

Abstract
Oxidation of LDL (oxLDL) is a crucial step in the development of cardiovascular disease. Treatment with antibodies directed against oxLDL can reduce atherosclerosis in rodent models through unknown mechanisms. We demonstrate that through a novel mechanism of immune complex formation and Fc-γ receptor (FcγR) engagement, antibodies targeting oxLDL (MLDL1278a) are anti-inflammatory on innate immune cells via modulation of Syk, p38 MAPK phosphorylation and NFκB activity. Subsequent administration of MLDL1278a in diet-induced obese (DIO) nonhuman primates (NHP) resulted in a significant decrease in pro-inflammatory cytokines and improved overall immune cell function. Importantly, MLDL1278a treatment improved insulin sensitivity independent of body weight change. This study demonstrates a novel mechanism by which an anti-oxLDL antibody improves immune function and insulin sensitivity independent of internalization of oxLDL. This identifies MLDL1278a as a potential therapy for reducing vascular inflammation in diabetic conditions.
Keywords: Nonhuman primate, Obesity, Atherosclerosis, ox-LDL, Inflammation, Diabetes
1. Introduction
Obesity is a major risk factor for the development of metabolic syndrome, which is comprised of pathophysiological conditions such as hypertension, hyperlipidemia, and insulin resistance [1]. Epidemiological studies have shown that metabolic syndrome greatly increases the risk for associated diseases such as type 2 diabetes mellitus (T2DM) and cardiovascular disease (CVD) [2,3]. A recent report examining data in the Framingham Heart Offspring Study demonstrated that, on average, metabolic syndrome increases the relative risk of CVD by 2.5 fold [4]. It is now well accepted that insulin resistance is a major risk factor for the development of cardiovascular complications like atherosclerosis, which account for the majority of the mortality and morbidity associated with metabolic syndrome [5–8].
The progression of atherogenesis has been well described and is comprised of several processes. LDL deposition and oxidation within vasculature is an initial step and oxidized LDL (oxLDL) activates endothelial cells and attracts monocyte infiltration into the subendothelial space. These events result in activation of macrophages, the generation of foam cells, and, ultimately, the formation of atherogenic plaques. Besides their role in plaque development, destabilization, and rupture [9], pro-inflammatory macrophages, in particular those dependent on the MCP-1/CCR2 chemokine-receptor axis, have been implicated in the development of metabolic syndrome [10–16]. Similarly, there is increasing evidence that oxLDL is involved not only in the pathogenesis of atherosclerosis and CVD, but also in the development of adiposity, insulin resistance, and even T2DM [17,18].
Native LDL, a lipoprotein that consists of phospholipids, cholesterol, cholesterol esters, triglycerides, and apolipoprotein B (apoB), circulates through the blood stream and shuttles lipid particles. The LDL-receptor mediates cellular uptake of native LDL. Oxidized LDL, however, is recognized by scavenging receptors, such as CD36 [19], present on macrophages and endothelial cells, which directly results in the formation of foam cells [20]. In addition, oxLDL itself is pro-atherogenic through its ability to act as a chemoattractant for macrophages [21,22], and induce the release of MCP-1 from endothelial cells and macrophages [23,24]. OxLDL is also very immunogenic as some people develop autoantibodies to oxLDL. Interestingly, several studies have suggested that increased oxLDL autoantibody levels might be protective against the development of cardiovascular disease [25–27] as well as metabolic disease [28].
Previous studies by Schiopu et al. have demonstrated that human oxLDL antibodies administered to atherosclerosis-prone apoE−/− mice can reduce the formation of atherosclerotic plaque [29], and, in the apoE−/− LDLR−/− mice, this antibody reduces plaque macrophage content and leads to plaque regression [30]. These same oxLDL antibodies potently inhibited release of the atherogenic M1 macrophage-tropic chemokine (MCP-1) from human monocytes and monocyte-derived macrophages cultured in the presence of oxLDL in vitro [31]. Together, these observations point to targeted inhibition of macrophage recruitment and inflammatory activity being important mechanisms underlying the anti-atherogenic activity of the anti-oxLDL antibody, but do not assess the molecular mechanisms through which the anti-oxLDL antibody inhibits inflammatory responses in isolated monocytes. Given that obesity and the metabolic syndrome are considered an inflammatory condition [32] involving both the adaptive and innate immune systems, further testing this anti-oxLDL antibody in a model of insulin resistance could provide insight into the possible contribution of inflammation through oxLDL to disrupt glucose homeostasis.
The role of the innate immune system in the development of atherosclerosis and insulin resistance has been well documented [32]. Recent work has reported a contribution of the adaptive immune system to both of these disease processes as well. T-cells in the atherosclerotic plaque [33,34] as well as in adipose tissue [35] are known to contribute to the development of atherosclerosis and insulin resistance, respectively. Thus, it is plausible that the underlying mechanisms for activating T-cells in both of these diseases are overlapping. For instance, adiponectin has been demonstrated to elicit a pro-inflammatory response in both human macrophages and CD4 positive T-cells [36]. Further, a recent study demonstrated that ablation of T-bet, a transcription factor important for proper CD4 positive T-cell lineage development, resulted in improved glucose tolerance in mice fed a high fat diet [37] by reducing the number of activated CD4+ T-cells. Given that activated T-cells are associated with the development of atherosclerosis [37], these findings would suggest that improvements in the immune system, adaptive or innate, could have a beneficial outcome on metabolism as well as atherosclerosis.
Here, we characterize the anti-oxLDL molecular mechanism of action, and assess its effects on metabolic and immunological function in a unique nonhuman primate model of diet-induced obesity (DIO-NHP). Unexpectedly, we demonstrate that the inhibitory effect of anti-oxLDL on the monocyte-mediated inflammation response is mediated through immune complex formation, and this effect is dependent on the antibody Fc fragment and FcγRII on responsive cells to transduce inhibitory intra-cellular signaling. We then confirm the anti-inflammatory action of anti-oxLDL in the DIO-NHP model and clearly demonstrate that treatment with anti-oxLDL in DIO-NHP decreased systemic pro-inflammatory markers, improved adaptive immunity and improved insulin sensitivity.
2. Materials and methods
2.1. oxLDL antibody
MLDL1278a is a fully human recombinant monoclonal antibody based on a human IgG1 framework directed against a specific oxidized low-density lipoprotein (oxLDL) epitope (malondialdehyde [MDA]-modified human ApoB-100). A high-affinity human IgG1 antibody was then developed with specificity to both MDA-LDL and the MDA-ApoB-100 epitope. Biacore analyses showed that this antibody binds to human MDA LDL (Academy Biomedical, Houston, TX) with high affinity, with a KD-value of 8±6 nM [29]. The binding epitope corresponds to the human ApoB100 (amino acids 661–680). This region is completely conserved in the Rhesus macaque (ApoB100 amino acids 684–703).
2.2. Monocyte isolation and culture
Blood samples were obtained from healthy subjects under approved protocols. Monocytes were isolated using CD14 microbeads (Miltenyi Biotec) from purified PBMC (Ficoll-Paque Plus, Amersham Biosciences, Piscataway, NJ), according to the manufacturer's instructions. Freshly isolated CD14-positive monocytes were resuspended in RPMI 1640 medium (Invitrogen, Carlsbad, CA) supplemented with 10% human AB-positive serum containing oxidized LDL (Sigma-Aldrich, St. Louis, MO), penicillin, and streptomycin. Cells were sown into 96-well high binding ELISA plates (Corning, Tewksbury, MA) pre-coated with 3 μg/ml MDA-LDL, at a density of 7.5×105 cells/ml. Lipopolysaccharide (LPS, Sigma-Aldrich, St. Louis, MO) at 0.3 ng/ml was added to the culturing medium to stimulate cytokine secretion from these cells, and the cells were concurrently treated with MLDL1278a or control IgG1. Function-blocking mouse IgG1 anti-human FcγRI, FcγRII and FcγRIII monoclonal antibodies were from AbD Serotech (Dusseldorf, Germany). TLR activators Pam3Csk4, R837, polyIc were purchased from InvivoGen (San Diego, CA), IL-1β and GM-CSF were from R&D systems (Minneapolis, MN). Culture media were collected 40 h after treatment to test MCP-1 concentration.
2.3. Cytokine measurements and antibody binding ELISA assays
MCP-1 protein was quantified in cell culture media using a commercially available ELISA kit (R&D Systems, Minneapolis, MN). Supernatants harvested at completion of the cell culture were saved and stored at −80 °C until the start of analysis. Cytokines and biomarker levels in both monocyte culturing medium and monkey plasma were determined using a commercially available high-throughput assay (Myriad RBM, Austin, TX). Binding of MLDL1278a and its mutants to immobilized LDL or MDL-LDL was tested with an ELISA assay as described previously [29].
2.4. Immunoblot analysis
Whole-cell lysates from primary human monocytes were extracted with RIPA buffer (150 mM NaCl, 1.0% IGEPAL CA-630, 0.5% [v/v] sodium deoxycholate, 0.1% [w/v] SDS, and 50 mM Tris–chloride, pH 8.0). After centrifugation to remove cell debris, aliquots of the 20,000 g supernatant were subjected to 10% SDS/PAGE, after which the proteins were transferred to Hybond-C extra nitrocellulose filters (Amersham Biosciences, Piscataway, NJ). The filters were incubated at room temperature with primary antibodies. Bound antibodies were visualized by chemiluminescence (Super Signal Substrate; Thermo Fisher Scientific, Waltham, MA) using a 1:5000 dilution of donkey anti-rabbit IgG, or donkey anti-mouse IgG (Jackson ImmunoResearch, West Grove, PA), conjugated to horseradish peroxidase. Filters were exposed to Kodak X-Omat Blue XB-1 film at room temperature for 1–60 s. Blotting intensities were quantified by ImageJ. All primary antibodies (purified commercially by protein A and peptide affinity chromatography) were obtained from Cell Signaling Technology (Danvers, MA).
2.5. Animals
The experimental group consisted of 6 adult monkeys (age 9–12) that have been exposed to a high-fat diet for 5 years, weighing from 14.4 to 21 kg at the start of the study. In addition, 20 control animals maintained on standard chow diet were analyzed for most of the measurements and experiments. All animal care and procedures were done according to the IACUC at the Oregon National Primate Research Center (ONPRC) at Oregon Health & Science University (OHSU). For all studies, food intake was carefully recorded every day and water was provided ad libitum. Lights were on from 7am–7pm.
2.6. Animal diet
Animals were fed a diet high in sucrose and fat, manufactured by Test Diet (Diet 5A1F, 31.6% of calories from fat, 52.3% calories from carbohydrates, 16.1% from protein; cholesterol 946 ppm; Purina Mills, Inc., St. Louis, MO). Twenty high-fat diet biscuits were provided to each animal and food intake was documented at the end of the day. The animals did not have access to food overnight. Animals on the high-fat diet were supplemented with 500 ml of Kool-Aid+20% fructose three times a week (HFFD). The control animals were fed regular chow (Test Diet 5049, 13.9% of calories from fat, 59.6% calories from carbohydrates, 26.5% from protein; Purina Mills, Inc.).
2.7. Animal study experimental design
The experiment was divided in three phases. In Phase I, or baseline period, animals were maintained on the HFFD, and food intake and fructose consumption were recorded on a daily basis. One week prior to the start of the MLDL1278a treatment, the animals were sedated with Telazol, and an intravenous glucose tolerance test (IVGTT) was performed to obtain baseline information on the glucose homeostasis and insulin resistance of the animals. In addition, the animals were analyzed for body composition using a DEXA scanner (Hologic Discovery A, Hologic, Inc., Bedford, MA) while the animals were still sedated. Blood samples were collected while the animals were sedated, immediately prior to the start of the IVGTT.
During treatment, MLDL1278a was administered at 10 mg/kg once a week intravenously for a total of 12 weeks, meaning all animals received a total of 13 doses. This dose was based on previous studies in mice (0.5–1 mg per mouse; [37]), taking into account that the Rhesus macaque has a slower clearance than mice. Three days after the final dose was administered, the animals underwent another DEXA scanning and an IVGTT. In addition, blood was collected for cytokine and metabolite measurements. During the washout period, animals received no drug. Food intake and fructose consumption were monitored daily. At the end of 12 weeks of washout, the animals underwent a final DEXA scan, IVGTT, and blood sample collection.
2.8. Intravenous glucose tolerance test
Intravenous glucose tolerance tests were performed by measuring blood glucose clearance after an intravenous bolus infusion of a sterile 50% dextrose solution (600 mg/kg) as previously described [38]. An IVGTT was performed prior to the drug (PRE), after 12 weeks of drug exposure (POST) and a final one after 12 weeks of drug washout (WASH). Animals were not fed on the morning of IVGTTs after an overnight fast. Blood glucose was measured immediately in whole blood with a glucometer (Onetouch Ultra Blood Glucose Monitor, LifeScan, Milpitas, CA, USA). Plasma insulin level was assayed by the ONPRC/OHSU Endocrine Services Laboratory using Immunolite 2000.
2.9. T cell phenotype and cytokine production
T-cell phenotype and T-cell responsiveness were determined as described previously [39,40]. PBMCs were isolated from whole blood following centrifugation over Ficoll-Histopaque (Sigma, St Louis, MO). Cells were either analyzed immediately or were frozen for subsequent analysis using a controlled cryopreservation apparatus procedure (CryoMed Freezer ♯7454, ThermoForma, Marietta, OH), with no significant differences between the analyzed properties of fresh and frozen/thawed cells. For phenotype analysis, PBMC were stained with antibodies directed against CD8b, CD4, CD28, and CD95. All antibodies were purchased from Biolegend (San Diego, CA) with the exception of the CD8b antibody, which was purchased from Beckman Coulter (Brea, CA) and used per manufacturer's recommendations. Samples were collected using FACSLSRII (BD, San Jose, CA), and data analyzed using FlowJo (Treestar, Ashland, OR), with a minimum of 106 events collected/sample.
For cytokine production, PBMC were stimulated with anti-CD3 (FN18 clone, Invitrogen, Carlsbad, CA) for 6 h in the presence of Brefeldin A (Sigma, St Louis, MO) to block cytokine secretion. At the end of the incubation, PBMC were stained with antibodies directed against CD8b, CD4, CD28, and CD95 as described above. The cells were then fixed and permeabilized using a kit (Biolegend, San Diego, CA), before the addition of antibodies directed against IFNγ and TNFα (Biolegend, San Diego, CA). Samples were collected using FACSLSRII (BD, San Jose, CA), and data analyzed using FlowJo (Treestar, Ashland, OR), with a minimum of 106 events collected/sample.
2.10. Body composition analysis
The monkeys were scanned in a DEXA-scanner (Hologic, Inc., Bedford, MA) for body composition. Animals were sedated with Telazol (3 mg/kg) and the sedation was maintained with ketamine (5 mg/kg). The animals were laid flat on the DEXA (Hologic Discovery A) and a whole body scan performed. Data were imported into an Excel file and analyzed with GraphPad Prism (GraphPad Software, La Jolla, CA).
2.11. Statistical analysis
Data are expressed as mean±s.e.m. A repeated measures ANOVA was used to test the significance of the outcomes utilizing a Dunnett's posthoc test to test for significant differences from the starting condition (GraphPad Software, La Jolla, CA). A p-value of 0.05 or lower was considered significant. In some instances, an ANOVA with Bonferroni posthoc testing was performed and is noted as such in the figure legends. A p-value lower than 0.05 was considered significant.
3. Results
3.1. MLDL1278a inhibits LPS stimulated MCP-1 release in primary monocytes in an antibody Fc-dependent manner
We have shown previously that MLDL1278a inhibits MCP-1 release from primary monocytes, a property that may contribute to the decreased macrophage content observed in antibody-treated atherosclerotic mice [30]. Comprehensive analysis of cytokines and biomarkers revealed that a series of LPS-stimulated inflammatory cytokines (CD40, CD40 ligand, IL-1a, Il-6, IL-7, IL12 p40) were significantly reduced by anti-oxLDL treatment in human primary monocytes (Supplementary Table 1). Therefore, the antibody exhibits a general anti-inflammatory effect on these stimulated immune cells. In an effort to dissect the molecular mechanism underlying the antibody's anti-inflammatory effect, we used MCP-1 as the primary readout for the in vitro monocyte assay because of its role in atherosclerosis progression, the robustness of the change and consistency among donor cells.
Macrophages express an array of cell surface receptors capable of binding and signaling in response to oxLDL [41,42]. Importantly, macrophages also express inhibitory antibody receptors (Fcγ receptors) that have been implicated in protection against atherosclerosis [43]. It is therefore conceivable that the anti-inflammatory effects of MLDL1278a may result from either scavenging of oxLDL and prevention of oxLDL-induced receptor signaling—a mechanism that would involve antibody variable domain (Fv) binding to oxLDL—and/or active inhibitory signaling involving antibody constant domain (Fc) interactions with macrophage Fc receptors following antibody (Fv-mediated) binding to oxLDL. We therefore characterized MLDL1278a's dependency on antibody Fv- and Fc-domains for blockade of monocyte MCP-1 release.
Monocytes were incubated with MLDL1278a antibody in the full-length IgG1 format or in the F(ab′)2 format lacking the FcγR binding antibody constant region in the presence of oxLDL-containing human serum. Consistent with previous findings, MLDL1278a IgG1 showed near complete inhibition of MCP-1 release, as compared to treatment with control (ctrl) IgG1 antibody. In contrast, treatment with MLDL1278a F(ab′)2 did not reduce MCP-1 concentrations significantly compared to treatment with ctrl IgG1 (Figure 1A). This result suggests that MLDL1278a confers its inhibitory activity through its constant domain (Fc fragment). Since monocytes and macrophages express multiple FcγRs capable of binding antibody Fc, we assessed roles of different FcγRs in MLDL1278a blocking of monocyte MCP-1 release. Monocytes were pre-incubated with function-blocking monoclonal antibodies to FcγRI, FcγRII, or FcγRIII before being treated with MLDL1278a. Antibodies to FcγRII, but not to FcγRI or FcγRIII, completely neutralized the inhibitory activity of MLDL1278a and restored culture supernatant MCP-1 concentrations to ctrl IgG1 treatment levels. FcγR function-blocking antibodies themselves had no effect on MCP-1 release from cells exposed to control IgG1 (Figure 1B). These results demonstrated that MLDL1278a blocking of MCP-1 was both antibody Fc-dependent and macrophage FcγR-dependent.
Figure 1.
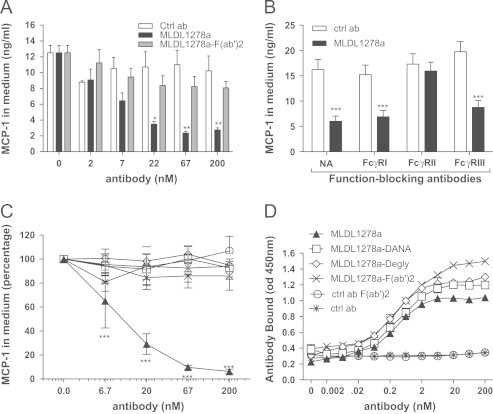
Inhibitory activity of MLDL1278a is antibody Fc and FcγRII dependent. Human monocytes are freshly purified from PBMC as described in Section 2, and cultured with addition of LPS (0.3 ng/ml) and indicated antibodies. MCP-1 protein concentrations in culturing medium are assessed with an ELISA kit after 40-hour incubation. Data presented are average of cells from 3 donors; error bars represent stand error of the mean. (A) MCP-1 protein concentration in cell culture supernatant after primary monocytes were treated with control antibody, wild-type MLDL1278a or MLDL1278a F(ab′)2, at increasing concentrations. (B) Cells were either pre-incubated or not for 15 min on ice with 133 nM of FcγRI (CD64), FcγRII (CD32), or FcγRIII (CD16) function blocking mAbs, prior to addition of 25 nM MLDL1278a (black bars) or control IgG1 (gray bars). (C) Relative MCP-1 protein level in cell culture supernatant after primary monocytes was treated with wild-type or Fc mutant MLDL1278a. Non-tissue culture treated plates were precoated with 3 µg/ml MDA-LDL and were then used for culturing monocytes. MCP-1 levels from cells with no antibody treatment were set as 100%. (D) Binding of wild-type and mutant MLDL1278s to immobilized MDA-LDL, at conditions corresponding to C. Values represent the mean±SEM. Values were analyzed in A and B with Multiple T-tests and two-way anova for C and D followed by Tukey multiple comparison test.
3.2. MLDL1278a forms immune complexes with aggregated oxLDL to inhibit monocyte MCP-1 release
FcγRs, with the exception of FcγRI, are low-affinity, high-avidity receptors that bind antibody constant domains only when they are presented in multimerized form, such as when antibodies form immune complexes following binding to aggregated or multivalent antigen [44]. The above data indicated that MLDL1278a exerted its MCP-1 blocking activity through antibody Fc interactions with FcγRs. To gain further evidence that MLDL1278 MPC-1 blocking activity was mediated through FcγR-signaling, we investigated if the blocking activity of MLDL1278a is dependent on formation of oxLDL:antibody immune complexes. We first examined if the oxidized LDLs that are present in human serum used for culturing these cells can mediate immune complex formation and are required for the antibody's blocking activity, by depleting lipids from the serum. Surprisingly, we continued to detect MCP-1 inhibition by MLDL1278a with delipidated serum, although the MCP-1 level is generally lower (Supplementary Figure 1A and B). Further investigation led to the observation that MLDL1278a spontaneously binds the surface of culturing plates, even in the presence of 10% serum (Supplementary Figure 1C). The immobilized antibody would mimic immune complex and could engage FcγRs on the surface of monocytes. To circumvent this problem and confirm oxLDL:MLDL1278a immune complex is indeed capable and required to mediate the blocking activity of MLDL1278a, we avoided spontaneous antibody binding to plate surface by using plates without tissue culture treatment (Supplementary Figure 1D). We then pre-coated MDA-LDL or native LDL at increasing density on untreated plates, before culturing and treating monocytes.
We confirmed Fc dependency using F(ab′)2 of MLDL1278a with the new culturing system. To rule out the possibility that decreased bulkiness of F(ab′)2 accounted for lost blocking activity, we also generated full-length IgG1 MLDL1278a Fc-mutants (deglycosylated MLDL1278a and MLDL1278a IgG1-DANA) with abolished binding to antibody FcγRs, and analyzed their MCP-1 blocking activity. The deglycosylated form and DANA mutant, as well as F(ab′)2 of MLDL1278a, showed no MCP-1 blocking activity, validating our data with the F(ab′)2 reagent and further demonstrating that MLDL1278a blocking effects were truly Fc-dependent (Figure 1C). Importantly, wild-type IgG1 and FcγR-binding deficient variants of MLDL1278a showed near identical binding to oxLDL, demonstrating that the loss of functional activity did not result from impaired (Fv-mediated) oxLDL binding (Figure 1D).
Consistent with an immune complex dependent mechanism, MLDL1278a blocked MCP-1 release only when MDA-LDL was coated, with the efficacy increasing in an MDA-LDL coating concentration dependent manner that reached saturation at approximately 3 μg/ml (Figure 2A and B). Similarly, when a fixed MDA-LDL coating concentration (3 μg/ml) was used, MLDL1278a dose-dependently blocked monocyte MCP-1 release (Figure 2C), in a manner that correlated with increasing density of antibody complexed with immobilized oxLDL (Figure 2D). MLDL1278a did not inhibit MCP-1 release when native LDL was coated at the same concentrations (Figure 2C), confirming that MLDL1278a's effect was oxLDL specific, and demonstrating that blocking activity could not be achieved by incubation with monovalent ligand but required immune complex formation between aggregated oxLDL and MLDL1278a.
Figure 2.
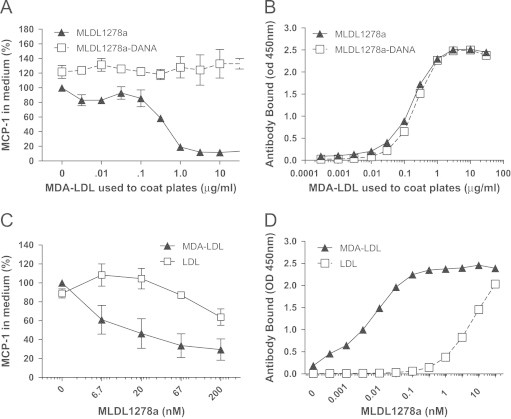
Inhibition of MCP-1 release from primary monocytes by MLDL1278a is mediated by antibody binding to immobilized antigen. (A) Relative MCP-1 protein level in cell culture supernatant after primary monocytes were treated with wild-type MLDL1278a or MLDL1278a DANA mutant, both at 67 nM. The plates were precoated with increasing concentrations of MDA-LDL, as indicated. MCP-1 level from cells with MLDL1278a treatment on non-coated plates was set as 100%. (B) Binding of wild-type and DANA mutant of MLDL1278a (67 nM) to immobilized MDL-LDL at increasing concentration, under the same condition the cells are treated as in A. (C) Plates are pre-coated with MDA-LDL or native LDL (3 µg/ml) in PBS before being used for culturing monocytes with increasing concentrations of MLDL1278a. MCP-1 level from cells with MLDL1278a treatment on non-coated plates was set as 100%. (D) Binding of MLDL1278a under culturing condition (in RPMI medium with 10% serum) to immobilized MDA-LDL or LDL were examined using HRP conjugated anti-human Fc as detection antibody. Values represent the mean±SEM. Values were analyzed using two-way anova.
3.3. MLDL1278a inhibits monocyte MCP-1 release induced by multiple different pro-inflammatory stimuli by negatively regulating NF-κB activity
The above data had unexpectedly demonstrated that MLDL1278a exerted its MCP-1 blocking effects by molecular mechanisms that involved antibody:oxLDL immune complex formation and antibody constant domain interactions with monocyte FcγRs. These observations are incompatible with a mechanism-of-action involving reduced pro-inflammatory signaling through blocking of oxLDL binding to monocyte oxLDL receptor, but instead implied that by binding to FcγRs, MLDL1278a triggered active inhibitory signaling that counteracted macrophage inflammatory responses induced by LPS stimulation. With such a mechanism of action, we might expect MLDL1278a to reduce monocyte MCP-1 release induced by other pro-inflammatory stimuli. To assess this intriguing possibility, we treated monocytes with multiple different potent pro-inflammatory stimuli known to trigger monocyte MCP-1 release and implicated as drivers of plaque and adipose tissue inflammation, and assessed MLDL1278a's MCP-1 blocking activity.
As expected, pro-inflammatory agents LPS, TLR-1/2 ligand Pam3CSK4, TLR-7 ligand R837, TLR-3 activator poly IC, as well as IL-1β and GM-CSF, which activate through IL-1 receptor and GM-CSF receptor, respectively, induced robust MCP-1 release in monocytes. Strikingly, MLDL1278a potently attenuated monocyte MCP-1 release induced by all stimuli, irrespective of what surface receptor they signaled through (Figure 3A). This is consistent with the notion that MLDL1278a, through immune complex formation and FcγR engagement triggers active inhibitory signaling, rather than blocking individual receptors for certain inflammatory stimulus.
Figure 3.
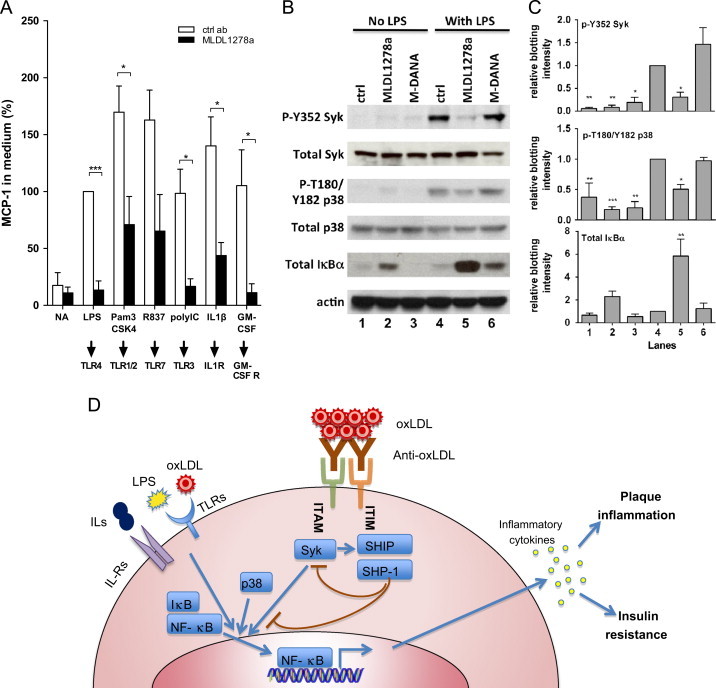
MLDL1278a modulates inflammatory signaling and inhibits various inflammation stimuli. (A) MCP-1 concentration from monocytes treated with control antibody and MLDL1278a in combination with a variety of inflammatory stimuli. The corresponding receptor for each of these stimuli is illustrated at the bottom. MCP-1 level from LPS stimulated cells with no antibody treatment was set as 100%. (B) Immunoblot analysis of phosphorylation or total amount of the indicated protein in the presence of control antibody, MLDL1278a or MLDL1278a DANA mutant (M-DANA), in the absence or presence of 0.3 ng/ml LPS. (C) Quantification of the western blotting. The intensity of the phosphor-protein bands is normalized against the corresponding total protein bands, and total IκBα is normalized to actin. Data shown are average+SEM from 3 independent experiments. (D) Schematic representation of the proposed mechanism for anti-oxLDL immune complex and FcγRs mediated anti-inflammatory effect.
Since the antibody effect on primary monocytes is mediated through antibody clustering and FcγRs engagement, we next surveyed the phosphorylation of FcγR downstream kinases, aiming to identify the signaling event mediating the inhibitory effects. We show in Figure 3B that LPS treatment increased phosphorylation of Syk, the kinase directly associated with FcγRIIa, as well as MAPK p38. More importantly, the addition of MLDL1278a dramatically decreased the phosphorylation of these two kinases caused by LPS. Increased activity of MAPK, in particular p38 MAPK, has been shown to increase synthesis of inflammation mediators at the level of transcription and translation [45–47]. We also examined the expression level and post-translational modification of regulatory proteins of NF-κB, the transcriptional activator of a spectrum of pro-inflammatory cytokines. IκBα the major negative regulator of NF-kB, is dramatically upregulated at the protein level by MLDL1278a treatment, and this effect is particularly profound in the presence of LPS. IκBα binds NF-κB and prevents it from translocating to the nucleus, thus preventing NF-κB activation. The ubiquitination and degradation of IκBα is dependent on its phosphorylation, so the elevated protein level of this protein is consistent with the attenuated phosphorylation observed (Figure 3B and C). Thus, increased IκBα level, which inhibits NF-κB nuclear translocation and activity, combined with reduced MAPK p38 activity, contributes to decreased cytokine production upon MLDL1278a treatment (Figure 3D).
3.4. Baseline characterization of the diet-induced obese macaque
Since the anti-inflammatory effect of MLDL1278a we observed on primary cells was impressive and significant, a subsequent study was performed to determine what the effect of this antibody was in an animal model. Diet-induced obese Rhesus macaques (DIO-NHP) were maintained on a high fat diet supplemented with fructose (HFFD) and have previously been demonstrated to develop the full spectrum of the metabolic diseases. The basic characterization of the HFFD animals is shown in Table 1, along with the relative comparison to 20 normal lean age matched macaques maintained on a standard chow diet. HFFD intake varied greatly between animals, ranging from 1657 kCal/day to 3246 kCal/day. On average, the animals on the highly palatable HFFD diet ate 3 times more calories than animals on a control diet (2323±223 for HFFD versus 718±26 kCal/day) (Table 1 and Figure 4A). Animals maintained on a HFFD had a higher body weight and adiposity, with the total percent fat mass being approximately 35% in HFFD animals and 20% in controls (Figure 4B and C). These animals also had a small but significant increase in lean mass (18%; Table 1 and Figure 4D), which is typically associated with obesity. Regarding glucose homeostasis, there was no significant difference in fasted blood glucose levels and HbA1c levels. However, the HFFD animals did demonstrate significant increases in fasting insulin and HOMA-IR (Figure 4E and F). In addition, in an IVGTT HFFD animals have impaired glucose clearance and require more insulin secretion, as measured by the area under the curve (Table 1 and Figure 4G). This indicates that the animals were insulin resistant, but not diabetic. As expected from animals being maintained on a HFFD, fasting circulating triglycerides (TGs) levels, HDL-C and total cholesterol were significantly elevated in the HFFD animals, compared to chow fed controls. Using NMR-based analysis of particle sizes, we showed that fractions containing VLDL+chylomicron particles and HDL particles were significantly elevated in the HFFD animals (Table 1 and Supplementary Figure 3).
Table 1.
Characteristics of diet-induced obese Rhesus Macaques.
| High fat fructose diet (n=6) | Chow (n=20) | |
|---|---|---|
| Body weight (kg) | 17.1±0.9 | 11.6±0.5⁎⁎⁎ |
| Lean mass (kg) | 10.5±1.4 | 8.9±0.3⁎⁎⁎ |
| Fat mass (kg) | 5.7±1.1 | 2.4±0.3⁎⁎⁎ |
| Trunk fat (%) | 41.7±5.2 | 21.9±2.0⁎⁎⁎ |
| BMC (g) | 466.9±70.2 | 439.3±10.4 |
| BMD (g/cm2) | 0.616±0.058 | 0.600±0.010 |
| Food intake (kCal/day) | 2323±223 | 718±25.7⁎⁎⁎ |
| Glucose (mg/dL) | 54.8±1.87 | 54.4±1.29 |
| Insulin (mIU/l) | 46.5±11.5 | 20.9±3.29⁎ |
| GTT-Glucose AUC | 12,034±484 | 9411±319⁎⁎⁎ |
| GTT-Insulin AUC | 16,539±4633 | 7681±1066⁎ |
| HbA1c (%) | 6.00±0.20 | 6.29±0.22 |
| HOMA-IR | 6.12±1.4 | 2.78±0.4⁎⁎ |
| VLDL particles (nM) | 48.6±3.06 | 3.6±1.08⁎⁎⁎ |
| Triglycerides (mg/dL) | 52.2±7.6 | 33.2±1.2⁎⁎⁎ |
| Cholesterol (mg/dL) | 196.8±20.1 | 110.0±4.5⁎⁎⁎ |
| HDL-C (mg/dL) | 104±13.1 | 70±3.8⁎⁎ |
Comparison of body weight, food intake, body composition and serum chemistry between diet-induced obese Rhesus Macaques and regular chow-fed Rhesus Macaques. Values represent the mean±SEM. Analysis ANOVA with Bonferroni posthoc.
p<0.05.
p<0.01.
p<0.001 denotes significant difference from HFFD.
Figure 4.
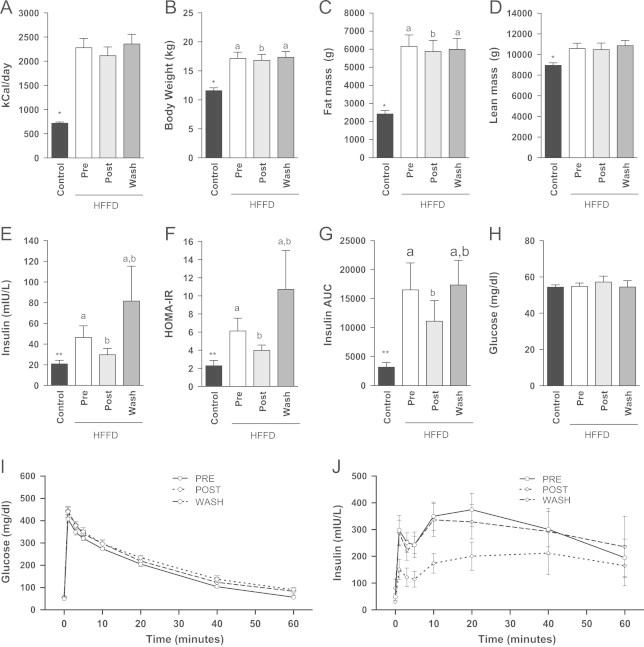
MLDL1278a has little impact on body weight homeostasis, but improves insulin sensitivity. (A) Animals eating a HFFD consume on average 3 fold more calories per day than control animals. MLDL1278a treatment did not affect HFFD food intake in obese rhesus macaques (pre versus post), nor was there an effect of drug washout on calorie intake (Wash). HFFD increased body weight (B), Fat mass (C) and Lean mass (D) (*p<0.001 unpaired two-tailed t-test between control and pre for all comparisons). MLDL1278a treatment has a small, but significant, effect on body weight (p=0.0037) and fat mass (p=0.019; repeated measures ANOVA, with letters a and b denoting significant differences of groups by Bonferroni posthoc). Animals fed a HFFD had significantly higher fasting insulin level (E), HOMA-IR (F) and insulin secretion during a glucose tolerance test (G). All increases were attenuated by treatment with MLDL1278a during the treatment period, and returned to baseline levels after treatment was stopped (E–G). Fasting blood glucose levels were not affected by diet or treatment (H). (I-J) Combined glucose clearance (I) and insulin secretion (J) during the ivGTT in HFFD animals. Two-tailed unpaired t-test where p<0.05 is considered significant. **p<0.01.
3.5. Effects of MLDL1278a treatment on metabolism and glucose homeostasis
MLDL1278A was administered to HFFD-fed rhesus monkeys, and MLDL1278A serum concentrations were measured at multiple time points, and the pharmacokinetic parameters were determined (Supplementary Figure 2 and Supplementary Table 3). While the HFFD animals ate substantially more than the control cohort, there was no effect on the food intake when animals were treated with MLDL1278a (Figure 4A). Body weight (Figure 4B), however, was slightly but significantly decreased, using repeated measures ANOVA during the treatment period (p=0.0037). The animals rebounded after treatment was stopped. There was no significant difference in body fat and lean mass (Figure 4C and D).
To determine whether MLDL1278a affected glucose homeostasis, we performed IVGTT prior to and at the end of a 12-week treatment period. An additional IVGTT was performed after 12 weeks of washout, to determine whether any changes would return to baseline. Treatment with MLDL1278a significantly reduced fasting insulin levels (Figure 4E), HOMA-IR (Figure 4F) and insulin secretion during the IVGTT (Figure 4G) in the HFFD-prediabetic animals over the 12 weeks, although their levels were not completely normalized to CTR levels. After the 12-week washout period, all values returned to pretreatment levels. Fasting glucose (Figure 4H) and the rate of glucose clearance during the IVGTT (Figure 4I) were not altered by 12 weeks of MLDL1278a treatment, which may not be surprising since these animals were not diabetic. We have graphed the glucose and insulin concentrations for the HFFD diet animals at the three different timepoints (Figure 4I and J). These data support the original finding that insulin secretion during a glucose tolerance is significantly reduced, most likely the result of improved insulin sensitivity.
While animals treated with MLDL1278a tended towards decreased HDL and TG levels closer to the range of the control animals, this effect was not significant. Furthermore, there was no effect of MLDL1278a treatment on systemic LDL or oxLDL levels. The finding that MLDL1278a did not affect systemic oxLDL levels is consistent with MLDL1278a's specificity for extensively modified (MDA-modified) oxLDL. MLDL1278a was raised against MDA-modified ApoB100 peptide [30]. MDA modification is a relatively advanced form of oxidation, and MDA-LDL is thought to be principally present in plaque and chronically inflamed tissue and only at low or undetectable levels in the systemic compartment [48]. Our previous findings are consistent with MLDL1278a being specific for such extensively modified oxLDL; MLDL1278a binds strongly to plaque oxLDL [49], but does not bind to or binds only weakly to oxLDL present in serum (unpublished data). Further, MLDL1278a does not interfere in herein used oxLDL quantification ELISA, as demonstrated in previous spiking experiments with MLDL1278a in human serum (data not shown). VLDL levels steadily declined throughout the treatment period with MLDL1278a; however, this effect was only significant in the post-treatment period (Supplementary Figure 3).
3.6. Effects of MLDL1278a treatment on the immune system
Many studies have now provided evidence for a strong correlation between increased cytokine levels and insulin resistance [50,51]. There is evidence to suggest that pro-inflammatory macrophages are causal and propagate insulin resistance by cytokine secretion [52]. Specifically, TNFα or molecules in the signaling pathway downstream of the TNFα receptor have been implicated in the development of DIO and several associated conditions, such as insulin resistance [53–55]. Using an ELISA-based analysis, we determined that plasma TNFα concentrations are indeed significantly elevated in the HFFD animals compared to the control animals (Figure 5A). Furthermore, these HFFD fed animals also had elevated basal levels of plasma IL1β, GM-CSF, and IFN-γ (Figure 5B–D). Using a commercially available high-throughput assay, we identified several cytokines and biomarkers associated with metabolic diseases in humans that were different between the HFFD and chow-fed animals (Supplementary Table 1). MCP-1, CD40, and sex hormone-binding globulin (SHBG) were elevated, according to the commercial cytokine-screening assay; elevations in all three are associated with an increased risk of cardiovascular disease [56–59].
Figure 5.
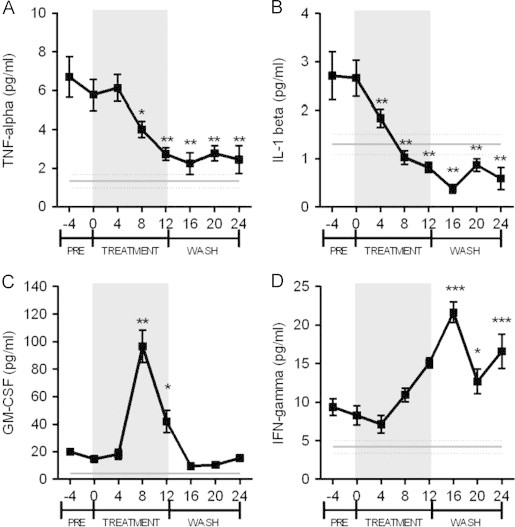
Changes in plasma inflammatory cytokines in response to diet and MLDL1278a antibody treatment. The gray straight line in each panel indicates the average plasma cytokine level in normal diet-fed control animals. (A) TNF-α was significantly increased in HFFD animals (two-tailed t-test; p<0.0001) compared to control animals when measured by ELISA. Treatment with MLDL1278a significantly reduced TNF-α levels compared to week 0 (repeated measures ANOVA with Dunnett's posthoc analysis) from week 8 to at least week 24. (B) HFFD significantly increased IL-1β levels (two-tailed t-test; p<0.001) and MLDL1278a treatment significantly improved IL-1β levels from 4 weeks after treatment was initiated. IL-1β levels were still significantly lower 24 weeks after the start of the treatment. (C) GM-CSF levels were not altered by HFFD, however a significant spike in levels was detected 8 weeks after the treatment was initiated. (D) IFN-γ levels were increased in response to the HFFD, and MLDL1278a treatment resulted in a significant rise in IFN-γ levels during the treatment period. Levels remained elevated during the washout period. Values represent the mean±SEM.
Consistent with the observation that MLDL1278a exhibits anti-inflammatory effect on purified monocytes, treatment with MLDL1278a resulted in a significant decrease in plasma TNFα and IL--1β levels by week 8 (Figure 5A and B). Surprisingly, neither of these cytokines returned to pretreatment levels, even after 12 weeks of washout. GM-CSF levels were relatively stable during the earlier and later course of the study, but peaked significantly after 8 weeks of MLDL1278a treatment in HFFD NHP. IFN-γ levels trended upwards during the 12-week treatment phase; however this did not reach significance until after the treatment was completed. Although MCP-1 was elevated with HFFD in these monkeys, its level was not reduced upon antibody treatment (Supplemental Figure 4). Nevertheless, we did note a significant decrease of En-Rage, which is also a potent chemoattractant for monocytes and able to stimulate monocyte cytokine release. In addition, CD40 ligand, βFGF, EGF and VEGF are down-regulated by MLDL1278a in both LPS-stimulated monocytes and HFFD-fed monkeys (Supplementary Table 1 and Figure 4), as determined by high-throughput screening analysis of the samples. Considering the difference between LPS-stimulated pure monocytes cultured in vitro, and the multiple cell types of the immune system under physiological conditions, it is not surprising that some cytokines differ between the two systems.
3.7. Effect of MLDL1278a treatment on T-cell populations
Recent studies have demonstrated that both the adaptive and innate immune systems play a role in diet-induced obesity [60,61]. As such, we analyzed the effect of the MLDL1278a treatment on the phenotype of the T-cell populations in the circulation. First, naïve, central memory and effector memory CD4 and CD8 T-cell subset populations were determined by fluorescence flow analysis, following combined staining with CD4/CD8, CD28, and CD95 mAb, as previously described [62]. The HFFD animals displayed a clear change in CD4 populations, significantly decreasing the pool of CD4 naïve T-cells (Figure 6A, control versus pre, p=0.05), in favor of CD-4 central memory T-cells (Figure 6B, control versus pre, ♯, p<0.05). More importantly, treatment of the animals with MLDL1278a resulted in a reversal of this shift (Figure 6A and B, pre versus post, ⁎p<0.05, ⁎⁎p<0.01). The CD4 effector memory pool was not affected by either the HFFD or the treatment (Figure 6C).
Figure 6.
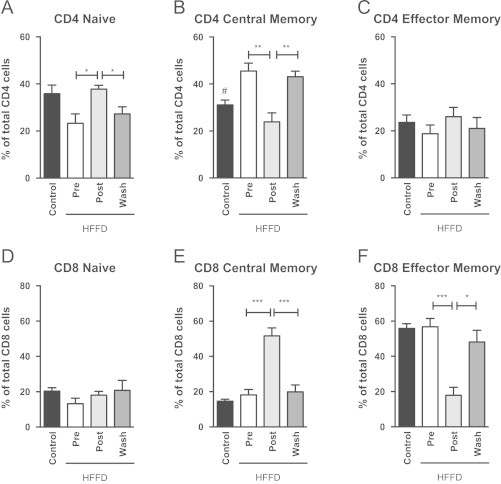
Changes in T-cell repertoire in response to HFFD and MLDL1278a treatment. (A) HFFD reduced populations of CD4 Naïve T-cells, which was reversed by treatment with MLDL1278a. (B) CD4 Central Memory T-cells were increased in HFFD animals, and returned to control levels when treated with MLDL1278a. CD4 effector memory T-cells (C) and CD8 Naïve T-cells (D) were not altered by diet or treatment. (E and F). Central and Effector memory T-cells were not affected by diet, however treatment induced a significant shift in T-cell population from Effector to Central Memory T-cells. Values represent the mean±SEM. Student t-test for diet effect (Pre versus control; ♯=p<0.05) and repeated measures ANOVA for effect of treatment (*=p<0.05, **=p<0.01, ***=p<0.001 with Bonferroni post hoc testing).
Although HFFD had no direct effect on the distribution of CD8 positive T-populations, MLDL1278a treatment resulted in a significant shift from CD8 effector memory to the central memory phenotype (Figure 6E and F), with no change in CD8 naïve cells (Figure 6D). These observations were reversed after the washout period. In all, these results argue that MLDL1278a has a positive outcome on the status of the adaptive immune system.
3.8. T-cell responsiveness is improved with MLDL1278a treatment
To determine whether the responsiveness of the T-cells has been affected by the HFFD or MLDL1278a treatment, we subjected isolated subsets of T-cells to T-cell receptor cross-linking, using agonistic anti-CD3 mAb and determined IFN-γ and TNF-α production. Following this immunological stimulus, T-cell responsiveness in all subsets was clearly diminished in HFFD animals (Figure 7A–D, ctrl versus pre). This decrease in T-cell responsiveness induced by HFFD was maintained over the duration of the study. However, within the HFFD diet group, MLDL1278a treatment significantly increased the percentage of responding cells in all of the subsets of T-cells (Figure 7A–D, pre versus post). In addition, this improvement was reversed when the treatment was stopped (Figure 7A–D, post versus wash).
Figure 7.
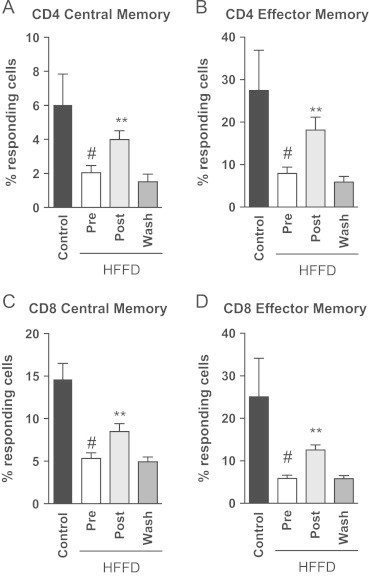
The ability of T-cells to respond to stimuli is improved with treatment. (A–D). The percentage of all CD4 and CD8 memory T-cells was significantly reduced in animals fed a HFFD (Student's t-test ♯=p<0.05). Repeated measures analysis with Bonferroni posthoc testing demonstrated significant improvements in the ability of T-cells to respond to CD3-mediated activation, which returned to pre-treatments levels after the washout period was completed (**p<0.01). Values represent the mean±SEM.
4. Discussion
In recent years, it has been recognized that inflammation is an important driving force for metabolic disorders, such as obesity, insulin resistance, and atherosclerosis [63–65]. Oxidative stress contributes to inflammation both locally and systemically. oxLDL is thought to be a key oxidatively-modified molecule that drives atherosclerosis, and it has been implicated in plaque initiation, progression, and destabilization. The strong association of oxLDL with atherosclerosis [66] and the ability of oxLDL particles to promote inflammation in different types of cells provided the rationale for development of a human anti-oxLDL antibody for treatment of atherosclerosis [29]. The molecular premise was that antibodies to oxLDL would neutralize and prevent oxLDL from binding to and signaling inflammation through oxLDL receptors present on inflammatory immune cells, most notably macrophages. In this study, we demonstrate that an anti-oxLDL antibody exhibits significant anti-inflammatory effects in a unique nonhuman primate diet-induced obesity model of human metabolic syndrome and cardiovascular disease, and that this antibody functions unexpectedly by a mechanism that does not involve inhibition of oxLDL:oxLDL receptor signaling.
The inhibitory signal delivered by MLDL1278a was co-dependent on antibody:antigen immune complex formation, requiring binding to high-density oxLDL- and antibody constant domain interactions with FcγR to attenuate immune cell inflammatory responses. It was known that oxLDL co-localizes with macrophages and is abundant in plaque tissue [49]. These observations are consistent with a unique mechanism-of-action of MLDL1278a involving (1) antibody localization to oxLDL rich plaque tissue; (2) immune complex formation through binding to densely aggregated antigen; (3) FcγR crosslinking and inhibitory signaling to deactivate inflammatory activity in macrophages. With such a mechanism, MLDL1278a would present a truly tissue specific anti-inflammatory drug, i.e., it would only exert its anti-inflammatory activity and modulate macrophage function in the relevant diseased (chronically inflamed, oxLDL and macrophage containing) tissue.
Although immune complexes are thought to drive inflammatory responses in several disease settings, including rheumatoid arthritis, it has also been reported that immune complexes can attenuate inflammatory cytokine release induced by LPS [67]. Monocytes and macrophages show remarkable plasticity and functional heterogeneity [68]. On the one extreme of this continuum are the classical pro-inflammatory M1 macrophages, which in response to exogenous or endogenous danger signals, e.g., toll-like receptor ligands, respond by vigorous release of pro-inflammatory cytokines. On the other side of the continuum are the regulatory M2 macrophages, which by production of IL-10 suppress immune responses and exert anti-inflammatory activity. Interestingly, previous studies had demonstrated that co-incubation of monocytes with M1-inducing pro-inflammatory ligands, e.g. toll-like receptor agonists or IL-1, with antibody immune complexes skews macrophages away from a pro-inflammatory M1 phenotype towards a regulatory anti-inflammatory phenotype [69]. The signaling pathways that mediate these changes, however, remain largely uncharacterized. Here, we further the understanding of cell signaling pathways involved in antibody immune complex-mediated skewing of macrophages by demonstrating that MLDL1278a—an antibody that is dependent on immune-complex formation and antibody interactions with macrophage FcγRs to exert its anti-inflammatory activity—modulates phosphorylation of several kinases downstream of FcγRs and TLRs, ultimately leading to decreased activation of the transcription factor NF-κB and decreased pro-inflammatory cytokine release. We propose that when clustered by high density of its antigen, MLDL1278a, through its Fc fragment, cross-links immunoreceptor tyrosine-based activation motif (ITAM) associated FcγRIIa and immunoreceptor tyrosine-based inhibitory motif (ITIM) associated FcγRIIb. This leads to dephosphorylation and inactivation of FcγRIIa associated Syk through the ITIM associated phosphatases SH2-Containing 5′-Inositol Phosphatase (SHIP) and SH2-Containing protein tyrosine phosphatase-1 (SHP-1), and subsequent inactivation of downstream signaling molecules including p38 MAPK, as has been previously described [70,71]. Although we have not detected consistent changes on the phosphorylation of SHP-1 and SHIP, which may not fully recapitulate their phosphatase activities, we did find phosphatase inhibitors could reverse the inhibitory effect of MLDL1278a on primary monocytes, indicating phosphatase activities are important for the observed effects in these cells (data not shown). Notably, anti-oxLDL immune complexes also significantly increased levels of the NF-κB inhibitory protein IκBα, which leads to decreased NF-κB activity in the nucleus. While more detailed characterization of the kinases that mediate these changes is needed, herein presented data demonstrate for the first time that antibody immune complexes through FcγRII engagement can modulate a variety of signaling and transcriptional events, which ultimately lead to the downregulation of inflammatory cytokine production.
Consistent with this proposed mechanism of action, administration of MLDL1278a to diet-induced obese monkeys decreased systemic levels of several macrophage-associated proinflammatory cytokines, including TNF-α and IL-1β (Figure 5). The decrease in IL1β levels may be especially important as this change would be predicted to be protective against CVD, since knockout of IL1β in the mouse model of CVD (apoE−/−) significantly reduced the severity and occurrences of atherosclerosis [72]. Similarly, anti-oxLDL antibody treatment also reduced several other cytokines, including EN-RAGE, CD40 ligand and bFGF, even though their levels were not initially different from control animals (Supplementary Figure 4 and Supplementary Table 1). EN-RAGE, like MCP-1, is a potent chemoattractant for monocytes and EN-RAGE induces monocyte IL-1β and TNF-α release [73]. Like oxLDL and pro-inflammatory macrophages, the pro-inflammatory EN-RAGE/RAGE axis has further been implicated in metabolic syndrome [74], T2DM [75], and atherosclerosis [76–78]. EN-RAGE is the ligand for RAGE that can be activated in response to CRP [79]. EN-RAGE is also thought to be involved in diabetic macro- and micro-angiopathy [80]. IL15 is expressed in atherosclerotic plaques in humans and apoE−/− mice, and is thought to be involved in T cell recruitment [81]. Finally, it has been shown that oxLDL increases the production and release of bFGF, which, in turn, directly stimulates smooth muscle cell proliferation in vasculature and promotes atherosclerosis [82].
However, not all cytokines were decreased by MLDL1278a treatment, as both IFNγ and GM-CSF were increased by this treatment (Figure 5). The increase in IFNγ was persistent beyond the cessation of oxLDL immunotherapy, while the increase in GM-CSF was only transient, peaking after 8 weeks of treatment and then declining by 12 weeks of treatment. The increase in IFNγ may appear counter intuitive; however this increase may be reflective of the adaptive nature of the immune system and could signal a change in other areas of the system. Follow-up studies are needed to fully understand the impact of the immune system, including innate and adaptive immune systems. The transient increase in GM-CSF in response to oxLDL immunotherapy may also be an indication of a reduced risk of cardiovascular complications, since GM-CSF deficiency increases the lesion size and macrophage infiltration in ApoE−/− mice [83]. Thus, GM-CSF may promote smaller stable atherosclerotic lesions. The reason for the only transient spike in GM-CSF is unknown. Although levels were still elevated by the end of the treatment period, the changes in GM-CSF levels with ox-LDL immunotherapy quickly returned to basal levels once treatment was stopped.
The finding that several pro-inflammatory cytokines were suppressed beyond the 12-week washout period suggests that oxLDL immunotherapy could have long-term beneficial effects. However, in our study, the beneficial effects of oxLDL immunotherapy on insulin sensitivity had returned to pretreatment levels after the 12-week washout period. These observations indicate that the insulin-sensitizing and the systemic cytokine-decreasing effects of oxLDL immunotherapy may result from targeting pro-inflammatory macrophages residing in different body compartments. The role of pro-inflammatory macrophages in development of metabolic syndrome, insulin-resistance, T2DM, as well as CVD, is now well-established, and recent studies using a system biology approach to interconnect genetic, genomic, and environmental data support a causal role for macrophages in disease [84,85]. Notably, tissue macrophages are known to be major producers of several of the pro-inflammatory cytokines shown herein to be down-regulated by anti-oxLDL-treatment, including TNF-α, IL-1β, and EN-RAGE [80]. It was, for example, reported that almost all of adipose tissue TNF-α in rodents is produced by adipose tissue macrophages [86]. OxLDL immunotherapy may further directly, or indirectly, through targeting of macrophages, block the action of oxLDL on the adipocyte or hepatocyte and improves insulin sensitivity independent of the circulating cytokine levels [11,12]. Based on our hypothesized mechanism-of-action, for the anti-oxLDL antibody to exert its effect in adipose tissue, the latter would need to contain oxLDL targeted by this antibody. According to current understanding and our own observations, extensively oxidatively modified LDL, including the MDA-LDL that is bound by MLDL1278A, is largely confined to plaque tissue where it is thought to perpetuate inflammation [48,49,87]. Interestingly, however, as greater adipose tissue increases lipoxygenase and decreases superoxide dismutase production, it has been hypothesized that adipose tissue contributes to LDL oxidation [48]. It is thus conceivable that inflamed adipose tissue, like inflamed plaque, contains oxLDL targeted by MLDL1278a. While further studies are needed to characterize in more detail the cellular and molecular interactions that underlie anti-oxLDL IgG effects on inflammation and insulin sensitization, herein presented and earlier reported [31,40] data are consistent with targeting of pro-inflammatory macrophages being an important mechanism underlying anti-oxLDL therapeutic activity.
Adaptive immune system abnormality has recently been proposed to promote insulin resistance and obesity [60,61]. Our DIO monkeys indeed show a skewed peripheral CD4 T cell compartment, and both CD4 and CD8 T cell responses to T cell receptor cross-linking were decreased in DIO compared to control monkeys, indicative of suppressed immune function. More importantly, treatment with MLDL1278a shifted the T-cells to a more naïve state and improves T-cell responsiveness. The underlying mechanism of these phenomena and whether these changes contribute to the beneficial effects on the metabolic parameter need to be further investigated.
In summary, this study demonstrated that anti-oxLDL IgG immune therapy is capable of modulating macrophage pro-inflammatory activity through delivery of dominant inhibitory FcγR-signaling in vitro, and that it reduces inflammation and improves insulin sensitivity in a unique nonhuman primate model, sharing several characteristics with human metabolic syndrome and cardiovascular disease. Impressively, these changes were observed independent of any changes in food intake or body composition. In addition to providing protection from cardiovascular disease [29], the insulin-sensitizing action of this therapy simultaneously targets two co-morbidities of obesity. Anti-oxLDL IgG treatment could provide a new tool in the fight against the morbidity of the obesity epidemic.
Conflict of interest
None declared.
Acknowledgments
This work was supported by a U.S. National Institutes of Health Grant, P51 OD011092, and funding from Genentech, USA. We thank the Division of Comparative Medicine at the Oregon National Primate Research Center at OHSU for their excellent care of the animals.
Footnotes
MLDL1278a is under clinical development by Genentech and BioInvent. S. Li, A. Robertson, G.K. Kolumam, X. Li, K. von Wachenfeldt, C. Valfridsson, S. Bullens, A. Joshi, N. van Bruggen, S. Bunting, and B. Frendéus were all paid employees of Genentech or Bioinvent International at the time of their contribution to the study.
Contributor Information
Shijie Li, Email: li.shijie@gene.com.
Kevin L. Grove, Email: grovek@ohsu.edu.
Appendix A. Supplementary material
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.molmet.2013.06.001.
Appendix A. Supplementary material
Supplementary material
Supplementary material
References
- 1.Gregor M.F., Hotamisligil G.S. Inflammatory mechanisms in obesity. Annual Review of Immunology. 2011;29:415–445. doi: 10.1146/annurev-immunol-031210-101322. [DOI] [PubMed] [Google Scholar]
- 2.Holvoet P., Lee D.H., Steffes M., Gross M., Jacobs D.R., Jr. Association between circulating oxidized low-density lipoprotein and incidence of the metabolic syndrome. Journal of the American Medical Association. 2008;299:2287–2293. doi: 10.1001/jama.299.19.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhou X., Robertson A.K., Hjerpe C., Hansson G.K. Adoptive transfer of CD4+ T cells reactive to modified low-density lipoprotein aggravates atherosclerosis. Arteriosclerosis, Thrombosis, and Vascular Biology. 2006;26:864–870. doi: 10.1161/01.ATV.0000206122.61591.ff. [DOI] [PubMed] [Google Scholar]
- 4.Ketelhuth D.F., Hansson G.K. Cellular immunity, low-density lipoprotein and atherosclerosis: break of tolerance in the artery wall. Thrombosis and Haemostasis. 2011;106:779–786. doi: 10.1160/TH11-05-0321. [DOI] [PubMed] [Google Scholar]
- 5.Feuerer M., Herrero L., Cipolletta D., Naaz A., Wong J., Nayer A. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nature Medicine. 2009;15:930–939. doi: 10.1038/nm.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nishimura S., Manabe I., Nagasaki M., Eto K., Yamashita H., Ohsugi M. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nature Medicine. 2009;15:914–920. doi: 10.1038/nm.1964. [DOI] [PubMed] [Google Scholar]
- 7.Morris D.L., Cho K.W., Delproposto J.L., Oatmen K.E., Geletka L.M., Martinez-Santibanez G. Adipose tissue macrophages function as antigen presenting cells and regulate adipose tissue CD4+ T cells in mice. Diabetes. 2013;62:2762–2772. doi: 10.2337/db12-1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stolarczyk E., Vong C.T., Perucha E., Jackson I., Cawthorne M.A., Wargent E.T. Improved insulin sensitivity despite increased visceral adiposity in mice deficient for the immune cell transcription factor T-bet. Cell Metabolism. 2013;17:520–533. doi: 10.1016/j.cmet.2013.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cornier M.A., Dabelea D., Hernandez T.L., Lindstrom R.C., Steig A.J., Stob N.R. The metabolic syndrome. Endocrine Reviews. 2008;29:777–822. doi: 10.1210/er.2008-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Malik S., Wong N.D., Franklin S.S., Kamath T.V., L'Italien G.J., Pio J.R. Impact of the metabolic syndrome on mortality from coronary heart disease, cardiovascular disease, and all causes in United States adults. Circulation. 2004;110:1245–1250. doi: 10.1161/01.CIR.0000140677.20606.0E. [DOI] [PubMed] [Google Scholar]
- 11.Sattar N., McConnachie A., Shaper A.G., Blauw G.J., Buckley B.M., de Craen A.J. Can metabolic syndrome usefully predict cardiovascular disease and diabetes? Outcome data from two prospective studies. Lancet. 2008;371:1927–1935. doi: 10.1016/S0140-6736(08)60602-9. [DOI] [PubMed] [Google Scholar]
- 12.Wilson P.W., D'Agostino R.B., Parise H., Sullivan L., Meigs J.B. Metabolic syndrome as a precursor of cardiovascular disease and type 2 diabetes mellitus. Circulation. 2005;112:3066–3072. doi: 10.1161/CIRCULATIONAHA.105.539528. [DOI] [PubMed] [Google Scholar]
- 13.Ginsberg H.N., Maccallum P.R. The obesity, metabolic syndrome, and type 2 diabetes mellitus pandemic: II. Therapeutic management of atherogenic dyslipidemia. Journal of Clinical Hypertension (Greenwich) 2009;11:520–527. doi: 10.1111/j.1559-4572.2009.00060.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ginsberg H.N., MacCallum P.R. The obesity, metabolic syndrome, and type 2 diabetes mellitus pandemic: Part I. Increased cardiovascular disease risk and the importance of atherogenic dyslipidemia in persons with the metabolic syndrome and type 2 diabetes mellitus. Journal of the Cardiometabolic Syndrome. 2009;4:113–119. doi: 10.1111/j.1559-4572.2008.00044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Song S.H., Hardisty C.A. Type 2 diabetes mellitus: a high-risk condition for cardiovascular disease irrespective of the different degrees of obesity. Quarterly Journal of Medicine. 2008;101:875–879. doi: 10.1093/qjmed/hcn109. [DOI] [PubMed] [Google Scholar]
- 16.Song S.H., Hardisty C.A. Early-onset Type 2 diabetes mellitus: an increasing phenomenon of elevated cardiovascular risk. Expert Review of Cardiovascular Therapy. 2008;6:315–322. doi: 10.1586/14779072.6.3.315. [DOI] [PubMed] [Google Scholar]
- 17.Li A.C., Glass C.K. The macrophage foam cell as a target for therapeutic intervention. Nature Medicine. 2002;8:1235–1242. doi: 10.1038/nm1102-1235. [DOI] [PubMed] [Google Scholar]
- 18.Arkan M.C., Hevener A.L., Greten F.R., Maeda S., Li Z.W., Long J.M. IKK-beta links inflammation to obesity-induced insulin resistance. Nature Medicine. 2005;11:191–198. doi: 10.1038/nm1185. [DOI] [PubMed] [Google Scholar]
- 19.Wellen K.E., Hotamisligil G.S. Inflammation, stress, and diabetes. Journal of Clinical Investigation. 2005;115:1111–1119. doi: 10.1172/JCI25102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Neels J.G., Olefsky J.M. Inflamed fat: what starts the fire? Journal of Clinical Investigation. 2006;116:33–35. doi: 10.1172/JCI27280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weisberg S.P., Hunter D., Huber R., Lemieux J., Slaymaker S., Vaddi K. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. Journal of Clinical Investigation. 2006;116:115–124. doi: 10.1172/JCI24335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lumeng C.N., Deyoung S.M., Saltiel A.R. Macrophages block insulin action in adipocytes by altering expression of signaling and glucose transport proteins. American Journal of Physiology-Endocrinology and Metabolism. 2007;292:E166–E174. doi: 10.1152/ajpendo.00284.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Odegaard J.I., Ricardo-Gonzalez R.R., Goforth M.H., Morel C.R., Subramanian V., Mukundan L. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature. 2007;447:1116–1120. doi: 10.1038/nature05894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qatanani M., Szwergold N.R., Greaves D.R., Ahima R.S., Lazar M.A. Macrophage-derived human resistin exacerbates adipose tissue inflammation and insulin resistance in mice. Journal of Clinical Investigation. 2009;119:531–539. doi: 10.1172/JCI37273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Njajou O.T., Kanaya A.M., Holvoet P., Connelly S., Strotmeyer E.S., Harris T.B. Association between oxidized LDL, obesity and type 2 diabetes in a population-based cohort, the health, aging and body composition study. Diabetes/Metabolism Research and Reviews. 2009;25:733–739. doi: 10.1002/dmrr.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Couillard C., Ruel G., Archer W.R., Pomerleau S., Bergeron J., Couture P. Circulating levels of oxidative stress markers and endothelial adhesion molecules in men with abdominal obesity. Journal of Clinical Endocrinology and Metabolism. 2005;90:6454–6459. doi: 10.1210/jc.2004-2438. [DOI] [PubMed] [Google Scholar]
- 27.Kashyap S.R., Ioachimescu A.G., Gornik H.L., Gopan T., Davidson M.B., Makdissi A. Lipid-induced insulin resistance is associated with increased monocyte expression of scavenger receptor CD36 and internalization of oxidized LDL. Obesity (Silver Spring) 2009;17:2142–2148. doi: 10.1038/oby.2009.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Henriksen T., Mahoney E.M., Steinberg D. Interactions of plasma lipoproteins with endothelial cells. Annals of the New York Academy of Sciences. 1982;401:102–116. doi: 10.1111/j.1749-6632.1982.tb25711.x. [DOI] [PubMed] [Google Scholar]
- 29.Quinn M.T., Parthasarathy S., Steinberg D. Endothelial cell-derived chemotactic activity for mouse peritoneal macrophages and the effects of modified forms of low density lipoprotein. Proceedings of the National Academy of Sciences of the United States of America. 1985;82:5949–5953. doi: 10.1073/pnas.82.17.5949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Quinn M.T., Parthasarathy S., Fong L.G., Steinberg D. Oxidatively modified low density lipoproteins: a potential role in recruitment and retention of monocyte/macrophages during atherogenesis. Proceedings of the National Academy of Sciences of the United States of America. 1987;84:2995–2998. doi: 10.1073/pnas.84.9.2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rajavashisth T.B., Andalibi A., Territo M.C., Berliner J.A., Navab M., Fogelman A.M. Induction of endothelial cell expression of granulocyte and macrophage colony-stimulating factors by modified low-density lipoproteins. Nature. 1990;344:254–257. doi: 10.1038/344254a0. [DOI] [PubMed] [Google Scholar]
- 32.Wang G.P., Deng Z.D., Ni J., Qu Z.L. Oxidized low density lipoprotein and very low density lipoprotein enhance expression of monocyte chemoattractant protein-1 in rabbit peritoneal exudate macrophages. Atherosclerosis. 1997;133:31–36. doi: 10.1016/s0021-9150(97)00109-3. [DOI] [PubMed] [Google Scholar]
- 33.Horkko S., Bird D.A., Miller E., Itabe H., Leitinger N., Subbanagounder G. Monoclonal autoantibodies specific for oxidized phospholipids or oxidized phospholipid-protein adducts inhibit macrophage uptake of oxidized low-density lipoproteins. Journal of Clinical Investigation. 1999;103:117–128. doi: 10.1172/JCI4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kobayashi K., Tada K., Itabe H., Ueno T., Liu P.H., Tsutsumi A. Distinguished effects of antiphospholipid antibodies and anti-oxidized LDL antibodies on oxidized LDL uptake by macrophages. Lupus. 2007;16:929–938. doi: 10.1177/0961203307084170. [DOI] [PubMed] [Google Scholar]
- 35.Nagarajan S. Anti-OxLDL IgG blocks OxLDL interaction with CD36, but promotes FcgammaR, CD32A-dependent inflammatory cell adhesion. Immunology Letters. 2007;108:52–61. doi: 10.1016/j.imlet.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 36.Schiopu A., Bengtsson J., Soderberg I., Janciauskiene S., Lindgren S., Ares M.P. Recombinant human antibodies against aldehyde-modified apolipoprotein B-100 peptide sequences inhibit atherosclerosis. Circulation. 2004;110:2047–2052. doi: 10.1161/01.CIR.0000143162.56057.B5. [DOI] [PubMed] [Google Scholar]
- 37.Schiopu A., Frendeus B., Jansson B., Soderberg I., Ljungcrantz I., Araya Z. Recombinant antibodies to an oxidized low-density lipoprotein epitope induce rapid regression of atherosclerosis in apobec-1(−/−)/low-density lipoprotein receptor(−/−) mice. Journal of the American College of Cardiology. 2007;50:2313–2318. doi: 10.1016/j.jacc.2007.07.081. [DOI] [PubMed] [Google Scholar]
- 38.McCurdy C.E., Bishop J.M., Williams S.M., Grayson B.E., Smith M.S., Friedman J.E. Maternal high-fat diet triggers lipotoxicity in the fetal livers of nonhuman primates. Journal of Clinical Investigation. 2009;119:323–335. doi: 10.1172/JCI32661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Messaoudi I., Fischer M., Warner J., Park B., Mattison J., Ingram D.K. Optimal window of caloric restriction onset limits its beneficial impact on T-cell senescence in primates. Aging Cell. 2008;7:908–919. doi: 10.1111/j.1474-9726.2008.00440.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Messaoudi I., Warner J., Fischer M., Park B., Hill B., Mattison J. Delay of T cell senescence by caloric restriction in aged long-lived nonhuman primates. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:19448–19453. doi: 10.1073/pnas.0606661103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nicolaou G., Erridge C. Toll-like receptor-dependent lipid body formation in macrophage foam cell formation. Current Opinion in Lipidology. 2010;21:427–433. doi: 10.1097/MOL.0b013e32833cacd5. [DOI] [PubMed] [Google Scholar]
- 42.Tsimikas S., Miller Y.I. Oxidative modification of lipoproteins: mechanisms, role in inflammation and potential clinical applications in cardiovascular disease. Current Pharmaceutical Design. 2011;17:27–37. doi: 10.2174/138161211795049831. [DOI] [PubMed] [Google Scholar]
- 43.Hernandez-Vargas P., Ortiz-Munoz G., Lopez-Franco O., Suzuki Y., Gallego-Delgado J., Sanjuan G. Fcgamma receptor deficiency confers protection against atherosclerosis in apolipoprotein E knockout mice. Circulation Research. 2006;99:1188–1196. doi: 10.1161/01.RES.0000250556.07796.6c. [DOI] [PubMed] [Google Scholar]
- 44.Fridman W.H. Fc receptors and immunoglobulin binding factors. FASEB Journal. 1991;5:2684–2690. doi: 10.1096/fasebj.5.12.1916092. [DOI] [PubMed] [Google Scholar]
- 45.Herlaar E., Brown Z. p38 MAPK signalling cascades in inflammatory disease. Molecular Medicine Today. 1999;5:439–447. doi: 10.1016/s1357-4310(99)01544-0. [DOI] [PubMed] [Google Scholar]
- 46.Hippenstiel S., Soeth S., Kellas B., Fuhrmann O., Seybold J., Krull M. Rho proteins and the p38-MAPK pathway are important mediators for LPS-induced interleukin-8 expression in human endothelial cells. Blood. 2000;95:3044–3051. [PubMed] [Google Scholar]
- 47.Kaminska B. MAPK signalling pathways as molecular targets for anti-inflammatory therapy—from molecular mechanisms to therapeutic benefits. Biochimica et Biophysica Acta-Proteins and Proteomics. 2005;1754:253–262. doi: 10.1016/j.bbapap.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 48.Goncalves I., Nitulescu M., Ares M.P., Fredrikson G.N., Jansson B., Li Z.C. Identification of the target for therapeutic recombinant anti-apoB-100 peptide antibodies in human atherosclerotic lesions. Atherosclerosis. 2009;205:96–100. doi: 10.1016/j.atherosclerosis.2008.11.020. [DOI] [PubMed] [Google Scholar]
- 49.Goldfine A.B., Fonseca V., Jablonski K.A., Pyle L., Staten M.A., Shoelson S.E. The effects of salsalate on glycemic control in patients with type 2 diabetes: a randomized trial. Annals of Internal Medicine. 2010;152:346–357. doi: 10.1059/0003-4819-152-6-201003160-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shoelson S.E., Goldfine A.B. Getting away from glucose: fanning the flames of obesity-induced inflammation. Nature Medicine. 2009;15:373–374. doi: 10.1038/nm0409-373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Berres M.L., Nellen A., Wasmuth H.E. Chemokines as immune mediators of liver diseases related to the metabolic syndrome. Digestive Diseases. 2010;28:192–196. doi: 10.1159/000282085. [DOI] [PubMed] [Google Scholar]
- 52.Fantuzzi G. Adipose tissue, adipokines, and inflammation. Journal of Allergy and Clinical Immunology. 2005;115:911–919. doi: 10.1016/j.jaci.2005.02.023. quiz 920. [DOI] [PubMed] [Google Scholar]
- 53.Abedini A., Shoelson S.E. Inflammation and obesity: STAMPing out insulin resistance? Immunology and Cell Biology. 2007;85:399–400. doi: 10.1038/sj.icb.7100107. [DOI] [PubMed] [Google Scholar]
- 54.Shoelson S.E., Herrero L., Naaz A. Obesity, inflammation, and insulin resistance. Gastroenterology. 2007;132:2169–2180. doi: 10.1053/j.gastro.2007.03.059. [DOI] [PubMed] [Google Scholar]
- 55.Pascual-Figal D.A., Tornel P.L., Nicolas F., Sanchez-Mas J., Martinez M.D., Gracia M.R. Sex hormone-binding globulin: a new marker of disease severity and prognosis in men with chronic heart failure. Revista española de cardiología. 2009;62:1381–1387. doi: 10.1016/s1885-5857(09)73532-7. [DOI] [PubMed] [Google Scholar]
- 56.Ballantyne C.M., Nambi V. Markers of inflammation and their clinical significance. Atherosclerosis Supplements. 2005;6:21–29. doi: 10.1016/j.atherosclerosissup.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 57.Di Benedetto A., Russo G.T., Corrado F., Di Cesare E., Alessi E., Nicocia G. Inflammatory markers in women with a recent history of gestational diabetes mellitus. Journal of Endocrinological Investigation. 2005;28:34–38. doi: 10.1007/BF03345527. [DOI] [PubMed] [Google Scholar]
- 58.Pawlak K., Pawlak D., Mysliwiec M. Possible new role of monocyte chemoattractant protein-1 in hemodialysis patients with cardiovascular disease. American Journal of Nephrology. 2004;24:635–640. doi: 10.1159/000082936. [DOI] [PubMed] [Google Scholar]
- 59.Sell H., Habich C., Eckel J. Adaptive immunity in obesity and insulin resistance. Nature Reviews Endocrinology. 2012;8:709–716. doi: 10.1038/nrendo.2012.114. [DOI] [PubMed] [Google Scholar]
- 60.Winer S., Winer D.A. The adaptive immune system as a fundamental regulator of adipose tissue inflammation and insulin resistance. Immunology and Cell Biology. 2012;90:755–762. doi: 10.1038/icb.2011.110. [DOI] [PubMed] [Google Scholar]
- 61.Winer S., Chan Y., Paltser G., Truong D., Tsui H., Bahrami J. Normalization of obesity-associated insulin resistance through immunotherapy. Nature Medicine. 2009;15:921–929. doi: 10.1038/nm.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Meisinger C., Baumert J., Khuseyinova N., Loewel H., Koenig W. Plasma oxidized low-density lipoprotein, a strong predictor for acute coronary heart disease events in apparently healthy, middle-aged men from the general population. Circulation. 2005;112:651–657. doi: 10.1161/CIRCULATIONAHA.104.529297. [DOI] [PubMed] [Google Scholar]
- 63.Gerber J.S., Mosser D.M. Stimulatory and inhibitory signals originating from the macrophage Fcgamma receptors. Microbes and Infection. 2001;3:131–139. doi: 10.1016/s1286-4579(00)01360-5. [DOI] [PubMed] [Google Scholar]
- 64.Mosser D.M., Edwards J.P. Exploring the full spectrum of macrophage activation. Nature Reviews Immunology. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mantovani A., Sica A., Sozzani S., Allavena P., Vecchi A., Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends in Immunology. 2004;25:677–686. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 66.He J.S., Takano T., Ding J.Y., Gao S.Y., Noda C., Sada K. Syk is required for p38 activation and G2/M arrest in B cells exposed to oxidative stress. Antioxidants & Redox Signaling. 2002;4:509–515. doi: 10.1089/15230860260196317. [DOI] [PubMed] [Google Scholar]
- 67.Lee H.M., Won K.J., Kim J., Park H.J., Kim H.J., Roh H.Y. Endothelin-1 induces contraction via a Syk-mediated p38 mitogen-activated protein kinase pathway in rat aortic smooth muscle. Journal of Pharmacological Sciences. 2007;103:427–433. doi: 10.1254/jphs.fp0070039. [DOI] [PubMed] [Google Scholar]
- 68.Kirii H., Niwa T., Yamada Y., Wada H., Saito K., Iwakura Y. Lack of interleukin-1beta decreases the severity of atherosclerosis in ApoE-deficient mice. Arteriosclerosis, Thrombosis, and Vascular Biology. 2003;23:656–660. doi: 10.1161/01.ATV.0000064374.15232.C3. [DOI] [PubMed] [Google Scholar]
- 69.Hofmann M.A., Drury S., Fu C., Qu W., Taguchi A., Lu Y. RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell. 1999;97:889–901. doi: 10.1016/s0092-8674(00)80801-6. [DOI] [PubMed] [Google Scholar]
- 70.Koyama H., Shoji T., Yokoyama H., Motoyama K., Mori K., Fukumoto S. Plasma level of endogenous secretory RAGE is associated with components of the metabolic syndrome and atherosclerosis. Arteriosclerosis, Thrombosis, and Vascular Biology. 2005;25:2587–2593. doi: 10.1161/01.ATV.0000190660.32863.cd. [DOI] [PubMed] [Google Scholar]
- 71.Kosaki A., Hasegawa T., Kimura T., Iida K., Hitomi J., Matsubara H. Increased plasma S100A12 (EN-RAGE) levels in patients with type 2 diabetes. Journal of Clinical Endocrinology and Metabolism. 2004;89:5423–5428. doi: 10.1210/jc.2003-032223. [DOI] [PubMed] [Google Scholar]
- 72.Harja E., Bu D.X., Hudson B.I., Chang J.S., Shen X., Hallam K. Vascular and inflammatory stresses mediate atherosclerosis via RAGE and its ligands in apoE-/- mice. Journal of Clinical Investigation. 2008;118:183–194. doi: 10.1172/JCI32703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Soro-Paavonen A., Watson A.M., Li J., Paavonen K., Koitka A., Calkin A.C. Receptor for advanced glycation end products (RAGE) deficiency attenuates the development of atherosclerosis in diabetes. Diabetes. 2008;57:2461–2469. doi: 10.2337/db07-1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mori Y., Kosaki A., Kishimoto N., Kimura T., Iida K., Fukui M. Increased plasma S100A12 (EN-RAGE) levels in hemodialysis patients with atherosclerosis. American Journal of Nephrology. 2009;29:18–24. doi: 10.1159/000148646. [DOI] [PubMed] [Google Scholar]
- 75.Mahajan N., Bahl A., Dhawan V. C-reactive protein (CRP) up-regulates expression of receptor for advanced glycation end products (RAGE) and its inflammatory ligand EN-RAGE in THP-1 cells: inhibitory effects of atorvastatin. International Journal of Cardiology. 2009;142:273–278. doi: 10.1016/j.ijcard.2009.01.008. [DOI] [PubMed] [Google Scholar]
- 76.Hasegawa T., Kosaki A., Kimura T., Matsubara H., Mori Y., Okigaki M. The regulation of EN-RAGE (S100A12) gene expression in human THP-1 macrophages. Atherosclerosis. 2003;171:211–218. doi: 10.1016/j.atherosclerosis.2003.08.021. [DOI] [PubMed] [Google Scholar]
- 77.Wuttge D.M., Eriksson P., Sirsjo A., Hansson G.K., Stemme S. Expression of interleukin-15 in mouse and human atherosclerotic lesions. American Journal of Pathology. 2001;159:417–423. doi: 10.1016/S0002-9440(10)61712-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cucina A., Scavo M.P., Muzzioli L., Coluccia P., Ceccarini S., Fuso A. High density lipoproteins downregulate basic fibroblast growth factor production and release in minimally oxidated-LDL treated smooth muscle cells. Atherosclerosis. 2006;189:303–309. doi: 10.1016/j.atherosclerosis.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 79.Ditiatkovski M., Toh B.H., Bobik A. GM-CSF deficiency reduces macrophage PPAR-gamma expression and aggravates atherosclerosis in ApoE-deficient mice. Arteriosclerosis, Thrombosis, and Vascular Biology. 2006;26:2337–2344. doi: 10.1161/01.ATV.0000238357.60338.90. [DOI] [PubMed] [Google Scholar]
- 80.Chen Y., Zhu J., Lum P.Y., Yang X., Pinto S., MacNeil D.J. Variations in DNA elucidate molecular networks that cause disease. Nature. 2008;452:429–435. doi: 10.1038/nature06757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Emilsson V., Thorleifsson G., Zhang B., Leonardson A.S., Zink F., Zhu J. Genetics of gene expression and its effect on disease. Nature. 2008;452:423–428. doi: 10.1038/nature06758. [DOI] [PubMed] [Google Scholar]
- 82.Xu H., Barnes G.T., Yang Q., Tan G., Yang D., Chou C.J. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. Journal of Clinical Investigation. 2003;112:1821–1830. doi: 10.1172/JCI19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nishi K., Itabe H., Uno M., Kitazato K.T., Horiguchi H., Shinno K. Oxidized LDL in carotid plaques and plasma associates with plaque instability. Arteriosclerosis, Thrombosis, and Vascular Biology. 2002;22:1649–1654. doi: 10.1161/01.atv.0000033829.14012.18. [DOI] [PubMed] [Google Scholar]
- 84.Schiopu A., Bengtsson J., Söderberg I., Janciauskiene S., Lindgren S., Ares M.P.S. Recombinant human antibodies against aldehyde-modified apolipoprotein B-100 peptide sequences inhibit atherosclerosis. Circulation. 2004;110:2047–2052. doi: 10.1161/01.CIR.0000143162.56057.B5. [DOI] [PubMed] [Google Scholar]
- 85.Cheng X., Folco E.J., Shimizu K., Libby P. Adiponectin induces pro-inflammatory programs in human macrophages and CD4+ T cells. Journal of Biological Chemistry. 2012;287:36896–36904. doi: 10.1074/jbc.M112.409516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Festa A., Kopp H.P., Schernthaner G., Menzel E.J. Autoantibodies to oxidised low density lipoproteins in IDDM are inversely related to metabolic control and microvascular complications. Diabetologia. 1998;41:350–356. doi: 10.1007/s001250050914. [DOI] [PubMed] [Google Scholar]
- 87.Ammirati E., Cianflone D., Vecchio V., Banfi M., Vermi A.C., De Metrio M. Effector memory T cells are associated with atherosclerosis in humans and animal models. Journal of American Heart Association. 2012;1:27–41. doi: 10.1161/JAHA.111.000125. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material
Supplementary material