

Abstract
Fibroblast growth factor 21 is an emerging metabolic regulator that was recently proposed to be a fed-state inducible factor in adipose tissue. As mice lacking FGF21 were refractory to treatment with rosiglitazone, FGF21 was suggested to underlie PPARγ-driven pharmacology and side effect profile (Dutchak et al., 2012 [12]). To evaluate FGF21/PPARγ cross-talk we conducted experiments in control and FGF21 null animals and found that rosiglitazone was equally efficacious in both strains. Specifically, diverse endpoints ranging from enhanced glycemic control, improved lipid homeostasis and side effects such as adipose accumulation were evident in both genotypes. Furthermore, the transcriptional signature and cytokine secretion profile of rosiglitazone action were maintained in our FGF21KO animals. Finally, we found that FGF21 in adipose was expressed at comparable levels in fasted and fed states. Thus, our data present a new viewpoint on the FGF21/PPARγ interplay whereby FGF21 is not necessary for the metabolic events downstream of PPARγ.
Keywords: FGF21, Rosiglitazone, PPARγ, Metabolism, Adiponectin
1. Introduction
Fibroblast growth factor 21 (FGF21) [35] has recently garnered significant attention as a novel metabolic regulator and a promising agent to treat diabetes and other metabolic diseases [23]. FGF21 is a member of the so called “endocrine” FGF sub-family, as unlike the canonical FGFs it does not possess a heparin binding domain, thus is able to escape into the circulation. When administered to animal models of diabetes and obesity native FGF21 and more recently engineered FGF21 variants cause dramatic and rapid improvements in overall metabolic health [17,25,33,42]. Mice overexpressing FGF21 are lean, resistant to the development of diet induced obesity [24] and exhibit significantly augmented lifespan [46].
Physiologically, FGF21 is regulated by a number of transcription factors in a complex and tissue specific manner [3]. The peroxisome proliferator-activated receptors (PPARs) were among the first molecular entities reported to modulate FGF21 expression [6,21,39,43]. To date, both PPARα and PPARγ agonists have been demonstrated to induce FGF21 secretion in vitro and in vivo [6,8,16,37]. However, while PPAR ligands such as the thiazoladinediones (TZDs) can modulate FGF21 levels, the functional contribution of FGF21 to the in vivo activity of the PPARs has yet to be established.
Recently it was proposed that FGF21 is an inducible, autocrine factor in white adipose tissue (WAT) and a critical element in a feed forward loop regulating the activity of PPARγ [12]. This novel paradigm represents a significant departure from the traditional viewpoint of FGF21 elevation in liver as a hallmark of the fasting response [6,21,31], and suggests that in addition to its endocrine function, FGF21 may also act in an autocrine manner in specific tissues such as adipose. Furthermore, it was also proposed that FGF21KO mice are refractory to treatment with the PPARγ agonist rosiglitazone [12]. The latter finding is of particular significance since it positions adipose tissue derived FGF21 downstream of PPARγ to serve as a necessary driver for the wide spectrum of beneficial as well as undesirable pharmacologic consequences associated with the use of the TZD class of therapeutics.
Given the potential for these findings to reshape the understanding of the in vivo mechanisms of action of both FGF21 and PPARγ, we sought to further explore them through a series of experiments designed to elucidate the interactions between these two important metabolic pathways.
First, we observed that our own strain of independently derived FGF21KO mice did not display a lipodsytrophic phenotype and instead presented with mild obesity, a condition which was exacerbated by feeding of a high fat diet. Furthermore, we found that rosiglitazone was equally efficacious in ameliorating hyperglycemia and insulin resistance and impacting other systemic measures of metabolic health in both WT and FGF21KO mice. Excess adiposity, increased circulating liver enzyme levels as well as elevations in adiponectin were also fully evident in rosiglitazone-treated FGF21 null animals.
At the molecular level the transcriptional signature of TZD administration in WAT was largely unchanged in mice lacking FGF21; however, we did observe increased expression of the cofactor critically required for FGF21 signaling, KLB [22,36,41]. Concordant with this finding, in mice treated with a combination of FGF21 and rosiglitazone we observed robust enhancement of FGF21s metabolic actions and the absence of side effects when rosiglitazone is administered alone. Finally, in an effort to further explore the differences between previously published findings [12] and our own data, we examined PPARγ in vivo sumoylation and FGF21 mRNA expression in various tissues. Under the conditions used in our study, we were unable to detect FGF21 mediated sumoylation of PPARγ and noted no substantial regulation of adipose FGF21 transcript upon fasting.
Taken together our data suggest that FGF21 does not play a key role downstream of PPARγ activation, and is not required for the in vivo efficacy of rosiglitazone. Thus, in our hands FGF21 and PPARγ appear to operate as potent metabolic regulators acting via functionally distinct and independent mechanisms.
2. Methods
2.1. Animals
All animals were individually housed in a temperature-controlled (24 °C) facility with 12 h/12 h light/dark cycle. The animal protocols in this study were approved by the Eli Lilly and Co. Animal Use and Care Committee (Protocol nos. 12225 and 13030).
2.2. Metabolic phenotype of chow and HFD fed WT and FGF21 null mice
For basal phenotype studies male WT (n=24) and FGF21KO (n=24) mice were maintained on a chow diet until 10 weeks of age. At this point mice were weighed and body composition was analyzed by qNMR. Development of diet induced obesity in 10 week old male WT (N=8) and FGF21KO (n=8) mice was evaluated following maintenance on a calorie-rich diet consisting of 40% fat, 39% carbohydrate, and 21% protein caloric content (TD95217; Harlan Teklad, Madison, WI) for a period of 12 weeks. Following the 12-week diet exposure animals were weighed and body composition was determined using qNMR.
2.3. Rosiglitazone treatment of DIO WT and FGF21KO mice
Male WT and FGF21KO mice (n=8 per group) were maintained on a calorie-rich diet consisting of 40% fat, 39% carbohydrate, and 21% protein caloric content (TD95217; Harlan Teklad, Madison, WI) for a period of 12 weeks prior to the study onset and had free access to food and water before randomization by weight. Mice were administered rosiglitazone at the doses indicated for a period of 14 days via oral gavage. Prior sacrifice glucose levels were determined using Precision G Blood Glucose Testing System (Abbott Laboratories, Abbott Park, IL). Following sacrifice tissues were rapidly dissected and flash frozen in liquid nitrogen.
2.4. FGF21/rosiglitazone combination treatment of WT DIO mice
Male WT mice (n=7 per group) were maintained on a calorie-rich diet consisting of 40% fat, 39% carbohydrate, and 21% protein caloric content (TD95217; Harlan Teklad, Madison, WI) for a period of 12 weeks prior to the study onset and had free access to food and water before randomization by weight. Mice were administered either FGF21 for a period of 10 days via continuous infusion using osmotic minipumps (ALZET, Cupertino, CA) at a dose of 1 mg/kg. Following recovery from surgery mice were administered either vehicle or rosiglitazone at the doses indicated for a period of 14 days via oral gavage. Prior sacrifice glucose levels were determined using Precision G Blood Glucose Testing System (Abbott Laboratories, Abbott Park, IL). Following sacrifice tissues were rapidly dissected and flash frozen in liquid nitrogen.
2.5. Analysis of metabolites and circulating factors
Blood samples were collected on ice temperature prior to storage of plasma at −80 °C. Serum metabolites were measured by small-scale enzymatic assays for glucose, triglycerides, and cholesterol (Stanbio Laboratory). Insulin (Crystal Chem Inc.), leptin (Crystal Chem Inc.) and Total Adiponectin (BioVendor Inc.) were measured by specific ELISA.
2.6. Body composition analysis
Body composition of mice was determined using Quantitative Nuclear Magnetic Resonance analysis (ECHO MRI, 3-1 Composition Analyzer; Echo Medical Systems, Houston, TX).
2.7. RNA isolation, RT and real-time quantitative PCR
RNA was isolated from tissues using TRIzol reagent (Invitrogen, Carlsbad, CA) or by homogenization of frozen samples in Lysing Matrix D shaker tubes (MP Biomedicals, Santa Ana, CA) and was reverse transcribed into cDNA using a High-Capacity cDNA Reverse Transcription Kit (PE Applied Biosystems, Foster City, CA). Reactions were performed in triplicate on an ABI Prism 7900 HT (PE Applied Biosystems) and were normalized to PPIA mRNA.
2.8. Statistical analysis
Data are presented as mean±SEM. Statistical analysis was performed using one-way ANOVA, followed by Dunnett's multiple comparisons test where appropriate. Differences were considered significant when P=<0.05.
3. Results
3.1. FGF21KO mice have mildly increased adiposity
We have previously reported that deletion of the FGF21 gene leads to a basal phenotype of mild obesity and glucose intolerance [5]. Given the lipodystrophy recently reported in a separate strain of FGF21KO animals [12] we re-examined a large age matched cohort of WT (n=30) and FGF21KO (n=24) mice. We found that WT mice when maintained on a chow diet had significantly lower body weight than their FGF21KO counterparts (Figure 1A). Using qNMR analysis of body composition we determined that the difference in body weight was largely due to significantly elevated adiposity in the FGF21KO mice (Figure 1B), similar to our previously reported data [5]. Fat free mass (Figure 1C) and total water (Figure 1D) were not different between WT and FGF21KO mice. When body composition was expressed as a percentage of total body mass we again found that there was significantly greater fat mass in the FGF21KO mice when compared to the WT animals (Figure 1E) while lean mass was not different between the cohorts (Figure 1F).
Figure 1.
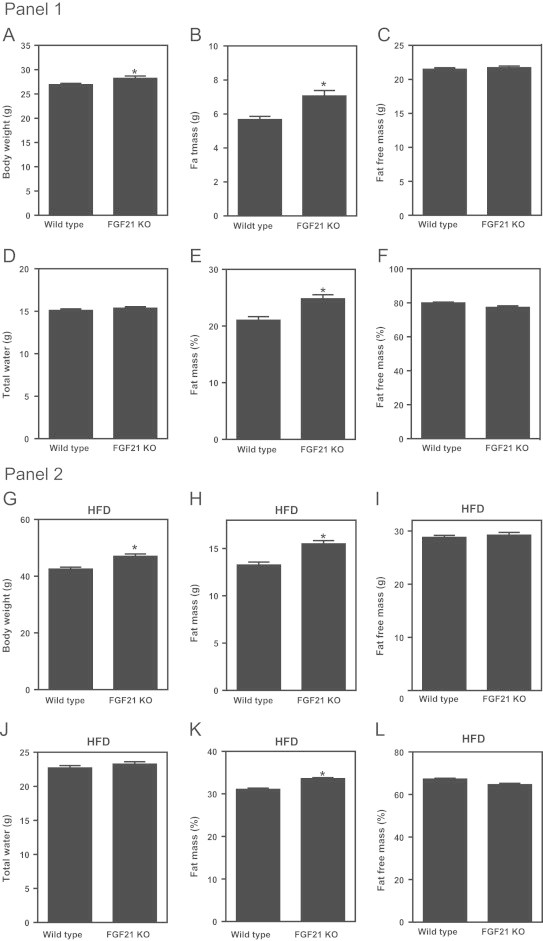
FGF21 null mice display mild obesity which is exacerbated by a high fat diet. Panel 1. Body weight was determined in a large cohort of chow fed, age matched male WT (n=30) and FGF21KO mice (n=24) (A). In the same cohort we also assessed body composition via qNMR and determined absolute fat mass (B), fat free mass (C) and water mass (D). The measures of both fat mass (E) and fat free mass (F) were then also expressed as a percentage of total body mass. Statistical significance is denoted by *. Differences were considered significant when P≤0.05.Panel 2. At 10 weeks of age WT and FGF21KO mice were fed a HFD for a period of 12 weeks. Following diet exposure we determined body weight (G), absolute fat mass (H), fat free mass (I) and water mass (J). The measures of both fat mass (K) and fat free mass (L) were then also expressed as percentage of total body mass. Statistical significance is denoted by *. Differences were considered significant when P≤0.05.
To determine if this mild basal phenotype would be altered following a dietary challenge we placed 10-week old WT and FGF21 null mice on a high fat diet for a period of 12 weeks. As on chow diet, the weights of the HFD fed FGF21KO mice were elevated over that of the WT animals (Figure 1G). This difference in body mass was again due to significantly higher adiposity in the FGF21 null mice (Figure 1H) since fat free mass was not altered between the genotypes (Figure 1I). There was also no difference in total water between the two HFD cohorts (Figure 1J). When expressed as a percentage of total body mass there remained a significant elevation in adiposity (Figure 1K) in the FGF21 null animals while fat free mass (Figure 1L) was again unchanged between the groups. Thus, on both normal chow and high fat diet, FGF21 null mice were obese in agreement with the phenotype of a third independent FGF21KO strain [20] and the ability of exogenous FGF21 to correct adiposity in mice (Coskun et al. [9]) but converse to the lipodystrophic state reported by Dutchak et al. [12] in their FGF21KO animals.
3.2. FGF21 null animals are sensitive to rosiglitazone treatment
If FGF21 is required for full PPARγ activity, then FGF21KO mice should be resistant to the actions of TZDs. Thus, we sought to further investigate the effects of chronic rosiglitazone administration in FGF21KO mice. In our experiments the rosiglitazone effect on hyperglycemia and insulin resistance was fully evident in FGF21KO mice when compared to their WT counterparts as plasma glucose and insulin lowering in the DIO setting was equivalent in both genotypes (Figure 2A & 2B). A comparable reduction of plasma lipids such as triglycerides (Figure 2C), cholesterol (Figure 2D) or free fatty acids (Figure 2E) was also observed in rosiglitazone-treated WT or FGF21KO animals.
Figure 2.
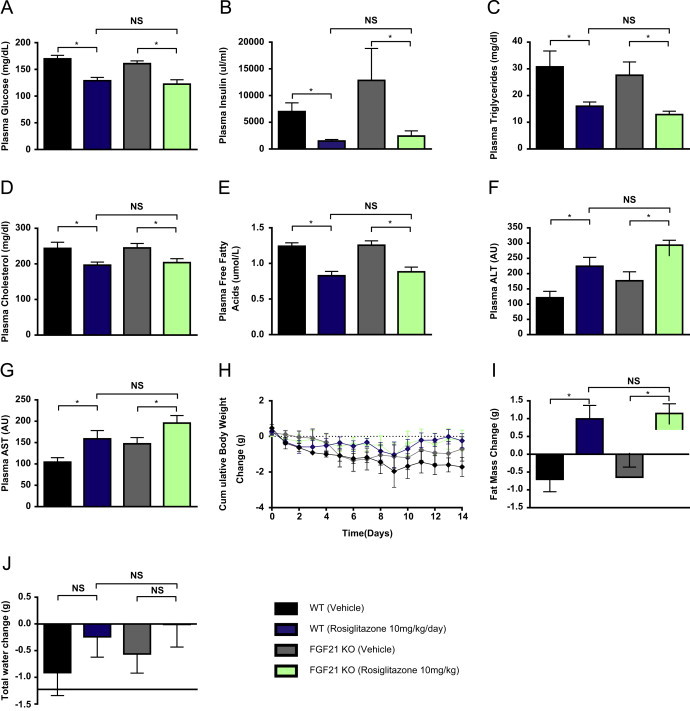
FGF21 is not required for the metabolic effects of TZD treatment in DIO mice. To determine the extent to which the effects of rosiglitazone are mediated by FGF21 we examined plasma glucose (A), insulin (B), triglycerides (C), Cholesterol (D) and free fatty acids (E), ALT (F) and AST (G) in WT and FGF21 null mice. We also examined the effects of chronic treatment on cumulative body weight change (H), change in fat mass (I) and change in total body water content (J).
Consistent with earlier reports [44] significant increases in plasma ALT (Figure 2F) and AST (Figure 2G) levels were also detected under rosiglitazone treatment in both strains of mice. Of potential interest, and opposite to the rosiglitazone mediated effect on liver enzymes, administration of 1 mg/kg of recombinant FGF21 led to an approximate reduction in ALT and AST of up to 50% in both WT and FGF21KO animals potentially attributable to FGF21-induced clearance of liver lipids (data not shown) and evidencing an improved hepatic health.
Rosiglitazone also caused a minor but equivalent body weight gain in WT and FGF21KO mice (Figure 2H) with increased adiposity present to a similar degree in both cohorts (Figure 2I). While fluid accumulation trended to be higher with rosiglitazone treatment this effect neither reached significance nor was different between the two genotypes (Figure 2J). Thus, the overall results of our study indicate that mice lacking FGF21 are fully responsive to TZD treatment with regard to multiple integrated metabolic endpoints.
We attempted to carry out our initial in vivo study to match as closely as possible the conditions which were employed in the previous report [12]. Specifically, we replicated time on diet, dosing regimen, and age & sex of animals. It is important howewer to note that factors such as strain of mice, backcrossing status and source of HFD were unable to be replicated and may contribute to the differences we observe compared to [12].
3.3. The transcriptional response to TZD treatment is largely preserved in FGF21KO mice
Given that significant alterations in the rosiglitazone gene expression signature were reported [12] we next examined rosiglitazone mediated transcriptional effects in FGF21 null animals. As with the integrated readouts at the whole body level, the induction of known PPARγ target genes in WAT such as adiponectin and UCP1 was fully apparent and reached a similar magnitude of elevation in WT and FGF21KO mice (Figure 3). Furthermore, the repression of FASN and TNFα gene expression was also equally robust regardless of genotype (Figure 3).
Figure 3.
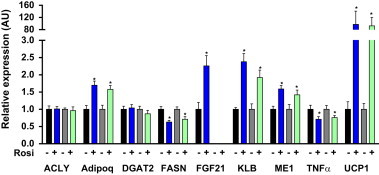
Mice lacking FGF21 preserve the transcriptional hallmarks of rosiglitazone treatment. Following treatment with either vehicle or rosiglitazone we assessed gene expression in WAT of WT and FGF21KO mice. Statistical analysis was performed using one-way ANOVA, followed by Dunnett's multiple comparisons test where appropriate. Statistical significance from vehicle was denoted by * while differences between genotypes are indicated with †. Differences were considered significant when P≤0.05.
Consistent with our previous report [32], expression of KLB, the FGF21 co-receptor [22,36,41], was up-regulated by rosiglitazone treatment in both genotypes suggesting a possible role for rosiglitazone as a sensitizer of FGF21 action (Figure 3). As previously communicated [12,34] we also observed a rosiglitazone-induced increase of FGF21 mRNA in the WAT of WT mice (Figure 3). It is important to note however that FGF21 expression in adipose tissue was extremely low when compared to that observed in the pancreas and liver.
3.4. Biomarkers of rosiglitazone treatment are equally induced in WT and FGF21KO mice
Our initial study was unable to replicate a lack of TZD efficacy in FGF21 null mice. Thus, we conducted an additional in vivo experiment in WT and FGF21KO animals with both the original 10 mg/kg and a sub-maximal 3 mg/kg dose of rosiglitazone. We reasoned that the high 10 mg/kg dose could be masking a possible contribution of FGF21 to the PPARγ-driven pharmacologic effect, and a lower dose may provide an opportunity to reveal the specific involvement of FGF21 in the mechanism of rosiglitazone action.
Confirming our earlier data detailed in Figure 2, we observed reductions in both plasma glucose (Figure 4B) and insulin (Figure 4C) in animals treated with the 10 mg/kg dose. Importantly, the equal efficacy in WT and FGF21KO cohorts was also detected even at the submaximal 3 mg/kg dose. At either dose and in both groups of rosiglitazone-treated animals we found significant increases in adiponectin, a classical biomarker of TZD action in vivo [27] (Figure 4D). Furthermore, since adiponectin levels in the basal state were significantly lower in FGF21KO mice as compared to their WT counterparts (Figure 4D), FGF21 is likely a critical physiological factor for maintaining adiponectin production in mice.
Figure 4.
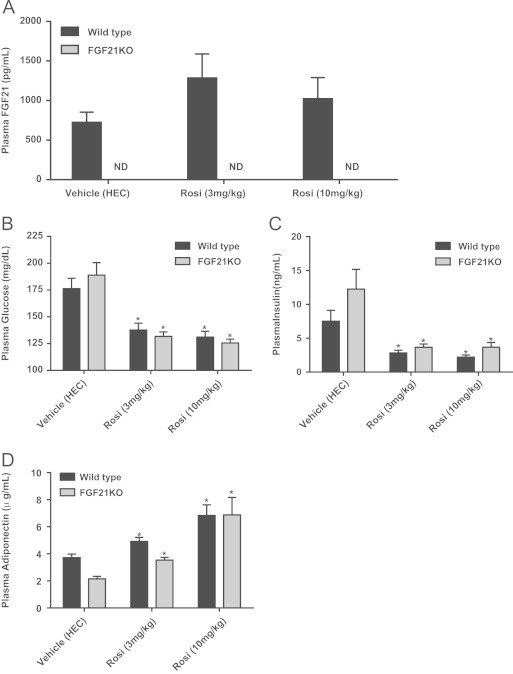
FGF21KO animals respond to both 3 mg/kg and 10 mg/kg doses of rosiglitazone. FGF21 levels were measured in plasma of WT and FGF21KO mice following treatment with either vehicle or rosiglitazone at either 3 mg/kg or 10 mg/kg using ELISA (A). Following sacrifice of WT and FGF21KO mice we examined plasma glucose (B), insulin (C) and total adiponectin (D). Statistical analysis was performed using one-way ANOVA, followed by Dunnett's multiple comparisons test where appropriate. Statistical significance from vehicle was denoted by *. Differences were considered significant when P≤0.05.
We also assessed the effect of rosiglitazone administration on FGF21 production and found no increase in FGF21 in the serum (Figure 4A) consistent with the hypothesis that adipose tissue-originated FGF21 does not function as an endocrine factor [12,15]. The data regarding the induction of circulating FGF21 by TZD treatment is unclear with conflicting evidence for Oishi and Tomita [38] and against Christodoulides et al. [8] a role for PPARγ in modulating FGF21 levels in blood. Of particular interest, chronic rosiglitazone treatment was reported to lower circulating FGF21 in man [28].
3.5. The metabolic response to FGF21 is amplified in rosiglitazone treated animals
Treatment with rosiglitazone led to an increase in KLB expression in adipose, a critical target tissue where the majority of FGF21s metabolic action is initiated [1,4]. Given high levels of FGFR1c expression in fat, elevation in KLB expression may invoke greater abundance of the FGF21 receptor complex thus leading to an augmented response to FGF21. To test this hypothesis we treated mice with FGF21 in combination with rosiglitazone and assessed systemic measures of metabolic health. Following chronic treatment with FGF21 we observed the expected reduction in body weight and increased fat mass in the rosiglitazone treated group. However, in the combination group that was dosed with both FGF21 and rosiglitazone, a trend toward exaggerated weight loss was observed (Figure 5A). Glucose levels were reduced by both FGF21 and rosiglitazone when either agent was administered alone with a similar reduction in glycemia detected in the combination treated group (Figure 5B). Concomitant with glucose lowering a significant reduction in insulin levels was also detected upon either FGF21 or rosiglitazone administration. However, in the case of ameliorating hyperinsulinemia, the combination of FGF21 and rosiglitazone led to a significantly greater efficacy than was found with either agent alone (Figure 5C). Lipid lowering followed a similar pattern with reductions in circulating cholesterol (Figure 5D) and FFA's (Figure 5E) observed with both agents and an increased efficacy in FGF21/rosiglitazone treated animals. Importantly, while the increased body water retention was fully evident in mice receiving rosiglitazone (Figure 5F), water mass loss was akin to that seen with FGF21 alone when both agents were co-administered.
Figure 5.
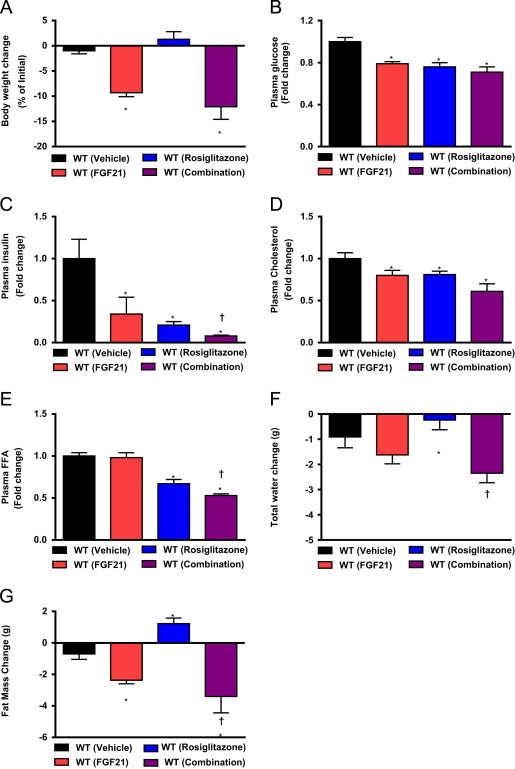
Treatment with rosiglitazone sensitizes to the metabolic effects of FGF21. To assess the potential for interaction between FGF21 and rosiglitazone we compared treatment with either agent alone versus a combination of the two. Following treatment we examine body mass change (A), plasma glucose (B), insulin (C), Cholesterol (D) and free fatty acids (E) in addition to change in total water (F) and fat mass gain/loss (G). Statistical significance from vehicle was denoted by * while differences from rosiglitazone alone are indicated with †. Differences were considered significant when P≤0.05.
Finally, while rosiglitazone caused the expected but undesirable increase in adipose tissue, FGF21 led a significant reduction in fat mass, and this loss of adipose was substantially more pronounced in animals treated with the FGF21/rosiglitazone combination (Figure 5G). Finally, while rosiglitazone caused the expected but undesirable increase in adipose tissue, FGF21 led a significant reduction in fat mass, and this loss of adipose was substantially more pronounced in animals treated with the FGF21/rosiglitazone combination (Figure 5G). Our findings indicate that TZD treatment appears to maintain or even enhance the physiological response to FGF21. The latter is suggestive that rosiglitazone sensitizes animals to FGF21 action. More critically, FGF21 is able to rescue some of the negative aspects of the TZD pharmacology in mice. While at the molecular level rosiglitazone induced KLB upregulation is likely to be a cause for the enhanced metabolic potency of FGF21/TZD combination, the ability of FGF21 to “brown” adipose [15] may also be a contributing mechanism at the tissue-specific level.
3.6. Expression of FGF21 in WAT is unaffected by the fed to fasted transition
Consistent with earlier observations [6,21], overnight fasting led to a significant increase in plasma FGF21 in WT mice (Figure 6A). To further delineate the tissue specific response to nutrient deprivation we evaluated FGF21 expression in animals transitioning from the fed to fasted state at the tissue specific level, and profiled FGF21 mRNA in WAT, liver and pancreas—the main sources for FGF21 production in rodents [23].
Figure 6.
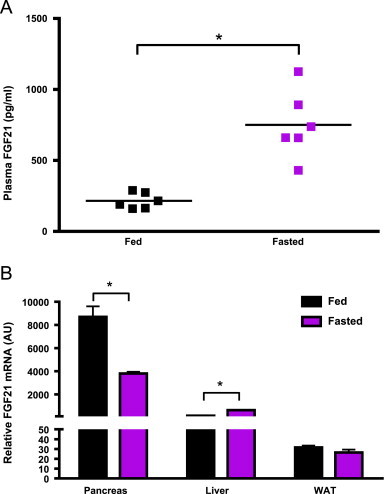
FGF21 expression is regulated by the fed to fasted transition in liver and pancreas but not in adipose. FGF21 levels were measured in plasma of fed and fasted mice using ELISA (A). Tissue specific expression of FGF21 was assessed in the pancreas, liver and WAT at the mRNA level (B). Statistical analysis was performed using one-way ANOVA, followed by Dunnett's multiple comparisons test where appropriate. Statistical significance from vehicle was denoted by *. Differences were considered significant when P≤0.05.
The highest relative expression in the fed state was observed in the pancreas followed by the liver (Figure 6B). In WAT, FGF21 gene transcript was approximately five fold lower than observed in liver and orders of magnitude below than that in the pancreas (Figure 6B). Interestingly, FGF21 mRNA was appreciably reduced in the pancreas upon fasting while the induction of FGF21 transcript in the liver was consistent with earlier studies [6,21,31]. However, in contrast to the result reported by Dutchak et al. [12], we detected no significant difference in FGF21 mRNA levels in white adipose from either fed or fasted mice (Figure 6B). While we measured FGF21 mRNA levels after an overnight fast and 24 hours after initiation of the dark cycle, previous reports [12,38] utilized a restricted feeding paradigm whereby animals are allowed access to food for only a short period of time, possibly explaining the observed differences. Further complicating the possible role of adipose derived FGF21 are data from cold exposed mice where FGF21 levels in circulation also did not change while mRNA in adipose was increased. Therefore, it was proposed that cold exposure elevates local WAT FGF21.
3.7. FGF21 does not alter the sumoylation state of PPARγ
The earlier reported effect of FGF21 to alter PPARγ sumoylation [12] is of importance as this observation was one of the first to demonstrate such modification of PPARγ in living animals. However, we were unable to detect sumoylated PPARγ in vivo and observed only excessive background staining in the area of 50kDa, likely due to detection of endogenous mouse antibodies present in tissue extracts (Supplemental Figure 1). Indeed, a previous publication demonstrated that sumoylation of PPARγ was not evident in the basal state and was only detectable following ligand stimulation of PPARγ [40].
4. Discussion
In this report we show that FGF21 is neither required for the beneficial metabolic outcomes of rosiglitazone treatment nor is a prerequisite for the side effects associated with the use of the TZD class of therapeutics. In our studies, all of the profiled responses to rosiglitazone were fully preserved in FGF21KO mice both at the whole animal and molecular level. Of note, the report by Dutchak et al. [12] also demonstrated that both circulating and liver triglyceride levels were lowered by rosiglitazone treatment regardless of genotype, and the attenuation of the ability of rosiglitazone to control glyceamia in FGF21KO mice was fairly subtle raising the question if FGF21 null mice are indeed refractory to TZD treatment. Taken together, our data demonstrate that the link between FGF21 and PPARγ is certainly not as simple as originally suggested.
As compared to WT animals our FGF21KO mice retained the full physiological response to rosiglitazone (Figures 2, 4 and 5). Indeed, rosiglitazone effects observed in FGF21 null animals spanned the traditional gamut of TZD pharmacologic actions—including insulin sensitization, glucose and lipid lowering, elevation of liver enzyme levels and accrual of adipose tissue mass. However, one potential concern we had was that the 10 mg/kg dose of rosiglitazone could overwhelm the impact of FGF21 ablation; therefore we conducted a follow-up experiment utilizing a lower dose of rosiglitazone. Nevertheless, even at the submaximal level of 3 mg/kg, comparable effects of rosiglitazone were observed regardless of genotype further indicative that FGF21 is not required for PPARγ action in vivo. The consistent rosiglitazone signature at the transcriptional level was also noted in both the WT and FGF21KO mice, lending added support to the lack of alterations of PPARγ-driven pharmacologic responses at the whole animal level. Finally, a dose dependent increase in circulating adiponectin was fully apparent in FGF21 null mice. The latter is important as recent reports show FGF21 is a potent adiponectin secretagogue and that this induction in adiponectin plasma levels is vital for the glycemic efficacy of FGF21 in vivo [18,29]. Given that following rosiglitazone stimulation adiponectin was also elevated in the FGF21KO mice our findings suggest that FGF21 and PPARγ regulate adiponectin levels via functionally distinct and independent pathways.
As we reported earlier, treatment of adipocytes with rosiglitazone did increase cell sensitivity to FGF21 [32]. This in vitro observation is suggestive of the possibility that rosiglitazone may act in vivo by inducing KLB expression in adipose tissue, a critical target organ for FGF21 in living animals [1,4,42]. Furthermore, given the apparent excess of FGFR1 expression over that of KLB in WAT [1] we thought that KLB induction may lead to a higher abundance of KLB/FGFR1 complex – FGF21 receptor in adipose tissue – thus providing molecular means for animals' increased response to FGF21. Indeed, when rosiglitazone and FGF21 were co-administered we noted a significant enhancement of the FGF21 action in DIO mice (Figure 5B and E). Importantly, in the FGF21/rosiglitazone treated group we did not find an intermediate phenotype of rosiglitazone side effects—such as adipose mass accrual and elevation in liver enzyme levels. Specifically, fat mass in the FGF21/rosiglitazone combination group was reduced more than with FGF21 alone (Figure 5G), supportive of a functional and rather synergistic relationship between the two factors. These data present the possibility that the PPARγ pathway may indeed intersect with the actions of FGF21, however, in a distinctly different manner than initially proposed.
Confirming earlier reports we show in our study that FGF21 in mouse plasma is increased following a 24h fast and that hepatic FGF21 mRNA was elevated by nutrient deprivation. Furthermore, while [12] observed elevated levels of FGF21 mRNA in WAT upon feeding, our data indicate that FGF21 in adipose tissue was not significantly altered between fed and 24h fasted mice. Finally, FGF21 transcript in pancreas (Figure 5B) was several orders of magnitude higher than in liver and WAT and was reduced upon fasting posing the question as to the role of pancreatic FGF21 in the overall physiology of this protein. Thus, future work with deletion of FGF21 in each of these three metabolically-relevant organs is required to determine the relative contribution of tissue-specific FGF21 to distinct facets of FGF21 physiology.
As communicated in Dutchak et al. [12], the FGF21 concentration in adipose tissue is particularly low compared to other organs. Indeed, the FGF21 protein level in WAT equates to approximately 1.1 ng/g, or 0.99 ng/ml when the average tissue density of 0.92 for WAT [30] is taken into account. While this finding aligns well with a recent report demonstrating the absence of FGF21 mRNA in human adipose tissue [11] it is unclear if such low FGF21 levels are able to induce a meaningful activation of the FGF21 receptor complex given dose response parameters for FGF21 activity in adipose derived cells [2,24,26,45]. Indeed, an FGF21 concentration of 0.99 ng/ml [12] is 10× lower than the EC50 of approximately 10 ng/ml established for FGF21 action in adipocytes [24]. While FGF21 protein reached 2ng/g (or 1.8 ng/ml) in WAT following rosiglitazone treatment [12], this level remains below the threshold for meaningful local signaling to occur, raising doubts as to the ability of WAT-produced FGF21 to efficiently function in adipose tissue in an autocrine manner.
While we believe it is unlikely that FGF21 originated from adipose tissue plays a major role downstream of PPARγ, there are states as prolonged cold exposure at which local FGF21 signaling may occur. One such state is FGF21-induced “browning” of white adipose tissue, a phenomenon which was initially shown in FGF21 transgenic animals [24] and was recently reported in FGF21 treated mice [15]. FGF21 also is able to activate extant brown adipose tissue [19] and is also induced in BAT following cold exposure [7] suggesting that the thermogenic response to FGF21 may result from both activation of BAT and “browning” of WAT. Indeed, adipose is a critical tissue for FGF21's metabolic mechanism of action as it carries FGFR1c and KLB, together comprising FGF21 receptor, and the deletion of either of these vital components of the FGF21 receptor machinery profoundly disrupts both pharmacological and physiological responses to FGF21 in vivo ([1,4,10]). Nevertheless, the contribution of liver and pancreas as other important target tissues for FGF21 action must also be considered [1,4,14].
The discrepancy between the fat mass phenotypes of the three FGF21 lines reported to date is puzzling with two lines presenting with mixed elevations in diet dependent adiposity [5,13,20] while the third line manifests with reduced fat mass [12]. It has been suggested that the differences between the three lines may lie in the presence or absence of soy in the diet used by the various research groups [12]. Nevertheless, the chow maintenance diet (Harlan Teklad, Rodent maintenance diet, No. 2014) utilized in the present study also does not contain soy. Lending further support to the obese phenotype of FGF21KO mice on a chow diet is the predisposition of these animals to gain more weight and specifically fat when placed on a high fat diet (Figure 5H).
While the exact reason for the difference in phenotype between the three FGF21KO lines is unknown [5,20,12] one possibility lies with the breeding strategy utilized by Dutchak and colleagues versus our own, the specific diets employed and the strain background of the study animals.
In conclusion our data neither support the requirement of FGF21 for the actions of rosiglitazone, nor do they corroborate the proposed role for FGF21 as an autocrine factor in adipose tissue. Rather, our studies provide a convincing case that FGF21 is extraneous to the functions of the TZD class of therapeutics in vivo with respect to both beneficial metabolic outcomes and some of the side effects associated with TZD treatment. Further studies are required to elucidate the complex and likely tissue specific relationship between the actions of PPARs and the FGF21 axis.
Conflict of interest
All authors are current employees of Eli Lilly and Company.
Appendix A. Supplementary materials
Supplementary Fig. 1.
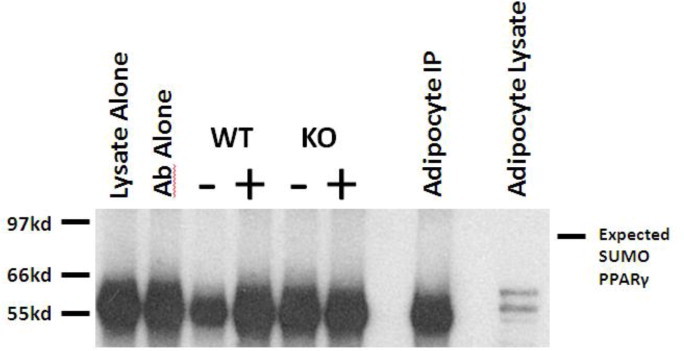
Using an immunoprecipitation of adipose tissue from both WT and FGF21KO mice treated with either vehicle (−) or rosiglitazone (+) we were unable to detect either basal or ligand stimulated sumoylation of PPARγ. Detection of PPARγ was hindered in in vivo samples by detection of endogenous mouse antibodies at the correct molecular weight for PPARγ.
References
- 1.Adams A.C., Cheng C.C., Coskun T., Kharitonenkov A. FGF21 requires betaklotho to act in vivo. PloS One. 2012;7:e49977. doi: 10.1371/journal.pone.0049977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adams A.C., Coskun T., Irizarry Rovira A.R., Schneider M.A., Raches D.W., Micanovic R., Bina H.A., Dunbar J.D., Kharitonenkov A. Fundamentals of FGF19 & FGF21 action in vitro and in vivo. PLoS One. 2012;7:e38438. doi: 10.1371/journal.pone.0038438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adams A.C., Kharitonenkov A. FGF21: the center of a transcriptional nexus in metabolic regulation. Current Diabetes Reviews. 2012;8:285–293. doi: 10.2174/157339912800840505. [DOI] [PubMed] [Google Scholar]
- 4.Adams A.C., Yang C., Coskun T., Cheng C.C., Gimeno R.E., Luo Y., Kharitonenkov A. The breadth of FGF21s metabolic actions are governed by FGFR1 in adipose tissue. Molecular Metabolism. 2013;2:31–37. doi: 10.1016/j.molmet.2012.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Badman M.K., Koester A., Flier J.S., Kharitonenkov A., Maratos-Flier E. Fibroblast growth factor 21-deficient mice demonstrate impaired adaptation to ketosis. Endocrinology. 2009;150:4931–4940. doi: 10.1210/en.2009-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Badman M.K., Pissios P., Kennedy A.R., Koukos G., Flier J.S., Maratos-Flier E. Hepatic fibroblast growth factor 21 is regulated by PPARalpha and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metabolism. 2007;5:426–437. doi: 10.1016/j.cmet.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 7.Chartoumpekis D.V., Habeos I.G., Ziros P.G., Psyrogiannis A.I., Kyriazopoulou V.E., Papavassiliou A.G. Brown adipose tissue responds to cold and adrenergic stimulation by induction of FGF21. Molecular Medicine. 2011;17:736–740. doi: 10.2119/molmed.2011.00075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Christodoulides C., Dyson P., Sprecher D., Tsintzas K., Karpe F. Circulating fibroblast growth factor 21 is induced by peroxisome proliferator-activated receptor agonists but not ketosis in man. Journal of Clinical Endocrinology and Metabolism. 2009;94:3594–3601. doi: 10.1210/jc.2009-0111. [DOI] [PubMed] [Google Scholar]
- 9.Coskun T., Bina H.A, Schneider M.A., Dunbar J.D., Hu C.C. Fibroblast growth factor 21 corrects obesity in mice. Endocrinology. 2008;149:6018–6027. doi: 10.1210/en.2008-0816. [DOI] [PubMed] [Google Scholar]
- 10.Ding X., Boney-Montoya J., Owen B.M., Bookout A.L., Coate K.C., Mangelsdorf D.J., Kliewer S.A. betaKlotho is required for fibroblast growth factor 21 effects on growth and metabolism. Cell Metabolism. 2012;16:387–393. doi: 10.1016/j.cmet.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dushay J., Chui P.C., Gopalakrishnan G.S., Varela-Rey M., Crawley M., Fisher F.M., Badman M.K., Martinez-Chantar M.L., Maratos-Flier E. Increased fibroblast growth factor 21 in obesity and nonalcoholic fatty liver disease. Gastroenterology. 2010;139:456–463. doi: 10.1053/j.gastro.2010.04.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dutchak P.A., Katafuchi T., Bookout A.L., Choi J.H., Yu R.T., Mangelsdorf D.J., Kliewer S.A. Fibroblast growth factor-21 regulates PPARgamma activity and the antidiabetic actions of thiazolidinediones. Cell. 2012;148:556–567. doi: 10.1016/j.cell.2011.11.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fisher F.M., Chui P.C., Antonellis P.J., Bina H.A., Kharitonenkov A., Flier J.S., Maratos-Flier E. Obesity is a fibroblast growth factor 21 (FGF21)-resistant state. Diabetes. 2010;59:2781–2789. doi: 10.2337/db10-0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fisher F.M., Estall J.L., Adams A.C., Antonellis P.J., Bina H.A., Flier J.S., Kharitonenkov A., Spiegelman B.M., Maratos-Flier E. Integrated regulation of hepatic metabolism by fibroblast growth factor 21 (FGF21) in vivo. Endocrinology. 2011;152:2996–3004. doi: 10.1210/en.2011-0281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fisher F.M., Kleiner S., Douris N., Fox E.C., Mepani R.J., Verdeguer F., Wu J., Kharitonenkov A., Flier J.S., Maratos-Flier E. FGF21 regulates PGC-1alpha and browning of white adipose tissues in adaptive thermogenesis. Genes & Development. 2012;26:271–281. doi: 10.1101/gad.177857.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Galman C., Lundasen T., Kharitonenkov A., Bina H.A., Eriksson M., Hafstrom I., Dahlin M., Amark P., Angelin B., Rudling M. The circulating metabolic regulator FGF21 is induced by prolonged fasting and PPARalpha activation in man. Cell Metabolism. 2008;8:169–174. doi: 10.1016/j.cmet.2008.06.014. [DOI] [PubMed] [Google Scholar]
- 17.Hale C., Chen M.M., Stanislaus S., Chinookoswong N., Hager T., Wang M., Veniant M.M., Xu J. Lack of overt FGF21 resistance in two mouse models of obesity and insulin resistance. Endocrinology. 2012;153:69–80. doi: 10.1210/en.2010-1262. [DOI] [PubMed] [Google Scholar]
- 18.Holland W.L., Adams A.C., Brozinick J.T., Bui H.H., Miyauchi Y., Kusminski C.M., Bauer S.M., Wade M., Singhal E., Cheng C.C. An FGF21–adiponectin-ceramide axis controls energy expenditure and insulin action in mice. Cell Metabolism. 2013;17:790–797. doi: 10.1016/j.cmet.2013.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hondares E., Rosell M., Gonzalez F.J., Giralt M., Iglesias R., Villarroya F. Hepatic FGF21 expression is induced at birth via PPARalpha in response to milk intake and contributes to thermogenic activation of neonatal brown fat. Cell Metabolism. 2010;11:206–212. doi: 10.1016/j.cmet.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hotta Y., Nakamura H., Konishi M., Murata Y., Takagi H., Matsumura S., Inoue K., Fushiki T., Itoh N. Fibroblast growth factor 21 regulates lipolysis in white adipose tissue but is not required for ketogenesis and triglyceride clearance in liver. Endocrinology. 2009;150:4625–4633. doi: 10.1210/en.2009-0119. [DOI] [PubMed] [Google Scholar]
- 21.Inagaki T., Dutchak P., Zhao G., Ding X., Gautron L., Parameswara V., Li Y., Goetz R., Mohammadi M., Esser V. Endocrine regulation of the fasting response by PPARalpha-mediated induction of fibroblast growth factor 21. Cell Metabolism. 2007;5:415–425. doi: 10.1016/j.cmet.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 22.Kharitonenkov A., Dunbar J.D., Bina H.A., Bright S., Moyers J.S., Zhang C., Ding L., Micanovic R., Mehrbod S.F., Knierman M.D. FGF-21/FGF-21 receptor interaction and activation is determined by betaKlotho. Journal of Cellular Physiology. 2008;215:1–7. doi: 10.1002/jcp.21357. [DOI] [PubMed] [Google Scholar]
- 23.Kharitonenkov A., Larsen P. FGF21 reloaded: challenges of a rapidly growing field. Trends in Endocrinology and Metabolism. 2011;22:81–86. doi: 10.1016/j.tem.2010.11.003. [DOI] [PubMed] [Google Scholar]
- 24.Kharitonenkov A., Shiyanova T.L., Koester A., Ford A.M., Micanovic R., Galbreath E.J., Sandusky G.E., Hammond L.J., Moyers J.S., Owens R.A. FGF-21 as a novel metabolic regulator. Journal of Clinical Investigation. 2005;115:1627–1635. doi: 10.1172/JCI23606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kharitonenkov A., Wroblewski V.J., Koester A., Chen Y.F., Clutinger C.K., Tigno X.T., Hansen B.C., Shanafelt A.B., Etgen G.J. The metabolic state of diabetic monkeys is regulated by fibroblast growth factor-21. Endocrinology. 2007;148:774–781. doi: 10.1210/en.2006-1168. [DOI] [PubMed] [Google Scholar]
- 26.Kurosu H., Choi M., Ogawa Y., Dickson A.S., Goetz R., Eliseenkova A.V., Mohammadi M., Rosenblatt K.P., Kliewer S.A., Kuro-o M. Tissue-specific expression of betaKlotho and fibroblast growth factor (FGF) receptor isoforms determines metabolic activity of FGF19 and FGF21. Journal of Biological Chemistry. 2007;282:26687–26695. doi: 10.1074/jbc.M704165200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kusminski C.M., Scherer P.E. The road from discovery to clinic: adiponectin as a biomarker of metabolic status. Clinical Pharmacology and Therapeutics. 2009;86:592–595. doi: 10.1038/clpt.2009.155. [DOI] [PubMed] [Google Scholar]
- 28.Li K., Li L., Yang M., Zong H., Liu H., Yang G. Effects of rosiglitazone on fasting plasma fibroblast growth factor-21 levels in patients with type 2 diabetes mellitus. European Journal of Endocrinology/European Federation of Endocrine Societies. 2009;161:391–395. doi: 10.1530/EJE-09-0335. [DOI] [PubMed] [Google Scholar]
- 29.Lin Z., Tian H., Lam K.S., Lin S., Hoo R.C., Konishi M., Itoh N., Wang Y., Bornstein S.R., Xu A. Adiponectin mediates the metabolic effects of FGF21 on glucose homeostasis and insulin sensitivity in mice. Cell Metabolism. 2013;17:779–789. doi: 10.1016/j.cmet.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 30.Liu H.H., Lu P., Guo Y., Farrell E., Zhang X., Zheng M., Bosano B., Zhang Z., Allard J., Liao G. An integrative genomic analysis identifies Bhmt2 as a diet-dependent genetic factor protecting against acetaminophen-induced liver toxicity. Genome Research. 2010;20:28–35. doi: 10.1101/gr.097212.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lundasen T., Hunt M.C., Nilsson L.M., Sanyal S., Angelin B., Alexson S.E., Rudling M. PPARalpha is a key regulator of hepatic FGF21. Biochemical and Biophysical Research Communications. 2007;360:437–440. doi: 10.1016/j.bbrc.2007.06.068. [DOI] [PubMed] [Google Scholar]
- 32.Moyers J.S., Shiyanova T.L., Mehrbod F., Dunbar J.D., Noblitt T.W., Otto K.A., Reifel-Miller A., Kharitonenkov A. Molecular determinants of FGF-21 activity-synergy and cross-talk with PPARgamma signaling. Journal of Cellular Physiology. 2007;210:1–6. doi: 10.1002/jcp.20847. [DOI] [PubMed] [Google Scholar]
- 33.Mu J., Pinkstaff J., Li Z., Skidmore L., Li N., Myler H., Dallas-Yang Q., Putnam A.M., Yao J., Bussell S. FGF21 analogs of sustained action enabled by orthogonal biosynthesis demonstrate enhanced antidiabetic pharmacology in rodents. Diabetes. 2012;61:505–512. doi: 10.2337/db11-0838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Muise E.S., Azzolina B., Kuo D.W., El-Sherbeini M., Tan Y., Yuan X., Mu J., Thompson J.R., Berger J.P., Wong K.K. Adipose fibroblast growth factor 21 is up-regulated by peroxisome proliferator-activated receptor gamma and altered metabolic states. Molecular Pharmacology. 2008;74:403–412. doi: 10.1124/mol.108.044826. [DOI] [PubMed] [Google Scholar]
- 35.Nishimura T., Nakatake Y., Konishi M., Itoh N. Identification of a novel FGF, FGF-21, preferentially expressed in the liver. Biochimica et Biophysica Acta. 2000;1492:203–206. doi: 10.1016/s0167-4781(00)00067-1. [DOI] [PubMed] [Google Scholar]
- 36.Ogawa Y., Kurosu H., Yamamoto M., Nandi A., Rosenblatt K.P., Goetz R., Eliseenkova A.V., Mohammadi M., Kuro-o M. BetaKlotho is required for metabolic activity of fibroblast growth factor 21. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:7432–7437. doi: 10.1073/pnas.0701600104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ohshima T., Koga H., Shimotohno K. Transcriptional activity of peroxisome proliferator-activated receptor gamma is modulated by SUMO-1 modification. Journal of Biological Chemistry. 2004;279:29551–29557. doi: 10.1074/jbc.M403866200. [DOI] [PubMed] [Google Scholar]
- 38.Oishi K., Tomita T. Thiazolidinediones are potent inducers of fibroblast growth factor 21 expression in the liver. Biological & Pharmaceutical Bulletin. 2011;34:1120–1121. doi: 10.1248/bpb.34.1120. [DOI] [PubMed] [Google Scholar]
- 39.Oishi K., Uchida D., Ishida N. Circadian expression of FGF21 is induced by PPARalpha activation in the mouse liver. FEBS Letters. 2008;582:3639–3642. doi: 10.1016/j.febslet.2008.09.046. [DOI] [PubMed] [Google Scholar]
- 40.Pascual G., Fong A.L., Ogawa S., Gamliel A., Li A.C., Perissi V., Rose D.W., Willson T.M., Rosenfeld M.G., Glass C.K. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature. 2005;437:759–763. doi: 10.1038/nature03988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Suzuki M., Uehara Y., Motomura-Matsuzaka K., Oki J., Koyama Y., Kimura M., Asada M., Komi-Kuramochi A., Oka S., Imamura T. betaKlotho is required for fibroblast growth factor (FGF) 21 signaling through FGF receptor (FGFR) 1c and FGFR3c. Molecular Endocrinology. 2008;22:1006–1014. doi: 10.1210/me.2007-0313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Veniant M.M., Hale C., Helmering J., Chen M.M., Stanislaus S., Busby J., Vonderfecht S., Xu J., Lloyd D.J. FGF21 promotes metabolic homeostasis via white adipose and leptin in mice. PLoS One. 2012;7:e40164. doi: 10.1371/journal.pone.0040164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang H., Qiang L., Farmer S.R. Identification of a domain within peroxisome proliferator-activated receptor gamma regulating expression of a group of genes containing fibroblast growth factor 21 that are selectively repressed by SIRT1 in adipocytes. Molecular and Cellular Biology. 2008;28:188–200. doi: 10.1128/MCB.00992-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Z.J., Liu Q., Li P.P., Zou C.H., Shen Z.F. Effect of GCP-02, a PPARalpha/gamma dual activator, on glucose and lipid metabolism in insulin-resistant mice. European Journal of Pharmacology. 2008;580:277–283. doi: 10.1016/j.ejphar.2007.10.042. [DOI] [PubMed] [Google Scholar]
- 45.Yang C., Jin C., Li X., Wang F., McKeehan W.L., Luo Y. Differential specificity of endocrine FGF19 and FGF21 to FGFR1 and FGFR4 in complex with KLB. PLoS One. 2012;7:e33870. doi: 10.1371/journal.pone.0033870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang Y., Xie Y., Berglund E.D., Coate K.C., He T.T., Katafuchi T., Xiao G., Potthoff M.J., Wei W., Wan Y. The starvation hormone, fibroblast growth factor-21, extends lifespan in mice. eLife. 2012;1:e00065. doi: 10.7554/eLife.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]