

Abstract
Peroxisome proliferator-activated receptorγ coactivators (PGC-1α and PGC-1β) play important roles in the transcriptional regulation of intermediary metabolism. To evaluate the effects of overexpressing PGC-1α or PGC-1β at physiologic levels in liver, we generated transgenic mice with inducible overexpression of PGC-1α or PGC-1β. Gene expression array profiling revealed that whereas both PGC-1 family proteins induced mitochondrial oxidative enzymes, the expression of several genes involved in converting glucose to fatty acid was induced by PGC-1β, but not PGC-1α. The increased expression of enzymes involved in carbohydrate utilization and de novo lipogenesis by PGC-1β required carbohydrate response element binding protein (ChREBP). The interaction between PGC-1β and ChREBP, as well as PGC-1β occupancy of the liver-type pyruvate kinase promoter, was influenced by glucose concentration and liver-specific PGC-1β−/− hepatocytes were refractory to the lipogenic response to high glucose conditions. These data suggest that PGC-1β-mediated coactivation of ChREBP is involved in the lipogenic response to hyperglycemia.
Keywords: Hepatic, Metabolism, ChREBP, PGC-1
1. Introduction
Derangements in hepatic intermediary metabolism can lead to systemic abnormalities in circulating metabolites and to contribute to the pathology of diabetes-related diseases. For example, defects in mitochondrial oxidative pathways in insulin-resistant liver are associated with hepatic insulin resistance and failure to suppress hepatic glucose production, which contributes to circulating hyperglycemia in diabetic subjects. Similarly, increased hepatic lipogenesis and overproduction of very low density lipoprotein (VLDL) particles contribute to nonalcoholic fatty liver disease and dyslipidemia, respectively. Although hepatic metabolism is controlled at a variety of levels, the transcriptional regulation of these metabolic pathways has emerged as an important regulatory nodal point.
The peroxisome proliferator-activated receptor γ coactivator (PGC-1) family plays important roles in regulating hepatic intermediary metabolism [1,2]. PGC-1α and PGC-1β play largely redundant roles in regulating mitochondrial oxidative metabolism [3] through coactivation of the peroxisome proliferator-activated receptors [4,5], estrogen-related receptors [6], and nuclear respiratory factors [7]. However, in liver, previous work has identified regulated pathways that are unique to each PGC-1 family member [8–10]. For example, PGC-1α is known to drive expression of enzymes involved in gluconeogenesis [8,10], while PGC-1β enhances lipogenesis and VLDL synthesis and secretion through coactivation of sterol regulatory element binding protein 1 (SREBP1) and liver X receptors (LXR) [9] and FOXA2 [11]. The liver-specific metabolic pathways uniquely regulated by the two PGC-1s make this organ an attractive model system to identify differential effects of the two PGC-1 proteins. However, much of the previous work in this system has utilized adenoviral overexpression of PGC-1s, which leads to expression levels far in excess of what is observed physiologically.
We aimed to evaluate the effects of chronic, physiologic levels of PGC-1 overexpression on hepatic intermediary metabolism by developing transgenic mice that overexpress either PGC-1α or β in an inducible manner using a tetracycline-regulated transgene. Interestingly, many of the genes found to be regulated by PGC-1β are known targets of carbohydrate response element binding protein (ChREBP), which is a glucose responsive transcription factor [12]. We present evidence that ChREBP is a novel transcription factor partner for PGC-1β and identify several PGC-1β target genes that require ChREBP for full activation. These data raise the intriguing possibility that PGC-1β plays an important role in coordinating the lipogenic response to nutritional inputs by activating ChREBP.
2. Materials and methods
2.1. Animal studies
To generate mice with liver-specific, tetracycline-regulated overexpression of PGC-1α or PGC-1β, two transgenic strains of mice were intercrossed. LAP-tTA mice (obtained from Dean Felsher, Stanford University) express tetracycline transactivator (tTA) protein, a transcription factor that is inactivated by the presence of tetracycline or its analogs, in a liver-specific manner [13]. Hemizygous TRE-PGC-1α and TRE-PGC-1β mice, which are transgenic for either a PGC-1α or β cDNA under the control of a tTA response element [14,15], were mated to homozygous LAP-tTA mice to generate double transgenic mice. All mice were generated in and maintained on a pure FVB background. Breeding pairs and weaned offspring were maintained on doxycycline-containing chow. At 6-weeks of age, study mice were placed on regular mouse chow for 3 weeks to allow for a doxycycline “wash out period” to ensure full transgene expression. The generation of mice harboring PGC-1β alleles containing LoxP sites flanking exons 4 through 6 has been described previously [3,16]. To drive liver-specific knockout of PGC-1β, PGC-1β flox/flox mice were crossed with hemizygous transgenic mice expressing Cre recombinase under control of the albumin promoter (Alb-Cre). The recombination generates a coding sequence frameshift, resulting in a premature stop codon in exon 7, and has previously been shown to result in a complete loss of PGC-1β protein [3]. ChREBP-/- mice in the C57/BL6 background were purchased from Jackson Labs and have been previously described [17]. For all experiments, transgenic mice were compared to sex-matched littermate wild-type control mice. Unless otherwise noted, mice were euthanized by carbon dioxide asphyxiation after a 4 h fast and plasma and liver tissue were harvested and snap frozen in liquid nitrogen for later analyses.
High fat diet feeding studies were conducted by giving PGC-1 overexpressing or WT control mice ad libitum access to high fat chow, which provided 43% of the calories from fat (TD 97268, Harlan Teklad, Madison, WI) [18,19], for 8 weeks. The high fat diet did not contain doxycycline and thus, the mice were induced to overexpress PGC-1 for the duration of the trial. For fasting-refeeding studies, mice were individually housed in cages containing aspen chip bedding and all groups were fasted for 24 h with ad libitum access to water. Following the fasting period, a subset of mice were refed standard chow for 16 h or remained fasting for a total of 40 h and then hepatocytes were isolated. All animal studies were approved by the Animal Studies Committee at Washington University School of Medicine.
2.2. Hepatocyte isolation, culture, and infection with adenovirus
Primary mouse hepatocytes were isolated from mice as previously described [20]. Briefly, mice were anesthetized with isofluorane and perfused through the portal vein with pre-warmed HBSS containing collagenase. Liver architecture was mechanically disrupted using forceps. Isolated hepatocytes were washed extensively and plated onto collagen coated plated and were further cultured in DMEM containing 10% FBS unless otherwise noted. To contrast the effects of glucose concentration, following plating, the hepatocytes were all cultured in low glucose DMEM (5.5 mM) supplemented with 10% FBS and penicillin/streptomycin, and fungizone for 24 h. Subsets of WT and LAP-tet-OFF-PGC-1β hepatocytes were then switched to high glucose DMEM (25 mM) or received fresh low glucose DMEM and were cultured for an additional 24 h. The cells were then used for metabolic assays (fatty acid synthesis and glycolysis) or were harvested for RNA isolation or co-immunoprecipitation or chromatin immunoprecipitation (ChIP) studies. For ChIP and co-immunoprecipitation studies, hepatocytes were dispersed and crosslinked or cells were plated. Hepatocytes were infected with previously described adenovirus constructs to overexpress PGC-1β [3] or GFP at time of plating, and endpoints were harvested 48 h post-infection.
To assess the effects of glucose concentration on ChREBP target gene expression in WT and LS-PGC-1β−/− isolated hepatocytes, cells were plated for 4 h in M199 media (23 mM HEPES, 26 mM sodium bicarbonate, 11 mM glucose, 10% FBS, 50 IU of each penicillin and streptomycin, 10 nM dexamethasone, and 10 nM insulin). Following 4 h, the hepatocytes were cultured overnight in the same media except the glucose concentration was reduced to 5.5 mM and insulin and FBS were not included in the media. The next morning, cells were cultured in fresh media (either containing 5.5 or 25 mM glucose) for 12 h and RNA was then collected or cells were fixed and immunoflurescent staining was performed [21].
2.3. Rates of palmitate oxidation, glycolysis, and fatty acid synthesis
Palmitate oxidation rates were assessed in isolated hepatocytes 2–3 h after cells were plated using [3H]palmitate as previously described [22]. Rates of glycolysis or fatty acid synthesis were assessed as described previously [23].
2.4. Microarray analysis
Total RNA was isolated using the RNAzol method (Tel-Test). Total RNA isolated from the livers of WT and LAP-tet-OFF-PGC-1 mice was used for microarray analysis. The hybridization to Agilent whole mouse genome oligo microarray was performed by the Digestive Diseases Research Core Center (DDRCC) at Washington University School of Medicine. Each Agilent array was co-hybridized to two hepatic RNA targets—one prepared from a PGC-1α hepatic overexpresser, and the other from a WT littermate control. This was repeated for hepatic RNAs prepared from PGC-1β overexpressers and WT littermate controls. Four (n=4) biological replicate arrays were generated for each comparison. Signals were acquired and normalized, and the ratios of trangenic/wild type signals were calculated for each Agilent ID. To generate lists of genes altered by hepatic overexpression of PGC-1α or PGC-1β, thresholds of >±1.5-fold difference versus WT were set. To limit false positives, all four co-hybridized arrays reached this threshold to be added to the list of genes regulated by PGC-1α or PGC-1β overexpression. Lists of mined genes were (i) uniquely regulated in PGC-1α overexpressing (versus WT), (ii) uniquely regulated in PGC-1β overexpressing (versus WT) liver, or (iii) altered in both PGC-1α versus WT and PGC-1β versus WT. Derived lists of genes were analyzed using Ingenuity Pathways Analysis to determine enriched gene ontology processes, using Fisher’s exact test [24]. The full results of the gene expression array are being deposited in the GEO database. Biological replicate hepatic samples were used for select genes to confirm changes in expression by RT-qPCR and immunoblotting.
2.5. Quantitative real time PCR analysis
Real-time PCR was performed using the ABI PRISM 7500 sequence detection system (Applied Biosystems, Foster City, CA) and the SYBR green kit. Arbitrary units of target mRNA were corrected by measuring the levels of 36B4 RNA.
2.6. Western blot analysis
Total cell proteins were collected using RIPA buffer containing protease and phosphatase inhibitors. Western blotting studies were performed using antibodies directed against PGC-1α (Calbiochem), PGC-1β (gift from A. Kralli), ChREBP (Novus Biologicals), fatty acid synthase (Abcam Inc.), acetyl-coA carboxylase (pan-specific; Cell Signaling), actin (Sigma-Aldrich), lamin A/C (Cell Signaling), and SREBP1 (Santa Cruz) as previously described [25]. Bands were imaged on the Li-COR Odyssey System (Li-COR, Biosciences Lincoln, NE).
2.7. Co-immunoprecipitation studies
Hepatocytes were isolated from WT and LAP-tet-OFF-PGC-1β mice under the conditions described in Section 3 and co-immunoprecipitation was performed as previously described [26] using an antibody against PGC-1β [3,27]. Co-immunoprecipitation products were subjected to SDS-PAGE, and western blot analysis was performed using a ChREBP antibody (Novus Biologicals).
2.8. Tissue and plasma metabolite analyses
Plasma triglyceride (Infinity, Thermo Scientific, Middletown, VA), free fatty acid (Wako Pure Chemical Industries, Osaka, Japan), and cholesterol (Infinity, Thermo Electron, Melbourne, Australia) concentrations were determined using colorimetric assays using plasma from ad libitum fed mice. Plasma glucose concentrations were determined in whole blood using a One Touch Ultra glucometer. Hepatic triglyceride content was determined using a colorimetric assay as described previously [28].
2.9. GST-pulldown assays
The glutathione S-transferase (GST) protein–protein interaction assay was performed as previously described [5]. Briefly, all 35S-labeled proteins were synthesized using a TNT Quick Coupled transcription-translation system (Promega). All GST fusion proteins were grown in BL21 competent bacterial cells (Stratagene) and purified on GSH Sepharose. In the pulldown reaction, 3 µg of the fusion protein was incubated with 10 µl of 35S labeled protein in 500 µl of binding buffer (20 mM Tris, pH 7.5, 100 mM KCl, 0.1 mM EDTA, 0.05% NP-40, 10% glycerol, 1 mg/ml BSA, 0.5 mM PMSF, and 1× Complete; Roche). The reactions were rotated for 1 h at room temperature. Samples were washed five times with ice cold binding buffer. The fusion proteins and their potential interacting partners were analyzed by SDS-PAGE.
2.10. Chromatin immunoprecipitation studies
Chromatin immunoprecipitation studies were performed using the Millipore EZ ChIP Kit according to the manufacturer’s protocol. Chromatin was analyzed using endpoint PCR or quantitative real time PCR analyses using primers designed to flank the ChoRE sequence within the Lpk or Acaca promoter as previously published [29] or to flank the nuclear receptor response element in the Acadm promoter [30]. For real time RT-PCR, SYBR Green was used and conditions immunoprecipitated with PGC-1β antibody were corrected to IgG immunoprecipitated samples.
2.11. Statistical analysis
Statistical comparisons were made using analysis of variance (ANOVA) or a Student’s t-test. All data are presented as means±SEM, with a statistically significant difference defined as a P value <0.05.
3. Results
3.1. Generation of mice overexpressing either PGC-1α or PGC-1β in a liver-specific manner
We sought to evaluate the effects of physiologic PGC-1α or β overexpression on hepatic gene expression profiles and intermediary metabolism. To this end, mice overexpressing PGC-1α or PGC-1β in a liver-specific manner were generated by using a tetracycline-regulated, double transgenic system as previously described ([31] and Supplemental Figure 1). Upon removal of doxycycline, and following a three week wash out period, PGC-1α or PGC-1β mRNA expression was increased 5- or 6-fold, respectively, in the livers of LAP-tet-OFF-PGC-1α or LAP-tet-OFF-PGC-1β mice compared to matched wild-type (WT) controls (Figure 1A). A corresponding increase in PGC-1 protein was also observed. Importantly, these levels of overexpression are comparable to the induction observed with fasting [32] and are therefore within the physiologically-relevant range. Overexpression of PGC-1α or β at this level had no effect on circulating concentrations of glucose, triglycerides, free fatty acids, or cholesterol or on glucose tolerance in mice fed normal chow (Supplemental Figure 2), which contrasts previous work using adenoviral technology to markedly overexpress the PGC-1 proteins [8–10]. Hepatic triglyceride content was also unaffected by PGC-1α or β overexpression (Supplemental Figure 2).
Figure 1.
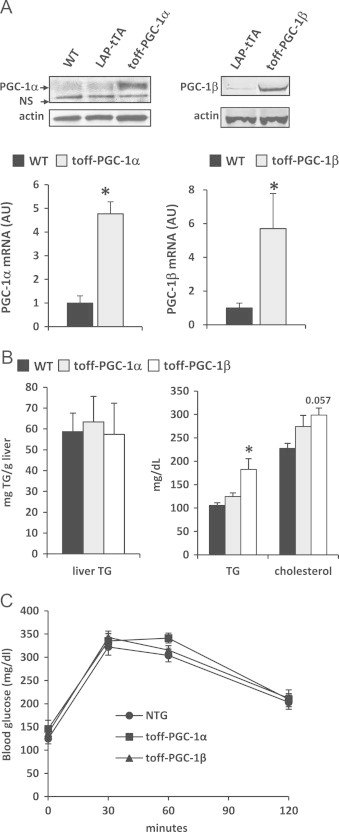
PGC-1α and PGC-1β regulate overlapping but distinct gene expression patterns: (A) LAP-tet-OFF-PGC-1α or LAP-tet-OFF-PGC-1β transgenic mice exhibit increased expression (5- or 6-fold, respectively) of PGC-1α or β in a liver specific manner as assessed by quantitative RT-PCR and Western blot analysis. (B) and (C) Graphs depict liver triglyceride content and plasma concentrations of triglyceride (TG) and cholesterol (B), and results of glucose tolerance tests (C) on WT, LAP-tet-OFF-PGC-1α, or LAP-tet-OFF-PGC-1β transgenic mice on high fat (HF) diet (n=6-7). *P<0.05 versus WT mice.
Previous work has linked PGC-1 activation to metabolic abnormalities of insulin-resistant liver. We therefore evaluated the effects of 8 weeks of feeding a high fat “Western” type diet. Mice given high fat diet gained more weight than standard chow-fed controls and mice gained weight similarly on the high fat diet regardless of genotype (Supplemental Figure 2). Though high fat diet increased liver TG content about 8-fold compared to chow-fed mice, no effect of PGC-1 overexpression was observed (Figure 1B). Consistent with previous work, PGC-1β overexpression increased plasma triglyceride concentration and tended to increase cholesterol concentration after 8 weeks on high fat diet, likely due to increased VLDL secretion as described [9,11]. No effect of PGC-1β overexpression on glucose tolerance was observed and PGC-1α overexpression was without significant effect on any measured parameter. These data suggest that physiologic PGC-1β overexpression in the context of high fat diet feeding may promote dyslipidemia, as previously suggested by supra-physiologic approaches.
3.2. Transcriptional profiling reveals a distinct role for PGC-1β in the regulation of hepatic glycolysis and lipogenesis
To evaluate the genomic transcriptional profile regulated by these transcriptional coactivators, microarray analysis was performed on liver RNA 3 weeks after transgene induction. The expression of 495 genes was concordantly up- or down-regulated by PGC-1α and PGC-1β (Figure 2A). These genes are overwhelmingly up-regulated and primarily involved in mitochondrial metabolism (Figure 2B). Quantitative real time RT-PCR confirmed that both PGC-1α and β induced expression of enzymes involved in mitochondrial metabolism, including electron transport chain components (Cytc, Cox2, Cox7a1, Sdha) and fatty acid oxidation (Acadm, Cpt1a) (Figure 3A). Consistent with the gene expression results, rates of palmitate oxidation were increased approximately 40% in hepatocytes isolated from LAP-tet-OFF-PGC-1α and LAP-tet-OFF-PGC-1β mice compared to WT littermate controls (Figure 3B). Thus, as anticipated, activation of either PGC-1α or β increased hepatic oxidative capacity.
Figure 2.
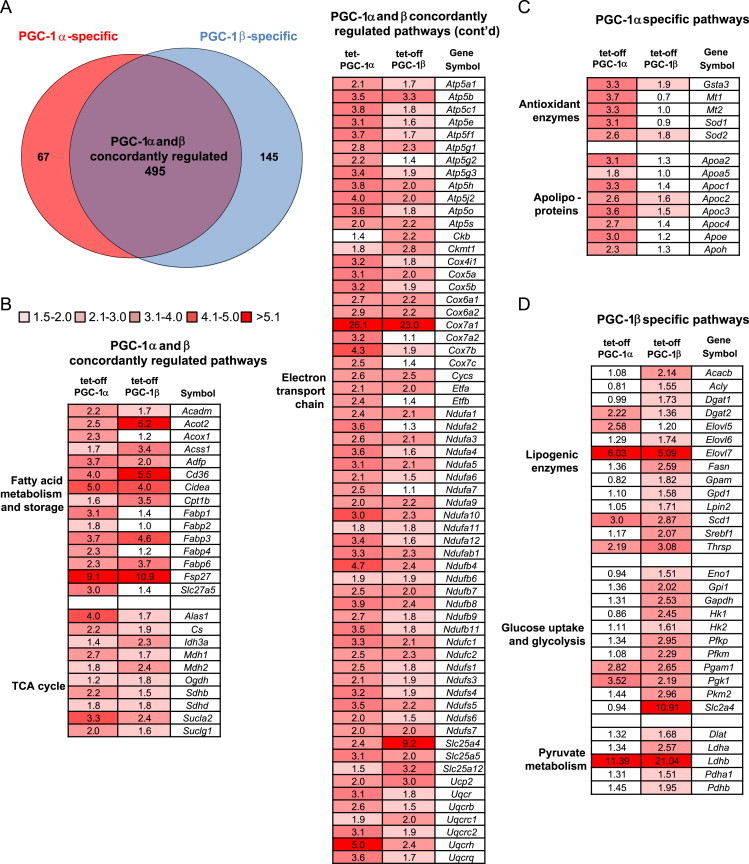
PGC-1α and PGC-1β regulate overlapping but distinct gene expression patterns: (A) The Venn diagram illustrates the number of genes that were regulated concordantly (induced or suppressed) or discordantly (PGC-1α-regulated pathways or PGC-1β-regulated pathways) in microarray studies by PGC-1α or PGC-1β overexpression in transgenic mice versus littermate controls (n=4). (B) The table lists regulated genes in pathways that are significantly regulated in a concordant manner by PGC-1α and PGC-1β. Shaded boxes denote genes that are regulated >1.5 fold and are significant (P<0.05) by Mann-Whitney U-test (n=4). (C) The table lists regulated genes in pathways that are significantly regulated only by PGC-1α overexpression. Shaded boxes denote genes that are regulated >1.5 fold and are significant (P<0.05) by Mann-Whitney U-test (n=4). (D) The table lists regulated genes in pathways that are significantly regulated only by PGC-1β overexpression. Shaded boxes denote genes that are regulated >1.5 fold and are significant (P<0.05) by Mann Whitney U-test (n=4).
Figure 3.
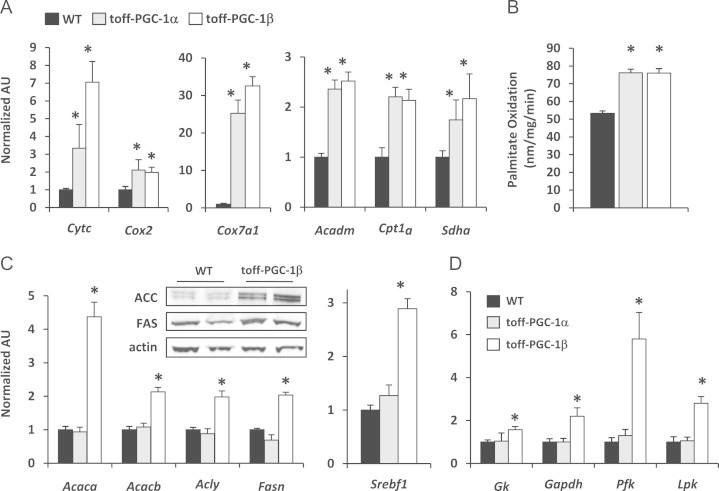
Concordant and discordant regulation of intermediary metabolism by PGC-1α and β: (A) The graphs depict expression of cytochrome C (CytC), cytochrome C oxidase 2 Cox2, cytochrome C oxidase subunit VIIa polypeptide 1 Cox7a1, medium chain acyl CoA dehydrogenase Acadm, muscle-type carnitine palmitoyltransferase (Cpt1a), or succinate dehydrogenase Sdha in LAP-tet-OFF-PGC-1α, LAP-tet-OFF-PGC-1β or matched littermate WT (LAP-tTA) control mice after a 3-week withdrawal from DOX to induce transgene expression (n=6). *P<0.05 versus WT control mice. (B) The graph represents mean rates of palmitate oxidation (nm/mg/min) in hepatocytes isolated from WT, LAP-tet-OFF-PGC-1α, or LAP-tet-OFF-PGC-1β mice. *P<0.05 versus WT control mice. (C) The graphs depict the expression of acetyl CoA carboxylase a, (Acaca), acetyl CoA carboxylase b (Acacb), ATP citrate lyase (Acly), fatty acid synthase (Fasn), and sterol regulatory element binding protein (Srebf1) in LAP-tet-OFF-PGC-1α, LAP-tet-OFF-PGC-1β, or matched littermate WT (LAP-tTA) control mice after a 3-week withdrawal from DOX chow to induce transgene expression (n=6). *P<0.05 versus WT control mice and LAP-tet-OFF-PGC-1α mice. Western blot inset depicts protein levels of ACC and FAS in livers harvested from WT and LAP-tet-OFF-PGC-1β mice. (D) The graph depicts the expression of genes encoding the glucose metabolic enzymes glucokinase (Gk), glyceraldehyde-3-phosphate dehydrogenase (Gapdh), phosphofructokinase (Pfk), and liver-type pyruvate kinase (Lpk). *P<0.05 versus WT control mice and LAP-tet-OFF-PGC-1α mice (n=6).
We also detected patterns of gene expression that were discordantly regulated by PGC-1α and PGC-1β. The expression of 67 genes was regulated by PGC-1α, but not PGC-1β (Figure 2A). These genes included several encoding anti-oxidant enzymes and apolipoproteins (Figure 1D). Additionally, 145 genes were uniquely influenced by PGC-1β overexpression (Figure 2A). Pathway analyses of the genes discordantly regulated by PGC-1β demonstrated a significant enrichment in genes encoding glucose transporters, glycolytic enzymes, and enzymes involved in lipogenesis. The lipogenic enzymes Acaca, Acacb, Acly, and Fasn were induced by PGC-1β, but not PGC-1α overexpression (Figure 3C). A number of these genes are known to be targets of SREBP1, which has previously been shown to be a binding partner of PGC-1β [9], and overexpression of PGC-1β resulted in increased expression of Srebf1 (Figure 3C). Among the genes induced discordantly by PGC-1β overexpression were several genes encoding proteins involved in glucose uptake, glucose intermediate phosphorylation, glycolysis, and glucose oxidation (Figure 2D). A subset of these genes were validated by qRT-PCR, and we found that Gk, Gapdh, Pfk, and Lpk were induced by PGC-1β, but not PGC-1α (Figure 3D). These unbiased gene expression profiling results demonstrate that PGC-1β overexpression in liver activates a paradoxical pattern of gene expression characterized by an induction of enzymes involved in both fatty acid synthesis and degradation.
3.3. The activation of lipogenic and glycolytic genes requires ChREBP
In addition to a number of known SREBP1 target genes, the pattern of gene expression also overlaps significantly with the transcriptional profile of genes regulated by carbohydrate response element binding protein (ChREBP), a glucose responsive transcription factor [12]. To determine whether ChREBP was required for the PGC-1β-mediated transcriptional activation of genes encoding glucose utilization and lipogenic factors, we utilized ChREBP−/− mice. Hepatocytes were isolated from wild type and ChREBP−/− mice and infected with adenovirus to overexpress PGC-1β. Compared to GFP control adenovirus, PGC-1β overexpression increased the expression of ChREBP and, as was observed in the transgenic mice, induced Lpk, Gckr, G6pdh, Me1, Acly, Fasn, and Acaca gene expression in wild type hepatocytes (Figure 4A). The transcriptional activation of these genes in response to PGC-1β overexpression was abolished in ChREBP−/− hepatocytes (Figure 4A). This occurred despite a significant increase in the active form of SREBP1 in response to PGC-1β overexpression in WT and ChREBP−/− hepatocytes (Figure 4B). The induction of Ldhb and Mdh1 and the oxidative enzymes Acadvl and Cox7a1 in response to PGC-1β overexpression was equivalent in WT and ChREBP−/− hepatocytes (Figure 4C), which verified that PGC-1β is active in these cells and is consistent with these genes being regulated by the PPARs [5,33]. These data indicate that the induction of genes encoding glycolytic and lipogenic enzymes by PGC-1β requires ChREBP.
Figure 4.
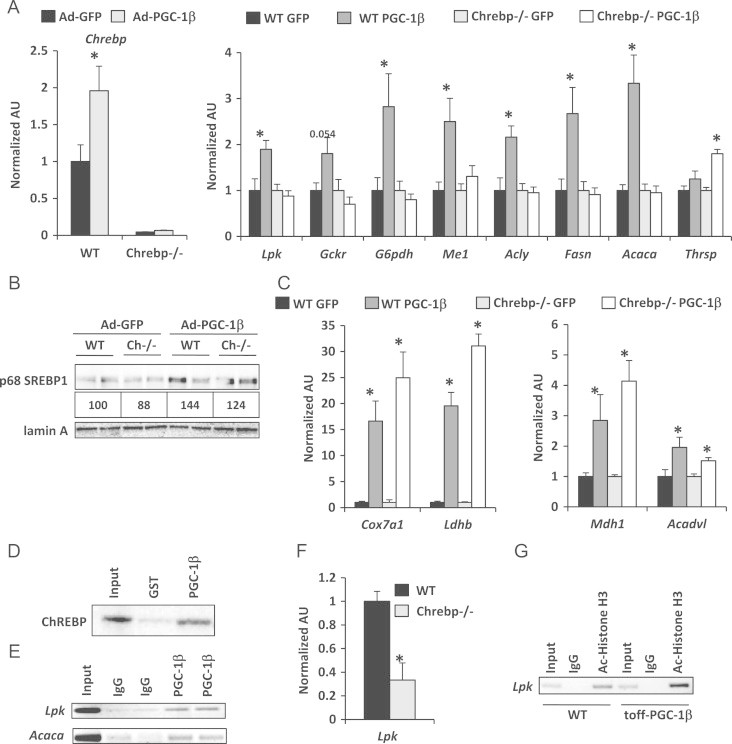
PGC-1β overexpression fails to induce a full glycolytic and lipogenic response in hepatocytes from ChREBP−/− mice: (A) The graphs depict Chrebp and ChREBP target gene expression, including glucokinase regulatory protein (Gckr), glucose-6-phosphate dehydrogenase (G6pdh), malic enzyme (Me1), Acly, Fasn, Acaca, and thyroid hormone-inducible hepatic protein (Thrsp) in hepatocytes isolated from WT and ChREBP−/− that were infected with either Ad-GFP or Ad-PGC-1β. (B) Western blot analysis demonstrating the levels of p68 SREBP1 in WT and ChREBP−/− hepatocytes infected with Ad-GFP or Ad-PGC-1β. (C) The expression of genes in other transcriptional pathways induced by PGC-1β, including Cox7a1, lactate dehydrogenase subunit b (Ldhb), malate dehydrogenase (Mdh1), and very long chain acyl CoA dehydrogenase (Acadvl), is preserved in ChREBP−/− and WT hepatocytes (n=8) *P<0.05 vs WT Ad-GFP. (D) The autoradiogram depicts the presence of ChREBP protein when GST-PGC-1β is used as bait. (E) ChIP analyses demonstrate that PGC-1β is enriched at ChoRE sites located within the Lpk and Acaca promoters in hepatocytes isolated from LAP-tet-OFF-PGC-1β mice. (F) ChIP analysis performed in WT and ChREBP−/− isolated hepatocytes infected with Ad-PGC-1β exhibits an ablation of PGC-1β enrichment at the ChoRE in the Lpk promoter. (G) ChIP analysis demonstrates increased levels of acetylated histone H3 associated with the ChoREs in the Lpk promoter in hepatocystes isolated from LAP-tet-OFF-PGC-1β mice compared to WT control isolated hepaocytes.
3.4. ChREBP is a novel transcription factor partner of PGC-1β
We hypothesized that PGC-1β might serve as a coactivator for ChREBP. Using GST-tagged full length PGC-1β as bait in a GST pulldown assay, we demonstrated a direct protein–protein interaction between ChREBP and PGC-1β (Figure 4D). ChIP analyses using an antibody directed against PGC-1β also detected an enrichment of PGC-1β to the carbohydrate response elements (ChoRE) found within the promoters of Lpk and Acaca, which are both previously identified target genes of ChREBP (Figure 4E). Lpk is a prototypical ChREBP target gene, and thus, this gene was used as a model system for a ChREBP target gene henceforth. Importantly, the enrichment of PGC-1β at Lpk promoter chromatin was significantly diminished in hepatocytes from ChREBP−/− mice (Figure 4F). Increased acetylation of histone H3 is associated with transcriptional activation by ChREBP [29]. Consistent with this, ChIP using an antibody directed against acetylated histone H3 pulled down increased amounts of chromatin near the ChoRE found within the Lpk promoter in the LAP-tet-OFF-PGC-1β hepatocytes compared to WT controls (Figure 4G). These data indicate that PGC-1β interacts with ChREBP at the promoters of known ChREBP target genes, and suggests that ChREBP is a novel transcription factor partner of PGC-1β.
3.5. The interaction between PGC-1β and ChREBP is influenced by glucose concentration
Interestingly, the quantity of PGC-1β that was associated with chromatin in the Lpk promoter was significantly increased under high glucose compared to low glucose conditions (Figure 5A). In contrast, PGC-1β association with the promoter of the Acadm gene, which encodes a fatty acid oxidation enzyme, was approximately 6-fold greater in low glucose compared to high glucose conditions (Figure 5A). PGC-1β occupancy at the Lpk locus was also markedly enhanced after fasting-refeeding compared to fasted liver (Figure 5B), but was enriched at the Acadm promoter in hepatocytes isolated from fasted mice (Figure 5B). Consistent with the data above, the quantity of ChREBP protein co-immunoprecipitated with the PGC-1β antibody was markedly enhanced by high glucose culture conditions in both WT and LAP-tet-OFF-PGC-1β hepatocytes (Figure 5C). A robust interaction between PGC-1β and ChREBP was detected under high glucose conditions in WT hepatocytes, demonstrating that the endogenous proteins physically interact (Figure 5C). Refeeding also strongly increased the interaction between ChREBP and PGC-1β in WT and LAP-tet-OFF-PGC-1β hepatocytes (Figure 5C). These data suggest that the physical and functional interactions between PGC-1β and ChREBP are influenced by glucose concentration. Furthermore, the ChIP data provide evidence that PGC-1β may switch promoters based on nutrient availability. Consistent with the increased interaction between PGC-1β and ChREBP under high glucose conditions, rates of glycolysis and fatty acid synthesis were increased in LAP-tet-OFF-PGC-1β hepatocytes only under high glucose conditions compared to WT controls (Figure 5D).
Figure 5.
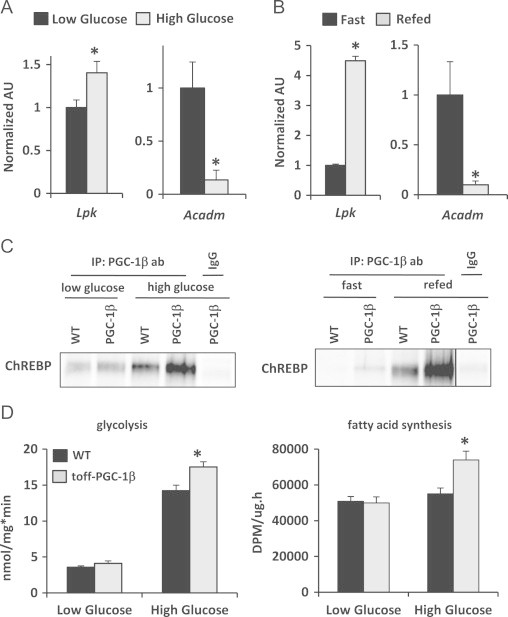
High glucose concentrations induce the interaction between ChREBP and PGC-1β: (A) ChIP analysis demonstrates an enrichment of PGC-1β at the ChoRE in the Lpk promoter under high glucose concentrations, while under low glucose conditions, PGC-1β is enriched at the Acadm promoter. (B) ChIP analysis demonstrates an enrichment of PGC-1β at the ChoRE in the Lpk promoter under refeeding conditions, while following a 24h fast, PGC-1β is located at the Acadm promoter. (C) Western blot analysis following co-immunoprecipitation reveals increased interaction between PGC-1β and ChREBP in WT and LAP-tet-OFF-PGC-1β hepatocytes cultured in high glucose conditions and refed conditions. (D) LAP-tet-OFF-PGC-1β hepatocytes exhibit increased rates of glycolysis and fatty acid synthesis under high glucose culture conditions. *P<0.05 versus WT control mice.
3.6. Liver-specific PGC-1β deficiency impairs the lipogenic response to high glucose conditions
We next examined the lipogenic response to high glucose conditions in hepatocytes of recently-characterized mice with liver-specific deletion of PGC-1β (LS-PGC-1β−/− mice) [16]. We have shown that under normal ad libitum conditions, there is no effect of liver-specific PGC-1β gene deletion on lipogenic gene expression, but that the lipogenic response to refeeding after fasting is attenuated [16].
The effects of low (5.5 mM) and high glucose (25 mM) culture conditions on lipogenic gene expression in WT and LS-PGC-1β−/− hepatocytes were examined. As expected, 12 h exposure to high glucose conditions induced the expression of several known ChREBP target genes (Lpk, Gckr, Fasn, Acaca, Me1, and Acly) (Figure 6A). However, the induction of ChREBP target genes by high glucose was completely abolished in LS-PGC-1β−/− hepatocytes (Figure 6A). The nuclear content of ChREBP as assessed by immunofluorescent staining (data not shown) and ChREBP occupancy of the Lpk and Acaca promoters (Figure 6C) was unaffected by loss of PGC-1β. On the other hand, histone H3 acetylation was diminished by loss of PGC-1β, which is consistent with the model that PGC-1β is recruiting coactivators with histone acetyltransferase activity to the Lpk promoter. These data demonstrate that PGC-1β is necessary for the induction of enzymes involved in carbohydrate metabolism and lipogenesis in response to increased glucose concentration.
Figure 6.
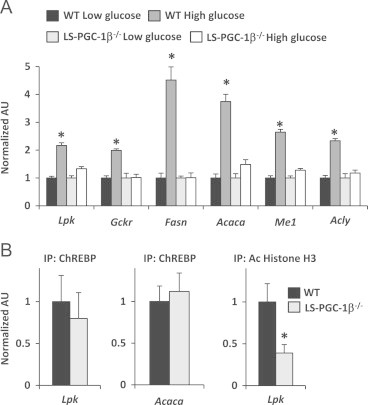
Hepatocytes isolated from LS-PGC-1β−/− mice exhibit a blunted lipogenic response under high glucose culture conditions: (A) Graphs depict the blunted response of several ChREBP target genes in LS-PGC-1β−/− hepatocytes under high glucose culture conditions that normally stimulate ChREBP activity (n=4/group) *P<0.05 versus WT low glucose. (B) ChIP analysis shows no difference in the presence of ChREBP protein at the ChoRE in the Lpk or Acaca promoter. ChIP analysis demonstrates that hepatocyte PGC-1β deficiency results in decreased acetylation of histone H3 at the Lpk promoter under high glucose conditions. *P<0.05 versus WT.
4. Discussion
PGC-1 transcriptional coactivators are important regulators of intermediary metabolism by controlling programs of gene transcription to affect changes in metabolic processes. In this work, we provide evidence that PGC-1β selectively enhances glycolysis and lipid synthesis in response to increased glucose availability through activation of ChREBP. This effect of PGC-1β and the interaction between these two transcriptional regulators was strongly modulated by the nutritional status of the hepatocyte. Collectively, our findings identify a novel partner for PGC-1β-mediated regulation of hepatic metabolism and suggest that PGC-1β coordinates hepatic lipogenic capacity via interactions with multiple lipogenic transcription factors.
ChREBP is a basic-helix-loop-helix leucine zipper transcription factor, like SREBP1, that was originally purified from rat liver nuclear extracts in a search for the transcription factors bound to the carbohydrate response elements (ChoRE) found in the promoters of genes encoding several lipogenic and glycolytic enzymes that are induced in response to high glucose conditions [17,34–38]. Loss of ChREBP activity in hepatocytes almost completely abolishes the lipogenic response to high glucose conditions, suggesting that this transcription factor is obligatory for this glucose-sensing response [17]. SREBP1 also regulates the expression of genes encoding a variety of lipogenic enzymes in liver [39] and SREBP1 is thought to mediate the lipogenic response to hyperinsulinemic conditions [40,41]. The promoters of many glycolytic and lipogenic genes contain both ChoREs and SREs [42,43], suggesting that these two factors work together to regulate the hepatic lipogenic response to hyperinsulinemia and hyperglycemia that occurs in conditions of nutrient excess [12,44]. PGC-1β has now been shown to coordinately activate both the insulin-responsive (SREBP1) and glucose-responsive (ChREBP) arms [9] (Figure 7). A coordinated response would be especially critical in physiologic conditions associated with hyperinsulinemia and hyperglycemia, such as during refeeding. Indeed, loss of PGC-1β in liver markedly blunted the expression of lipogenic genes that are induced by refeeding [16,45]. However, even with complete loss of PGC-1β in liver, a significant induction in the hepatic response to refeeding was noted [16], suggesting that other transcriptional coactivators are sufficient to mount at least a partial response.
Figure 7.
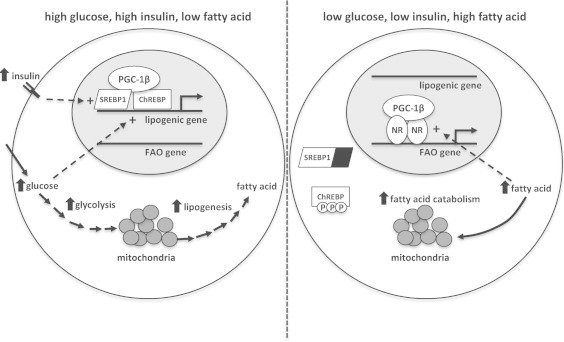
Schematic depicting the differential roles of PGC-1β on hepatic intermediary metabolism depending upon nutrient supply. (left) When glucose and insulin concentrations are elevated, as occurs in the fed state, the lipogenic transcription factors ChREBP and SREBP1 are activated and nuclear-localized. PGC-1β coactivates these factors to increase expression of genes encoding glycolytic and lipogenic enzymes. (right) When glucose and insulin concentrations are low and fatty acid availability is high, such as occurs during fasting conditions, SREBP1 and ChREBP are cytosolic and inactive. PGC-1β is associated with promoters of genes encoding fatty acid oxidation (FAO) enzymes to enhance the capacity for fatty acid catabolism through mitochondrial pathways.
Glucose concentration had a strong effect on the physical interaction between PGC-1β and ChREBP in co-immunoprecipitation and ChIP experiments. Carbohydrate availability clearly regulates ChREBP nuclear localization and DNA-binding activity, but the molecular mechanisms remain controversial [46–51]. Recent work has also demonstrated the existence of a ChREBP splice variant encoding a smaller ChREBP protein that is induced by carbohydrate abundance and is predominantly nuclear (ChREBPβ) [52]. Our data are also consistent with promoter switching by PGC-1β in response to nutrient availability (Figure 7). Under fasting or low glucose conditions, when fatty acid oxidation predominates, PGC-1β was associated with a nuclear receptor response element in the Acadm gene promoter. Conversely, under high glucose or refed conditions, the association of PGC-1β with Acadm was reduced and the association of PGC-1β with the ChoRE in the Lpk promoter was increased. The regulatory mechanisms that control PGC-1β promoter preference remain to be elucidated, but could be as straightforward as altered nuclear abundance of ChREBPα or ChREBPβ and the ligand availability or activity of the nuclear receptors that control Acadm expression during these conditions, since PGC-1β is thought to be constitutively nuclear.
It is unclear why PGC-1β promotes both fatty acid synthesis and oxidation, which would seem to be a futile cycle. There are physiologic and pathophysiologic conditions characterized by increased lipogenesis and fatty acid oxidation, including the response to high fat diet and obesity-related fatty liver disease [53]. The aforementioned promoter switching of PGC-1β between the promoters of oxidative and lipogenic enzymes depending upon nutrient availability might explain this observation (Figure 7). It should also be noted that the conversion of glucose to de novo fatty acid requires the carbon contained in glucose to enter the TCA cycle as acetyl-CoA, which might explain why PGC-1β also activates the expression of TCA cycle enzymes to enhance citrate synthesis. Enhanced fatty acid oxidation may help to drive the TCA flux or prevent the accumulation of downstream intermediates.
Previous work using adenoviral approaches demonstrated that overexpression of PGC-1α or PGC-1β in liver drove marked changes in circulating concentrations of glucose or lipids, respectively [8–11]. Although we found similar transcriptional responses, we did not observe changes in circulating glucose or lipids in the present studies except in experiments conducted with the mice on a “Western” diet, when PGC-1β overexpression increased plasma TG concentration and tended to increase plasma cholesterol concentration compared to NTG control mice on the high fat diet. There are a few likely explanations for the lack of effect on metabolic homeostasis in our normal chow-fed mice. There could be chronic compensatory counter-regulatory responses that dampen expression of these genes or suppress glucose or lipoprotein release by the liver. Alternatively, the magnitude of overexpression may not be sufficient to induce some key genes involved in these processes to affect blood or hepatic metabolite concentrations. For example, previous work has suggested that the hyperlipidemia in response to PGC-1β was associated with an induction in microsomal triglyceride transfer protein [9,11], which is obligatory for VLDL secretion. The increased hepatic glucose output after PGC-1α overexpression involved marked activation of phosphoenolpyruvate carboxykinase and glucose-6-phosphatase [8,10]. However, in our transgenic mice, the expression of these genes was not significantly altered (data not shown). Altogether, these findings suggest that modest enhancements in PGC-1 activity may predispose to metabolic abnormalities when nutrients are in excess, such as in response to high fat diet, but in isolation, this level of overexpression is not sufficient to drive changes in circulating metabolites.
5. Conclusions
In summary, our unbiased gene expression profiling studies confirmed an important role for PGC-1β in regulating hepatic lipogenesis, and have identified new mechanisms that mediate this response. We provide evidence that ChREBP is a novel transcription factor partner of PGC-1β and that the interaction and promoter association of the PGC-1β/ChREBP complex is regulated by glucose availability. These findings could have important implications for our understanding of the pathologic abnormalities in hepatic metabolism that occur in obesity and type 2 diabetes.
Conflict of interest
None declared.
Acknowledgements
The authors thank Dr. Douglas Mashek for technical advice on the high/low glucose studies with mouse hepatocytes and Dr. Geoffrey Girnun for helpful discussions. K.T.C. is supported by a Liver Scholar Award from the American Liver Foundation and a pilot and feasibility award from the Nutrition Obesity Research Center at Washington University (P30 DK056341). The project was also supported by R01 DK078187 (to B.N.F.), R01 DK091538 (to P.A.C.), and DK064951 (to A.K.). The Core Centers of Washington University also supported these studies (P30 DK056341, P60 DK020579, P30 DK052574).
Appendix A. Supporting information
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.molmet.2013.05.001.
Appendix A. Supplementary materials
Supplementary material
References
- 1.Finck B.N., Kelly D.P. PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. Journal of Clinical Investigation. 2006;116:615–622. doi: 10.1172/JCI27794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Handschin C., Spiegelman B.M. Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocrine Reviews. 2006;27:728–735. doi: 10.1210/er.2006-0037. [DOI] [PubMed] [Google Scholar]
- 3.Lai L., Leone T.C., Zechner C., Schaeffer P.J., Kelly S.M. Transcriptional coactivators PGC-1alpha and PGC-lbeta control overlapping programs required for perinatal maturation of the heart. Genes & Development. 2008;22:1948–1961. doi: 10.1101/gad.1661708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Puigserver P., Wu Z., Park C.W., Graves R., Wright M. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–839. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- 5.Vega R.B., Huss J.M., Kelly D.P. The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Molecular and Cellular Biology. 2000;20:1868–1876. doi: 10.1128/mcb.20.5.1868-1876.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huss J.M., Kopp R.P., Kelly D.P. Peroxisome proliferator-activated receptor coactivator-1alpha (PGC-1alpha) coactivates the cardiac-enriched nuclear receptors estrogen-related receptor-alpha and -gamma. Identification of novel leucine-rich interaction motif within PGC-1alpha. Journal of Biological Chemistry. 2002;277:40265–40274. doi: 10.1074/jbc.M206324200. [DOI] [PubMed] [Google Scholar]
- 7.Wu Z., Puigserver P., Andersson U., Zhang C., Adelmant G. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–124. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- 8.Herzig S., Long F., Jhala U.S., Hedrick S., Quinn R. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature. 2001;413:179–183. doi: 10.1038/35093131. [DOI] [PubMed] [Google Scholar]
- 9.Lin J., Yang R., Tarr P.T., Wu P.H., Handschin C. Hyperlipidemic effects of dietary saturated fats mediated through PGC-1beta coactivation of SREBP. Cell. 2005;120:261–273. doi: 10.1016/j.cell.2004.11.043. [DOI] [PubMed] [Google Scholar]
- 10.Yoon J.C., Puigserver P., Chen G., Donovan J., Wu Z. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature. 2001;413:131–138. doi: 10.1038/35093050. [DOI] [PubMed] [Google Scholar]
- 11.Wolfrum C., Stoffel M. Coactivation of Foxa2 through Pgc-1beta promotes liver fatty acid oxidation and triglyceride/VLDL secretion. Cell Metabolism. 2006;3:99–110. doi: 10.1016/j.cmet.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 12.Towle H.C. Glucose as a regulator of eukaryotic gene transcription. Trends in Endocrinology & Metabolism. 2005;16:489–494. doi: 10.1016/j.tem.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 13.Gossen M., Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:5547–5551. doi: 10.1073/pnas.89.12.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Russell L.K., Mansfield C.M., Lehman J.J., Kovacs A., Courtois M. Cardiac-specific induction of the transcriptional coactivator peroxisome proliferator-activated receptor gamma coactivator-1alpha promotes mitochondrial biogenesis and reversible cardiomyopathy in a developmental stage-dependent manner. Circulation Research. 2004;94:525–533. doi: 10.1161/01.RES.0000117088.36577.EB. [DOI] [PubMed] [Google Scholar]
- 15.Schilling J., Lai L., Sambandam N., Dey C.E., Leone T.C. Toll-like receptor-mediated inflammatory signaling reprograms cardiac energy metabolism by repressing peroxisome proliferator-activated receptor gamma coactivator-1 signaling. Circulation: Heart Failure. 2011;4:474–482. doi: 10.1161/CIRCHEARTFAILURE.110.959833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chambers K.T., Chen Z., Crawford P.A., Fu X., Burgess S.C. Liver-specific PGC-1beta deficiency leads to impaired mitochondrial function and lipogenic response to fasting-refeeding. PLoS One. 2012;7:e52645. doi: 10.1371/journal.pone.0052645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iizuka K., Bruick R.K., Liang G., Horton J.D., Uyeda K. Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:7281–7286. doi: 10.1073/pnas.0401516101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chambers K.T., Leone T.C., Sambandam N., Kovacs A., Wagg C.S. Chronic inhibition of pyruvate dehydrogenase in heart triggers an adaptive metabolic response. Journal of Biological Chemistry. 2011;286:11155–11162. doi: 10.1074/jbc.M110.217349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Finck B.N., Han X., Courtois M., Aimond F., Nerbonne J.M. A critical role for PPARalpha-mediated lipotoxicity in the pathogenesis of diabetic cardiomyopathy: modulation by dietary fat content. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:1226–1231. doi: 10.1073/pnas.0336724100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen Z., Fitzgerald R.L., Averna M.R., Schonfeld G. A targeted apolipoprotein B-38.9-producing mutation causes fatty livers in mice due to the reduced ability of apolipoprotein B-38.9 to transport triglycerides. Journal of Biological Chemistry. 2000;275:32807–32815. doi: 10.1074/jbc.M004913200. [DOI] [PubMed] [Google Scholar]
- 21.Skinner J.R., Shew T.M., Schwartz D.M., Tzekov A., Lepus C.M. Diacylglycerol enrichment of endoplasmic reticulum or lipid droplets recruits perilipin 3/TIP47 during lipid storage and mobilization. Journal of Biological Chemistry. 2009;284:30941–30948. doi: 10.1074/jbc.M109.013995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen Z., Gropler M.C., Norris J., Lawrence J.C., Jr., Harris T.E. Alterations in hepatic metabolism in fld mice reveal a role for lipin 1 in regulating VLDL-triacylglyceride secretion. Arteriosclerosis, Thrombosis, and Vascular Biology. 2008;28:1738–1744. doi: 10.1161/ATVBAHA.108.171538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin X., Schonfeld G., Yue P., Chen Z. Hepatic fatty acid synthesis is suppressed in mice with fatty livers due to targeted apolipoprotein B38.9 mutation. Arteriosclerosis, Thrombosis, and Vascular Biology. 2002;22:476–482. doi: 10.1161/hq0302.105271. [DOI] [PubMed] [Google Scholar]
- 24.Wentz A.E., d’Avignon D.A., Weber M.L., Cotter D.G., Doherty J.M. Adaptation of myocardial substrate metabolism to a ketogenic nutrient environment. Journal of Biological Chemistry. 2010;285:24447–24456. doi: 10.1074/jbc.M110.100651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mitra M.S., Schilling J.D., Wang X., Jay P.Y., Huss J.M. Cardiac lipin 1 expression is regulated by the peroxisome proliferator activated receptor gamma coactivator 1alpha/estrogen related receptor axis. Journal of Molecular and Cellular Cardiology. 2011;51:120–128. doi: 10.1016/j.yjmcc.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Finck B.N., Gropler M.C., Chen Z., Leone T.C., Croce M.A. Lipin 1 is an inducible amplifier of the hepatic PGC-1alpha/PPARalpha regulatory pathway. Cell Metabolism. 2006;4:199–210. doi: 10.1016/j.cmet.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 27.Chang C.Y., Kazmin D., Jasper J.S., Kunder R., Zuercher W.J. The metabolic regulator ERRalpha, a downstream target of HER2/IGF-1R, as a therapeutic target in breast cancer. Cancer Cell. 2011;20:500–510. doi: 10.1016/j.ccr.2011.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schwartz D.M., Wolins N.E. A simple and rapid method to assay triacylglycerol in cells and tissues. Journal of Lipid Research. 2007;48:2514–2520. doi: 10.1194/jlr.D700017-JLR200. [DOI] [PubMed] [Google Scholar]
- 29.Bricambert J., Miranda J., Benhamed F., Girard J., Postic C. Salt-inducible kinase 2 links transcriptional coactivator p300 phosphorylation to the prevention of ChREBP-dependent hepatic steatosis in mice. Journal of Clinical Investigation. 2010;120:4316–4331. doi: 10.1172/JCI41624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kamei Y., Ohizumi H., Fujitani Y., Nemoto T., Tanaka T. PPARgamma coactivator 1beta/ERR ligand 1 is an ERR protein ligand, whose expression induces a high-energy expenditure and antagonizes obesity. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:12378–12383. doi: 10.1073/pnas.2135217100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kistner A., Gossen M., Zimmermann F., Jerecic J., Ullmer C. Doxycycline-mediated quantitative and tissue-specific control of gene expression in transgenic mice. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:10933–10938. doi: 10.1073/pnas.93.20.10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lin J., Tarr P.T., Yang R., Rhee J., Puigserver P. PGC-1beta in the regulation of hepatic glucose and energy metabolism. Journal of Biological Chemistry. 2003;278:30843–30848. doi: 10.1074/jbc.M303643200. [DOI] [PubMed] [Google Scholar]
- 33.Gan Z., Burkart-Hartman E.M., Han D.H., Finck B., Leone T.C. The nuclear receptor PPARbeta/delta programs muscle glucose metabolism in cooperation with AMPK and MEF2. Genes & Development. 2011;25:2619–2630. doi: 10.1101/gad.178434.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ma L., Tsatsos N.G., Towle H.C. Direct role of ChREBP.Mlx in regulating hepatic glucose-responsive genes. Journal of Biological Chemistry. 2005;280:12019–12027. doi: 10.1074/jbc.M413063200. [DOI] [PubMed] [Google Scholar]
- 35.Ma L., Robinson L.N., Towle H.C. ChREBP⁎Mlx is the principal mediator of glucose-induced gene expression in the liver. Journal of Biological Chemistry. 2006;281:28721–28730. doi: 10.1074/jbc.M601576200. [DOI] [PubMed] [Google Scholar]
- 36.Ishii S., Iizuka K., Miller B.C., Uyeda K. Carbohydrate response element binding protein directly promotes lipogenic enzyme gene transcription. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:15597–15602. doi: 10.1073/pnas.0405238101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dentin R., Girard J., Postic C. Carbohydrate responsive element binding protein (ChREBP) and sterol regulatory element binding protein-1c (SREBP-1c): two key regulators of glucose metabolism and lipid synthesis in liver. Biochimie. 2005;87:81–86. doi: 10.1016/j.biochi.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 38.Dentin R., Pegorier J.P., Benhamed F., Foufelle F., Ferre P. Hepatic glucokinase is required for the synergistic action of ChREBP and SREBP-1c on glycolytic and lipogenic gene expression. Journal of Biological Chemistry. 2004;279:20314–20326. doi: 10.1074/jbc.M312475200. [DOI] [PubMed] [Google Scholar]
- 39.Horton J.D., Shah N.A., Warrington J.A., Anderson N.N., Park S.W. Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:12027–12032. doi: 10.1073/pnas.1534923100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li S., Brown M.S., Goldstein J.L. Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:3441–3446. doi: 10.1073/pnas.0914798107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Peterson T.R., Sengupta S.S., Harris T.E., Carmack A.E., Kang S.A. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell. 2011;146:408–420. doi: 10.1016/j.cell.2011.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koo S.H., Dutcher A.K., Towle H.C. Glucose and insulin function through two distinct transcription factors to stimulate expression of lipogenic enzyme genes in liver. Journal of Biological Chemistry. 2001;276:9437–9445. doi: 10.1074/jbc.M010029200. [DOI] [PubMed] [Google Scholar]
- 43.Poupeau A., Postic C. Cross-regulation of hepatic glucose metabolism via ChREBP and nuclear receptors. Biochimica et Biophysica Acta. 2011;1812:995–1006. doi: 10.1016/j.bbadis.2011.03.015. [DOI] [PubMed] [Google Scholar]
- 44.Girard J., Ferre P., Foufelle F. Mechanisms by which carbohydrates regulate expression of genes for glycolytic and lipogenic enzymes. Annual Review of Nutrition. 1997;17:325–352. doi: 10.1146/annurev.nutr.17.1.325. [DOI] [PubMed] [Google Scholar]
- 45.Nagai Y., Yonemitsu S., Erion D.M., Iwasaki T., Stark R. The role of peroxisome proliferator-activated receptor gamma coactivator-1 beta in the pathogenesis of fructose-induced insulin resistance. Cell Metabolism. 2009;9:252–264. doi: 10.1016/j.cmet.2009.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dentin R., Tomas-Cobos L., Foufelle F., Leopold J., Girard J. Glucose 6-phosphate, rather than xylulose 5-phosphate, is required for the activation of ChREBP in response to glucose in liver. Journal of Hepatology. 2011 doi: 10.1016/j.jhep.2011.07.019. [DOI] [PubMed] [Google Scholar]
- 47.Kabashima T., Kawaguchi T., Wadzinski B.E., Uyeda K. Xylulose 5-phosphate mediates glucose-induced lipogenesis by xylulose 5-phosphate-activated protein phosphatase in rat liver. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:5107–5112. doi: 10.1073/pnas.0730817100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li M.V., Chang B., Imamura M., Poungvarin N., Chan L. Glucose-dependent transcriptional regulation by an evolutionarily conserved glucose-sensing module. Diabetes. 2006;55:1179–1189. doi: 10.2337/db05-0822. [DOI] [PubMed] [Google Scholar]
- 49.Li M.V., Chen W., Harmancey R.N., Nuotio-Antar A.M., Imamura M. Glucose-6-phosphate mediates activation of the carbohydrate responsive binding protein (ChREBP) Biochemical and Biophysical Research Communications. 2010;395:395–400. doi: 10.1016/j.bbrc.2010.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tsatsos N.G., Towle H.C. Glucose activation of ChREBP in hepatocytes occurs via a two-step mechanism. Biochemical and Biophysical Research Communications. 2006;340:449–456. doi: 10.1016/j.bbrc.2005.12.029. [DOI] [PubMed] [Google Scholar]
- 51.Yamashita H., Takenoshita M., Sakurai M., Bruick R.K., Henzel W.J. A glucose-responsive transcription factor that regulates carbohydrate metabolism in the liver. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:9116–9121. doi: 10.1073/pnas.161284298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Herman M.A., Peroni O.D., Villoria J., Schon M.R., Abumrad N.A. A novel ChREBP isoform in adipose tissue regulates systemic glucose metabolism. Nature. 2012;484:333–338. doi: 10.1038/nature10986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ryu M.H., Cha Y.S. The effects of a high-fat or high-sucrose diet on serum lipid profiles, hepatic acyl-CoA synthetase, carnitine palmitoyltransferase-I, and the acetyl-CoA carboxylase mRNA levels in rats. Journal of Biochemistry and Molecular Biology. 2003;36:312–318. doi: 10.5483/bmbrep.2003.36.3.312. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material