

Abstract
Proper development and function of white adipose tissue (WAT), which are regulated by multiple transcription factors and coregulators, are crucial for glucose homeostasis. WAT is also the main target of thiazolidinediones, which are thought to exert their insulin-sensitizing effects by promoting mitochondrial biogenesis in adipocytes. Besides being expressed in WAT, the role of the coactivator PGC-1β in this tissue has not been addressed. To study its function in WAT, we have generated mice that lack PGC-1β in adipose tissues. Gene expression profiling analysis of WAT reveals that PGC-1β regulates mitochondrial genes involved in oxidative metabolism. Furthermore, lack of PGC-1β prevents the induction of mitochondrial genes by rosiglitazone in WAT without affecting the capacity of thiazolidinediones to enhance insulin sensitivity. Our findings indicate that PGC-1β is important for basal and rosiglitazone-induced mitochondrial function in WAT, and that induction of mitochondrial oxidative capacity is not essential for the insulin-sensitizing effects of thiazolidinediones.
Keywords: Peroxisome proliferator-activated receptor γ coactivator-1, Mitochondrial biogenesis, Adipocytes, Thiazolidinediones, Type 2 diabetes
1. Introduction
White adipose tissue (WAT) plays a crucial role in the regulation of glucose homeostasis and whole body energy balance. In accordance with this central regulatory role, alterations in WAT lipid-storage capacity or endocrine function, as observed in obese or lipodystrophic patients, are associated to the development of diverse metabolic disorders, including insulin resistance and type 2 diabetes [1].
WAT function depends on the differentiation of precursor cells into mature adipocytes and their ability to sense the energetic status of the organism and elicit appropriate responses, including the storage or release of fatty acids and the secretion of adipokines, thereby enabling adaptation to different physiological states. The adipogenic process is regulated by a complex network of transcription factors that coordinately control the expression of a broad set of genes involved in the acquisition of adipocyte traits and functions. The core of this regulatory network is formed by the transcription factors CCAAT/enhancer binding protein (C/EBP) β, peroxisome proliferator-activated receptor γ (PPARγ) and C/EBPα, which act sequentially to control the adipogenic program [2]. Among these, PPARγ is considered to be the master regulator of adipogenesis due to its unique capacity to induce adipocyte differentiation in the absence of any other adipogenic factor [3]. Noteworthy, PPARγ is the target of thiazolidinediones (TZDs), a family of antidiabetic drugs whose insulin-sensitizing properties depend on their capacity to promote adipogenesis and prevent lipotoxicity in insulin target tissues [4]. It has been suggested that the capacity of TZDs to ameliorate insulin resistance also depends on their ability to promote mitochondrial biogenesis and oxidative metabolism in adipocytes. In addition of producing most of the ATP required by adipocytes, adipose tissue mitochondria play a fundamental role in the synthesis of intermediate metabolites required for lipogenesis and are also crucial for the synthesis and secretion of adipokines [5,6]. Thus, by increasing mitochondrial biogenesis, TZDs would enhance fatty acid oxidation and lipogenesis in adipocytes, contributing to lipid clearance from the blood and preventing their toxic effects in insulin sensitive tissues, like muscle and liver [7]. However, the mechanisms by which TZDs induce mitochondrial biogenesis are not fully understood [8–11].
The activity of DNA-binding transcription factors is modulated in response to different metabolic signals or in a time- and tissue-specific manner by their interaction with regulatory cofactors. Some of these transcriptional cofactors modulate adipocyte differentiation and function positively (e. g. SRC-3, CBP/p300 or TRAP220) [12–14], or negatively (e.g. NCoR/SMRT or SIRT1) [15,16]. Many of these cofactors exert their activity through their interaction with PPARγ, facilitating or repressing its transcriptional activity. By regulating adipogenesis and adipocyte function, these coregulators play important roles in whole body energy homeostasis [13,14].
The coactivators of the PGC-1 (PPARγ coactivator-1) family have emerged as key players in the control of energy homeostasis. PGC-1α, the first and best-characterized member of the family, was originally identified as a PPARγ-interacting protein in brown adipose tissue (BAT), where it regulates non-shivering adaptive thermogenesis [17]. PGC-1α also regulates mitochondrial biogenesis and oxidative metabolism in a wide variety of tissues, including brain, skeletal muscle or heart [18]. PGC-1β, the closest homolog to PGC-1α, follows an expression pattern similar to PGC-1α, with highest levels in tissues with elevated oxidative capacity [19,20]. Accordingly, PGC-1β function has been studied mostly in tissues like BAT, skeletal muscle or heart, where it regulates mitochondrial gene expression and cell respiration [21–24]. In at least some of these tissues, PGC-1α and PGC-1β coactivators seem to carry redundant roles in the control of mitochondrial oxidative capacity [24,25]. In addition, both PGC-1α and PGC-1β carry distinct and non-redundant roles in the regulation of glucose and lipid metabolism in liver, with PGC-1α controlling hepatic gluconeogenesis in response to fasting [26] and PGC-1β regulating triglyceride synthesis and VLDL secretion [27,28]. The role of PGC-1β in the regulation of lipid metabolism in liver together with the fact that PGC-1β is expressed at moderate levels in WAT [19] suggest that PGC-1β could play a role in adipocyte biology. However, the in vivo function of PGC-1β in WAT has not yet been addressed.
To gain insights into the gene networks and processes regulated by PGC-1β in WAT, we have generated a mouse model that lacks PGC-1β in adipocytes. Our results indicate that PGC-1β regulates basal and rosiglitazone-induced expression of mitochondrial genes involved in ATP production. Moreover, we show that enhanced mitochondrial activity is not essential for the insulin sensitizing effects of rosiglitazone.
2. Material and methods
2.1. Animals
To generate mice with floxed Ppargc1b alleles, a targeting vector was constructed by subcloning a Sac II–Bgl II (8294 bp, containing exons 3, 4 and 5) and a Bgl II–Sma I (3102 bp, containing exons 6, 7 and 8) DNA fragment of a BAC genomic DNA clone carrying the murine Ppargc1b gene locus (Incyte Genomics, Palo Alto, USA) upstream and downstream, respectively, of a PGK-neomycin cassette flanked by two FRT sites and one LoxP site. An additional LoxP site was introduced upstream of exon 4. The linearized targeting vector (Figure 1A) was electroporated into E14TG2a embryonic stems cells, and a G418-resistant clone with the correct targeting event was injected into C57BL/6 blastocysts. Germline-transmitting mice were mated with FLP deleter mice to remove the PGK-neomycin selection cassette, generating mice with floxed exons 4 and 5 of the Ppargc1b gene. Mice with Ppargc1b floxed alleles (Ppargc1bflox/flox) were crossed for at least 10 generations with C57BL/6J mice and maintained in the C57BL/6J genetic background. Ppargc1bflox/flox mice were then crossed to aP2-Cre [B6.Cg-Tg(Fabp4-cre)1rev/J] mice (Jackson Laboratory, Bar Harbor, USA) to generate mice with exons 4 and 5 of the Ppargc1b gene deleted in adipose tissues (PGC1β-FAT-KO mice). The deletion introduces a translation stop codon after exon 3. The efficient deletion of the region containing exons 4 and 5 flanked by the loxP sites was assessed by PCR analysis of genomic DNA isolated from different WAT depots and BAT, using primers F (5′-gaaagcctgggctacatgtga-3′) and R (5′-aggacagatgccctttaaggtgacata-3′) (Figure 1A).
Figure 1.
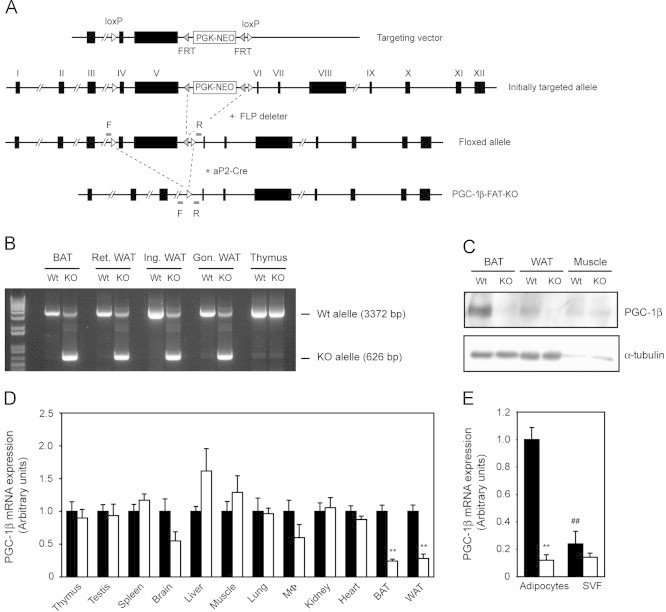
Generation of PGC1β-FAT-KO mice. (A) A targeting vector, containing a PGK-NEO selection cassette flanked by flippase-specific FRT sites in intron 5, and having exons 4 and 5 of Ppargc1b gene flanked by loxP sites, was used to generate mice with floxed Ppargc1b alleles. To generate PGC1β-FAT-KO mice, mice with floxed Ppargc1b alleles were crossed to aP2-Cre mice that overexpress Cre recombinase in adipose tissues. (B) The efficiency of genomic recombination in adipose tissues of PGC1β-FAT-KO mice was analyzed by PCR using primers F and R (see A) located upstream and downstream, respectively, of the 5′ and 3′ loxP sites flanking exons 4 and 5 of the Ppargc1b gene. Amplification of the Wt allele yields a band of 3372 bp, while the recombined allele yields a shorter band of 626 bp. (Ret., retroperitoneal; Ing., inguinal; Gon., gonadal). (C) Western blot analysis of PGC-1β protein levels in interscapular BAT, retroperitoneal WAT and gastrocnemius muscle of Wt and PGC1β-FAT-KO mice. (D) Expression of PGC-1β mRNA was analyzed by real-time quantitative RT-PCR in several tissues of Wt (black bars) and PGC1β-FAT-KO (open bars) mice. (E) PGC-1β expression was assessed by quantitative RT-PCR in white adipocytes and the SVF isolated from the inguinal WAT of Wt and PGC1β-FAT-KO mice. Results are expressed as mean±SEM (n=4–7 animals/group, **, ## P≤0.01).
To minimize the potential defects in adaptive thermogenesis due to lack of PGC-1β in BAT and their influence on whole body energy homeostasis, mice were raised and housed at thermoneutrality (30 °C) throughout the study, unless otherwise specified. Food intake was measured daily over a period of 5 days in wild type (Wt) and PGC1β-FAT-KO mice fed a chow diet. For the rosiglitazone treatment, 6-week old mice were fed a high fat (HF) diet (45% kcal fat, 35% kcal carbohydrates, 20% kcal protein) (Research Diets Inc., New Brunswich, USA) for a period of 13 weeks. On week 11 on HF diet, mice were started on treatment with 10 mg/kg rosiglitazone maleate (Selleck Chemicals, Houston, USA) or vehicle by oral gavage, twice daily, for 2 weeks.
All procedures involving animals were performed in accordance with the institutional animal care guidelines of the Vall d'Hebron-Institut de Recerca and approved by the Animal Experimentation and Ethics Committee of the institution (ID 5/07 and 12/11 CEEA).
2.2. Isolation of mature adipocytes and stromal vascular fraction (SVF)
To isolate adipocytes and SVF from WAT, inguinal white adipose depots were collected from C57BL/6J mice and digested with collagenase A (2 mg/ml) in Dulbecco's Modified Eagle's Medium (DMEM) containing 2% bovine serum albumin during 20–30 min at 37 °C. Once digested, cell suspension was filtered through a nylon 100 μm-mesh cell strainer to remove undigested tissue and let stand for 20 min to allow flotation of adipocytes. Floating adipocytes were collected with a pipette while the remaining digestion solution was centrifuged at 500g for 10 min to collect SVF cells. Both, adipocytes and SVF were immediately processed for RNA isolation.
2.3. 3T3-L1 adipocyte culture
3T3-L1 preadipocyte culture and differentiation have been described elsewhere [29]. For PGC-1α and PGC-1β knockdown, adipocytes were transfected on day 6 of differentiation with 50 nM of ON-TARGETplus SMART pool siRNAs specifically targeting PGC-1α, PGC-1β or both simultaneously using DharmaFECT 4 reagent (Thermo Fisher Scientific, Waltham, USA). ON-TARGETplus Non-Targeting siRNA#2 was used as negative control. Transfection was carried in suspension onto collagen-coated cell culture dishes following the protocol described by Kilroy et al. [30] with minor modifications. Briefly, 3T3-L1 adipocytes that had been differentiated for 6 days were trypsinized, resuspended in differentiation medium (DMEM, 10% fetal calf serum, 100 nM insulin) and collected by centrifugation at 500g during 5 min. The pelleted adipocytes were resuspended in a small volume of differentiation media and counted. Then, a transfection mix was prepared by combining siRNAs (50 nM) and DharmaFECT 4 reagent (2.8 μl) in a final volume of 200 μl of OPTIMEM media. Each transfection mix was added to a well of a collagen-coated 12-well plate and incubated for 20 min at room temperature to allow the formation of siRNA complexes. Next, 4.5×105 adipocytes in a total volume of 800 μl were added to the well and the mixture adipocytes:siRNA complexes were incubated for 24 h, allowing cells to get transfected while getting attached to the surface of the plate. Twenty four hours after transfection, media were replaced and cells were treated with 1 μM of rosiglitazone or vehicle (DMSO) for 48 h before being harvested to analyze gene expression or oxygen consumption.
For the PGC-1β overexpression studies, 3T3-L1 mature adipocytes were transduced at a multiplicity of infection (MOI) of 100 with recombinant adenoviruses containing the cDNAs encoding for the green fluorescent protein (GFP) or a Flag-tagged version of human PGC-1β (2xFlag-PGC1β). Forty eight hours after transduction, cell were harvested and processed for RNA or protein isolation. Adenoviruses expressing human PGC-1β and GFP were generated as previously described [31].
2.4. Gene expression profiling
For gene expression profiling, RNA was isolated from retroperitoneal WAT of PGC1β-FAT-KO mice and Wt littermates (n=5) using the RNeasy Lipid Tissue Kit (Qiagen, Hilden, Germany). Sense ssDNA was first synthesized from total RNA using the Ambion WT Expression Kit (Life Technologies, Paisley, UK) and then fragmented, labeled and hybridized onto Mouse Gene 1.0 ST arrays (Affymetrix, UK) following the manufacturer's instructions. The array images were processed with the Microarray Analysis Suite 5.0 software and the microarray data were analyzed using the open source software Bioconductor. Data analysis was performed by the Statistics and Bioinformatics Unit of the Vall d'Hebron-Institut de Recerca.
2.5. Gene expression
Total RNA was isolated from tissues or cultured cells by using TRIzol reagent (Life Technologies, Paisley, UK) according to the manufacturer's instructions, and 400 ng of RNA were used to synthesize cDNA with SuperScript II reverse transcriptase (Life Technologies, Paisley, UK) and oligo(dT). Gene expression was assessed by real-time quantitative PCR using SYBR Green dye and gene-specific primers in an ABI PRISM 7500 Sequence Detection System. Relative expression was calculated according to the 2−ΔΔCT threshold method using cyclophilin as a reference gene.
2.6. Western blot
Retroperitoneal WAT protein extracts were prepared in homogenization buffer containing 50 mM Tris-HCl, 150 mM NaCl, 1% NP-40, 5 mM EGTA, 5 mM EDTA, 1 mM PMSF and protease inhibitors. 30 μg of proteins were resolved in a 15% SDS/PAGE and transferred to a PVDF membrane. Immunodetection was performed with specific antibodies against ACO2, SDHB, NDUFB9, UCP1 (Abcam, Cambridge, UK), COXIV, CYCS and α-tubulin (Merck Millipore, USA). To detect endogenous PGC-1β, 100 μg of interscapular BAT, retroperitoneal WAT and gastrocnemious muscle protein extracts were resolved in a 7% SDS/PAGE, transferred to a PVDF membrane and probed with an antibody against PGC-1β. The antibody was generated by immunizing rabbits with a bacterially expressed protein having aminoacids 91–426 of PGC-1β fused to GST.
2.7. Mitochondrial DNA quantification
To determine mtDNA content, total DNA was first isolated from inguinal WAT by proteinase K digestion followed by phenol/chloroform extraction and precipitation with ethanol. Relative amounts of mitochondrial DNA (mtDNA) and nuclear DNA (nDNA) were determined by quantitative real-time PCR using 2 ng of total DNA as a template and specific primers to detect COXII (mtDNA) and RIP140 (nDNA), as previously described [32].
2.8. Citrate synthase (CS) activity
Inguinal WAT was first homogenized in extraction buffer (0.25 M sucrose, 1 mM EGTA, 10 mM Hepes) using a Dounce hand homogenizer. Tissue homogenates were then centrifuged at 600g for 30 min at 4 °C to pellet nuclei and large cellular debris. After removal of the top triglyceride layer, the supernatant, containing mitochondria, was recovered and subjected to three cycles of freezing and thawing to disrupt mitochondrial integrity. CS activity was measured as described by Srere [33].
2.9. Oxygen consumption
Oxygen consumption was measured in 3T3-L1 adipocytes using a Clark-type oxygen electrode (Hansatech Instruments Ltd, Norfolk, UK). Briefly, adipocytes were first transfected with specific siRNAs to knockdown PGC-1α, PGC-1β or both simultaneously and treated with 1 μM rosiglitazone or DMSO, as described in Section 2.3. Forty eight hours after transfection, cells were harvested by trypsinization in DMEM media and counted. Basal oxygen consumption of 2.5×105 cells was measured over a period of 5 min prior to the addition to the respiration media of 10 μM of the CCCP uncoupler to determine maximal respiration. Background oxygen consumption, measured after the inhibition of mitochondrial respiration with 1 mM of KCN, was subtracted from basal and maximal oxygen consumption values to obtain net respiration rates.
2.10. Glucose and insulin tolerance tests
Glucose tolerance test was performed on mice fasted for 12 h. Blood glucose levels were determined at 0, 15, 30, 60, 90 and 120 min after an intraperitoneal injection of 2 g/kg of glucose. For the insulin tolerance test, animals fasted for 5 h were injected intraperitoneally with 0.9 U/kg of insulin and glucose levels were measured at 0, 15, 30, 60, 90 and 120 min post-injection. Glucose levels were measured using an ELITE glucometer (Bayer, Barcelona, Spain).
2.11. Insulin signaling
After an overnight fast, mice were given an intravenous injection of saline or insulin (5 U/kg) and 3 min later, liver, muscle and WAT depots were rapidly removed and frozen. Protein extracts from liver, gastrocnemius muscle and inguinal WAT were obtained as described above in Section 2.6 and Akt and phospho-Akt were detected by western blot using specific antibodies (Merck Millipore, USA).
2.12. Serological parameters
Blood from Wt and PGC1β-FAT-KO mice that were fed a HF diet and treated with rosiglitazone or vehicle (see Section 2.1) was collected from the saphenous vein after a 5 h fast and then centrifuged at 3000 rpm during 5 min to obtain serum. Triglycerides and total cholesterol were determined using commercial kits based on the Trinder colorimetric method (FAR Diagnostics, Italy). Free fatty acids were measured with the NEFA-C kit (Wako Chemicals GmbH, Germany). Insulin and leptin were determined by immunoassay using a MILLIPLEXMAG magnetic bead-based assay and the Luminex MAGPIX system (Merck Millipore, USA).
2.13. Statistical analysis
All values are presented in figures and tables as mean±SEM. Where appropriate, unpaired Student's t test or analysis of the variance (ANOVA) followed by post hoc analysis using the Tukey's multiple comparison test were used. Differences were considered significant when P≤0.05.
3. Results
3.1. Generation of PGC1β-FAT-KO mice
Besides BAT, heart and skeletal muscle, PGC-1β is expressed at moderate levels in WAT [19], [20]. Isolation of the adipocyte and stromal vascular fractions has allowed the identification of adipocytes as the major contributors to the expression of PGC-1β mRNA in WAT (Figure 1E and [34]). Similarly, PGC-1β mRNA levels in 3T3-L1 cells have been shown to increase dramatically during differentiation [35]. This suggests that PGC-1β could play a role in the acquisition of mature adipocyte functions.
To gain insights into the function of PGC-1β in white adipocytes, we generated a mouse model in which the Ppargc1b gene was disrupted by homologous recombination in adipose tissues (PGC1β-FAT-KO mice) (Figure 1A). For this, mice with Ppargc1b floxed alleles were crossed with transgenic mice expressing the Cre recombinase under the aP2 promoter, which is strongly expressed in adipocytes. Efficient disruption of the Ppargc1b gene was verified by PCR to occur in interscapular BAT and different depots of WAT at similar levels, while no recombination was detected in tissues that do not express, or express very low levels of aP2, such as thymus (Figure 1B). The disruption of the Ppargc1b gene resulted in a substantial decrease in PGC-1β mRNA and protein levels in WAT and BAT, but not in tissues like skeletal muscle (Figure 1C–D). The reduction in the PGC-1β mRNA levels was more dramatic in the purified adipocyte fraction than in whole WAT of PGC1β-FAT-KO mice (Figure 1D–E), suggesting that the remaining PGC-1β mRNA expression in WAT was mostly due to the contribution of other non-adipocyte cell types present in SVF of WAT (Figure 1E). However, we found that PGC1β-FAT-KO mice exhibited a noticeable, although not statistically significant, decrease in the expression of PGC-1β mRNA in brain and peritoneal macrophages, a finding that is consistent with the recently reported expression of aP2 in these non-adipose tissues and cells [36].
Initial characterization of PGC1β-FAT-KO mice housed at 21 °C did not reveal any gross abnormality or differences in body weight or in the mass of major organs (Supplemental Figure A.1A–B). The weight of main WAT depots was also similar between Wt and PGC1β-FAT-KO mice, but an approximately 50% increase in the mass of interscapular BAT was observed in knockout mice (Supplemental Figure A.1C). A histological analysis of interscapular BAT revealed that brown adipocytes of PGC1β-FAT-KO mice that had been housed at 21 °C exhibit a white adipocyte-like appearance, accumulating large amount of triglycerides in a single big vacuole (Supplemental Figure A.1D). The excessive accumulation of lipids in brown adipocytes is indicative of a poorly active BAT, which is consistent with the reduced expression of mitochondrial genes, especially UCP1, observed in BAT of PGC1β-FAT-KO mice (Supplemental Figure A.1E).
To focus on the role of PGC-1β in WAT, we raised and maintained mice throughout the study at thermoneutrality (30 °C). This minimizes the influence that BAT dysfunction and alterations in adaptive thermogenesis could have on whole body energy homeostasis, and renders mouse physiology closer to that of humans [37]. Under these housing conditions, no differences in body weight were found between Wt and PGC1β-FAT-KO male littermates when fed a standard chow diet or a HF diet (Figure 2A). Consistent with the lack of differences in body weight, adult Wt and PGC1β-FAT-KO mice exhibited similar fat pads mass (Figure 2B) and white adipocyte size (Figure 2C). Also, when housed at thermoneutrality, brown adipocytes from Wt and PGC1β-FAT-KO mice adopt a similar appearance, indicating that in the absence of thermogenic stimuli BAT from Wt and PGC1β-FAT-KO behave similarly (Figure 2D). No significant differences in food intake were observed between Wt and PGC1β-FAT-KO mice (Wt=0.195±0.022 Kcal/g. day vs PGC1β-FAT-KO=0.179±0.013 Kcal/g. day; n=5–6 animals/group).
Figure 2.
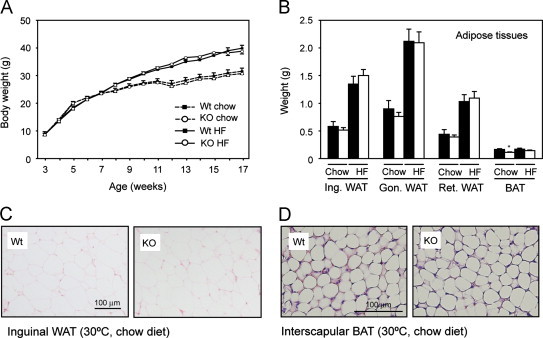
Body weight and adipose tissue mass of PGC1β-FAT-KO mice at thermoneutrality. (A) Body weight of Wt (black squares) and PGC1β-FAT-KO (open circles) male littermates fed a standard chow diet (dashed line) or a high fat diet (solid line) and housed at thermoneutrality. (B) Weight of the major adipose tissue depots from Wt (black bars) and PGC1β-FAT-KO (white bars) fed a standard chow diet or a high fat (HF) diet (Ing.: inguinal; Gon.: gonadal; Ret.: retroperitoneal). (C) Histological sections of inguinal WAT stained with hematoxylin/eosin from mice housed at thermoneutrality and fed a chow diet. (D) Histological sections of interscapular BAT stained with hematoxylin/eosin from mice housed at thermoneutrality and fed a chow diet. Results are expressed as mean±SEM, n=7–9 animals/group, *P≤0.05.
3.2. PGC-1β regulates mitochondrial function in WAT
To identify the genes and cellular processes regulated by PGC-1β in white adipocytes, we used DNA microarrays to compare the gene expression profiles of WAT from Wt and PGC1β-FAT-KO mice. After filtering for non-annotated and redundant genes, we found that a total of 351 genes were differentially regulated (P<0.05) in retroperitoneal WAT of PGC1β-FAT-KO mice compared to Wt littermates. Of these, 133 genes were down-regulated and 218 up-regulated. To analyze the biological function of the differentially expressed genes, we performed a Gene Enrichment Analysis. Interestingly, Gene Ontology (GO) terms significantly over-represented among the down-regulated genes corresponded to categories exclusively related to mitochondrial substrate oxidation and ATP production, including the respiratory chain/oxidative phosphorylation (OxPhos) system and the tricarboxylic acid (TCA) cycle (Table 1). Up to 48% of all down-regulated genes fitted into oxidative pathways. Fewer GO terms with small number of genes in each category and lower statistical significance were associated with the genes up-regulated, providing no insights into pathways up-regulated in the absence of PGC-1β (Table 1).
Table 1.
Gene enrichment analysis of differentially expressed genes in WAT of PGC1β-FAT-KO mice (CC, cellular component; BP, biological process; MF, molecular function).
| Ontology | GO ID | Gene ontology term | # of genes | P value (<1.0E-11) |
|---|---|---|---|---|
| Down-regulated genes | ||||
| CC | GO:0005739 | Mitochondrion | 35 | 1.13E−28 |
| BP | GO:0006091 | Generation of precursor metabolites and energy | 21 | 3.33E−24 |
| CC | GO:0044429 | Mitochondrial part | 24 | 6.77E−24 |
| CC | GO:0005740 | Mitochondrial envelope | 22 | 1.06E−21 |
| CC | GO:0005743 | Mitochondrial inner membrane | 20 | 1.06E−20 |
| CC | GO:0031966 | Mitochondrial membrane | 21 | 1.61E−20 |
| CC | GO:0019866 | Organelle inner membrane | 20 | 2.50E−20 |
| CC | GO:0031967 | Organelle envelope | 22 | 6.03E−20 |
| BP | GO:0045333 | Cellular respiration | 12 | 4.06E−18 |
| CC | GO:0044444 | Cytoplasmic part | 37 | 2.24E−15 |
| BP | GO:0015980 | Energy derivation by oxidation of organic compounds | 12 | 7.47E−15 |
| BP | GO:0006084 | Acetyl-CoA metabolic processes | 9 | 1.31E−13 |
| BP | GO:0022900 | Electron transport chain | 11 | 3.27E−13 |
| BP | GO:0006732 | Coenzyme metabolic process | 12 | 6.70E−13 |
| BP | GO:0006099 | Tricarboxylic acid cycle | 7 | 2.72E−12 |
| BP | GO:0046356 | Acetyl-CoA catabolic process | 7 | 2.72E−12 |
| BP | GO:0051186 | Cofactor metabolic process | 12 | 2.91E−12 |
| BP | GO:0006119 | Oxidative phosphorylation | 8 | 5.04E−12 |
| BP | GO:0009060 | Aerobic respiration | 7 | 5.06E−12 |
| MF | GO:0015078 | Hydrogen ion transmembrane transporter activity | 8 | 5.22E−11 |
| Up-regulated genes | ||||
| CC | GO:0005576 | Extracellular region | 10 | 2.77E−4 |
| BP | GO:0030513 | Positive regulation of BMP signaling pathway | 2 | 1.21E−4 |
| MF | GO:0004181 | Metallocarboxypeptidase activity | 2 | 2.34E−4 |
To confirm the extent to which PGC-1β regulates mitochondrial oxidative pathways in white adipocytes, we first measured mRNA levels of the PGC-1β targets identified in the gene expression profiling study by real-time quantitative PCR. In retroperitoneal WAT of mice kept at thermoneutrality, genes encoding for proteins of the five complexes of the OxPhos system and the TCA cycle were down-regulated by 20–70% in PGC1β-FAT-KO mice (Figure 3A–B). Likewise, genes involved in fatty acid oxidation (FAO), including the nuclear receptor PPARα, were also decreased (Figure 3C). Similar results were observed in inguinal WAT and interscapular BAT (Supplemental Figure A.2).
Figure 3.
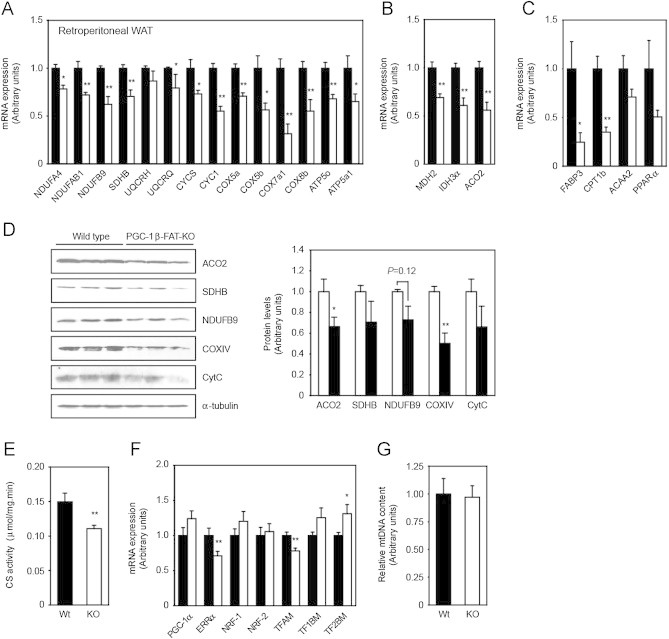
Decreased gene expression and mitochondrial function in WAT of PGC1β-FAT-KO mice. Expression (mRNA) levels of mitochondrial genes involved in oxidative phosphorylation (A), tricarboxylic acid cycle (B) or lipid oxidation (C) in retroperitoneal WAT of Wt (black bars) and PGC1β-FAT-KO littermates (open bars) housed at thermoneutrality and fed a regular chow diet were determined by real-time quantitative RT-PCR. Data are expressed relative to levels in Wt mice. (D) Levels of mitochondrial proteins encoded by PGC-1β target genes were determined by western blot in retroperitoneal WAT of PGC1β-FAT-KO and Wt mice. Protein expression levels normalized by α-tubulin expression were determined with the Image J software. (E) Citrate synthase activity was measured in crude extracts of inguinal WAT as an estimation of mitochondrial oxidative function. (F) mRNA levels of well-established transcriptional regulators of mitochondrial gene expression were assessed in retroperitoneal WAT by real-time quantitative RT-PCR. (G) Relative mitochondrial DNA copy number was determined by real-time quantitative PCR in inguinal WAT. Results are expressed as mean±SEM, n=5–8 animals/group, *P≤0.05 **P≤0.01.
Western blot analysis of protein extracts from retroperitoneal WAT showed that decreased mRNA levels of PGC-1β target genes correlated with lower protein levels (Figure 3D). Furthermore, lack of PGC-1β resulted in a 25–30% decrease in the activity of CS in inguinal WAT (Figure 3E), a widely recognized marker of mitochondrial oxidative function [38]. Impaired mitochondrial gene expression in WAT of PGC1β-FAT-KO mice occurred in the absence of major changes in the expression of other well-established transcriptional regulators of mitochondrial biogenesis, such as PGC-1α, NRF-1 or NRF-2, although a slight decrease in ERRα and TFAM mRNA and a minor increase in TF1BM and TF2BM mRNA levels was observed (Figure 3F). Interestingly, reduction of mitochondrial gene expression or activity in WAT of PGC1β-FAT-KO mice was not accompanied by a reduction in mtDNA content (Figure 3G).
3.3. PGC-1β regulates basal and rosiglitazone-induced mitochondrial gene expression and activity in 3T3-L1 adipocytes
To verify that altered gene expression in WAT of PGC1β-FAT-KO mice was a primary defect due to lack of PGC-1β and not the result of an adaptive process or a developmental defect, we acutely knocked down PGC-1β expression in 3T3-L1 white adipocytes. For this, differentiated 3T3-L1 adipocytes were transfected in suspension with siRNAs specifically targeting PGC-1β or PGC-1α. The use of siRNA against PGC-1β reduced PGC-1β mRNA expression by 60–70% (Figure 4A). Consistent with the in vivo results, knockdown of PGC-1β resulted in a significant decrease in the expression of the genes identified in the microarray study as targets of PGC-1β (Figure 4B). No effect on the expression of these same genes was observed in adipocytes transfected with siRNAs targeting PGC-1α. Furthermore, simultaneous knockdown of PGC-1α and PGC-1β in adipocytes had little or no effect on gene expression compared to cells in which only PGC-1β was knocked down, suggesting that PGC-1β plays a preponderant role in the regulation of mitochondrial genes in white adipocytes. In agreement with a direct role of PGC-1β in the regulation of mitochondrial gene expression in white adipocytes, the adenoviral-mediated overexpression of PGC-1β in 3T3-L1 adipocytes was sufficient to induce the expression of PGC-1β target genes (Figure 4C).
Figure 4.
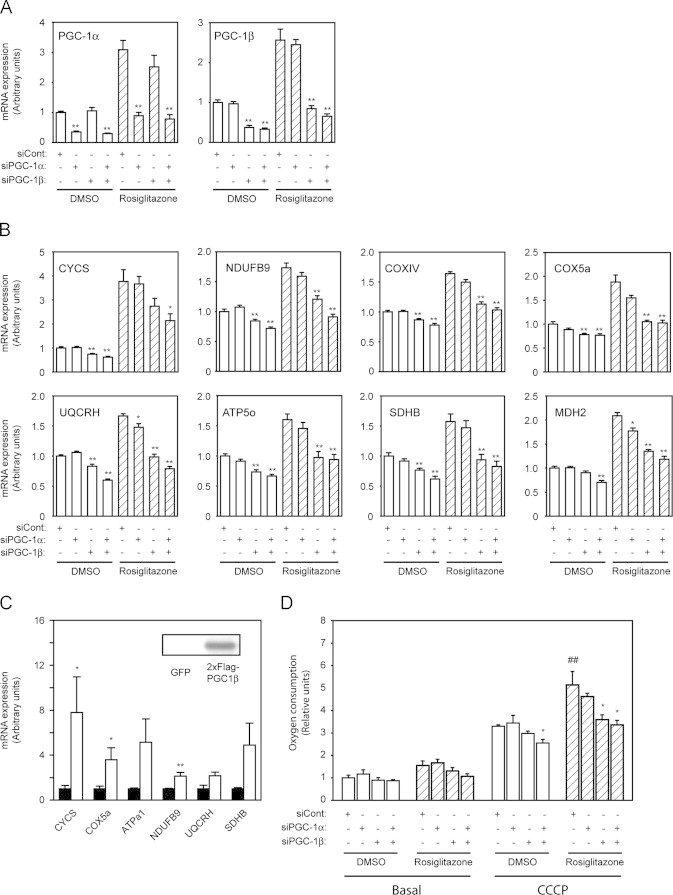
PGC-1β regulates basal and rosiglitazone-induced mitochondrial gene expression and cell respiration in 3T3-L1 adipocytes. 3T3-L1 adipocytes were transfected with siRNAs specifically targeting PGC-1α and/or PGC-1β and then treated with vehicle (open bars) or 1 µM rosiglitazone (hatched bars) for 48 h. Expression of PGC-1α and PGC-1β mRNA (A) and mitochondrial PGC-1β target genes mRNA levels (B) were measured by real-time quantitative RT-PCR. (C) Expression of mitochondrial genes was assessed by real-time quantitative PCR in 3T3-L1 adipocytes transduced with adenoviral vectors to overexpress GFP (black bars) or 2xFlag-PGC-1β (open bars). The box inside the graph indicates the expression level of 2xFlag-PGC1β that was detected by western blot using an specific antibody to detected the Flag antigen. Results are expressed as mean±SEM of 2–3 independent experiments with duplicates. (D) Basal and maximal (CCCP) cell respiration rates were measured in 3T3-L1 adipocytes using a Clark-type oxygen electrode. Results are expressed as mean±SEM of 3–4 independent experiments with triplicates. * Indicates statistical significance of the comparison between control adipocytes (siCont) and adipocytes in which any of the PGC-1s have been knocked down; # indicates the statistical significance of the comparison between vehicle- and rosiglitazone-treated cells.*, #P≤0.05, **,##P≤0.01.
We and others have found that PGC-1β expression is increased in WAT in response to TZD treatment, suggesting that PGC-1β could mediate the effects of TZDs on mitochondrial function in adipocytes [11,34,39]. To address this question, 3T3-L1 adipocytes were transfected with siRNA targeting PGC-1β, PGC-1α or both simultaneously and then treated with rosiglitazone for 48 h. As expected, in control adipocytes, rosiglitazone increased the expression of both PGC-1α and PGC-1β by ∼3-fold, as well as the expression of genes from the OxPhos system and the TCA cycle (Figure 4A–B). However, knockdown of PGC-1β prevented the effect of rosiglitazone on gene expression, while knockdown of PGC-1α had no or little effect (Figure 4B). Furthermore, only knockdown of PGC-1β in cultured adipocytes significantly decreased cell respiratory capacity and prevented the induction in oxygen consumption elicited by rosiglitazone treatment (Figure 4D).
3.4. Effects of lack of PGC-1β in WAT on rosiglitazone-induced expression of mitochondrial genes and insulin sensitivity in vivo
The promotion of mitochondrial oxidative metabolism by TZDs in WAT has been suggested as part of the mechanism by which TZDs enhance insulin sensitivity. To investigate the contribution of PGC-1β to the TZD-induced mitochondrial function and improvement of insulin sensitivity, Wt and PGC1β-FAT-KO mice were fed a HF diet to induce obesity and insulin resistance, and then treated with rosiglitazone. The HF diet resulted in similar body weight gain and adipose tissue accumulation in Wt and PGC1β-FAT-KO mice (Figure 2A–B). Consistent with findings in mice fed a standard chow diet, PGC1β-FAT-KO mice on HF diet and treated with vehicle exhibited a decrease in mitochondrial gene expression, protein levels and CS activity in WAT, compared to Wt (Figure 5A–C). Interestingly, lack of PGC-1β prevented the rosiglitazone-induced expression of mitochondrial genes, rise of mitochondrial protein levels and the increase in CS activity, indicating that PGC-1β mediates the effects of rosiglitazone on mitochondrial gene expression and function in WAT (Figure 5A–C). Of note, the increase in mtDNA content induced by rosiglitazone was not dependent on PGC-1β (Figure 5D).
Figure 5.
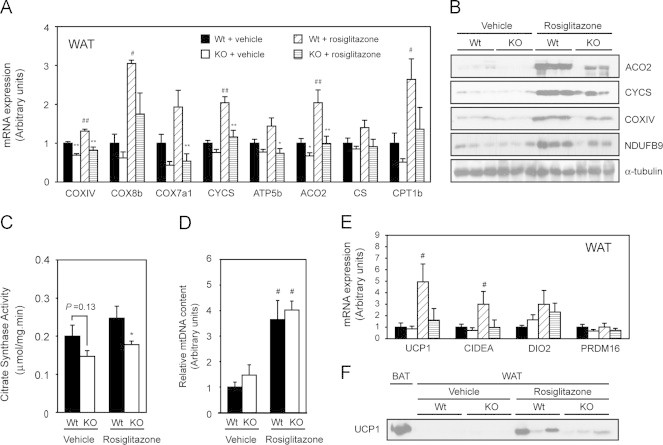
Rosiglitazone-induced expression of mitochondrial genes and mitochondrial function is dependent on PGC-1β. Expression (mRNA) levels of mitochondrial genes (A), protein levels (B), citrate synthase activity (C) and relative mtDNA content (D) were analyzed in retroperitoneal WAT of Wt and PGC1β-FAT-KO mice housed at thermoneutrality that have been subjected to vehicle or rosiglitazone (10 mg/kg) treatment for 15 days after a period of 12 weeks of feeding with a high fat diet. Mice were raised and housed at thermoneutrality (30 °C) throughout the duration of the experiment. Expression of brown adipocyte-specific genes (E) was also determined by real-time quantitative RT-PCR in retroperitoneal WAT. UCP1 protein levels (F) were detected by western blot in retroperitoneal WAT. As a positive control for UCP1 protein expression, a BAT protein extract from Wt mice housed at 21 °C was used. Results are expressed as mean±SEM, n=7–9 animals/group. * Indicates statistical significance of the comparison between Wt and PGC1β-FAT-KO mice; # indicates the statistical significance of the comparison between vehicle- and rosiglitazone-treated groups. *, #P≤0.05; **, ## P≤0.01.
Rosiglitazone and other PPARγ agonists have been shown to induce the expression of brown adipocyte-specific makers, such as UCP1, in WAT. Since PGC-1β, together with PGC-1α, is required for full differentiation of brown adipocytes in BAT [24], we asked if PGC-1β could participate in the recruitment of rosiglitazone-inducible brown adipocytes in WAT. As expected, rosiglitazone treatment increased the expression of UCP1 mRNA and other brown adipocyte markers, like CIDEA or DIO2, in WAT of Wt mice (Figure 5E). However, we observed that the induction by rosiglitazone of UCP1 and CIDEA was blunted in PGC1β-FAT-KO mice. Consistent with the mRNA expression data, the induction by rosiglitazone of UCP1 protein levels in retroperitoneal WAT was reduced in PGC1β-FAT-KO mice (Figure 5F). These results suggest that PGC-1β is required to achieve maximal recruitment of inducible brown adipocytes by rosiglitazone in WAT.
Impaired mitochondrial function in WAT has been linked to increased insulin resistance. To assess if glucose homeostasis was altered in PGC1β-FAT-KO mice we first performed a GTT. For this, mice were fasted overnight to lower circulating glucose and insulin to basal levels and then glucose was administrated intraperitoneally. The clearance of glucose from blood was followed during a 2-h period. Despite the impaired mitochondrial oxidative capacity, mice lacking PGC-1β in adipose tissues exhibited similar degrees of glucose tolerance as Wt, independently of whether they had been fed a high fat diet (Figure 6A) or a regular chow diet (data not shown). To analyze whole body insulin sensitivity, an ITT was performed in mice fasted for 5 h. As shown in Figure 6B, both Wt and PGC1β-FAT-KO mice fed a diabetogenic diet responded to insulin administration by similarly reducing blood glucose levels, indicating a comparable degree of insulin sensitivity. Analogous results were obtained when mice were fed a regular diet (data not shown). Consistent with similar insulin sensitivity, glucose and insulin levels were similar in Wt and PGC1β-FAT-KO mice (Table 2). Moreover, both Wt and PGC1β-FAT-KO mice responded similarly to rosiglitazone treatment, improving whole body glucose tolerance and insulin sensitivity to the same extent (Figure 6A–B), and similarly reducing glucose, insulin and lipid levels (Table 2).
Figure 6.
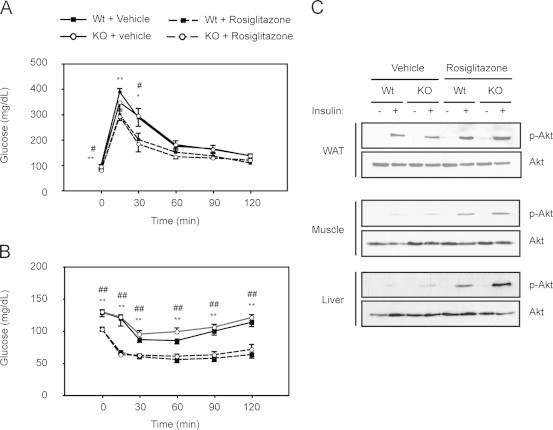
Glucose homeostasis is not altered in PGC1β-FAT-KO mice. (A) Glucose tolerance tests (GTTs) were performed on 12-h fasted mice. Blood glucose levels were measured at 0, 15, 30, 60, 90 and 120 min after an intraperitoneal injection of glucose (2 g/kg). (B) Insulin tolerance tests (ITTs), performed after a 5 h-fast. Glucose levels in blood were measured at 0, 15, 30, 60, 90 and 120 min after an intraperitoneal injection of insulin (0.9 U/kg) (n=6–9 animals/group). (C) Total and phosphorylated Akt were detected by western blot in protein lysates of liver, inguinal WAT and gastrocnemius muscle from mice that were treated with an insulin bolus after an overnight fast. Results are expressed as mean±SEM, n=5–7 animals/group. * Indicates statistical significance of the comparison between vehicle- and rosiglitazone-treated Wt mice; # indicates the statistical significance of the comparison between vehicle- and rosiglitazone-treated PGC1β-FAT-KO mice. *, #P≤0.05; **, ## P≤0.01.
Table 2.
Serum metabolites and hormone levels in Wt and PGC1β-FAT-KO mice fed a high fat diet and treated with rosiglitazone or vehicle for two weeks. Data are presented as mean±SEM; *P≤0.05, **P≤0.01 (* indicate statistical significance of the comparison between vehicle- and rosiglitazone-treated groups).
| Vehicle |
Rosiglitazone |
|||
|---|---|---|---|---|
| Wild type | PGC1β-FAT-KO | Wild type | PGC1β-FAT-KO | |
| Glucose (mg/dl) | 129.7±6.9 | 130.0±3.1 | 103.2±2.5⁎⁎ | 103.0±3.4⁎⁎ |
| FFA (mmol/l) | 0.440±0.05 | 0.446±0.04 | 0.314±0.04 | 0.341±0.05 |
| Triglycerides (mg/dl) | 56.1±3.7 | 54.7±5.3 | 58.1±3.8 | 61.5±3.8 |
| Cholesterol (mg/dl) | 200.0±3.7 | 188.4±6.7 | 162.2±8.1⁎⁎ | 151.0±7.9⁎⁎ |
| Insulin (ng/ml) | 3.34±0.36 | 3.38±0.33 | 1.88±0.20⁎⁎ | 2.13±0.29⁎ |
| Leptin (ng/ml) | 15.4±3.4 | 17.9±2.7 | 13.2±2.2 | 16.2±2.5 |
Even though whole body glucose homeostasis was not altered in PGC1β-FAT-KO mice, we further tested whether impaired mitochondrial function in white adipocytes could affect insulin signaling in different tissues. For this, mice were fasted overnight to reduced endogenous insulin signaling and then administered with an intravenous insulin bolus to activate insulin signaling in peripheral tissues. As shown in Figure 6C, insulin administration similarly stimulated the phosphorylation of Akt in inguinal WAT, liver and skeletal muscle of Wt and PGC1β-FAT-KO mice. As expected, treatment with rosiglitazone notably enhanced insulin-dependent phosphorylation of Akt in all tissues, including WAT, but no significant differences were observed between Wt and PGC1β-FAT-KO mice (Figure 6C).
4. Discussion
Our results provide in vivo evidence that PGC-1β in WAT plays a primary role in the regulation of mitochondrial function by regulating the expression of genes involved in oxidative metabolism. These findings are in agreement with previous studies carried out in a variety of non-adipose cell lines showing that adenoviral overexpression of PGC-1β increases mitochondrial gene expression and oxidative function [40–42]. Also consistent with our results, mice devoid of PGC-1β in all tissues exhibit reduced expression of mitochondrial genes in muscle, liver, heart and BAT that results in an impairment of mitochondrial activity similar to that observed in WAT of PGC1β-FAT-KO mice [21–23].
Proper mitochondrial activity is crucial to maintain WAT function; they play roles in both adipocyte differentiation and key adipocyte metabolic processes, such as lipogenesis and fatty acid re-esterification [6,8]. However, the normal fat accretion and white adipocyte size observed in PGC1β-FAT-KO mice indicate that adipogenesis and lipogenesis occur normally, despite the lack of PGC-1β. These observations are consistent with our gene profiling study in which expression of terminal markers of adipocyte differentiation or genes encoding for proteins involved in lipid synthesis did not appear differentially regulated in WAT of PGC1β-FAT-KO mice. The possibility that some compensatory mechanisms may have been set in WAT of PGC1β-FAT-KO mice in order to maintain lipid and energy homeostasis cannot be ruled out. Several studies have shown that PGC-1β and PGC-1α carry redundant roles regarding the regulation of mitochondrial genes [21–25,43,44]. In addition, it has been shown in cultured myotubes that both PGC-1 coactivators can modulate lipid synthesis [45]. Therefore, it is possible that PGC-1α present in adipocytes can compensate for the loss of PGC-1β in PGC1β-FAT-KO mice, preventing alterations in lipid metabolism and/or adipogenesis. Nevertheless, our results in mice and 3T3-L1 cells clearly show that PGC-1α activity in adipocytes devoid of PGC-1β does not prevent a decline in mitochondrial oxidative function, suggesting that PGC-1β is likely to be more important for mitochondrial gene expression and function than PGC-1α in white adipocytes. In support of this notion, we have recently shown that mice devoid of PGC-1α specifically in adipocytes exhibit normal mitochondrial gene expression and function in WAT [11].
PGC-1α and PGC-1β coregulators have been suggested to modulate gene expression in distinct physiological contexts. Based on its induction by different signals, (e.g. by exercise in muscle, TZDs in WAT, fasting in liver or cold in BAT), PGC-1α is thought to play roles in the adaptation to situations of altered energy demand [46]. Contrary, PGC-1β, which is not induced by classical signals that enhance mitochondrial biogenesis (e.g. exercise or cold), is thought of as a regulator of basal gene expression. However, our studies provide in vivo evidence that PGC-1β is induced by TZDs and is a key mediator of the TZD-induced adaptations in WAT mitochondrial oxidative metabolism. In contrast, mice lacking PGC-1α in WAT respond normally to rosiglitazone by increasing mitochondrial gene expression and function [11]. The mechanism by which TZDs induce Ppargc1b gene transcription appears to involve the binding of ligand-activated PPARγ to functional PPRE sites located in the first intron of the gene [34].
In addition of promoting mitochondrial biogenesis in white adipocytes, TZDs are known to activate the recruitment of inducible brown adipocytes in WAT [10,39,47,48], through mechanisms that strongly depend on PGC-1α and PRDM16 [11,49]. Our results indicate that PGC-1β also contributes to the induction of UCP1 and other brown-fat specific markers in WAT in response to TZD treatment, even in the absence of adrenergic stimulation, since our studies have been conducted at thermoneutrality. The requirement of both PGC-1α and PGC-1β for the full induction of brown adipoyte-specific markers by TZDs in WAT is consistent with their complementary role in BAT differentiation [24].
Numerous studies have shown correlations between mitochondrial function and insulin sensitivity. Indeed, impaired mitochondrial gene expression and oxidative capacity in skeletal muscle and WAT has been observed in humans with insulin resistance or type 2 diabetes, as well as in rodent models for the disease [10,39,50–52]. This has led to the notion that decreased mitochondrial function could be an underlying cause of the development of insulin resistance. A reduction in the capacity of mitochondria to oxidize lipids has been suggested to contribute to the intracellular accumulation of lipid intermediates, such as diacylglycerols and ceramides, which suppress insulin signaling by activating novel protein kinases C (nPKC) [7]. An impaired mitochondrial function has also been proposed to compromise the endocrine and lipogenic functions of white adipocytes, and by this to contribute to the development of systemic insulin resistance [5], [6]. Our findings in PGC1β-FAT-KO mice show that decreased oxidative capacity of adipocyte mitochondria is not sufficient for insulin resistance to develop. Although it could be claimed that, as it occurs for most mitochondrial pathologies, a critical mitochondrial dysfunction threshold has not been reached for insulin resistance to appear, it has to be noted that the 25–30% decrease in mitochondrial activity found in WAT of PGC1β-FAT-KO mice is within the range reported in patients with insulin resistance [53], [54]. Our findings join those by other authors using genetically-engineered mouse models with global or tissue-specific impairment of mitochondrial oxidative capacity to support the lack of a causative role of mitochondrial dysfunction in the onset of insulin resistance [43,44,55–57]. Furthermore, although the insulin sensitizing effects of TZDs and other PPARγ agonists have been linked to their capacity to promote oxidative metabolism in WAT by efficiently increasing expression of mitochondrial genes [8,9], our results suggest that increased mitochondrial gene expression and oxidative metabolism in WAT is not required for the insulin sensitizing capacity of TZDs. Of note, rosiglitazone treatment also induced PGC-1α expression and, consequently, a partial compensation by this coactivator cannot be excluded. Therefore, studies of adipose-specific PGC-1α/β double knockout mice will be required to test such compensation.
In conclusion, the present study demonstrates that PGC-1β regulates mitochondrial gene expression in WAT, and provides first in vivo evidence that this coactivator is important for the induction of genes involved in mitochondrial oxidative metabolism by TZDs. Furthermore, we have shown that improvement of mitochondrial oxidative capacity in WAT after treatment with rosiglitazone is not essential for the full insulin-sensitizing effects of TZDs. Notably, our results support a dissociation between mitochondrial dysfunction in WAT and the development of insulin resistance.
Conflict of interest
None.
Acknowledgments
The authors thank L. Dolfini, J. Cardenas and B. Hazen for the generation of targeting vector, ES cells screening and mouse colony management and S. Schenk for technical advice for the analysis of the insulin signaling pathway. This work was supported by grants SAF2008-03644, SAF2011-23886 and RYC-2006-002622 from Ministerio de Ciencia e Innovación (MICINN, Spain) to J.A.V. and R01DK95686 to A.K. J.A.V is a researcher of the “Ramon y Cajal” program (MICINN). N.E. is a FPI predoctoral fellow (BES-2009-013411) from MICINN.
Appendix A. Supporting information
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.molmet.2013.05.004.
Appendix A. Supporting information
Supplementary Figure A.1.
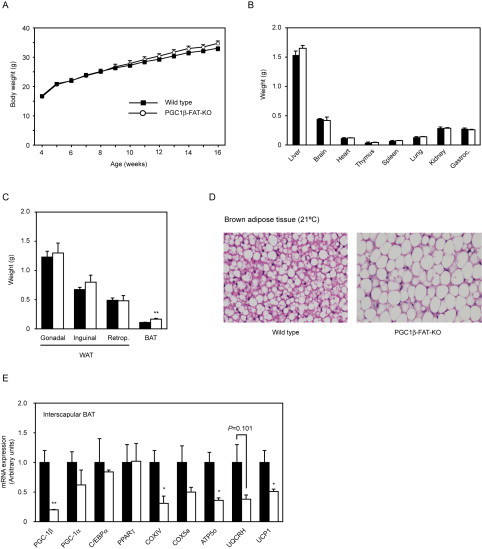
Analysis of PGC1β-FAT-KO mice housed at 21 °C. (A) Body weight of Wt and PGC1β-FAT-KO mice fed a regular chow diet and raised at 21 °C. (B) Weight of major organs of Wt (black bars) and PGC1β-FAT-KO (open bars). (C) Weight of major adipose tissue depots (retrop., retroperitoneal). (D) Histological sections of interscapular BAT of Wt and PGC1β-FAT-KO mice housed at 21 °C. (E) Gene expression in interscapular BAT was assessed by real-time quantitative PCR.
Supplementary Figure A.2.
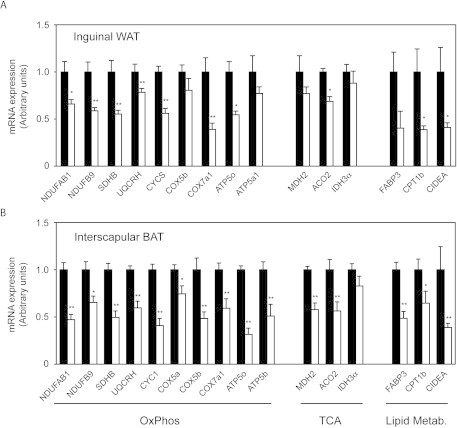
Decreased mitochondrial gene expression in inguinal WAT and interscapular BAT of PGC1β-FAT-KO mice. Expression (mRNA) levels of mitochondrial genes involved in oxidative phosphorylation (OxPhos), tricarboxylic acid cycle (TCA) or lipid metabolism in inguinal WAT (A) and interscapular BAT (B) of Wt (black bars) and PGC1β-FAT-KO mice (open bars) housed at thermoneutrality and fed a regular chow diet were determined by real-time quantitative RT-PCR. Data are expressed relative to levels in Wt mice. Results are expressed as mean±SEM, n=5–8 animals/group, *P≤0.05 **P≤0.01.
References
- 1.Guilherme A., Virbasius J.V., Puri V., Czech M.P. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nature Reviews Molecular Cell Biology. 2008;9(5):367–377. doi: 10.1038/nrm2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosen E.D., Spiegelman B.M. Molecular regulation of adipogenesis. Annual Review of Cell and developmental Biology. 2000;16:145–171. doi: 10.1146/annurev.cellbio.16.1.145. [DOI] [PubMed] [Google Scholar]
- 3.Rosen E.D., Sarraf P., Troy A.E., Bradwin G., Moore K., Milstone D.S. PPAR gamma is required for the differentiation of adipose tissue in vivo and in vitro. Molecular Cell. 1999;4(4):611–617. doi: 10.1016/s1097-2765(00)80211-7. [DOI] [PubMed] [Google Scholar]
- 4.Rosen E.D., Spiegelman B.M. PPARgamma: a nuclear regulator of metabolism, differentiation, and cell growth. Journal of Biological Chemistry. 2001;276(41):37731–37734. doi: 10.1074/jbc.R100034200. [DOI] [PubMed] [Google Scholar]
- 5.De Pauw A., Tejerina S., Raes M., Keijer J., Arnould T. Mitochondrial (dys)function in adipocyte (de)differentiation and systemic metabolic alterations. American Journal of Pathology. 2009;175(3):927–939. doi: 10.2353/ajpath.2009.081155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kusminski C.M., Scherer P.E. Mitochondrial dysfunction in white adipose tissue. Trends in Endocrinology & Metabolism. 2012;23(9):435–443. doi: 10.1016/j.tem.2012.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Samuel V.T., Shulman G.I. Mechanisms for insulin resistance: common threads and missing links. Cell. 2012;148(5):852–871. doi: 10.1016/j.cell.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wilson-Fritch L., Nicoloro S., Chouinard M., Lazar M.A., Chui P.C., Leszyk J. Mitochondrial remodeling in adipose tissue associated with obesity and treatment with rosiglitazone. Journal of Clinical Investigation. 2004;114(9):1281–1289. doi: 10.1172/JCI21752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boden G., Homko C., Mozzoli M., Showe L.C., Nichols C., Cheung P. Thiazolidinediones upregulate fatty acid uptake and oxidation in adipose tissue of diabetic patients. Diabetes. 2005;54(3):880–885. doi: 10.2337/diabetes.54.3.880. [DOI] [PubMed] [Google Scholar]
- 10.Bogacka I., Xie H., Bray G.A., Smith S.R. Pioglitazone induces mitochondrial biogenesis in human subcutaneous adipose tissue in vivo. Diabetes. 2005;54(5):1392–1399. doi: 10.2337/diabetes.54.5.1392. [DOI] [PubMed] [Google Scholar]
- 11.Pardo R., Enguix N., Lasheras J., Feliu J.E., Kralli A., Villena J.A. Rosiglitazone-induced mitochondrial biogenesis in white adipose tissue is independent of peroxisome proliferator-activated receptor gamma coactivator-1alpha. PLoS One. 2011;6(11):e26989. doi: 10.1371/journal.pone.0026989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ge K., Guermah M., Yuan C.X., Ito M., Wallberg A.E., Spiegelman B.M. Transcription coactivator TRAP220 is required for PPAR gamma 2-stimulated adipogenesis. Nature. 2002;417(6888):563–567. doi: 10.1038/417563a. [DOI] [PubMed] [Google Scholar]
- 13.Yamauchi T., Oike Y., Kamon J., Waki H., Komeda K., Tsuchida A. Increased insulin sensitivity despite lipodystrophy in Crebbp heterozygous mice. Nature Genetics. 2002;30(2):221–226. doi: 10.1038/ng829. [DOI] [PubMed] [Google Scholar]
- 14.Louet J.F., Coste A., Amazit L., Tannour-Louet M., Wu R.C., Tsai S.Y. Oncogenic steroid receptor coactivator-3 is a key regulator of the white adipogenic program. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(47):17868–17873. doi: 10.1073/pnas.0608711103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Picard F., Kurtev M., Chung N., Topark-Ngarm A., Senawong T., Machado De Oliveira R. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature. 2004;429(6993):771–776. doi: 10.1038/nature02583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu C., Markan K., Temple K.A., Deplewski D., Brady M.J., Cohen R.N. The nuclear receptor corepressors NCoR and SMRT decrease peroxisome proliferator-activated receptor gamma transcriptional activity and repress 3T3-L1 adipogenesis. Journal of Biological Chemistry. 2005;280(14):13600–13605. doi: 10.1074/jbc.M409468200. [DOI] [PubMed] [Google Scholar]
- 17.Puigserver P., Wu Z., Park C.W., Graves R., Wright M., Spiegelman B.M. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92(6):829–839. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- 18.Puigserver P., Spiegelman B.M. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): transcriptional coactivator and metabolic regulator. Endocrine Reviews. 2003;24(1):78–90. doi: 10.1210/er.2002-0012. [DOI] [PubMed] [Google Scholar]
- 19.Lin J., Puigserver P., Donovan J., Tarr P., Spiegelman B.M. Peroxisome proliferator-activated receptor gamma coactivator 1beta (PGC-1beta ), a novel PGC-1-related transcription coactivator associated with host cell factor. Journal of Biological Chemistry. 2002;277(3):1645–1648. doi: 10.1074/jbc.C100631200. [DOI] [PubMed] [Google Scholar]
- 20.Kressler D., Schreiber S.N., Knutti D., Kralli A. The PGC-1-related protein PERC is a selective coactivator of estrogen receptor alpha. Journal of Biological Chemistry. 2002;277(16):13918–13925. doi: 10.1074/jbc.M201134200. [DOI] [PubMed] [Google Scholar]
- 21.Vianna C.R., Huntgeburth M., Coppari R., Choi C.S., Lin J., Krauss S. Hypomorphic mutation of PGC-1beta causes mitochondrial dysfunction and liver insulin resistance. Cell Metabolism. 2006;4(6):453–464. doi: 10.1016/j.cmet.2006.11.00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lelliott C.J., Medina-Gomez G., Petrovic N., Kis A., Feldmann H.M., Bjursell M. Ablation of PGC-1beta results in defective mitochondrial activity, thermogenesis, hepatic function, and cardiac performance. PLoS Biology. 2006;4(11):e369. doi: 10.1371/journal.pbio.0040369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sonoda J., Mehl I.R., Chong L.W., Nofsinger R.R., Evans R.M. PGC-1beta controls mitochondrial metabolism to modulate circadian activity, adaptive thermogenesis, and hepatic steatosis. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(12):5223–5228. doi: 10.1073/pnas.0611623104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Uldry M., Yang W., St-Pierre J., Lin J., Seale P., Spiegelman B.M. Complementary action of the PGC-1 coactivators in mitochondrial biogenesis and brown fat differentiation. Cell Metabolism. 2006;3(5):333–341. doi: 10.1016/j.cmet.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 25.Lai L., Leone T.C., Zechner C., Schaeffer P.J., Kelly S.M., Flanagan D.P. Transcriptional coactivators PGC-1alpha and PGC-lbeta control overlapping programs required for perinatal maturation of the heart. Genes & Development. 2008;22(14):1948–1961. doi: 10.1101/gad.1661708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoon J.C., Puigserver P., Chen G., Donovan J., Wu Z., Rhee J. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature. 2001;413(6852):131–138. doi: 10.1038/35093050. [DOI] [PubMed] [Google Scholar]
- 27.Lin J., Yang R., Tarr P.T., Wu P.H., Handschin C., Li S. Hyperlipidemic effects of dietary saturated fats mediated through PGC-1beta coactivation of SREBP. Cell. 2005;120(2):261–273. doi: 10.1016/j.cell.2004.11.043. [DOI] [PubMed] [Google Scholar]
- 28.Wolfrum C., Stoffel M. Coactivation of Foxa2 through Pgc-1beta promotes liver fatty acid oxidation and triglyceride/VLDL secretion. Cell Metabolism. 2006;3(2):99–110. doi: 10.1016/j.cmet.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 29.Villena J.A., Roy S., Sarkadi-Nagy E., Kim K.H., Sul H.S. Desnutrin, an adipocyte gene encoding a novel patatin domain-containing protein, is induced by fasting and glucocorticoids: ectopic expression of desnutrin increases triglyceride hydrolysis. Journal of Biological Chemistry. 2004;279(45):47066–47075. doi: 10.1074/jbc.M403855200. [DOI] [PubMed] [Google Scholar]
- 30.Kilroy G., Burk D.H., Floyd Z.E. High efficiency lipid-based siRNA transfection of adipocytes in suspension. PLoS One. 2009;4(9):e6940. doi: 10.1371/journal.pone.0006940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schreiber S.N., Knutti D., Brogli K., Uhlmann T., Kralli A. The transcriptional coactivator PGC-1 regulates the expression and activity of the orphan nuclear receptor estrogen-related receptor alpha (ERRalpha) Journal of Biological Chemistry. 2003;278(11):9013–9018. doi: 10.1074/jbc.M212923200. [DOI] [PubMed] [Google Scholar]
- 32.Villena J.A., Hock M.B., Chang W.Y., Barcas J.E., Giguere V., Kralli A. Orphan nuclear receptor estrogen-related receptor alpha is essential for adaptive thermogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(4):1418–1423. doi: 10.1073/pnas.0607696104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Srere P.A. Methods in Enzymology. 1969;13:3–11. doi: 10.1016/s0076-6879(76)44004-1. [DOI] [PubMed] [Google Scholar]
- 34.Deng T., Sieglaff D.H., Zhang A., Lyon C.J., Ayers S.D., Cvoro A. A peroxisome proliferator-activated receptor gamma (PPARgamma)/PPARgamma coactivator 1beta autoregulatory loop in adipocyte mitochondrial function. Journal of Biological Chemistry. 2011;286(35):30723–30731. doi: 10.1074/jbc.M111.251926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kamei Y., Ohizumi H., Fujitani Y., Nemoto T., Tanaka T., Takahashi N. PPARgamma coactivator 1beta/ERR ligand 1 is an ERR protein ligand, whose expression induces a high-energy expenditure and antagonizes obesity. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(21):12378–12383. doi: 10.1073/pnas.2135217100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martens K., Bottelbergs A., Baes M. Ectopic recombination in the central and peripheral nervous system by aP2/FABP4-Cre mice: implications for metabolism research. FEBS Letters. 2010;584(5):1054–1058. doi: 10.1016/j.febslet.2010.01.061. [DOI] [PubMed] [Google Scholar]
- 37.Lodhi I.J., Semenkovich C.F. Why we should put clothes on mice. Cell Metabolism. 2009;9(2):111–112. doi: 10.1016/j.cmet.2009.01.004. [DOI] [PubMed] [Google Scholar]
- 38.Wang H., Hiatt W.R., Barstow T.J., Brass E.P. Relationships between muscle mitochondrial DNA content, mitochondrial enzyme activity and oxidative capacity in man: alterations with disease. European Journal of Applied Physiology and Occupational Physiology. 1999;80(1):22–27. doi: 10.1007/s004210050553. [DOI] [PubMed] [Google Scholar]
- 39.Rong J.X., Qiu Y., Hansen M.K., Zhu L., Zhang V., Xie M. Adipose mitochondrial biogenesis is suppressed in db/db and high-fat diet-fed mice and improved by rosiglitazone. Diabetes. 2007;56(7):1751–1760. doi: 10.2337/db06-1135. [DOI] [PubMed] [Google Scholar]
- 40.Lin J., Tarr P.T., Yang R., Rhee J., Puigserver P., Newgard C.B. PGC-1beta in the regulation of hepatic glucose and energy metabolism. Journal of Biological Chemistry. 2003;278(33):30843–30848. doi: 10.1074/jbc.M303643200. [DOI] [PubMed] [Google Scholar]
- 41.Shao D., Liu Y., Liu X., Zhu L., Cui Y., Cui A. PGC-1 beta-regulated mitochondrial biogenesis and function in myotubes is mediated by NRF-1 and ERR alpha. Mitochondrion. 2010;10(5):516–527. doi: 10.1016/j.mito.2010.05.012. [DOI] [PubMed] [Google Scholar]
- 42.Gao C.L., Liu G.L., Liu S., Chen X.H., Ji C.B., Zhang C.M. Overexpression of PGC-1beta improves insulin sensitivity and mitochondrial function in 3T3-L1 adipocytes. Molecular and Cellular Biochemistry. 2011;353(1-2):215–223. doi: 10.1007/s11010-011-0789-2. [DOI] [PubMed] [Google Scholar]
- 43.Lin J., Wu P.H., Tarr P.T., Lindenberg K.S., St-Pierre J., Zhang C.Y. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell. 2004;119(1):121–135. doi: 10.1016/j.cell.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 44.Leone T.C., Lehman J.J., Finck B.N., Schaeffer P.J., Wende A.R., Boudina S. PGC-1alpha deficiency causes multi-system energy metabolic derangements: muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Biology. 2005;3(4):e101. doi: 10.1371/journal.pbio.0030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Espinoza D.O., Boros L.G., Crunkhorn S., Gami H., Patti M.E. Dual modulation of both lipid oxidation and synthesis by peroxisome proliferator-activated receptor-gamma coactivator-1alpha and -1beta in cultured myotubes. FASEB Journal. 2010;24(4):1003–1014. doi: 10.1096/fj.09-133728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hock M.B., Kralli A. Transcriptional control of mitochondrial biogenesis and function. Annual Review of Physiology. 2009;71:177–203. doi: 10.1146/annurev.physiol.010908.163119. [DOI] [PubMed] [Google Scholar]
- 47.Petrovic N., Walden T.B., Shabalina I.G., Timmons J.A., Cannon B., Nedergaard J. Chronic peroxisome proliferator-activated receptor gamma (PPARgamma) activation of epididymally derived white adipocyte cultures reveals a population of thermogenically competent, UCP1-containing adipocytes molecularly distinct from classic brown adipocytes. Journal of Biological Chemistry. 2010;285(10):7153–7164. doi: 10.1074/jbc.M109.053942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sell H., Berger J.P., Samson P., Castriota G., Lalonde J., Deshaies Y. Peroxisome proliferator-activated receptor gamma agonism increases the capacity for sympathetically mediated thermogenesis in lean and ob/ob mice. Endocrinology. 2004;145(8):3925–3934. doi: 10.1210/en.2004-0321. [DOI] [PubMed] [Google Scholar]
- 49.Ohno H., Shinoda K., Spiegelman B.M., Kajimura S. PPARgamma agonists induce a white-to-brown fat conversion through stabilization of PRDM16 protein. Cell Metabolism. 2012;15(3):395–404. doi: 10.1016/j.cmet.2012.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kelley D.E., He J., Menshikova E.V., Ritov V.B. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes. 2002;51(10):2944–2950. doi: 10.2337/diabetes.51.10.2944. [DOI] [PubMed] [Google Scholar]
- 51.Mootha V.K., Lindgren C.M., Eriksson K.F., Subramanian A., Sihag S., Lehar J. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nature Genetics. 2003;34(3):267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 52.Patti M.E., Butte A.J., Crunkhorn S., Cusi K., Berria R., Kashyap S. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: potential role of PGC1 and NRF1. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(14):8466–8471. doi: 10.1073/pnas.1032913100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Petersen K.F., Dufour S., Befroy D., Garcia R., Shulman G.I. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. New England Journal of Medicine. 2004;350(7):664–671. doi: 10.1056/NEJMoa031314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Befroy D.E., Petersen K.F., Dufour S., Mason G.F., de Graaf R.A., Rothman D.L. Impaired mitochondrial substrate oxidation in muscle of insulin-resistant offspring of type 2 diabetic patients. Diabetes. 2007;56(5):1376–1381. doi: 10.2337/db06-0783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wredenberg A., Freyer C., Sandstrom M.E., Katz A., Wibom R., Westerblad H. Respiratory chain dysfunction in skeletal muscle does not cause insulin resistance. Biochemical and Biophysical Research Communications. 2006;350(1):202–207. doi: 10.1016/j.bbrc.2006.09.029. [DOI] [PubMed] [Google Scholar]
- 56.Pospisilik J.A., Knauf C., Joza N., Benit P., Orthofer M., Cani P.D. Targeted deletion of AIF decreases mitochondrial oxidative phosphorylation and protects from obesity and diabetes. Cell. 2007;131(3):476–491. doi: 10.1016/j.cell.2007.08.047. [DOI] [PubMed] [Google Scholar]
- 57.Vernochet C., Mourier A., Bezy O., Macotela Y., Boucher J., Rardin M.J. Adipose-specific deletion of TFAM increases mitochondrial oxidation and protects mice against obesity and insulin resistance. Cell Metabolism. 2012;16(6):765–776. doi: 10.1016/j.cmet.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]