

Abstract
The effect of peptide tyrosine–tyrosine (PYY) on feeding is well established but currently its role in glucose homeostasis is poorly defined. Here we show in mice, that intraperitoneal (ip) injection of PYY3-36 or Y2R agonist improves nutrient-stimulated glucose tolerance and enhances insulin secretion; an effect blocked by peripheral, but not central, Y2R antagonist administration. Studies on isolated mouse islets revealed no direct effect of PYY3-36 on insulin secretion. Bariatric surgery in mice, enterogastric anastomosis (EGA), improved glucose tolerance in wild-type mice and increased circulating PYY and active GLP-1. In contrast, in Pyy-null mice, post-operative glucose tolerance and active GLP-1 levels were similar in EGA and sham-operated groups. PYY3-36 ip increased hepato-portal active GLP-1 plasma levels, an effect blocked by ip Y2R antagonist. Collectively, these data suggest that PYY3-36 therefore acting via peripheral Y2R increases hepato-portal active GLP-1 plasma levels and improves nutrient-stimulated glucose tolerance.
Abbreviations: aCSF, artificial cerebrospinal fluid; AUC, area under the curve; CNS, central nervous system; EGA, entero-gastric anastomosis; DPP-4, di-peptidyl peptidase-4; active GLP-1, glucagon-like peptide-1(7-36)amide; ICV, intracerebroventricular; ip, intraperitoneal; IPGTT, intraperitoneal glucose tolerance test; HFD, high-fat diet; PYY, peptide tyrosine–tyrosine; T2DM, type 2 diabetes mellitus; Y2R, Y2-receptor; WT, wild-type
Keywords: Glucose homeostasis, PYY, Y2-receptor, GLP-1
1. Introduction
A number of gut-derived hormones that regulate bodyweight and glucose homeostasis have been identified [1,2]. Recently, the marked effects of bariatric surgery, which alters nutrient flow through the gastrointestinal (GI) tract, on weight-loss and glucose homeostasis has intensified research efforts aimed at understanding gut hormone biology [3]. Gastric bypass surgery, the current gold standard bariatric procedure, leads to marked concomitant increases in nutrient-stimulated circulating levels of the L-cell products peptide YY (PYY) and glucagon-like peptide-1 (GLP-1), which occur early post-surgery and independently of weight-loss [4]. Increased PYY is thought to reduce appetite and drive weight-loss [5], whilst increased GLP-1 mediates improved glycemia post-surgery [5].
GLP-1 is synthesised by post-translational processing of pro-glucagon as two major active molecular forms; GLP-1(7–36) amide, the predominant circulating form, and GLP-1(7–37). In response to nutrient-ingestion, circulating active GLP-1 levels increase rapidly and remain elevated for several hours. The greatest circulating active GLP-1 concentrations are found in the hepato-portal circulation, with only a small proportion of active GLP-1 reaching the systemic circulation [6]. The main biological actions of active GLP-1 are on glucose homeostasis; increased glucose-stimulated insulin release and insulin biosynthesis as well as suppressed glucagon production. These effects are limited by its short half-life, due to rapid degradation by dipeptidyl peptidase-4 (DPP-4). Hence, DPP-4 resistant GLP-1 analogs/receptor agonists and DPP-4 inhibitors have been developed for treating patients with type 2 diabetes mellitus (T2DM) [7].
PYY circulates as two major forms, PYY1-36 and PYY3-36, which exhibit differential Y-receptor (YR) subtype affinity and biological actions. PYY1-36 activates all YR subtypes whereas PYY3-36, generated by cleavage of two N-terminal amino acids by DPP-4 [8], displays Y2R selectivity [9,10]. PYY3-36 is the predominant circulating form and its role in appetite control has been well-characterised; exogenous administration of PYY3-36 reduces food intake in both animals and humans [10] whilst Pyy-null mice are hyperphagic and develop obesity [11,12]. However, the exact function of PYY in energy balance is disputed. Anatomical and electrophysiological studies in rodents together with functional magnetic resonance imaging studies in humans indicate that PYY3-36 acts in key homeostatic and reward brain regions to regulate feeding behavior. Central nervous system (CNS) pharmacological blockade, using a highly selective Y2R antagonist (BIIE0246) [13] abrogates the anorexigenic action of peripherally administered PYY3-36 [14], suggesting that the primary activity of PYY, with respect to appetite regulation, is mediated by central Y2R.
The role of PYY in glucose homeostasis remains controversial [15]. However, several lines of evidence suggest a potential role for PYY in glucose regulation. Pyy is expressed in pancreatic islets where it is thought to regulate insulin secretion; in vitro studies show that PYY1-36 causes a dose-dependent inhibition of insulin secretion [16] and islets of Pyy-null mice hypersecrete insulin [12]. Evidence for a potential gluco-regulatory role of circulating PYY is suggested by studies in rodents showing that intravenous PYY3-36 infusion, to high-fat diet fed mice during a hyperinsulinaemic–euglycaemic clamp improved glucose disposal and that repeated peripheral administration of PYY3-36 or Y2R agonists to rodents improves glycemic indices [17–19]. However, the concomitant effects of PYY3-36/Y2R on feeding and adiposity observed in these studies confounds the interpretation of these findings. In humans, PYY3-36 infusion studies have reported no effect on glucose homeostasis [20–22]. However, circulating PYY3-36 levels are greatest in the post-prandial state and these studies examined glucose and insulin concentrations in the fasted state. Thus, we undertook a series of studies in mice to examine the effects of peripheral PYY on nutrient-stimulated glucose tolerance and investigated the potential mechanisms underlying the gluco-regulatory actions of peripheral PYY3-36.
2. Materials and methods
2.1. Peptides and compounds
Murine PYY1-36 (Bachem, UK), murine PYY3-36 (Bachem, UK), Y2R agonist (N-Acetyl-[Leu28,Leu31]-neuropeptide Y24–36 (Sigma, UK), exendin9-39 (Bachem, UK), angiontensin II (Sigma, UK) were dissolved in saline (0.9% NaCl) and used at the doses indicated in individual experiments. The Y2R antagonist, (S)-N2-[[1-[2-[4-[(R,S)-5,11-dihydro-6(6H)-oxodibenz[b,e]azepin-11-yl]-1-piperazinyl]-2-oxoethyl]cyclopentyl]acetyl]-N-[2-[1,2-dihydro-3,5(2H)-dioxo-1,2-diphenyl-3H-1,2,4-triazol-4-yl]ethyl]-argininamide, BIIE0246 formate, (Tocris, UK) was dissolved in 5% DMSO. For intracerebroventricular (ICV) studies, compounds were further diluted in artificial cerebrospinal fluid (aCSF) as required. Animals not receiving specific compounds for each experiment were administered vehicle as appropriate for each study.
2.2. Mice
Male adult mice were maintained on a 12 h light/12 h dark cycle under constant temperature, free access to water and allowed access to standard mouse chow (Teklad Global 2018) or high-fat diet (HFD) chow (D12451, Research Diets Inc., USA) as outlined for each study. Following acclimatisation and handling, mice were studied aged between 10 and 16 weeks. C57BL/6 mice were purchased (Charles River Ltd., UK). The generation and genotyping of Pyy-null mice has been previously described [11]. Male Pyy-null and WT mice were bred and genotyped in-house. All studies were performed in accordance to the Home Office Animal Procedures Act (1986) and European Convention for the Protection of Laboratory Animals. Food/mouse weights were measured using a Sartorius BP610 balance.
2.3. Assessment of glucose homeostasis in vivo
Blood samples were collected from mice via tail vein bleeds. Glucose phenotyping studies were carried out as previously described [23]. Intraperitoneal glucose-tolerance tests (IPGTT) were performed after an overnight fast. A baseline (0 min) blood sample was taken. Mice were then injected ip with D-glucose at a dose of 2 g/kg of mouse bodyweight. Glucose concentrations were then measured at 15 min, 30 min, 60 min, 90 min and 120 min following injection. Antagonists were administered as described 30 min prior to feeding/glucose injections. For assessment of post-prandial glucose and hormone responses mice were trained to eat 1 g of a HFD (4.73 kcal) within a 60 min period. Mice were exposed to this procedure on four occasions each separated by 5 days of normal feeding days prior to experiments being carried out to allow habituation to protocol.
2.4. Intracerebroventricular (ICV) cannulation and Y2R antagonist administration
Under isoflurane anaesthesia a stainless steel cannulae was inserted into the third ventricle (midline 0 mm, 0.82 mm posterior from bregma, depth 4.8 mm from skull surface). Post-operatively, mice were singly-housed and given 1 week to recover. Cannula placement was confirmed by a positive dipsogenic response (persistent drinking response within 2 min) following ICV administration of 1 μl angiotensin II (10 ng/μl) [24]. ICV injection of the Y2R antagonist or vehicle was administered 30 min before an IPGTT test was performed with either glucose or glucose containing PYY3-36 (5 μg/100 g of bodyweight).
2.5. Islet isolation and in vitro studies
Islet isolation and static incubation studies were carried out as previously described [25]. Briefly, mice were sacrificed by cervical dislocation and a laparotomy performed. The common bile duct was clamped and a Liberase solution (Roche, UK) solution was injected into the pancreatic duct resulting in pancreatic disruption. The pancreas was excised, and mechanically disrupted. Tissue components were collected and washed before Ficoll gradient centrifugation. Islets were hand-picked. For insulin secretion studies, isolated islets were cultured overnight in DMEM with 11 mmol/l glucose. Static insulin release was assessed using batch cultures of 10 islets with additional d-glucose and/or compounds as indicated.
2.6. Studies on Pyy-null mice and wild-type mice
Male Pyy-null mice (n=30) and their wild-type (WT) littermates (n=30) were weaned at 3 weeks of age. Pyy-null mice gain more weight on control and HFD than their WT littermates [11]. Thus, to enable weight-matching, 6-week old WT mice were commenced on HFD whilst the Pyy-null mice continued on control-diet until aged 14 weeks when they were switched to HFD [26]. At 24 weeks of age, 16 weight-matched mice from each group were transported to facilities where the surgery was to be performed. Mice were then acclimatised for 1 week prior to randomisation to bypass (n=8 per genotype) or sham surgery (n=8 per genotype). The surgery group underwent EGA under isoflurane anesthesia as previously described [26]. In brief, a midline laparotomy was performed and the pyloric sphincter ligatured, followed by anastomosis between the mid-jejunum and stomach fundus, excluding the duodenum and the proximal jejunum from nutrient flow. Sham-operated counterparts were anesthetized for similar time periods; a midline incision was made, and the stomach and intestines exposed and manipulated. Previous studies by Troy et al., demonstrated that food intake returned to pre-surgery levels in the sham-operated group and had stabilised in the EGA group by 3 days post operatively [27]. At 5 days post-surgery, a time period chosen to allow recovery from surgery but to minimize the weight differences between groups, mice underwent an IPGTT. Mice were then sacrificed 10 days post-operatively following an overnight fast and blood collected by cardiac puncture as described below.
2.7. Terminal procedures
Mice were sacrificed by terminal anaesthesia following a 16 h fast. Blood was immediately collected by cardiac puncture or from the hepatic portal vein. For the active GLP-1 time course experiment, overnight fasted mice were injected ip with glucose 2 g/kg. Mice were then anaesthetised and blood collected from the hepatic portal vein at 0, 5, 15, 20, 25 and 30 min post-injection. For measurement of active GLP-1, DPP-4 inhibitor [10 μl/ml of blood] (Millipore, Watford, UK), was drawn into a chilled syringe before blood collection. Blood was transferred from syringes to tubes containing 0.5 M EDTA [50 μl/ml of blood], and aprotinin, [5000 Kallikrein inhibitor units (KIU)/ml of blood] (Trasylol; Bayer, UK). All samples were kept on ice until processing. Blood tubes were centrifuged for 15 min at 10,000 rpm at 4 °C. Plasma was then separated and stored at −80 °C until assayed.
2.8. Quantitative PCR analyses of gene expression
Islet total RNA was extracted using RNAeasy kit as per manufactures instructions (Qiagen, UK). Total RNA was also extracted from homogenised brainstem using TRIzol reagent (Invitrogen UK). Brainstem blocks were composed of pons and medulla regions of the hind brain and excluded cerebellum; dissection of the block started cranially at the level of intersection between midbrain and pons, and finished caudally at the level of intersection between pons and spinal cord [28]. Total RNA was quantified using a NanoDrop Bioanalyzer ND1000 (NanoDrop UK) and cDNA synthesised using a TaqMan Retrotranscription kit (Applied Biosystems, UK). Real-time quantitative PCR was performed using proprietary sequence Taqman Gene Expression assay FAM/TAMRA probes with Hprt1 as the housekeeping gene (Applied Biosystems, UK).
2.9. Hormone assays
Plasma insulin and active GLP-1 were measured with ELISA kits (Millipore, Watford, UK), and total plasma PYY was measured by radioimmunoassay (Millipore, Watford, UK) according to manufacturer specifications. To reduce inter-assay variation, samples from each experiment were assayed together.
2.10. Statistical analysis
Results are expressed as mean±standard error of mean (SEM). Values for integrated area under the curve (AUC) were calculated using the trapezoid rule. Between treatments comparisons were made using t-test or analysis of variance (ANOVA) with Newman–Keul or Dunnett post-hoc tests where appropriate. P≤0.05 was considered significant.
3. Results
3.1. Effect of peripheral PYY administration on glucose tolerance and insulin secretion
To evaluate the acute effect of PYY1-36 and PYY3-36 on glucose homeostasis we undertook IPGTT in adult male C57Bl/6 mice in combination with exogenous PYY1-36 or PYY3-36 administration. PYY1-36 administration (5 μg/100 g of bodyweight) had no significant effect on glucose excursions or glucose AUC0–120 (glucose AUC0–120 [mmol/L×min]; glucose alone=1517±46, glucose plus PYY1-36=1441±33, n=10 per group, P=0.15). In contrast, ip PYY3-36 administration (5 μg/100g of bodyweight) significantly reduced glucose concentrations at 30 min, 60 min and 90 min post-injection (Figure 1A) with reduced glucose AUC0–120 (Figure 1B). Assessment of plasma insulin showed increased insulin secretion at 15 min post-glucose injection following PYY3-36 administration compared to glucose alone with increased change in insulin concentration from baseline (Figure 1C) and increased actual insulin levels (insulin concentration at 15 min post-glucose injection (ng/ml); glucose alone=0.34±0.04, glucose with PYY3-36=0.52±0.05, n=8, P=0.02). However, by 30 min post-injection there was no difference in plasma insulin secretion between glucose alone and glucose with PYY3-36 groups (data not shown). Injection of PYY3-36 (5 μg/100 g of bodyweight) ip in the fasted state had no effect on glucose concentrations (data not shown). However, when administered in the post-prandial state, after a fixed-meal, PYY3-36 led to reduced glucose levels at 15 min, 30 min and 45 min post-injection (Figure 1D); (glucose concentration at baseline (mmol/L) ; saline alone=6.2±0.2, saline with PYY3-36=6.3±0.2, n=10, P=0.87).
Figure 1.
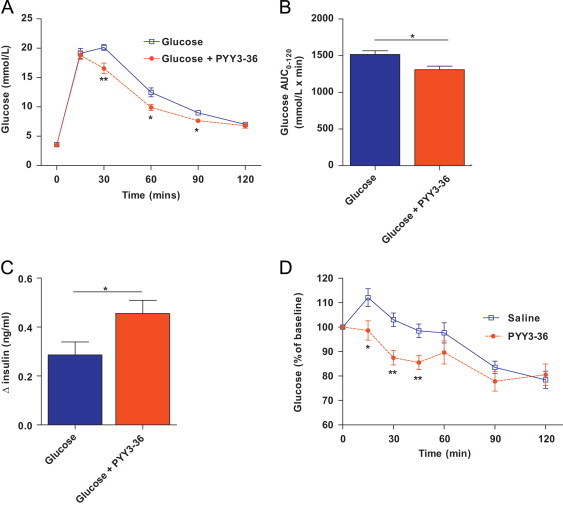
Effect of peripheral PYY3-36 administration on glucose tolerance and insulin release in 12–14 week-old male C57Bl/6 mice. (A) Time course of blood glucose concentrations during intraperitoneal glucose tolerance test (IPGTT), (B) glucose AUC0–120 during IPGTT and (C) delta plasma insulin concentration t0–t15 following ip injection of glucose or glucose in combination with PYY3-36. (D) Percentage change in blood glucose concentrations following ip injection of saline or PYY3-36 following a food intake (1 g of HFD consumed within 1 h). Data are expressed as mean±SEM. N=10 mice per group. * P<0.05, **P<0.01 vs. control as indicated.
3.2. PYY3-36 alters glucose tolerance via peripheral Y2 receptor activation
Next, to further evaluate the role of the Y2R in regulating glucose, we undertook IPGTT using either d-glucose alone (2 g/kg bodyweight) or a combination of the Y2R specific agonist, N-acetyl-[Leu28, Leu31]-NPY 24–36 at a dose of 5 μg/100 g bodyweight dissolved in d-glucose. Similar to our findings with PYY3-36, we found that co-administration of Y2R agonist with glucose significantly reduced blood glucose concentrations at 30 min, 60 min and 90 min compared to mice injected with glucose alone (Figure 2A), resulting in an overall reduction in glucose AUC0–120 (Figure 2B). Furthermore, blood samples collected at 0 min and 15 min for insulin measurement revealed an enhanced insulin secretion response with Y2R administration (Figure 2C).
Figure 2.
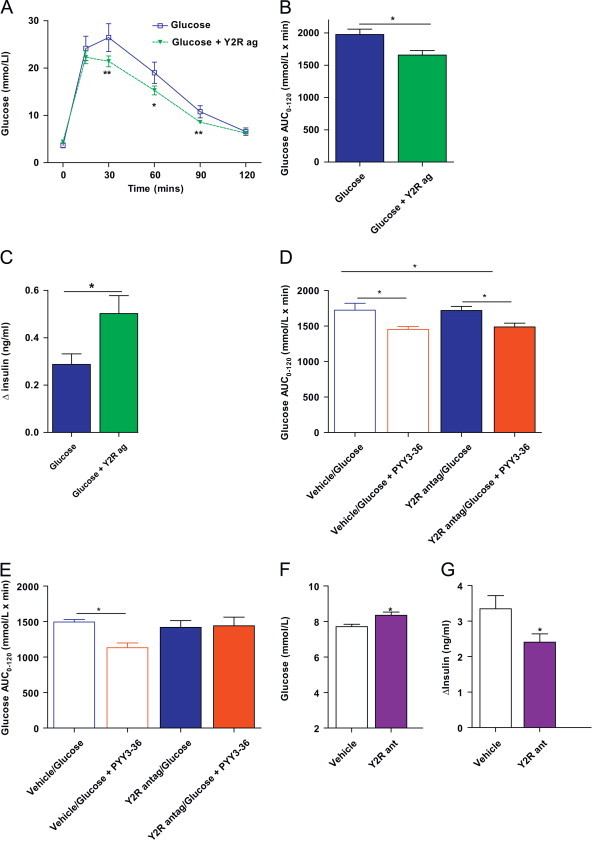
PYY3-36 alters glucose tolerance via peripheral Y2 receptor activation. (A) Time course of blood glucose concentrations during IPGTT, (B) glucose AUC0–120 during IPGTT and (C) delta plasma insulin concentration t0–t15 following ip injection of glucose alone or in combination with synthetic Y2 receptor agonist (Y2Rag). (D) Glucose AUC0–120 during IPGTT with glucose alone or glucose with PYY3-36 in mice pre-treated with ICV vehicle or Y2R antagonist BIIE0246 (Y2R antag). (E) Glucose AUC0–120 during IPGTT with glucose alone or glucose with PYY3-36 in mice pre-treated with ip vehicle or Y2R antagonist BIIE0246 (Y2R antag). (F and G) Adult C57Bl/6 mice were trained to eat 1 g of high-fat diet within a 1 h period in the early light phase after an overnight fast. Vehicle or BIIE0246 (Y2R ant) were injected prior to refeeding. Blood glucose (F) and plasma insulin (G) concentrations were measured 1 h post-refeed. Data are expressed as mean±SEM. N=8–14 mice per group. * P<0.05, **P<0.01 vs. control as indicated.
The anorectic effects of peripherally administered PYY3-36 are blocked by prior central administration of the Y2R specific antagonist, BIIE0246 [14] suggesting a central site of action of PYY3-36. Thus, we undertook studies to assess whether PYY3-36 also elicits its effects on glucose tolerance via central sites. First, we tested the dose and efficacy of ICV BIIE0246 (1 μg in 1 μl) dissolved in artificial cerebrospinal fluid, by emulating the feeding studies previously published [14]; antagonism of Y2R in the fed state resulted in a significant increase in food intake (2 h food intake (g): vehicle=1.3±0.1, BIIE0246=1.6±0.1, n=12 per group, P=0.003). However, the improved glucose tolerance observed with peripheral PYY3-36 administration was unaltered by CNS Y2R blockade with BIIE0246 (Figure 2D). In contrast, peripheral Y2R blockade through administration of ip BIIE0246 (2 mg/kg of bodyweight), abolished the glucose-lowering effects of PYY3-36 (Figure 2E).
Circulating PYY3-36 levels increase post-prandially, thus we examined the effect of peripheral Y2R blockade on post-meal glucose and insulin concentrations. Adult C57Bl/6 mice were trained to eat 1 g of HFD within a 1 h period in the early light phase after an overnight fast. Vehicle or BIIE0246 (2 mg/kg bodyweight) were injected ip prior to refeeding and blood was collected 1 h post-refeed for assessment of glucose and insulin concentration. BIIE0246 administration resulted in significantly elevated blood glucose concentrations and reduced insulin levels (Figure 2F and G). Taken together, these findings suggest that peripheral Y2R activation by PYY3-36 plays a role in regulating post-prandial glucose and insulin.
3.3. Effect of PYY1-36 and PYY3-36 on insulin secretion from isolated islets
To investigate whether PYY3-36 could directly modulate insulin secretion from islets, we undertook studies on islets isolated from adult male C57BL/6 mice. Islets were incubated with PYY1-36 or PYY3-36 and the effect on insulin secretion assessed. In agreement with previous reports [16,29,30], we found that PYY1-36 significantly reduced insulin secretion from islets (Figure 3A). In contrast, we observed no effect of PYY3-36 on islet insulin secretion (Figure 3B). Next, we extracted mRNA from isolated islets and undertook qPCR analysis for y1r, y2r, y4r and y5r expression. In agreement with published studies we detected y1r and y4r expression [31]. However, whilst we were able to detect y2r and y5r in our positive control, brainstem tissue, we were unable to detect y2r or y5r in isolated islets (Figure 3C). These results indicate that the effects of PYY3-36 on stimulating insulin secretion in vivo are mediated indirectly.
Figure 3.
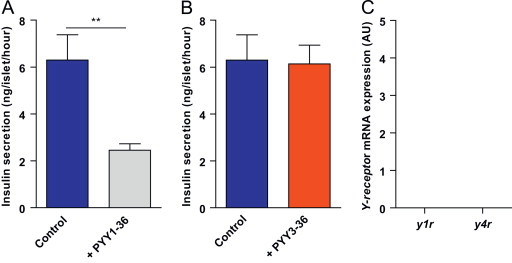
Effect of PYY on insulin secretion from isolated islets. (A and B) Secretion of insulin from islets isolated from adult C576Bl6 mice when incubated with medium containing glucose (Control) or with addition of (A) PYY3-36 or with addition of (B) PYY1-36. Relative expression of Y-receptor subtypes from mRNA extracted from isolated islets. Data are expressed as mean±SEM. ** P<0.01.
3.4. Effect of bariatric surgery on glucose tolerance in WT and Pyy-null mice
Bariatric surgery increases circulating PYY levels in rodents and humans and is associated with improved glucose tolerance [3]. Thus, we examined the effect of global Pyy deletion on glucose tolerance (IPGTT) following modified gastric bypass (EGA) surgery in mice. Previously, we have shown that Pyy-null mice lose less weight in the early post-operative phase than their WT littermates [26]. Thus, in order to minimise the effect of weight-loss differences on glucose homeostasis we undertook IPGTT at 5 days post-surgery when there were no significant bodyweight differences between groups (mouse bodyweight (g): WT sham 31.6±1.3, WT EGA=30.1±0.9, Pyy-null sham=34.4±2.2, Pyy-null EGA=32.5±1.0, P>0.05). Following EGA surgery, WT mice displayed improved glucose tolerance with reduced glucose AUC0–120 compared to WT sham mice (Figure 4A) whereas no significant differences were observed in Pyy-null mice (Figure 4A). Between post-operative day 5 and day 10 WT EGA lost weight, whereas WT sham-operated mice, Pyy-null EGA and Pyy-null sham-operated mice were weight-stable. Mice were sacrificed at day 10 post-surgery at which point WT EGA mice weighed less than WT sham-operated mice (bodyweight (g): WT sham=32.7±1.3, WT EGA=25.7±1.3, P<0.01) whilst EGA and sham-operated Pyy-null mice exhibited similar bodyweights (bodyweight (g): Pyy-null sham=33.6±0.9 and Pyy-null EGA=35.8±2.4). Fasting PYY concentrations were increased in WT EGA mice compared to WT sham mice (Figure 4B). Interestingly, fasted active GLP-1 levels were increased in WT EGA mice compared to WT sham (Figure 4C) but in Pyy-null mice active GLP-1 levels were similar in EGA and sham groups (Figure 4C). This observation suggested that endogenous PYY levels may regulate active GLP-1 levels.
Figure 4.
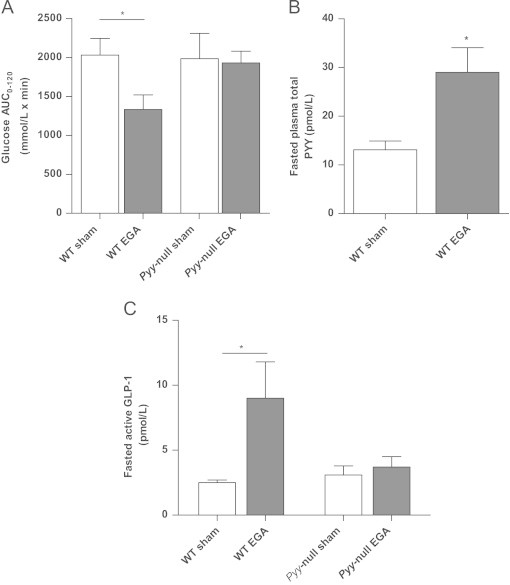
Effect of bariatric surgery on glucose tolerance in WT and Pyy-null mice. (A) Glucose AUC0–120 during IPGTT in WT and Pyy-null mice 5 days following either sham procedure or entero-gastric anastomosis surgery (EGA). (B) Fasting total PYY concentrations in WT sham and WT EGA mice. (C) Fasting active GLP-1 concentrations in WT and Pyy-null mice following sham and EGA surgery. Data are expressed as mean±SEM. N=7/8 mice per group. *P<0.05.
3.5. PYY3-36 increases nutrient-stimulated hepatic portal vein active GLP-1 concentrations via the Y2R
In overnight fasted adult C57Bl/6 male mice we undertook an active GLP-1 time course study to evaluate the temporal change in plasma active GLP-1 levels in the hepatic portal vein following ip glucose (2 g/kg) injection. Peak active GLP-1 levels were observed at 15 min post-injection (Figure 5A). Thus, we examined the effect of acute of ip PYY3-36 administration (5 μg/100 g bodyweight) on glucose-stimulated active GLP-1 levels in the systemic circulation and the hepatic portal vein at 15 min post-glucose injection. We found that whilst systemic active GLP-1 concentrations were unaltered by PYY3-36 co-administration (systemic active GLP-1 concentration (pmol/L): glucose=2.8±0.4, glucose plus PYY3-36=2.8±0.5, n=12–14 per group), hepatic portal vein active GLP-1 concentrations were significantly elevated (Figure 5B). Additionally, pre-administration of the Y2R antagonist BIIE0246 eliminated this enhanced hepato-portal vein active GLP-1 response to exogenously administered PYY3-36 (Figure 5C). These findings suggest that exogenous PYY3-36 acting via the Y2R increases active GLP-1 levels in the hepato-portal circulation.
Figure 5.
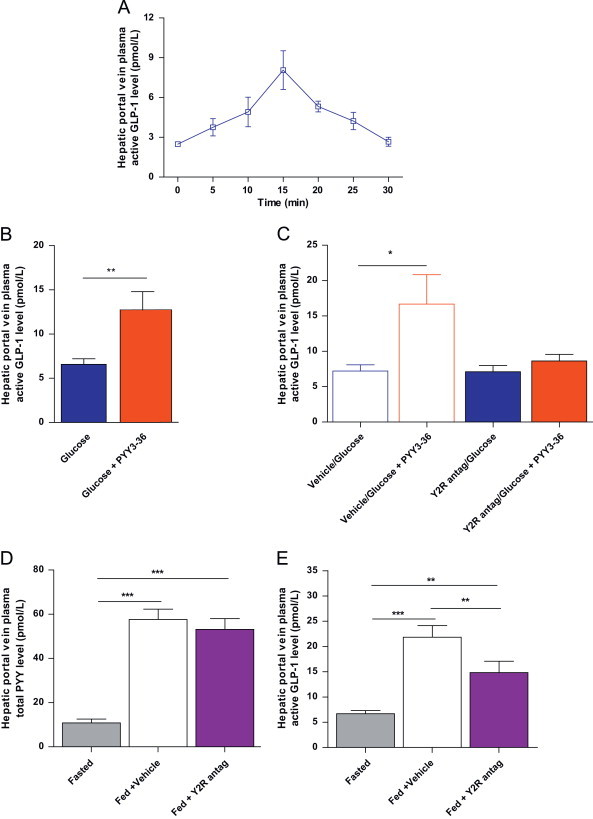
PYY3-36 increases nutrient-stimulated portal vein active GLP-1 concentrations via the Y2R. (A) Plasma concentration of active GLP-1 in the hepatic portal vein following ip injection of glucose. (B) Plasma concentration of active GLP-1 in the hepatic portal vein 15 min following ip injection of glucose or glucose in combination with PYY3-36. (C) Plasma hepatic portal vein concentration of active GLP-1 with pre-dosing of vehicle or Y2R antagonist (BIIE0246) (Y2R antag) before glucose or glucose combined with PYY3-36. (D and E) Hepatic portal vein plasma concentrations of (D) PYY and (E) active GLP-1 in the fasted state and following a 1 g HFD meal following ip injection with vehicle or Y2R antagonist (Y2R antag). Data are expressed as mean±SEM. N=8–14 mice per group. * P<0.05, **P<0.01, ***P<0.001 as indicated.
Next, we investigated whether endogenous post-prandial PYY acting via the Y2R regulated hepatic portal vein plasma active GLP-1 concentrations. Adult male C57Bl/6 mice were trained to eat 1 g of HFD within a 1 h period in the early light phase after a 16 h fast. On the study day, mice were randomised to three cohorts all matched for bodyweight. One cohort was sacrificed in the fasted state and the remaining two cohorts of mice were treated with either vehicle or Y2R antagonist, BIIE0246 administered ip (2 mg/kg bodyweight) immediately before food was returned to the mice. One hour following re-feeding, mice were sacrificed by terminal anaesthesia and blood was collected from the hepatic portal vein for measurement of PYY and active GLP-1 concentrations. Mice that failed to eat the entire 1 g HFD were excluded. Hepatic portal vein plasma PYY levels were significantly higher in the fed state in both the vehicle and BIIE0246 cohorts compared to the fasted cohort with no difference observed between treatment groups (Figure 5D). However, BIIE0246 administration led to a significant reduction in hepato-portal plasma active GLP-1 concentrations compared to the vehicle treated group (Figure 5E). Taken together, these findings suggest that endogenous PYY3-36 acting via the Y2R leads to increased hepatic portal vein plasma active GLP-1.
3.6. GLP-1 receptor blockade with exendin 9-39 abolishes the effects of PYY3-36 on glucose tolerance
We then investigated whether the improvement in glucose tolerance after PYY3-36 was altered by blockage of the GLP-1R with the GLP-1R antagonist, exendin 9-39 (Ex9-39). We found that ip injection of Ex9-39 (30 μg/100 g bodyweight) increased glucose levels and glucose AUC0–120 following IPGTT compared to glucose alone. Prior ip treatment with Ex9-39 (30 μg/100 g bodyweight) abolished the improved glucose tolerance (Figure 6A and B) observed in the PYY3-36 group. Glucose AUC0–120 in mice treated with Ex9-39 and PYY3-36 was not significantly different from glucose alone or with Ex9-39 plus glucose. Collectively, these findings suggest that the effects of PYY3-36 on glucose homeostasis are, in part, mediated by GLP-1.
Figure 6.
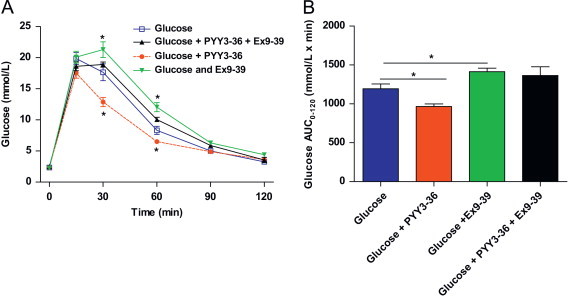
Exendin9-39 attenuates the effect of PYY3-36 on glucose tolerance. (A) Time course of blood glucose concentrations and (B) glucose AUC0–120 during IPGTT in C57Bl/6 mice injected (IP) with glucose, glucose and PYY3-36 or glucose, PYY3-36 and Ex9-39. Data are expressed as mean±SEM. N=8–14 mice per group. * P<0.05 compared to glucose alone.
4. Discussion
Our studies suggest that PYY1-36 and PYY3-36 exert differential effects on glucose homeostasis through different YR subtypes and sites of action. PYY is known to be co-expressed in pancreatic islets with glucagon, somatostatin and pancreatic polypeptide [16]. Previous studies undertaken on isolated islets have shown that PYY1-36 inhibits glucose-stimulated insulin secretion [29,32], that PYY immunoneutralisation enhances islet insulin release [30] and that islets lacking either PYY [12] (global Pyy-null mice) or Y1R [31] (y1r-null mice) exhibit enhanced glucose-stimulated insulin secretion. In agreement with these studies, we also found that PYY1-36 inhibited glucose-stimulated insulin secretion from isolated islets whilst, PYY3-36 had no effect. Our YR subtype expression studies on isolated islets revealed high abundance of the y1r with lower y4r abundance but no detectable y2r or y5r. The actions of PYY1-36 on insulin secretion appear to be predominantly mediated by intra-islet PYY, as we observed no effect of exogenously administered PYY1-36 on glucose handling in mice. Collectively, these data suggest that intra-islet PYY1-36 acts through either the Y1R or Y4R to inhibit glucose-stimulated insulin secretion and that PYY3-36 has no direct effect on islet insulin secretion. In contrast, we found that exogenously administered PYY3-36 significantly improved glucose tolerance and stimulated insulin secretion when co-administered with glucose or in the fed state. Exogenous PYY3-36 administration had no effect on glucose concentrations in the fasted state suggesting that the effect of PYY3-36 on glucose homeostasis is nutrient-dependent. In addition, our studies employing either Y2R agonist or Y2R antagonist implicate the Y2R as the key receptor mediating the actions of exogenous PYY3-36 on glucose homeostasis. The Y2R is widely distributed throughout the periphery and CNS [33] with central Y2R implicated in mediating the anorectic effects of PYY3-36 [14]. In contrast, our studies with peripheral and ICV Y2R antagonist (BIIE0246) administration suggest that exogenous PYY3-36 acts mainly through peripheral Y2R activation to modulate glucose homeostasis. Further studies are now warranted in mice with tissue-specific Y2R deletion in order to delineate the main tissue(s) responsible for the effect of PYY3-36 on glucose homeostasis. Our isolated islet studies examining the effects of PYY1-36 and PYY3-36 on insulin secretion and YR expression were undertaken on islets from normal weight C57BL/6 mice on normal chow. However, given that high-fat feeding and DIO have been shown to alter hypothalamic and brainstem YR expression [34,35] these studies should be extended to mice receiving HFD and with DIO.
Bariatric surgery is currently the most effective treatment for obesity resulting in sustained weight-loss, a reduction in obesity-related co-morbidities and reduced mortality [3]. Additionally, the majority of patients with T2DM that undergo GBP show immediate glycemic improvements prior to any significant weight-loss post-surgery [36]. Post-operative changes in circulating gut hormone concentrations are thought to play a key role in mediating changes in appetite/food intake and glucose homeostasis. Following GBP, circulating PYY and GLP-1 increase concomitantly and independently of weight-loss [36]. A key role for GLP-1 in mediating post-surgery glycemic improvement is supported by the finding that GLP-1R blockade with Ex9-39 attenuates surgically-induced improvement in glucose excursion and insulin secretion [5,27]. However, the role of GLP-1R in mediating post-operative changes in insulin sensitivity are less clear as Ex9-39 administration fails to reverse the effect of EGA surgery on insulin sensitivity [27]. Indeed, a number of interacting mechanisms; reduced nutrient intake, reduced hepatic gluconeogenesis, improved pancreatic beta cell function, increased peripheral insulin sensitivity and increased insulin secretion have all been suggested to play a role in the improvement in glucose homeostasis post-operatively [27,36,37].
Previously, we have shown that Pyy-null mice lose less weight in the early post-operative phase than WT mice, implicating a role for PYY in contributing to post-operative weight-loss [26]. In light of our finding, that exogenous PYY3-36 improved glucose tolerance, we next examined the effect of EGA surgery on post-operative glucose handling in WT and Pyy-null mice. Whilst several groups have reported that gut hormone changes and glycemic improvements occur independently of weight-loss [37], in order to diminish the potential confounding effects of weight-loss difference between groups we undertook IPGTT 5 days post-surgery. At this time-point glucose tolerance was significantly improved in the WT EGA mice compared to WT sham-operated mice but no difference in glucose tolerance was apparent between Pyy-null EGA and Pyy-null sham-operated groups. Moreover, while WT EGA mice exhibited increased fasting PYY and active GLP-1 concentrations compared to WT sham-operated mice, no differences in active GLP-1 levels were apparent between Pyy-null EGA mice and Pyy-null sham-operated mice. These results coupled with our findings that exogenous PYY3-36 increased glucose-stimulated insulin secretion, with no direct effect on isolated islets, suggested a potential regulatory effect of PYY3-36 on circulating active GLP-1 concentrations. A potential limitation of our studies is the early post-surgery time point at which we undertook our glucose studies, as at this stage mice may not have fully recovered. However, given that the surgical procedures undertaken were identical in Pyy-null and Pyy WT mice and that we have no evidence to suspect that post-operative recovery would be affected by genotype, the difference in glycemic response following EGA between Pyy-null are Pyy-WT mice are likely to be real. Our studies suggest that PYY impacts upon both weight-loss and glucose homeostasis in the early post-operative period. However, additional studies are needed to examine the longer term effects of Pyy-deletion on energy and glucose homeostasis following bariatric surgery.
Pyy-null mice exhibit hyperphagia and increased adiposity [11]. Thus, in order to achieve weight-matching of Pyy-null and WT mice at the time of surgery at 25 weeks of age, we commenced WT mice on HFD at 6 weeks of age and Pyy-null mice at 14 weeks of age. However, recent studies have shown that HFD exposure causes hypothalamic injury, a response which evolves over time [38]. Thus, our protocol of differentially exposing WT and Pyy-null mice to HFD may have impacted upon hypothalamic inflammation/injury and the subsequent responses of these two groups. This limitation of our experimental paradigm needs to be borne in mind when interpreting our findings.
Our studies with ip administration of PYY3-36, in combination with a Y2R antagonist revealed that PYY3-36, acting via the Y2R, enhanced nutrient-stimulated plasma active GLP-1 levels in the hepatic portal vein. Moreover, our Y2R antagonist studies undertaken in the post-prandial state following a fixed-meal, leading to high endogenous PYY3-36 levels, showed that Y2R blockade increased glucose excursions and reduced active GLP-1 hepatic portal vein plasma levels. These data strongly suggest that endogenous PYY3-36 acting through the Y2R increases nutrient-stimulated active GLP-1 hepatic portal vein plasma levels. The secretion of active GLP-1 from L-cells is regulated by an ever increasing number of different stimuli including direct nutrient contact, paracrine and endocrine regulation and neural mechanisms [39,40]. The widespread distribution of Y2R throughout the small and large intestine [41], on vagal afferents and within the enteric nervous system [42] means that the effects of PYY3-36 on active GLP-1 could be due to a direct effect on L-cells or an indirect effect either hormonal or neural and additional studies are now warranted to further delineate the underlying mechanism.
In keeping with the established actions of GLP-1 on glucose homeostasis, we found that Ex9-39 administration impaired glucose homeostasis post-IPGTT compared to glucose alone. Combined administration of PYY3-36 and Ex9-39 resulted in glucose handing that was not significantly different to mice injected with glucose alone and markedly impaired compared to mice treated with PYY3-36 alone. Interestingly, the adverse effect of Ex9-39 on glucose tolerance was partially attenuated by PYY3-36 co-administration. However, there was no significant difference in glucose AUC0–120 between Ex9-39 alone and Ex9-39 plus PYY3-36 groups. These findings taken together with our data showing that exogenous PYY3-36 increased glucose-stimulated insulin secretion but that PYY3-36 exerted no direct effect on insulin secretion from isolated islets, suggest that increased hepato-portal active GLP-1 levels mediate the effects of PYY3-36 on insulin secretion in vivo. Our finding that PYY3-36 administration partially attenuated the effect adverse effect of Ex9-39 of glucose tolerance suggest that additional non-GLP-1R dependent mechanisms, either humoral or neural, mediated by the Y2R, may also be involved in mediating the effects of PYY3-36 on glucose homeostasis.
Collectively, our studies demonstrate the complex interplay between PYY, the Y2R and GLP-1. This situation is potentially further complicated by DPP-4, which terminates the biological action of active GLP-1 and is also thought to be responsible for converting PYY1-36 to PYY3-36. Whilst PYY3-36 exhibits relative specificity for the Y2R, PYY1-36 binds to all known YR including the Y2R. Thus, one might have expected that exogenous administration PYY1-36 would have also improved glucose homeostasis acting through the Y2R. However, we observed no effect of PYY1-36 on glucose tolerance. Interestingly, studies comparing the effects of PYY1-36 and PYY3-36 on food intake in both humans and rodents also found that whilst PYY3-36 decreased food intake, a similar dose of PYY1-36 was ineffective [22,43]. Moreover, Chelikani et al., undertook a dosage ranging study with PYY1-36 and PYY3-36 in rats and found that PYY1-36 was an order of magnitude less potent in reducing food intake [43]. The mechanisms underlying the differential effects of peripheral PYY1-36 and PYY3-36 on both feeding and glucose homeostasis are unclear but may relate to the additional actions of PYY1-36 exerted via Y1R, Y4R or Y5R. DPP-4 inhibitors are currently being used to treat patients with T2DM based on their ability to increase active GLP-1 concentrations. However, our data suggest that their efficacy may be limited by their effects on PYY. DPP-4 inhibition may lead to increased intra-islet PYY1-36 with reduced glucose-stimulated insulin release whilst also reducing circulating PYY3-36 with potential effects on appetite and glucose homeostasis. These interactions warrant further investigation.
In agreement with previous studies, we found that active GLP-1 and PYY concentrations were higher in hepatic portal vein plasma than systemic plasma [44–46]. These findings highlight the importance of examining gut hormone and metabolic changes within the hepato-portal system rather than just the systemic circulation. Indeed, the marked immediate effect of bariatric surgery on glucose homeostasis compared to the lesser effects of GLP-1 analogs may, in part, be due to the disparity between the profiles and concentrations of active GLP-1 within the hepato-portal system and lymphatics post-surgery compared to those achieved with systemic administration. Several research groups are aspiring to medically mimic the gut hormone milieu of bariatric surgery by systemic administration of a combination of gut hormones [4]. However, the key to developing a medical equivalent of bariatric surgery may lie in mimicking the post-operative hormonal changes that occur within the hepato-portal system. In order to achieve these, we first need to gain a greater understanding of the complex interplay between gut hormones together with a greater understanding of the biology of enteroendocrine cells.
Conflict of interest
No potential conflicts of interest relevant to this article were reported.
Acknowledgments
This work was funded by the Rosetrees Trust. K.C., E.I.E., A.I.C., and D.J.W were funded by the MRC. K.C. researched data and wrote the article. C.G., E.E.I., A.I.C., C.A., F.A researched data and reviewed the article. D.J.W. researched data, contributed to discussion, and reviewed the article. R.L.B. researched data and wrote the article. We would like to thank Fiona Gribble for helpful discussions and critical appraisal of the manuscript.
References
- 1.Drucker D.J. The role of gut hormones in glucose homeostasis. Journal of Clinical Investigation. 2007;117:24–32. doi: 10.1172/JCI30076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Williams K.W., Elmquist J.K. From neuroanatomy to behavior: central integration of peripheral signals regulating feeding behavior. Nature Neuroscience. 2012;15:1350–1355. doi: 10.1038/nn.3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chandarana K., Batterham R.L. Shedding pounds after going under the knife: metabolic insights from cutting the gut. Nature Medicine. 2012;18:668–669. doi: 10.1038/nm.2748. [DOI] [PubMed] [Google Scholar]
- 4.Tam C.S., Berthoud H.R., Bueter M., Chakravarthy M.V., Geliebter A., Hajnal A. Could the mechanisms of bariatric surgery hold the key for novel therapies? report from a Pennington Scientific Symposium. Obesity Reviews. 2011;12:984–994. doi: 10.1111/j.1467-789X.2011.00902.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chambers A.P., Jessen L., Ryan K.K., Sisley S., Wilson-Perez H.E., Stefater M.A. Weight-independent changes in blood glucose homeostasis after gastric bypass or vertical sleeve gastrectomy in rats. Gastroenterology. 2011;141:950–958. doi: 10.1053/j.gastro.2011.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hansen L., Deacon C.F., Orskov C., Holst J.J. Glucagon-like peptide-1-(7-36)amide is transformed to glucagon-like peptide-1-(9-36)amide by dipeptidyl peptidase IV in the capillaries supplying the L cells of the porcine intestine. Endocrinology. 1999;140:5356–5363. doi: 10.1210/endo.140.11.7143. [DOI] [PubMed] [Google Scholar]
- 7.Asmar M., Hojberg P.V., Deacon C.F., Hare K., Holst J.J., Madsbad S. Pancreatic beta-cell responses to GLP-1 after near-normalization of blood glucose in patients with type 2 diabetes. Regulatory Peptides. 2010;160:175–180. doi: 10.1016/j.regpep.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 8.Mentlein R., Dahms P., Grandt D., Kruger R. Proteolytic processing of neuropeptide Y and peptide YY by dipeptidyl peptidase IV. Regulatory Peptides. 1993;49:133–144. doi: 10.1016/0167-0115(93)90435-b. [DOI] [PubMed] [Google Scholar]
- 9.Blomqvist A.G., Herzog H. Y-receptor subtypes—how many more? Trends in Neuroscience. 1997;20:294–298. doi: 10.1016/s0166-2236(96)01057-0. [DOI] [PubMed] [Google Scholar]
- 10.Chandarana K., Batterham R., Peptide Y.Y. Current Opinion in Endocrinology, Diabetes, and Obesity. 2008;15:65–72. doi: 10.1097/MED.0b013e3282f3f4b1. [DOI] [PubMed] [Google Scholar]
- 11.Batterham R.L., Heffron H., Kapoor S., Chivers J.E., Chandarana K., Herzog H. Critical role for peptide YY in protein-mediated satiation and body-weight regulation. Cell Metabolism. 2006;4:223–233. doi: 10.1016/j.cmet.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 12.Boey D., Lin S., Karl T., Baldock P., Lee N., Enriquez R. Peptide YY ablation in mice leads to the development of hyperinsulinaemia and obesity. Diabetologia. 2006;49:1360–1370. doi: 10.1007/s00125-006-0237-0. [DOI] [PubMed] [Google Scholar]
- 13.Doods H., Gaida W., Wieland H.A., Dollinger H., Schnorrenberg G., Esser F. BIIE0246: a selective and high affinity neuropeptide Y Y(2) receptor antagonist. European Journal of Pharmacology. 1999;384:R3–R5. doi: 10.1016/s0014-2999(99)00650-0. [DOI] [PubMed] [Google Scholar]
- 14.Abbott C.R., Small C.J., Kennedy A.R., Neary N.M., Sajedi A., Ghatei M.A. Blockade of the neuropeptide Y Y2 receptor with the specific antagonist BIIE0246 attenuates the effect of endogenous and exogenous peptide YY(3-36) on food intake. Brain Research. 2005;1043:139–144. doi: 10.1016/j.brainres.2005.02.065. [DOI] [PubMed] [Google Scholar]
- 15.Boey D., Sainsbury A., Herzog H. The role of peptide YY in regulating glucose homeostasis. Peptides. 2007;28:390–395. doi: 10.1016/j.peptides.2006.07.031. [DOI] [PubMed] [Google Scholar]
- 16.Bottcher G., Ahren B., Lundquist I., Sundler F. Peptide YY: intrapancreatic localization and effects on insulin and glucagon secretion in the mouse. Pancreas. 1989;4:282–288. [PubMed] [Google Scholar]
- 17.van den Hoek A.M., Heijboer A.C., Corssmit E.P., Voshol P.J., Romijn J.A., Havekes L.M. PYY3-36 reinforces insulin action on glucose disposal in mice fed a high-fat diet. Diabetes. 2004;53:1949–1952. doi: 10.2337/diabetes.53.8.1949. [DOI] [PubMed] [Google Scholar]
- 18.Ortiz A.A., Milardo L.F., DeCarr L.B., Buckholz T.M., Mays M.R., Claus T.H. A novel long-acting selective neuropeptide Y2 receptor polyethylene glycol-conjugated peptide agonist reduces food intake and body weight and improves glucose metabolism in rodents. Journal of Pharmacology and Experimental Therapeutics. 2007;323:692–700. doi: 10.1124/jpet.107.125211. [DOI] [PubMed] [Google Scholar]
- 19.Pittner R.A., Moore C.X., Bhavsar S.P., Gedulin B.R., Smith P.A., Jodka C.M. Effects of PYY[3-36] in rodent models of diabetes and obesity. International Journal of Obesity and Related Metabolic Disorders. 2004;28:963–971. doi: 10.1038/sj.ijo.0802696. [DOI] [PubMed] [Google Scholar]
- 20.Batterham R.L., Cowley M.A., Small C.J., Herzog H., Cohen M.A., Dakin C.L. Gut hormone PYY3-36 physiologically inhibits food intake. Nature. 2002;418:650–654. doi: 10.1038/nature00887. [DOI] [PubMed] [Google Scholar]
- 21.Batterham R.L., Cohen M.A., Ellis S.M., Le Roux C.W., Withers D.J., Frost G.S. Inhibition of food intake in obese subjects by peptide YY3-36. New England Journal of Medicine. 2003;349:941–948. doi: 10.1056/NEJMoa030204. [DOI] [PubMed] [Google Scholar]
- 22.Sloth B., Holst J.J., Flint A., Gregersen N.T., Astrup A. Effects of PYY1-36 and PYY3-36 on appetite, energy intake, energy expenditure, glucose and fat metabolism in obese and lean subjects. American Journal of Physiology—Endocrinology and Metabolism. 2007;292:E1062–E1068. doi: 10.1152/ajpendo.00450.2006. [DOI] [PubMed] [Google Scholar]
- 23.Choudhury A.I., Heffron H., Smith M.A., Al-Qassab H., Xu A.W., Selman C. The role of insulin receptor substrate 2 in hypothalamic and beta cell function. Journal of Clinical Investigation. 2005;115:940–950. doi: 10.1172/JCI24445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chu C.P., Kato K., Kunitake T., Watanabe S., Qiu D.L., Ueta Y. Enhanced effects of central angiotensin II on cardiovascular and drinking responses in inbred polydipsic (STR/N) mice. Brain Research. 2003;962:129–134. doi: 10.1016/s0006-8993(02)03981-1. [DOI] [PubMed] [Google Scholar]
- 25.Cantley J., Burchfield J.G., Pearson G.L., Schmitz-Peiffer C., Leitges M., Biden T.J. Deletion of PKC{varepsilon} selectively enhances the amplifying pathways of glucose-stimulated insulin secretion via increased lipolysis in mouse {beta}-cells. Diabetes. 2009 doi: 10.2337/db09-0132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chandarana K., Gelegen C., Karra E., Choudhury A.I., Drew M.E., Fauveau V. Diet and gastrointestinal bypass-induced weight loss: the roles of ghrelin and peptide YY. Diabetes. 2011;60:810–818. doi: 10.2337/db10-0566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Troy S., Soty M., Ribeiro L., Laval L., Migrenne S., Fioramonti X. Intestinal gluconeogenesis is a key factor for early metabolic changes after gastric bypass but not after gastric lap-band in mice. Cell Metabolism. 2008;8:201–211. doi: 10.1016/j.cmet.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 28.Gelegen C., Chandarana K., Choudhury A.I., Al-Qassab H., Evans I.M., Irvine E.E. Regulation of hindbrain Pyy expression by acute food deprivation, prolonged caloric restriction, and weight loss surgery in mice. American Journal of Physiology—Endocrinology and Metabolism. 2012;303:E659–E668. doi: 10.1152/ajpendo.00033.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bertrand G., Gross R., Roye M., Ahren B., Ribes G. Evidence for a direct inhibitory effect of PYY on insulin secretion in rats. Pancreas. 1992;7:595–600. doi: 10.1097/00006676-199209000-00013. [DOI] [PubMed] [Google Scholar]
- 30.Karlsson S., Ahren B. A role for islet peptide YY in the regulation of insulin secretion. Acta Physiologica Scandinavica. 1996;157:305–306. doi: 10.1046/j.1365-201X.1996.501245000.x. [DOI] [PubMed] [Google Scholar]
- 31.Burcelin R., Brunner H., Seydoux J., Thorensa B., Pedrazzini T. Increased insulin concentrations and glucose storage in neuropeptide Y Y1 receptor-deficient mice. Peptides. 2001;22:421–427. doi: 10.1016/s0196-9781(01)00357-6. [DOI] [PubMed] [Google Scholar]
- 32.Nieuwenhuizen A.G., Karlsson S., Fridolf T., Ahren B. Mechanisms underlying the insulinostatic effect of peptide YY in mouse pancreatic islets. Diabetologia. 1994;37:871–878. doi: 10.1007/BF00400941. [DOI] [PubMed] [Google Scholar]
- 33.Dumont Y., Moyse E., Fournier A., Quirion R. Distribution of peripherally injected peptide YY ([125I] PYY (3-36)) and pancreatic polypeptide ([125I] hPP) in the CNS: enrichment in the area postrema. Journal of Molecular Neuroscience. 2007;33:294–304. doi: 10.1007/s12031-007-9007-9. [DOI] [PubMed] [Google Scholar]
- 34.Huang X.F., Han M., Storlien L.H. The level of NPY receptor mRNA expression in diet-induced obese and resistant mice. Brain Research Molecular Brain Research. 2003;115:21–28. doi: 10.1016/s0169-328x(03)00174-8. [DOI] [PubMed] [Google Scholar]
- 35.Rahardjo G.L., Huang X.F., Tan Y.Y., Deng C. Decreased plasma peptide YY accompanied by elevated peptide YY and Y2 receptor binding densities in the medulla oblongata of diet-induced obese mice. Endocrinology. 2007;148:4704–4710. doi: 10.1210/en.2007-0107. [DOI] [PubMed] [Google Scholar]
- 36.Laferrere B. Diabetes remission after bariatric surgery: is it just the incretins? International Journal of Obesity (London) 2011;35(Suppl 3):S22–S25. doi: 10.1038/ijo.2011.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scott W.R., Batterham R.L. Roux-en-Y gastric bypass and laparoscopic sleeve gastrectomy: understanding weight loss and improvements in type 2 diabetes after bariatric surgery. American Journal of Physiology—Regulatory, Integrative and Comparative Physiology. 2011;301:R15–R27. doi: 10.1152/ajpregu.00038.2011. [DOI] [PubMed] [Google Scholar]
- 38.Thaler J.P., Yi C.X., Schur E.A., Guyenet S.J., Hwang B.H., Dietrich M.O. Obesity is associated with hypothalamic injury in rodents and humans. Journal of Clinical Investigation. 2012;122:153–162. doi: 10.1172/JCI59660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Holst J.J. The physiology of glucagon-like peptide 1. Physiological Reviews. 2007;87:1409–1439. doi: 10.1152/physrev.00034.2006. [DOI] [PubMed] [Google Scholar]
- 40.Tolhurst G., Reimann F., Gribble F.M. Nutritional regulation of glucagon-like peptide-1 secretion. Journal of Physiology. 2009;587:27–32. doi: 10.1113/jphysiol.2008.164012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goumain M., Voisin T., Lorinet A.M., Laburthe M. Identification and distribution of mRNA encoding the Y1, Y2, Y4, and Y5 receptors for peptides of the PP-fold family in the rat intestine and colon. Biochemical and Biophysical Research Communications. 1998;247:52–56. doi: 10.1006/bbrc.1998.8647. [DOI] [PubMed] [Google Scholar]
- 42.Wang L., Gourcerol G., Yuan P.Q., Wu S.V., Million M., Larauche M. Peripheral peptide YY inhibits propulsive colonic motor function through Y2 receptor in conscious mice. American Journal of Physiology—Gastrointestinal and Liver Physiology. 2010;298:G45–G56. doi: 10.1152/ajpgi.00349.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chelikani P.K., Haver A.C., Reidelberger R.D. Comparison of the inhibitory effects of PYY(3-36) and PYY(1-36) on gastric emptying in rats. American Journal of Physiology—Regulatory, Integrative and Comparative Physiology. 2004;287:R1064–R1070. doi: 10.1152/ajpregu.00376.2004. [DOI] [PubMed] [Google Scholar]
- 44.D'Alessio D., Lu W., Sun W., Zheng S., Yang Q., Seeley R. Fasting and postprandial concentrations of GLP-1 in intestinal lymph and portal plasma: evidence for selective release of GLP-1 in the lymph system. American Journal of Physiology—Regulatory, Integrative and Comparative Physiology. 2007;293:R2163–R2169. doi: 10.1152/ajpregu.00911.2006. [DOI] [PubMed] [Google Scholar]
- 45.Ruttimann E.B., Arnold M., Hillebrand J.J., Geary N., Langhans W. Intrameal hepatic portal and intraperitoneal infusions of glucagon-like peptide-1 reduce spontaneous meal size in the rat via different mechanisms. Endocrinology. 2009;150:1174–1181. doi: 10.1210/en.2008-1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stadlbauer U., Arnold M., Weber E., Langhans W. Possible mechanisms of circulating PYY-induced satiation in male rats. Endocrinology. 2013;154:193–204. doi: 10.1210/en.2012-1956. [DOI] [PubMed] [Google Scholar]