time ago. They have recently been shown to have anti-HIV-1 activity, with EC50 values of 4.0–36.3 µM and TI values of >20 in different assay systems.236 Further mechanism of action study revealed that they can block the transmission of virus through cell-to-cell fusion, but cannot inhibit the activities of HIV-1 RT, PR, NCp7 zinc ejection, or viral replication in chronically infected H9 lymphocytes.236 Thus, it is likely that 239 and 240 function as HIV-1 entry inhibitors.
3.4 The dammarane and euphane groups
Compounds 116–118, together with acutissimatriterpenes C (241) and D (242), were isolated from Phyllanthus acutissima.150 The anti-HIV-1 activity of the newly discovered compounds was tested by using a cell-based antiviral replication assay and HIV-1 reverse transcriptase (RT) assay. Compounds 118 and 242 exhibited moderate antiviral replication activity, with EC50 values of <5.8 and 8.3 µM, respectively, while 116, 117, and 241 showed weak anti-HIV activity (106.1–130.9 µM). Interestingly, 116 and 117 inhibited the RT by 55.5% and 65.9% at the tested concentration (^300 µM), while 241 and 242 showed only 11.0% and 37.8% inhibition against RT. In comparison, compound 242 did not inhibit RT, suggesting that RT may not be the compound’s target.150 The anti-HIV-1 activity and mechanism of action of C-31-secodammarane triterpenoids isolated from Alnus firma were reviewed by Yu et al.237 Among them, alnustic acid methyl ester (243) exhibited anti-HIV protease activity, with an EC50 value of 15.8 µM. Other similar compounds showed no effect against RT or protease.

3.5 Other triterpenes
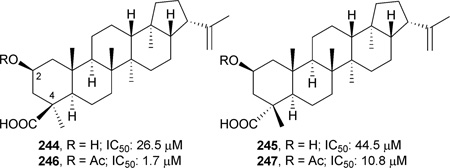
Two hopane triterpenes, dryopteric acids A (244) and B (245), were isolated from the rhizome of Dryopteris crassirhizoma (Aspidiaceae), and tested against recombinant HIV-1 protease (PR). Both compounds exhibited moderate anti-HIV protease activity, with EC50 values of 26.5 and 44.5 µM, respectively. In comparison, ursolic acid (46) has an EC50 of 8.9 µM. Acetylation of the C-2 hydroxy group of 244 and 245 yielded compounds 246 and 247, which showed enhanced anti-protease activity compared with the parent compounds.238 These results suggested that the carboxylic group at the C-4β (C-24) position and acetyl group at the C-2 position could play key roles in the inhibition of HIV-1 protease in this hopane series of triterpene acids.238
4 Conclusion
Triterpenoids are an important group for biologically active natural compounds, particularly with approximately 300 new triterpenoids identified each year. Since 1985, Connolly and Hill have regularly reviewed the scientific literature dealing with triterpenoid characterization from natural resources.239–256 Although numerous triterpenoids possessing various scaffolds have been reported, only a small portion of them were explored systematically for their pharmacological activities. Of those so explored, most showed only marginal potency in preliminary screening. However, this fact does not mean that they should be eliminated from further investigation. For instance, while betu-linic acid (1) exhibited only moderate anti-HIV activity, with an EC50 of 1.4 µM, modification of 1 furnished the clinical trial agent, bevirimat (161), soon to be a phase III anti-HIV drug. Another example is CDDO (63), a promising anticancer candidate in phase I clinical trials, which was derived from oleanolic acid (62), a weak antitumor compound. Generally speaking, bioactivity-directed fractionation and isolation (BDFI) is a major approach for new lead generation, which is then followed by lead optimization based on rational drug design, synthesis, and bioevaluation, as well as structure–activity relationship (SAR) and quantitative-SAR (QSAR) studies. Selection of a preclinical candidate also involves rapid, efficient assays to gather absorption, distribution, metabolism, excretion, and toxicity (ADMET) data, permitting an effective evaluation of drug candidates in the early stages of the drug discovery process. Although there are still some concerns, especially regarding structure complexity and non-specific pharmacological effects of naturally occurring bioactive triterpenoids, their diversity and uniqueness make them worthy of further pursuit for the discovery of new drugs.
Acknowledgements
This investigation was supported by NIH grants AI-077417 and AI-33066 from the National Institute of Allergy and Infectious Diseases (NIAID) and CA-17625 from the National Cancer Institute (NCI) awarded to K. H. Lee.
Biographies

Reen-Yen Kuo was born in Yi-lan, Taiwan, in 1975. He received his B.S. degree (1997) in Pharmacy and his Ph.D. degree (2003) in Pharmaceutical Science from Kaohsiung Medical University, Taiwan. He then worked as a researcher at the Institute for Information Industry, Taiwan, and in the government sector at the Science and Technology Advisory Group, Taiwan. He is currently a Visiting Scholar at the University of North Carolina at Chapel Hill. His research interests are focused on the isolation, structural elucidation, and total synthesis of biologically active natural products.

Keduo Qian was born in Beijing, China, in 1982. She received her B.S. degree in Pharmacy from Beijing University (2004), and her Ph. D. degree in Pharmaceutical Sciences from the University of North Carolina at Chapel Hill (2008). She is currently a Research Assistant Professor at UNC Chapel Hill. Her current research is focused on discovery, drug metabolism, and pharmacokinetic studies of natural product-derived anti-AIDS agents.

Susan L: Morris-Natschke received her B.S. in chemistry from the University of Maryland at College Park (1975) and her Ph.D. in organic chemistry from the University of North Carolina at Chapel Hill (1982). She is currently Research Professor in the Division of Medicinal Chemistry and Natural Products, in the .UNC Eshelman School of Pharmacy, where she has been on the faculty since 1983. Her interests include scientific writing/editing, as well as the synthesis and structure–activity relationships of bioactive natural products.

Kuo-Hsiung Lee received his B.S. in pharmacy at Kaohsiung Medical University, Taiwan (1961), his M.S. in pharmaceutical chemistry at Kyoto University, Japan (1965), and his Ph.D. in medicinal chemistry at the University of Minnesota, Minneapolis (1968). Since 1970, he has been on the faculty of the UNC Eshelman School of Pharmacy, and is now Kenan Professor of Medicinal Chemistry and Director of the Natural Products Research Laboratories. He has published over 640 research articles, been granted over 70 patents, and received numerous awards, including most recently the Norman R. Farnsworth Research Achievement Award, American Society of Pharmacognosy (2009).
Footnotes
Antitumor agents: Part 267, and Anti-AIDS agents: Part 77.
References
- 1.Chang CW, Wu TS, Hsieh YS, Kuo SC, Chao PD. J. Nat. Prod. 1999;62:327–328. doi: 10.1021/np980313w. [DOI] [PubMed] [Google Scholar]
- 2.Bringmann G, Saeb W, Assi LA, Francois G, Sankara Narayanan AS, Peters K, Peters EM. Planta Med. 1997;63:255–257. doi: 10.1055/s-2006-957666. [DOI] [PubMed] [Google Scholar]
- 3.Bae KH, Lee SM, Lee ES, Lee JS, Kang JS. Yakhak Hoechi. 1996;40:558–562. [Google Scholar]
- 4.Birgit DG, Neubert R, Wohlrab W. US Pat. 2001 6,175,035. [Google Scholar]
- 5.Kratsutsky PA, Carlson RM, Nesturenko VV, Kolomitsyn IV, Edwardson CF. WO Pat. 2001 2,001,010,885. [Google Scholar]
- 6.Trumbull ER, Bianchi E, Eckert DJ, Wiedhopf RM, Cole JR. J. Pharm. Sci. 1976;65:1407–1408. doi: 10.1002/jps.2600650938. [DOI] [PubMed] [Google Scholar]
- 7.Pisha E, Chai H, Lee IS, Chagwedera TE, Farnsworth NR, Cordell GA, Beecher CW, Fong HH, Kinghorn AD, Brown DM, Wani MC, Wall ME, Hieken TJ, Das Gupta TK, Pezzuto JM. Nat. Med. 1995;1:1046–1051. doi: 10.1038/nm1095-1046. [DOI] [PubMed] [Google Scholar]
- 8.Mukherjee R, Kumar V, Srivastava SK, Agarwal SK, Burman AC. Anticancer Agents Med. Chem. 2006;6:271–279. doi: 10.2174/187152006776930846. [DOI] [PubMed] [Google Scholar]
- 9.Cichewicz RH, Kouzi SA. Med. Res. Rev. 2004;24:90–114. doi: 10.1002/med.10053. [DOI] [PubMed] [Google Scholar]
- 10.Alakurtti S, Makela T, Koskimies S, Yli-Kauhaluoma J. Eur. J. Pharm. Sci. 2006;29:1–13. doi: 10.1016/j.ejps.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 11.Yogeeswari P, Sriram D. Curr. Med. Chem. 2005;12:657–666. doi: 10.2174/0929867053202214. [DOI] [PubMed] [Google Scholar]
- 12.Amico V, Barresi V, Condorelli D, Spatafora C, Tringali C. J. Agric. Food Chem. 2006;54:810–814. doi: 10.1021/jf052812q. [DOI] [PubMed] [Google Scholar]
- 13.Rzeski W, Stepulak A, Szymanski M, Sifringer M, Kaczor J, Wejksza K, Zdzisinska B, Kandefer-Szerszen M. Naunyn-Schmiedebergs Arch. Pharmacol. 2006;374:11–20. doi: 10.1007/s00210-006-0090-1. [DOI] [PubMed] [Google Scholar]
- 14.Kessler JH, Mullauer FB, de Roo GM, Medema JP. Cancer Lett. 2007;251:132–145. doi: 10.1016/j.canlet.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 15.Jung GR, Kim KJ, Choi CH, Lee TB, Han SI, Han HK, Lim SC. Basic Clin. Pharmacol. Toxicol. 2007;101:277–285. doi: 10.1111/j.1742-7843.2007.00115.x. [DOI] [PubMed] [Google Scholar]
- 16.Schmidt ML, Kuzmanoff KL, Ling-Indeck L, Pezzuto JM. Eur. J. Cancer. 1997;33:2007–2010. doi: 10.1016/s0959-8049(97)00294-3. [DOI] [PubMed] [Google Scholar]
- 17.Zuco V, Supino R, Righetti SC, Cleris L, Marchesi E, Gambacorti-Passerini C, Formelli F. Cancer Lett. 2002;175:17–25. doi: 10.1016/s0304-3835(01)00718-2. [DOI] [PubMed] [Google Scholar]
- 18.Fulda S, Jeremias I, Debatin KM. Oncogene. 2004;23:7611–7620. doi: 10.1038/sj.onc.1207970. [DOI] [PubMed] [Google Scholar]
- 19.Rabi T, Shukla S, Gupta S. Mol. Carcinog. 2008;47:964–973. doi: 10.1002/mc.20447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang L, Ho P, Chen CH. FEBS Lett. 2007;581:4955–4959. doi: 10.1016/j.febslet.2007.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chintharlapalli S, Papineni S, Ramaiah SK, Safe S. Cancer Res. 2007;67:2816–2823. doi: 10.1158/0008-5472.CAN-06-3735. [DOI] [PubMed] [Google Scholar]
- 22.Syrovets T, Buchelle B, Gedig E, Slupsky JR, Simmet T. Mol. Pharmacol. 2000;58:71–81. doi: 10.1124/mol.58.1.71. [DOI] [PubMed] [Google Scholar]
- 23.Chowdhury AR, Mandal S, Mittra B, Sharma S, Mukhopadhyay S, Majumder HK. Med. Sci. Monit. 2002;8:BR254–BR265. [PubMed] [Google Scholar]
- 24.Ganguly A, Das B, Roy A, Sen N, Dasgupta SB, Mukhopadhayay S, Majumder HK. Cancer Res. 2007;67:11848–11858. doi: 10.1158/0008-5472.CAN-07-1615. [DOI] [PubMed] [Google Scholar]
- 25.Rieber M, Rieber MS. Int. J. Cancer. 2006;118:1135–1143. doi: 10.1002/ijc.21478. [DOI] [PubMed] [Google Scholar]
- 26.Choi SZ, Yang MC, Choi SU, Lee KR. Arch. Pharmacal Res. 2006;29:203–208. doi: 10.1007/BF02969394. [DOI] [PubMed] [Google Scholar]
- 27.Chen W, Tang W, Lou L, Zhao W. Phytochemistry. 2006;67:1041–1047. doi: 10.1016/j.phytochem.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 28.Dinda B, Ghosh B, Arima S, Sato N, Harigaya Y. J. Indian Chem. Soc. 2006;83:1044–1046. [Google Scholar]
- 29.Lenta BN, Ngouela S, Boyom FF, Tantangmo F, Tchouya GRF, Tsamo E, Gut J, Rosenthal PJ, Connolly JD. Chem. Pharm. Bull. 2007;55:464–467. doi: 10.1248/cpb.55.464. [DOI] [PubMed] [Google Scholar]
- 30.Suksamrarn S, Panseeta P, Kunchanawatta S, Distaporn T, Ruktasing S, Suksamrarn A. Chem. Pharm. Bull. 2006;54:535–537. doi: 10.1248/cpb.54.535. [DOI] [PubMed] [Google Scholar]
- 31.Chung MY, Rho M-C, Lee SW, Park HR, Kim K, Lee IA, Kim DH, Jeune KH, Lee HS, Kim YK. Planta Med. 2006;72:267–269. doi: 10.1055/s-2005-916178. [DOI] [PubMed] [Google Scholar]
- 32.Bastos DZL, Pimentel IC, d’Jesus DA, d’Oliveira BH. Phytochemistry. 2007;68:834–839. doi: 10.1016/j.phytochem.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 33.Shi H-M, Yao Z-R, Chai X-Y, Xu Z-R, Zhou Y-H, Wen J, Tu P-F. Nat. Prod. Res. 2008;22:633–637. doi: 10.1080/14786410701614325. [DOI] [PubMed] [Google Scholar]
- 34.Yang Y, Deng Z, Proksch P, Lin W. Pharmazie. 2006;61:365–366. [PubMed] [Google Scholar]
- 35.Fang L, Ito A, Chai HB, Mi Q, Jones WP, Madulid DR, Oliveros MB, Gao Q, Orjala J, Farnsworth NR, Soejarto DD, Cordell GA, Swanson SM, Pezzuto JM, Kinghorn AD. J. Nat. Prod. 2006;69:332–337. doi: 10.1021/np058083q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tangmouo JG, Meli AL, Komguem J, Kuete V, Ngounou FN, Lontsi D, Beng VP, Choudhary MI, Sondengam BL. Tetrahedron Lett. 2006;47:3067–3070. [Google Scholar]
- 37.Nareeboon P, Kraus W, Beifuss U, Conrad J, Klaiber I, Sutthivaiyakit S. Tetrahedron. 2006;62:5519–5526. [Google Scholar]
- 38.Mbaze LMa, Poumale HMP, Wansi JD, Lado JA, Khan SN, Iqbal MC, Ngadjui BT, Laatsch H. Phytochemistry. 2007;68:591–595. doi: 10.1016/j.phytochem.2006.12.015. [DOI] [PubMed] [Google Scholar]
- 39.Lenta BN, Devkota KP, Ngouela S, Boyom FF, Naz Q, Choudhary MI, Tsamo E, Rosenthal PJ, Sewald N. Chem. Pharm. Bull. 2008;56:222–226. doi: 10.1248/cpb.56.222. [DOI] [PubMed] [Google Scholar]
- 40.Wansi JD, Lallemand M-C, Chiozem DD, Toze FAA, Mbaze LMa, Naharkhan S, Iqbal MC, Tillequin F, Wandji J, Fomum ZT. Phytochemistry. 2007;68:2096–2100. doi: 10.1016/j.phytochem.2007.02.020. [DOI] [PubMed] [Google Scholar]
- 41.Mohamed SAN, Gandhidasan R, Charles JA. Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem. 2006;45:1544–1546. [Google Scholar]
- 42.Giacomelli SR, Maldaner G, Stuücker C, Marasciulo C, Schmidt J, Wessjohann L, Dalcol II, Morel AF. Planta Med. 2007;73:499–501. doi: 10.1055/s-2007-967166. [DOI] [PubMed] [Google Scholar]
- 43.Ono M, Tsuru T, Abe H, Eto M, Okawa M, Abe F, Kinjo J, Ikeda T, Nohara T. J. Nat. Prod. 2006;69:1417–1420. doi: 10.1021/np068010m. [DOI] [PubMed] [Google Scholar]
- 44.Siddiqui BS, Aslam H, Ali ST, Begum S, Khatoon N. Chem. Pharm. Bull. 2007;55:516–519. doi: 10.1248/cpb.55.516. [DOI] [PubMed] [Google Scholar]
- 45.Khaliq S, Volk F-J, Frahm AW. Planta Med. 2007;73:77–83. doi: 10.1055/s-2006-951766. [DOI] [PubMed] [Google Scholar]
- 46.Phan MG, Phan TS, Matsunami K, Otsuka H. Chem. Pharm. Bull. 2006;54:546–549. doi: 10.1248/cpb.54.546. [DOI] [PubMed] [Google Scholar]
- 47.Sun J-M, Yang J-S, Zhang H. Chem. Pharm. Bull. 2007;55:474–476. doi: 10.1248/cpb.55.474. [DOI] [PubMed] [Google Scholar]
- 48.Woo SL, Im K-R, Park Y-D, Sung N-D, Jeong T-S. Biol. Pharm. Bull. 2006;29:382–384. doi: 10.1248/bpb.29.382. [DOI] [PubMed] [Google Scholar]
- 49.Zhang Y, Nakamura S, Wang T, Matsuda H, Yoshikawa M. Tetrahedron. 2008;64:7347–7352. [Google Scholar]
- 50.Aguirre MC, Delporte C, Backhouse N, Erazo S, Letelier ME, Cassels BK, Silva X, Alegria S, Negrete R. Bioorg. Med. Chem. 2006;14:5673–5677. doi: 10.1016/j.bmc.2006.04.021. [DOI] [PubMed] [Google Scholar]
- 51.Guo H, Li B-G, Wu Z-J, Zhang G-L. Planta Med. 2006;72:180–183. doi: 10.1055/s-2005-873187. [DOI] [PubMed] [Google Scholar]
- 52.Reutrakul V, Chanakul W, Pohmakotr M, Jaipetch T, Yoosook C, Kasisit J, Napaswat C, Santisuk T, Prabpai S, Kongsaeree P, Tuchinda P. Planta Med. 2006;72:1433–1435. doi: 10.1055/s-2006-951725. [DOI] [PubMed] [Google Scholar]
- 53.Isobe T, Doe M, Morimoto Y, Nagata K, Masuoka N, Ohsaki A. Yakugaku Zasshi. 2007;127:389–395. doi: 10.1248/yakushi.127.389. [DOI] [PubMed] [Google Scholar]
- 54.Li G-P, Zhao J-F, Yang L-J, Yang X-D, Li L. Helv. Chim. Acta. 2006;89:3018–3022. [Google Scholar]
- 55.Uchiyama N, Kiuchi F, Ito M, Honda G, Takeda Y, Khodzhimatov OK, Ashurmetov OA. Tetrahedron. 2006;62:4355–4359. [Google Scholar]
- 56.Chen IH, Du Y-C, Lu M-C, Lin A-S, Hsieh P-W, Wu C-C, Chen S-L, Yen H-F, Chang F-R, Wu Y-C. J. Nat. Prod. 2008;71:1352–1357. doi: 10.1021/np800093a. [DOI] [PubMed] [Google Scholar]
- 57.Ma CY, Musoke SF, Tan GT, Sydara K, Bouamanivong S, Southavong B, Soejarto DD, Fong HH, Zhang HJ. Chem. Biodiversity. 2008;5:2442–2448. doi: 10.1002/cbdv.200890209. [DOI] [PubMed] [Google Scholar]
- 58.Gauthier C, Legault J, Lebrun M, Dufour P, Pichette A. Bioorg. Med. Chem. 2006;14:6713–6725. doi: 10.1016/j.bmc.2006.05.075. [DOI] [PubMed] [Google Scholar]
- 59.Cmoch P, Pakulski Z, Swanczynova J, Strnad M. Carbohydr. Res. 2008;343:995–1003. doi: 10.1016/j.carres.2008.02.011. [DOI] [PubMed] [Google Scholar]
- 60.Chintharlapalli S, Papineni S, Liu S, Jutooru I, Chadalapaka G, Cho SD, Murthy RS, You Y, Safe S. Carcinogenesis. 2007;28:2337–2346. doi: 10.1093/carcin/bgm189. [DOI] [PubMed] [Google Scholar]
- 61.Liby K, Honda T, Williams CR, Risingsong R, Royce DB, Suh N, Dinkova-Kostova AT, Stephenson KK, Talalay P, Sundararajan C, Gribble GW, Sporn MB. Mol. Cancer Ther. 2007;6:2113–2119. doi: 10.1158/1535-7163.MCT-07-0180. [DOI] [PubMed] [Google Scholar]
- 62.Urban M, Sarek J, Kvasnica M, Tislerova I, Hajduch M. J. Nat. Prod. 2007;70:526–532. doi: 10.1021/np060436d. [DOI] [PubMed] [Google Scholar]
- 63.Kumar V, Rani N, Aggarwal P, Sanna VK, Singh AT, Jaggi M, Joshi N, Sharma PK, Irchhaiya R, Burman AC. Bioorg. Med. Chem. Lett. 2008;18:5058–5062. doi: 10.1016/j.bmcl.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 64.Urban M, Klinot J, Tislerova I, Biedermann D, Hajduch M, Cisarova I, Sarek J. Synthesis. 2006:3979–3986. [Google Scholar]
- 65.Steinkamp-Fenske K, Bollinger L, Xu H, Yao Y, Horke S, Forstermann U, Li H. J. Pharmacol. Exp. Ther. 2007;322:836–842. doi: 10.1124/jpet.107.123356. [DOI] [PubMed] [Google Scholar]
- 66.Bureeva S, Andia-Pravdivy J, Symon A, Bichucher A, Moskaleva V, Popenko V, Shpak A, Shvets V, Kozlov L, Kaplun A. Bioorg. Med. Chem. 2007;15:3489–3498. doi: 10.1016/j.bmc.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 67.Reyes CP, Nunez MJ, Jimenez IA, Busserolles J, Alcaraz MJ, Bazzocchi IL. Bioorg. Med. Chem. 2006;14:1573–1579. doi: 10.1016/j.bmc.2005.10.063. [DOI] [PubMed] [Google Scholar]
- 68.Wen C-C, Kuo Y-H, Jan J-T, Liang P-H, Wang S-Y, Liu H-G, Lee C-K, Chang S-T, Kuo C-J, Lee S-S, Hou C-C, Hsiao PW, Chien SC, Shyur LF, Yang NS. J. Med. Chem. 2007;50:4087–4095. doi: 10.1021/jm070295s. [DOI] [PubMed] [Google Scholar]
- 69.Tanachatchairatana T, Bremner JB, Chokchaisiri R, Suksamrarn A. Chem. Pharm. Bull. 2008;56:194–198. doi: 10.1248/cpb.56.194. [DOI] [PubMed] [Google Scholar]
- 70.Kuiate JR, Mouokeu S, Wabo HK, Tane P. Phytother. Res. 2007;21:149–152. doi: 10.1002/ptr.2039. [DOI] [PubMed] [Google Scholar]
- 71.Ziegler HL, Staalso T, Jaroszewski JW. Planta Med. 2006;72:640–642. doi: 10.1055/s-2006-931575. [DOI] [PubMed] [Google Scholar]
- 72.Chen JC, Chang YS, Wu SL, Chao DC, Chang CS, Li CC, Ho TY, Hsiang CY. J. Ethnopharmacol. 2007;113:233–239. doi: 10.1016/j.jep.2007.05.031. [DOI] [PubMed] [Google Scholar]
- 73.Chaturvedi PK, Bhui K, Shukla Y. Cancer Lett. 2008;263:1–13. doi: 10.1016/j.canlet.2008.01.047. [DOI] [PubMed] [Google Scholar]
- 74.Prasad S, Kalra N, Shukla Y. Nutr. Cancer. 2008;60:120–130. doi: 10.1080/01635580701613772. [DOI] [PubMed] [Google Scholar]
- 75.Prasad S, Nigam N, Kalra N, Shukla Y. Mol. Carcinogen. 2008;47:916–924. doi: 10.1002/mc.20442. [DOI] [PubMed] [Google Scholar]
- 76.Saleem M, Maddodi N, Abu Zaid M, Khan N, bin Hafeez B, Asim M, Suh Y, Yun JM, Setaluri V, Mukhtar H. Clin. Cancer Res. 2008;14:2119–2127. doi: 10.1158/1078-0432.CCR-07-4413. [DOI] [PubMed] [Google Scholar]
- 77.Nigam N, Prasad S, Shukla Y. Food Chem. Toxicol. 2007;45:2331–2335. doi: 10.1016/j.fct.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 78.Hata K, Mukaiyama T, Tsujimura N, Sato Y, Kosaka Y, Sakamoto K, Hori K. Cytotechnology. 2007;52:151–158. doi: 10.1007/s10616-007-9069-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Laszczyk M, Jaeger S, Simon-Haarhaus B, Scheffler A, Schempp CM. Planta Med. 2006;72:1389–1395. doi: 10.1055/s-2006-951723. [DOI] [PubMed] [Google Scholar]
- 80.Akihisa T, Tabata K, Banno N, Tokuda H, Nishihara R, Nakamura Y, Kimura Y, Yasukawa K, Suzuki T. Biol. Pharm. Bull. 2006;29:1976–1979. doi: 10.1248/bpb.29.1976. [DOI] [PubMed] [Google Scholar]
- 81.Kashiwada Y, Sekiya M, Yamazaki K, Ikeshiro Y, Fujioka T, Yamagishi T, Kitagawa S, Takaishi Y. J. Nat. Prod. 2007;70:623–627. doi: 10.1021/np060631s. [DOI] [PubMed] [Google Scholar]
- 82.Ammon HP. Planta Med. 2006;72:1100–1116. doi: 10.1055/s-2006-947227. [DOI] [PubMed] [Google Scholar]
- 83.Poeckel D, Werz O. Curr. Med. Chem. 2006;13:3359–3369. doi: 10.2174/092986706779010333. [DOI] [PubMed] [Google Scholar]
- 84.Naz Fadimatou Atta-ur-Rahman H, Makhmoor T, Yasin A, Fatima N, Ngounou FN, Kimbu SF, Sondengam BL, Choudhary MI. J. Nat. Prod. 2005;68:189–193. doi: 10.1021/np040142x. [DOI] [PubMed] [Google Scholar]
- 85.Shen T, Lou HX. Chem. Biodiversity. 2008;5:540–553. doi: 10.1002/cbdv.200890051. [DOI] [PubMed] [Google Scholar]
- 86.Siemoneit U, Hofmann B, Kather N, Lamkemeyer T, Madlung J, Franke L, Schneider G, Jauch J, Poeckel D, Werz O. Biochem. Pharmacol. 2008;75:503–513. doi: 10.1016/j.bcp.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 87.Burlando B, Parodi A, Volante A, Bassi AM. Toxicol. Lett. 2008;177:144–149. doi: 10.1016/j.toxlet.2008.01.008. [DOI] [PubMed] [Google Scholar]
- 88.Lu M, Xia L, Hua H, Jing Y. Cancer Res. 2008;68:1180–1186. doi: 10.1158/0008-5472.CAN-07-2978. [DOI] [PubMed] [Google Scholar]
- 89.Singh SK, Bhusari S, Singh R, Saxena A, Mondhe D, Qazi GN. Vasc. Pharmacol. 2007;46:333–337. doi: 10.1016/j.vph.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 90.Buechele B, Zugmaier W, Estrada A, Genze F, Syrovets T, Paetz C, Schneider B, Simmet T. Planta Med. 2006;72:1285–1289. doi: 10.1055/s-2006-951680. [DOI] [PubMed] [Google Scholar]
- 91.Singh S, Khajuria A, Taneja SC, Johri RK, Singh J, Qazi GN. Phytomedicine. 2008;15:400–407. doi: 10.1016/j.phymed.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 92.Singh S, Khajuria A, Taneja SC, Khajuria RK, Singh J, Johri RK, Qazi GN. Phytomedicine. 2008;15:408–415. doi: 10.1016/j.phymed.2008.02.017. [DOI] [PubMed] [Google Scholar]
- 93.Bishnoi M, Patil CS, Kumar A, Kulkarni SK. Pharmacology. 2007;79:34–41. doi: 10.1159/000097627. [DOI] [PubMed] [Google Scholar]
- 94.Singh S, Khajuria A, Taneja SC, Khajuria RK, Singh J, Qazi GN. Bioorg. Med. Chem. Lett. 2007;17:3706–3711. doi: 10.1016/j.bmcl.2007.04.034. [DOI] [PubMed] [Google Scholar]
- 95.Shah BA, Kumar A, Gupta P, Sharma M, Sethi VK, Saxena AK, Singh J, Qazi GN, Taneja SC. Bioorg. Med. Chem. Lett. 2007;17:6411–6416. doi: 10.1016/j.bmcl.2007.10.011. [DOI] [PubMed] [Google Scholar]
- 96.Subba Rao GSR, Kondaiah P, Singh SK, Ravanan P, Sporn MB. Tetrahedron. 2008;64:11541–11548. doi: 10.1016/j.tet.2008.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ikeda Y, Murakami A, Ohigashi H. Mol. Nutr. Food Res. 2008;52:26–42. doi: 10.1002/mnfr.200700389. [DOI] [PubMed] [Google Scholar]
- 98.Achiwa Y, Hasegawa K, Udagawa Y. Biosci., Biotechnol., Biochem. 2007;71:31–37. doi: 10.1271/bbb.60288. [DOI] [PubMed] [Google Scholar]
- 99.Andersson D, Cheng Y, Duan RD. J. Cancer Res. Clin. Oncol. 2008;134:101–107. doi: 10.1007/s00432-007-0255-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bonaccorsi I, Altieri F, Sciamanna I, Oricchio E, Grillo C, Contartese G, Galati EM. Cancer Lett. 2008;263:130–139. doi: 10.1016/j.canlet.2007.12.026. [DOI] [PubMed] [Google Scholar]
- 101.Cipak L, Grausova L, Miadokova E, Novotny L, Rauko P. Arch. Toxicol. 2006;80:429–435. doi: 10.1007/s00204-006-0072-6. [DOI] [PubMed] [Google Scholar]
- 102.Ovesńa Z, Kozics K, Slamenova D. Mutat. Res. 2006;600:131–137. doi: 10.1016/j.mrfmmm.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 103.Liu D, Meng Y-Q, Zhao J, Chen L-G. Chem. Res. Chin. Univ. 2008;24:42–46. [Google Scholar]
- 104.Chadalapaka G, Jutooru I, McAlees A, Stefanac T, Safe S. Bioorg. Med. Chem. Lett. 2008;18:2633–2639. doi: 10.1016/j.bmcl.2008.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Park BC, Paek S-H, Lee Y-S, Kim S-J, Lee E-S, Choi HG, Yong CS, Kim J-A. Biol. Pharm. Bull. 2007;30:176–179. doi: 10.1248/bpb.30.176. [DOI] [PubMed] [Google Scholar]
- 106.Cho CW, Choi DS, Cardone MH, Kim CW, Sinskey AJ, Rha C. Cell Biol. Toxicol. 2006;22:393–408. doi: 10.1007/s10565-006-0104-2. [DOI] [PubMed] [Google Scholar]
- 107.Chen IH, Chang F-R, Wu C-C, Chen S-L, Hsieh P-W, Yen H-F, Du Y-C, Wu Y-C. J. Nat. Prod. 2006;69:1543–1546. doi: 10.1021/np060369n. [DOI] [PubMed] [Google Scholar]
- 108.Wang ZY, Nixon DW. Nutr. Cancer. 2001;39:1–11. doi: 10.1207/S15327914nc391_1. [DOI] [PubMed] [Google Scholar]
- 109.Asl MN, Hosseinzadeh H. Phytother. Res. 2008;22:709–724. doi: 10.1002/ptr.2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Liu D, Song D, Guo G, Wang R, Lv J, Jing Y, Zhao L. Bioorg. Med. Chem. 2007;15:5432–5439. doi: 10.1016/j.bmc.2007.05.057. [DOI] [PubMed] [Google Scholar]
- 111.Chadalapaka G, Jutooru I, McAlees A, Stefanac T, Safe S. Bioorg. Med. Chem. Lett. 2008;18:2633–2639. doi: 10.1016/j.bmcl.2008.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Chintharlapalli S, Papineni S, Jutooru I, McAlees A, Safe S. Mol. Cancer Ther. 2007;6:1588–1598. doi: 10.1158/1535-7163.MCT-07-0022. [DOI] [PubMed] [Google Scholar]
- 113.Papineni S, Chintharlapalli S, Safe S. Mol. Pharmacol. 2008;73:553–565. doi: 10.1124/mol.107.041285. [DOI] [PubMed] [Google Scholar]
- 114.Nabekura T, Yamaki T, Ueno K, Kitagawa S. Cancer Chemother. Pharmacol. 2008;62:867–873. doi: 10.1007/s00280-007-0676-4. [DOI] [PubMed] [Google Scholar]
- 115.Nakagawa-Goto K, Nakamura S, Bastow KF, Nyarko A, Peng C-Y, Lee F-Y, Lee F-C, Lee K-H. Bioorg. Med. Chem. Lett. 2007;17:2894–2898. doi: 10.1016/j.bmcl.2007.02.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sultana N, Ata A. J. Enzyme Inhib. Med. Chem. 2008;23:739–756. doi: 10.1080/14756360701633187. [DOI] [PubMed] [Google Scholar]
- 117.Nishino H, Nishino A, Takayasu J, Hasegawa T, Iwashima A, Hirabayashi K, Iwata S, Shibata S. Cancer Res. 1988;48:5210–5215. [PubMed] [Google Scholar]
- 118.Honda T, Rounds BV, Gribble GW, Suh N, Wang Y, Sporn MB. Bioorg. Med. Chem. Lett. 1998;8:2711–2714. doi: 10.1016/s0960-894x(98)00479-x. [DOI] [PubMed] [Google Scholar]
- 119.Honda T, Honda Y, Favaloro FG, Gribble GW, Suh N, Place AE, Rendi MH, Sporn MB. Bioorg. Med. Chem. Lett. 2002;12:1027–1030. doi: 10.1016/s0960-894x(02)00105-1. [DOI] [PubMed] [Google Scholar]
- 120.Liby KT, Yore MM, Sporn MB. Nat. Rev. Cancer. 2007;7:357–369. doi: 10.1038/nrc2129. [DOI] [PubMed] [Google Scholar]
- 121.Sogno I, Vannini N, Lorusso G, Cammarota R, Noonan DM, Generoso L, Sporn MB, Albini A. Recent Results Cancer Res. 2009;181:209–212. doi: 10.1007/978-3-540-69297-3_19. [DOI] [PubMed] [Google Scholar]
- 122.Chan PK. Biochem. Pharmacol. 2007;73:341–350. doi: 10.1016/j.bcp.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 123.Tschesche R, Fugmann R. Chem. Ber. 1951;84:810–826. [Google Scholar]
- 124.He X, Liu RH. J. Agric. Food Chem. 2007;55:4366–4370. doi: 10.1021/jf063563o. [DOI] [PubMed] [Google Scholar]
- 125.Juan ME, Planas JM, Ruiz-Gutierrez V, Daniel H, Wenzel U. Br. J. Nutr. 2008;100:36–43. doi: 10.1017/S0007114508882979. [DOI] [PubMed] [Google Scholar]
- 126.Ramachandran C, Nair PK, Alamo A, Cochrane CB, Escalon E, Melnick SJ. Int. J. Cancer. 2006;119:2443–2454. doi: 10.1002/ijc.22174. [DOI] [PubMed] [Google Scholar]
- 127.Tian J-K, Xu L-Z, Zou Z-M, Yang S-L. Chem. Pharm. Bull. 2006;54:567–569. doi: 10.1248/cpb.54.567. [DOI] [PubMed] [Google Scholar]
- 128.Thibeault D, Gauthier C, Legault J, Bouchard J, Dufour P, Pichette A. Bioorg. Med. Chem. 2007;15:6144–6157. doi: 10.1016/j.bmc.2007.06.033. [DOI] [PubMed] [Google Scholar]
- 129.Thibeault D, Legault J, Bouchard J, Pichette A. Tetrahedron Lett. 2007;48:8416–8419. [Google Scholar]
- 130.Chen CR, Chen HW, Chang CI. Chem. Pharm. Bull. 2008;56:385–388. doi: 10.1248/cpb.56.385. [DOI] [PubMed] [Google Scholar]
- 131.Yue QX, Cao ZW, Guan SH, Liu XH, Tao L, Wu WY, Li YX, Yang PY, Liu X, Guo DA. Mol. Cell Proteomics. 2008;7:949–961. doi: 10.1074/mcp.M700259-MCP200. [DOI] [PubMed] [Google Scholar]
- 132.Chang UM, Li CH, Lin LI, Huang CP, Kan LS, Lin SB. Life Sci. 2006;79:1129–1139. doi: 10.1016/j.lfs.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 133.Kang HM, Lee SK, Shin DS, Lee MY, Han DC, Baek NI, Son KH, Kwon BM. Life Sci. 2006;78:607–613. doi: 10.1016/j.lfs.2005.05.066. [DOI] [PubMed] [Google Scholar]
- 134.Akihisa T, Nakamura Y, Tokuda H, Uchiyama E, Suzuki T, Kimura Y, Uchikura K, Nishino H. J. Nat. Prod. 2007;70:948–953. doi: 10.1021/np0780001. [DOI] [PubMed] [Google Scholar]
- 135.Sun P, Liu BS, Yi YH, Li L, Gui M, Tang HF, Zhang DZ, Zhang SL. Chem. Biodiversity. 2007;4:450–457. doi: 10.1002/cbdv.200790037. [DOI] [PubMed] [Google Scholar]
- 136.Chen JC, Chiu MH, Nie RL, Cordell GA, Qiu SX. Nat. Prod. Rep. 2005;22:386–399. doi: 10.1039/b418841c. [DOI] [PubMed] [Google Scholar]
- 137.Liu T, Zhang M, Zhang H, Sun C, Deng Y. Eur. Arch. Otorhinolaryngol. 2008;265:1225–1232. doi: 10.1007/s00405-008-0625-9. [DOI] [PubMed] [Google Scholar]
- 138.Yang L, Wu S, Zhang Q, Liu F, Wu P. Cancer Lett. 2007;256:267–278. doi: 10.1016/j.canlet.2007.06.018. [DOI] [PubMed] [Google Scholar]
- 139.Kitts DD, Popovich DG, Hu C. Can. J. Physiol. Pharmacol. 2007;85:1173–1183. doi: 10.1139/Y07-099. [DOI] [PubMed] [Google Scholar]
- 140.Chen J, Peng H, Ou-Yang X, He X. Melanoma Res. 2008;18:322–329. doi: 10.1097/CMR.0b013e32830b3536. [DOI] [PubMed] [Google Scholar]
- 141.Li G, Wang Z, Sun Y, Liu K. Basic Clin. Pharmacol. Toxicol. 2006;98:588–592. doi: 10.1111/j.1742-7843.2006.pto_415.x. [DOI] [PubMed] [Google Scholar]
- 142.Usami Y, Liu YN, Lin AS, Shibano M, Akiyama T, Itokawa H, Morris-Natschke SL, Bastow K, Kasai R, Lee KH. J. Nat. Prod. 2008;71:478–481. doi: 10.1021/np070613q. [DOI] [PubMed] [Google Scholar]
- 143.Wang W, Zhao Y, Rayburn ER, Hill DL, Wang H, Zhang R. Cancer Chemother. Pharmacol. 2007;59:589–601. doi: 10.1007/s00280-006-0300-z. [DOI] [PubMed] [Google Scholar]
- 144.Kwon HY, Kim EH, Kim SW, Kim SN, Park JD, Rhee DK. Arch. Pharmacal Res. 2008;31:171–177. doi: 10.1007/s12272-001-1137-y. [DOI] [PubMed] [Google Scholar]
- 145.Zhao Y, Wang W, Han L, Rayburn ER, Hill DL, Wang H, Zhang R. Med. Chem. 2007;3:51–60. doi: 10.2174/157340607779317508. [DOI] [PubMed] [Google Scholar]
- 146.Wang W, Wang H, Rayburn ER, Zhao Y, Hill DL, Zhang R. Br. J. Cancer. 2008;98:792–802. doi: 10.1038/sj.bjc.6604227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Lei J, Li X, Gong XJ, Zheng YN. Molecules. 2007;12:2140–2150. doi: 10.3390/12092140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Hong J, Min HY, Xu GH, Lee JG, Lee SH, Kim YS, Kang SS, Lee SK. Planta Med. 2008;74:151–155. doi: 10.1055/s-2008-1034286. [DOI] [PubMed] [Google Scholar]
- 149.Sekiya M, Kashiwada Y, Nabekura T, Kitagawa S, Yamagishi T, Yokozawa T, Ichiyanagi T, Ikeshiro Y, Takaishi Y. Planta Med. 2007;73:1558–1562. doi: 10.1055/s-2007-993745. [DOI] [PubMed] [Google Scholar]
- 150.Tuchinda P, Kornsakulkarn J, Pohmakotr M, Kongsaeree P, Prabpai S, Yoosook C, Kasisit J, Napaswad C, Sophasan S, Reutrakul V. J. Nat. Prod. 2008;71:655–663. doi: 10.1021/np7007347. [DOI] [PubMed] [Google Scholar]
- 151.Lee JH, Koo TH, Yoon H, Jung HS, Jin HZ, Lee K, Hong YS, Lee JJ. Biochem. Pharmacol. 2006;72:1311–1321. doi: 10.1016/j.bcp.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 152.Liao LM, Silva GA, Monteiro MR, Albuquerque S, Naturforsch Z. C: J. Biosci. 2008;63:207–210. doi: 10.1515/znc-2008-3-408. [DOI] [PubMed] [Google Scholar]
- 153.Morita H, Hirasawa Y, Muto A, Yoshida T, Sekita S, Shirota O. Bioorg. Med. Chem. Lett. 2008;18:1050–1052. doi: 10.1016/j.bmcl.2007.12.016. [DOI] [PubMed] [Google Scholar]
- 154.Nam NH. Mini-Rev. Med. Chem. 2006;6:945–951. doi: 10.2174/138955706777934937. [DOI] [PubMed] [Google Scholar]
- 155.Allison AC, Cacabelos R, Lombardi VR, Alvarez XA, Vigo C. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2001;25:1341–1357. doi: 10.1016/s0278-5846(01)00192-0. [DOI] [PubMed] [Google Scholar]
- 156.Brown IR. Ann. N. Y. Acad. Sci. 2007;1113:147–158. doi: 10.1196/annals.1391.032. [DOI] [PubMed] [Google Scholar]
- 157.Traynor BJ, Bruijn L, Conwit R, Beal F, O’Neill G, Fagan SC, Cudkowicz ME. Neurology. 2006;67:20–27. doi: 10.1212/01.wnl.0000223353.34006.54. [DOI] [PubMed] [Google Scholar]
- 158.Kutney JP, Hewitt GM, Kurihara T, Salisbury PJ, Sindelar RD, Stuart KL, Townsley PM, Chalmers WT, Jacoli GG. Can. J. Chem. 1981;59:2677–2683. [Google Scholar]
- 159.Zhang T, Hamza A, Cao X, Wang B, Yu S, Zhan CG, Sun D. Mol. Cancer Ther. 2008;7:162–170. doi: 10.1158/1535-7163.MCT-07-0484. [DOI] [PubMed] [Google Scholar]
- 160.Sethi G, Ahn KS, Pandey MK, Aggarwal BB. Blood. 2007;109:2727–2735. doi: 10.1182/blood-2006-10-050807. [DOI] [PubMed] [Google Scholar]
- 161.Zhang YQ, Sarge KD. J. Mol. Med. 2007;85:1421–1428. doi: 10.1007/s00109-007-0251-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Hieronymus H, Lamb J, Ross KN, Peng XP, Clement C, Rodina A, Nieto M, Du J, Stegmaier K, Raj SM, Maloney KN, Clardy J, Hahn WC, Chiosis G, Golub TR. Cancer Cell. 2006;10:321–330. doi: 10.1016/j.ccr.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 163.Hughes D, Guttenplan JB, Marcus CB, Subbaramaiah K, Dannenberg AJ. Cancer Prev. Res. 2008;1:485–493. doi: 10.1158/1940-6207.CAPR-08-0149. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 164.Huang Y, Zhou Y, Fan Y, Zhou D. Cancer Lett. 2008;264:101–106. doi: 10.1016/j.canlet.2008.01.043. [DOI] [PubMed] [Google Scholar]
- 165.Yang H, Chen D, Cui QC, Yuan X, Dou QP. Cancer Res. 2006;66:4758–4765. doi: 10.1158/0008-5472.CAN-05-4529. [DOI] [PubMed] [Google Scholar]
- 166.Abbas S, Bhoumik A, Dahl R, Vasile S, Krajewski S, Cosford ND, Ronai ZA. Clin. Cancer Res. 2007;13:6769–6778. doi: 10.1158/1078-0432.CCR-07-1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Roy A, Saraf S. Biol. Pharm. Bull. 2006;29:191–201. doi: 10.1248/bpb.29.191. [DOI] [PubMed] [Google Scholar]
- 168.Hasegawa S. ACS Symp. Ser. 2000;758:9–30. [Google Scholar]
- 169.Hasegawa S, Miyake M. Food Rev. Int. 1996;12:413–435. [Google Scholar]
- 170.Hasegawa S, Berhow MA, Manners GD. ACS Symp. Ser. 2000;758:1–8. [Google Scholar]
- 171.Manners GD. J. Agric. Food Chem. 2007;55:8285–8294. doi: 10.1021/jf071797h. [DOI] [PubMed] [Google Scholar]
- 172.Poulose SM, Harris ED, Patil BS. Nutr. Cancer. 2006;56:103–112. doi: 10.1207/s15327914nc5601_14. [DOI] [PubMed] [Google Scholar]
- 173.Zhang H, Wang X, Chen F, Androulakis XM, Wargovich MJ. Phytother. Res. 2007;21:731–734. doi: 10.1002/ptr.2148. [DOI] [PubMed] [Google Scholar]
- 174.Roy MK, Kobori M, Takenaka M, Nakahara K, Shinmoto H, Isobe S, Tsushida T. Phytother. Res. 2007;21:245–250. doi: 10.1002/ptr.2058. [DOI] [PubMed] [Google Scholar]
- 175.Anitha G, Raj JJ, Krishnan VR, Narasimhan S, Solomon KA, Rajan SS. J. Asian Nat. Prod. Res. 2007;9:73–78. doi: 10.1080/10286020500383742. [DOI] [PubMed] [Google Scholar]
- 176.Uddin SJ, Nahar L, Shilpi JA, Shoeb M, Borkowski T, Gibbons S, Middleton M, Byres M, Sarker SD. Phytother. Res. 2007;21:757–761. doi: 10.1002/ptr.2159. [DOI] [PubMed] [Google Scholar]
- 177.Mitsui K, Saito H, Yamamura R, Fukaya H, Hitotsuyanagi Y, Takeya K. J. Nat. Prod. 2006;69:1310–1314. doi: 10.1021/np068021f. [DOI] [PubMed] [Google Scholar]
- 178.Brandt GE, Schmidt MD, Prisinzano TE, Blagg BS. J. Med. Chem. 2008;51:6495–6502. doi: 10.1021/jm8007486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Chiruvella KK, Mohammed A, Dampuri G, Ghanta RG, Raghavan SC. Int. J. Biomed. Sci. 2007;3:269–278. [PMC free article] [PubMed] [Google Scholar]
- 180.Chiruvella KK, Kari V, Choudhary B, Nambiar M, Ghanta RG, Raghavan SC. FEBS Lett. 2008;582:4066–4076. doi: 10.1016/j.febslet.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 181.Vieira IJC, Braz-Filho R. Stud. Nat. Prod. Chem. 2006;33:433–492. [Google Scholar]
- 182.Seida AA, Kinghorn AD, Cordell GA, Farnsworth NR. Lloydia. 1978;41:584–587. [PubMed] [Google Scholar]
- 183.Nguyen-Cong V, Rode BM. Quant. Struct.-Act. Relat. 1995;14:512–517. [Google Scholar]
- 184.Polonsky J, Varon Z, Moretti C, Pettit GR, Herald CL, Rideout JA, Saha SB, Khastgir HN. J. Nat. Prod. 1980;43:503–509. doi: 10.1021/np50010a012. [DOI] [PubMed] [Google Scholar]
- 185.Jin X, Jin HR, Lee D, Lee JH, Kim SK, Lee JJ. Eur. J. Pharmacol. 2008;592:41–47. doi: 10.1016/j.ejphar.2008.06.104. [DOI] [PubMed] [Google Scholar]
- 186.Hitotsuyanagi Y, Kim IH, Hasuda T, Yamauchi Y, Takeya K. Tetrahedron. 2006;62:4262–4271. [Google Scholar]
- 187.Dai J, Fishback JA, Zhou YD, Nagle DG. J. Nat. Prod. 2006;69:1715–1720. doi: 10.1021/np060278q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Jain S, Shirode A, Yacoub S, Barbo A, Sylvester PW, Huntimer E, Halaweish F, El Sayed KA. Planta Med. 2007;73:591–596. doi: 10.1055/s-2007-967188. [DOI] [PubMed] [Google Scholar]
- 189.Shi Z, Jain S, Kim IW, Peng XX, Abraham I, Youssef DT, Fu LW, El Sayed K, Ambudkar SV, Chen ZS. Cancer Sci. 2007;98:1373–1380. doi: 10.1111/j.1349-7006.2007.00554.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Tang S, Pei Y, Fu H, Deng Z, Li J, Proksch P, Lin W. Chem. Pharm. Bull. 2006;54:4–8. doi: 10.1248/cpb.54.4. [DOI] [PubMed] [Google Scholar]
- 191.Li M, Wei S-Y, Tang S-A, Lin W-H, Cui J-R. J. Chin. Pharm. Sci. 2008;17:11–15. [Google Scholar]
- 192.Wei SY, Li M, Tang SA, Sun W, Xu B, Cui JR, Lin WH. Cancer Lett. 2008;262:114–122. doi: 10.1016/j.canlet.2007.11.039. [DOI] [PubMed] [Google Scholar]
- 193.Fujioka T, Kashiwada Y, Kilkuskie RE, Cosentino LM, Ballas LM, Jiang JB, Janzen WP, Chen IS, Lee KH. J. Nat. Prod. 1994;57:243–247. doi: 10.1021/np50104a008. [DOI] [PubMed] [Google Scholar]
- 194.Yu D, Morris-Natschke SL, Lee KH. Med. Res. Rev. 2007;27:108–132. doi: 10.1002/med.20075. [DOI] [PubMed] [Google Scholar]
- 195.Qian K, Morris-Natschke SL, Lee KH. Med. Res. Rev. 2009;29:369–393. doi: 10.1002/med.20138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Kashiwada Y, Hashimoto F, Cosentino LM, Chen CH, Garrett PE, Lee KH. J. Med. Chem. 1996;39:1016–1017. doi: 10.1021/jm950922q. [DOI] [PubMed] [Google Scholar]
- 197.Qian K, Nakagawa-Goto K, Yu D, Morris-Natschke SL, Nitz TJ, Kilgore N, Allaway GP, Lee KH. Bioorg. Med. Chem. Lett. 2007;17:6553–6557. doi: 10.1016/j.bmcl.2007.09.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Li F, Goila-Gaur R, Salzwedel K, Kilgore NR, Reddick M, Matallana C, Castillo A, Zoumplis D, Martin DE, Orenstein JM, Allaway GP, Freed EO, Wild CT. Proc. Natl. Acad. Sci. U. S. A. 2003;100:13555–13560. doi: 10.1073/pnas.2234683100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Soler F, Poujade C, Evers M, Carry JC, Henin Y, Bousseau A, Huet T, Pauwels R, De Clercq E, Mayaux JF, Le Pecq JB, Dereu N. J. Med. Chem. 1996;39:1069–1083. doi: 10.1021/jm950669u. [DOI] [PubMed] [Google Scholar]
- 200.Mayaux JF, Bousseau A, Pauwels R, Huet T, Henin Y, Dereu N, Evers M, Soler F, Poujade C, De Clercq E, Le Pecq JB. Proc. Natl. Acad. Sci. U. S. A. 1994;91:3564–3568. doi: 10.1073/pnas.91.9.3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Sun IC, Chen CH, Kashiwada Y, Wu JH, Wang HK, Lee KH. J. Med. Chem. 2002;45:4271–4275. doi: 10.1021/jm020069c. [DOI] [PubMed] [Google Scholar]
- 202.Huang L, Ho P, Lee KH, Chen CH. Bioorg. Med. Chem. 2006;14:2279–2289. doi: 10.1016/j.bmc.2005.11.016. [DOI] [PubMed] [Google Scholar]
- 203.Huang L, Lai W, Ho P, Chen CH. AIDS Res. Hum. Retrovir. 2007;23:28–32. doi: 10.1089/aid.2006.0137. [DOI] [PubMed] [Google Scholar]
- 204.Lai W, Huang L, Ho P, Li Z, Montefiori D, Chen CH. Antimicrob. Agents Chemother. 2008;52:128–136. doi: 10.1128/AAC.00737-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Qian K, Yu D, Chen CH, Huang L, Morris-Natschke SL, Nitz TJ, Salzwedel K, Reddick M, Allaway GP, Lee KH. J. Med. Chem. 2009 doi: 10.1021/jm900136j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Gerrish D, Kim In C, Kumar Dange V, Austin H, Garrus Jennifer E, Baichwal V, Saunders M, McKinnon Rena S, Anderson Mark B, Carlson R, Arranz-Plaza E, Yager Kraig M. Bioorg. Med. Chem. Lett. 2008;18:6377–6380. doi: 10.1016/j.bmcl.2008.10.098. [DOI] [PubMed] [Google Scholar]
- 207.Li WH, Zhang XM, Tian RR, Zheng YT, Zhao WM, Qiu MH. J. Asian Nat. Prod. Res. 2007;9:551–555. doi: 10.1080/10286020600883419. [DOI] [PubMed] [Google Scholar]
- 208.Wei Y, Ma CM, Chen DY, Hattori M. Phytochemistry. 2008;69:1875–1879. doi: 10.1016/j.phytochem.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 209.Parra A, Rivas F, Lopez PE, Garcia-Granados A, Martinez A, Albericio F, Marquez N, Munoz E. Bioorg. Med. Chem. 2009;17:1139–1145. doi: 10.1016/j.bmc.2008.12.041. [DOI] [PubMed] [Google Scholar]
- 210.Huang L, Yu D, Ho P, Lee K-H, Chen C-H. Lett. Drug Des. Discovery. 2007;4:471–478. [Google Scholar]
- 211.Yu D, Sakurai Y, Chen CH, Chang FR, Huang L, Kashiwada Y, Lee KH. J. Med. Chem. 2006;49:5462–5469. doi: 10.1021/jm0601912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Baltina LA, Jr, Kondratenko RM, Plyasunova OA, Baltina LA, Pokrovskii AG, Khalilov LM, Galin FZ, Tolstikov GA. Pharm. Chem. J. 2008;42:64–67. [Google Scholar]
- 213.Kashiwada Y, Wang HK, Nagao T, Kitanaka S, Yasuda I, Fujioka T, Yamagishi T, Cosentino LM, Kozuka M, Okabe H, Ikeshiro Y, Hu CQ, Yeh E, Lee KH. J. Nat. Prod. 1998;61:1090–1095. doi: 10.1021/np9800710. [DOI] [PubMed] [Google Scholar]
- 214.Ito J, Chang FR, Wang HK, Park YK, Ikegaki M, Kilgore N, Lee KH. J. Nat. Prod. 2001;64:1278–1281. doi: 10.1021/np010211x. [DOI] [PubMed] [Google Scholar]
- 215.Ho JC, Chen CM, Row LC. Phytochemistry. 2007;68:631–635. doi: 10.1016/j.phytochem.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 216.Kashiwada Y, Nagao T, Hashimoto A, Ikeshiro Y, Okabe H, Cosentino LM, Lee KH. J. Nat. Prod. 2000;63:1619–1622. doi: 10.1021/np990633v. [DOI] [PubMed] [Google Scholar]
- 217.Deng S-L, Baglin I, Nour M, Flekhter O, Vita C, Cave C. Phosphorus, Sulfur Silicon Relat. Elem. 2007;182:951–967. [Google Scholar]
- 218.Li RT, Han QB, Zheng YT, Wang RR, Yang LM, Lu Y, Sang SQ, Zheng QT, Zhao QS, Sun HD. Chem. Commun. 2005:2936–2938. doi: 10.1039/b501932j. [DOI] [PubMed] [Google Scholar]
- 219.Xiao WL, Li RT, Li SH, Li XL, Sun HD, Zheng YT, Wang RR, Lu Y, Wang C, Zheng QT. Org. Lett. 2005;7:1263–1266. doi: 10.1021/ol0473187. [DOI] [PubMed] [Google Scholar]
- 220.Xiao WL, Zhu HJ, Shen YH, Li RT, Li SH, Sun HD, Zheng YT, Wang RR, Lu Y, Wang C, Zheng QT. Org. Lett. 2005;7:2145–2148. doi: 10.1021/ol050502n. [DOI] [PubMed] [Google Scholar]
- 221.Xiao WL, Yang LM, Gong NB, Wu L, Wang RR, Pu JX, Li XL, Huang SX, Zheng YT, Li RT, Lu Y, Zheng QT, Sun HD. Org. Lett. 2006;8:991–994. doi: 10.1021/ol060062f. [DOI] [PubMed] [Google Scholar]
- 222.Xiao WL, Li XL, Wang RR, Yang LM, Li LM, Huang SX, Pu JX, Zheng YT, Li RT, Sun HD. J. Nat. Prod. 2007;70:1056–1059. doi: 10.1021/np0700927. [DOI] [PubMed] [Google Scholar]
- 223.Xiao W-L, Pu J-X, Wang R-R, Yang L-M, Li X-L, Li S-H, Li R-T, Huang S-X, Zheng Y-T, Sun H-D. Helv. Chim. Acta. 2007;90:1505–1513. [Google Scholar]
- 224.Xiao WL, Pu JX, Chang Y, Li XL, Huang SX, Yang LM, Li LM, Lu Y, Zheng YT, Li RT, Zheng QT, Sun HD. Org. Lett. 2006;8:1475–1478. doi: 10.1021/ol060324d. [DOI] [PubMed] [Google Scholar]
- 225.Xiao W-L, Pu J-X, Chang Y, Li X-L, Huang S-X, Yang L-M, Li L-M, Lu Y, Zheng Y-T, Li R-T, Zheng Q-T, Sun H-D. Org. Lett. 2006;8:4669. doi: 10.1021/ol060324d. [DOI] [PubMed] [Google Scholar]
- 226.Xiao WL, Huang SX, Wang RR, Zhong JL, Gao XM, He F, Pu JX, Lu Y, Zheng YT, Zheng QT, Sun HD. Phytochemistry. 2008;69:2862–2866. doi: 10.1016/j.phytochem.2008.09.010. [DOI] [PubMed] [Google Scholar]
- 227.Xiao W-L, Yang L-M, Li L-M, Pu J-X, Huang S-X, Weng Z-Y, Lei C, Liu J-P, Wang R-R, Zheng Y-T, Li R-T, Sun H-D. Tetrahedron Lett. 2007;48:5543–5546. [Google Scholar]
- 228.Huang SX, Yang J, Huang H, Li LM, Xiao WL, Li RT, Sun HD. Org. Lett. 2007;9:4175–4178. doi: 10.1021/ol701679n. [DOI] [PubMed] [Google Scholar]
- 229.Huang S-X, Han Q-B, Lei C, Pu J-X, Xiao W-L, Yu J-L, Yang L-M, Xu H-X, Zheng Y-T, Sun H-D. Tetrahedron. 2008;64:4260–4267. [Google Scholar]
- 230.Yang G-Y, Xiao W-L, Chang Y, Wang R-R, Pu J-X, Gao X-M, Lei C, Lu Y, Zheng Y-T, Sun H-D. Helv. Chim. Acta. 2008;91:1871–1878. [Google Scholar]
- 231.Xiao WL, Tian RR, Pu JX, Li X, Wu L, Lu Y, Li SH, Li RT, Zheng YT, Zheng QT, Sun HD. J. Nat. Prod. 2006;69:277–279. doi: 10.1021/np0503303. [DOI] [PubMed] [Google Scholar]
- 232.Pu JX, Yang LM, Xiao WL, Li RT, Lei C, Gao XM, Huang SX, Li SH, Zheng YT, Huang H, Sun HD. Phytochemistry. 2008;69:1266–1272. doi: 10.1016/j.phytochem.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 233.El Dine RS, El Halawany AM, Ma CM, Hattori M. J. Nat. Prod. 2008;71:1022–1026. doi: 10.1021/np8001139. [DOI] [PubMed] [Google Scholar]
- 234.Chen JC, Zhang GH, Zhang ZQ, Qiu MH, Zheng YT, Yang LM, Yu KB. J. Nat. Prod. 2008;71:153–155. doi: 10.1021/np0704396. [DOI] [PubMed] [Google Scholar]
- 235.Chen J, Tian R, Qiu M, Lu L, Zheng Y, Zhang Z. Phytochemistry. 2008;69:1043–1048. doi: 10.1016/j.phytochem.2007.10.020. [DOI] [PubMed] [Google Scholar]
- 236.Tian R-R, Chen J-C, Zhang G-H, Qiu M-H, Wang Y-H, Du L, Shen X, Liu N-F, Zheng Y-T. Zhongguo Tianran Yaowu. 2008;6:214–218. [Google Scholar]
- 237.Yu YB, Miyashiro H, Nakamura N, Hattori M, Park JC. Arch. Pharmacal Res. 2007;30:820–826. doi: 10.1007/BF02978831. [DOI] [PubMed] [Google Scholar]
- 238.Lee JS, Miyashiro H, Nakamura N, Hattori M. Chem. Pharm. Bull. 2008;56:711–714. doi: 10.1248/cpb.56.711. [DOI] [PubMed] [Google Scholar]
- 239.Connolly JD, Hill RA. Nat. Prod. Rep. 1985;2:1–17. doi: 10.1039/b003456h. [DOI] [PubMed] [Google Scholar]
- 240.Connolly JD, Hill RA. Nat. Prod. Rep. 1986;3:421–442. [Google Scholar]
- 241.Connolly JD, Hill RA. Nat. Prod. Rep. 1989;6:475–501. doi: 10.1039/np9890600475. [DOI] [PubMed] [Google Scholar]
- 242.Connolly JD, Hill RA. Nat. Prod. Rep. 1994;11:91–117. doi: 10.1039/np9941100091. [DOI] [PubMed] [Google Scholar]
- 243.Connolly JD, Hill RA. Nat. Prod. Rep. 1994;11:467–492. doi: 10.1039/np9941100467. [DOI] [PubMed] [Google Scholar]
- 244.Connolly JD, Hill RA. Nat. Prod. Rep. 1995;12:609–638. [Google Scholar]
- 245.Connolly JD, Hill RA. Nat. Prod. Rep. 1996;13:151–169. [Google Scholar]
- 246.Connolly JD, Hill RA. Nat. Prod. Rep. 1997;14:661–679. [Google Scholar]
- 247.Connolly JD, Hill RA. Nat. Prod. Rep. 1999;16:221–240. [Google Scholar]
- 248.Connolly JD, Hill RA. Nat. Prod. Rep. 2000;17:463–482. doi: 10.1039/b003456h. [DOI] [PubMed] [Google Scholar]
- 249.Connolly JD, Hill RA. Nat. Prod. Rep. 2001;18:131–147. doi: 10.1039/a807252c. [DOI] [PubMed] [Google Scholar]
- 250.Connolly JD, Hill RA. Nat. Prod. Rep. 2001;18:560–578. doi: 10.1039/b104602k. [DOI] [PubMed] [Google Scholar]
- 251.Connolly JD, Hill RA. Nat. Prod. Rep. 2002;19:494–513. doi: 10.1039/b110404g. [DOI] [PubMed] [Google Scholar]
- 252.Connolly JD, Hill RA. Nat. Prod. Rep. 2003;20:640–659. doi: 10.1039/b204068a. [DOI] [PubMed] [Google Scholar]
- 253.Connolly JD, Hill RA. Nat. Prod. Rep. 2005;22:230–248. doi: 10.1039/b500575m. [DOI] [PubMed] [Google Scholar]
- 254.Connolly JD, Hill RA. Nat. Prod. Rep. 2005;22:487–503. doi: 10.1039/b501301c. [DOI] [PubMed] [Google Scholar]
- 255.Connolly JD, Hill RA. Nat. Prod. Rep. 2007;24:465–486. doi: 10.1039/b507872p. [DOI] [PubMed] [Google Scholar]
- 256.Connolly JD, Hill RA. Nat. Prod. Rep. 2008;25:794–830. doi: 10.1039/b718038c. [DOI] [PubMed] [Google Scholar]