

Abstract
This review discusses recent progress in the development of anti-HIV agents targeting the viral entry process. The three main classes (attachment inhibitors, co-receptor binding inhibitors, and fusion inhibitors) are further broken down by specific mechanism of action and structure. Many of these inhibitors are in advanced clinical trials, including the HIV maturation inhibitor bevirimat, from the authors’ laboratories. In addition, the CCR5 inhibitor maraviroc has recently been FDA-approved. Possible roles for these agents in anti-HIV therapy, including treatment of virus resistant to current drugs, are also discussed.
Keywords: HIV entry inhibitors, attachment inhibitors, co-receptor binding inhibitors, fusion inhibitors, maraviroc (MVC; UK-427, 857), enfuvirtide (T20;fuzeon), bevirimat (DSB;PA-457)
1. INTRODUCTION
An estimated 33.2 million people are infected worldwide with human immunodeficiency virus (HIV), the etiologic cause of acquired immunodeficiency syndrome (AIDS). In 2007, 2.5 million people were newly infected with the virus, and 2.1 million people died of AIDS-related illnesses,1 making this pandemic the fourth leading cause of mortality globally. The successful development of a safe and effective HIV vaccine is still in the future.2 Therefore, research continues to focus on disease treatment by chemical anti-HIV agents.
Significant progress has been made in the development of antiretroviral therapy (ART). ART can successfully delay destruction of the immune system, reduce severity and frequency of opportunistic infections, and consequently delay AIDS progression. Introduction of highly active antiretroviral therapy (HAART), which employs a combination of nucleoside/nucleotide reverse transcriptase inhibitors (NRTIs), non-nucleoside reverse transcriptase inhibitors (NNRTIs), and/or protease inhibitors (PIs), can reduce viral load to below detectable levels in patient plasma, resulting in improved patient health and life span.3–6 However, the virus is suppressed rather than eradicated by HAART.7–9 On HAART regimens, multiple drug therapies can lead to increased adverse effects and toxicities due to long-term use and drug–drug interactions.10,11 Moreover, inadequate clinical viral suppression of HIV-1 due to various reasons and the high error rate of the reverse transcriptase lead to the emergence of drug-resistant and multi-drug-resistant viral strains.12 Drug-resistant virus is then involved in HIV transmission, and more than 25% of newly infected individuals harbor HIV-1 isolates that are resistant to at least one ART.13,14 Therefore, novel potent antiretroviral agents, which have simplified treatment regimens (fewer pills and less-frequent administration), and target not only reverse transcriptase and protease but also other viral targets, may hold particular promise in addressing issues of current therapies.
Besides the viral enzyme reverse transcriptase (RT) and protease (PR), the life cycle of HIV presents several potential targets for chemotherapeutic attack, including the viral entry pathway, nuclear import of the pre-integration complex, integration, viral transcription, processing of viral transcripts and nuclear export, and maturation.15,16 Among these targets, viral entry is one of the most promising for HIV drug development. Indeed, enfuvirtide (T-20, Fuzeon, Trimeris), which is a HIV fusion inhibitor, was the first drug with a target other than RT and PR to be approved by the US FDA.17 Because the peptidic nature of enfuvirtide does not permit its oral administration, the drug is currently administrated by subcutaneous injection twice daily, which limits its application. Therefore, continued effort in discovering new HIV entry inhibitors, especially potent, orally bioavailable small molecules, is still needed. Herein, we review the development of new HIV entry inhibitors, their mechanisms of action, and their possible role in anti-HIV therapy.
2. VIRAL ENTRY INHIBITORS
HIV entry into host cells is a complicated process that involves at least three steps: (1) an attachment step that requires CD4 receptor binding, (2) co-receptor binding, and (3) fusion process. The envelope glycoprotein (Env) on the surface of virions mediates entry. Heterotrimeric Env spikes are composed of three surface glycoproteins (SU, gp120) atop three transmembrane glycoproteins (TM, gp41).18 The gp41 subunits, which assemble into a trimer, comprise the “stalk” of Env that anchors the Env into the viral membrane. The gp120 subunits comprise the “head” of Env that is able to bind to cellular receptors.19,20 Virions can first attach to target cells in a relatively nonspecific manner, followed by specific binding of HIV gp120 to the CD4 receptor on the cellular membrane. This binding induces a conformational change in gp120 that opens up a high-affinity binding site located within the third variable loop (V3) and surrounding surfaces for the chemokine co-receptors (primarily CCR5 and CXCR4).21 Co-receptor binding results in further conformational rearrangements of gp120 that expose the fusion-peptide domain of gp41. Insertion of this hydrophobic domain into the cell membrane leads to the formation of a hairpin-like fold in the gp41 subunits.22 The heptad repeat (HR) regions, HR1 and HR2 of the three subunits of gp41, fold and pack into a six-helix bundle, which brings the viral and cell membranes into juxtaposition and creates pores in the target cell membrane, enabling the release of viral capsid into the cytoplasm.23–26 Different entry inhibitors that target one or more of these steps are now in preclinical and clinical trials.
A. Attachment Inhibitors
1. Nonspecific Attachment Inhibitors
Before gp120 and CD4 receptor binding firmly attaches HIV onto the target cells, less specific adsorption and attachment occur between the virions and cell membrane due to the interaction of the positively charged regions of Env with oppositely charged proteoglycans of the cell surface.27,28 Studies have reported that soluble polyanions, such as dextran sulfate, cyclodextrin sulfate and heparin, can block this nonspecific attachment of HIV virions.29
Cyanovirin-N (Cellegy Pharmaceuticals*), an 11 kDa protein isolated from the cyanobacterium (blue-green algae) Nostoc ellipsosporum, specifically binds to a conserved high-mannose carbohydrate region on gp120, which prevents the attachment of virus to target cells at low nanomolar concentrations.30–32 Another example is PRO 2000 (Indevus Pharmaceuticals), a naphthalene sulfonate polymer (1) (Fig. 1), which binds nonspecifically to CD4 receptor.33 Vaginal PRO 2000 gel has been effective and well tolerated in Phase I/II clinical trials.34,35 However, because of their mechanism of action and polymer properties, these two compounds are mainly being developed as topical microbicides.
Figure 1.

Structures of some selected sulfated compounds as HIV nonspecific attachment inhibitors.
Glycyrrhizin (2) (Fig. 1), a triterpenoid saponin isolated from licorice root inhibits HIV-1 replication by partially blocking viral adsorption to CD4+ cells.36 Sulfation of glycyrrhizin (3), amphotericin B, and lentinan, as well as polysulfonates [e.g., suramin (4)] and polyhydroxycarboxylates [e.g., aurintricarboxylic acid (5)] also interfere with the nonspecific viral attachment process (Fig. 1).
2. CD4-gp120 Binding Inhibitors
Many molecules can inhibit CD4 and gp120 binding via different modes of action, including inhibition of attachment by targeting Env gp120 or CD4 receptor and prevention of necessary conformational rearrangement.
PRO 542 (Progenics Pharmaceuticals) is a recombinant tetrameric CD4-IgG2 antibody-like fusion protein, where the heavy and light chain Fv portions of human IgG2 have been replaced by the D1 and D2 domains of human CD4.37 It targets the CD4 binding site on gp120 by mimicking the CD4 receptor. In Phase I clinical trials, PRO 542 reduced viral load after a single intravenous dose and was well tolerated in infected adults and children.38,39
TNX 355 (Tanox, Inc.) is a humanized IgG4 monoclonal antibody against CD4, which binds to the D2 domain of CD4 and inhibits CD4-induced post-binding conformational changes.40 Phase I clinical studies concluded that TNX 355 reduced plasma HIV-1 RNA loads and increased CD4+ T-cells.41 However, the need for infusion limits its clinical use.
BMS-378806 (6) and BMS-488043 (7) (Bristol-Myers Squibb) (Fig. 2) are novel, small-molecule CD4-attachment inhibitors that specifically block HIV-1 entry by targeting Env gp120 with an IC50 value of around 5 nM.42,43 Mechanism of action study revealed that both compounds selectively bind to gp120 and lead to conformational changes in gp120 at both the CD4 and CCR5 binding regions, which blocks CD4-gp120 interactions. Prior addition of soluble CD4 to the assay system before BMS-488043 treatment negates the inhibition activity of the compound, indicating that inhibition of CD4-gp120 binding is the primary mode of action.44 An earlier report by Si et al.45 suggested that BMS analogs function by blocking conformational changes of gp120 after CD4 binding rather than by directly inhibiting CD4 binding; however, a significantly altered Env structure and high concentrations of soluble CD4 may have led to this conclusion.44 Drug development of BMS-378806 was discontinued after Phase I clinical study, because target exposure was not achieved. BMS-488043 is currently in Phase II clinical trials.46 It is orally bioavailable with superior pharmacokinetic properties and good safety profiles.
Figure 2.
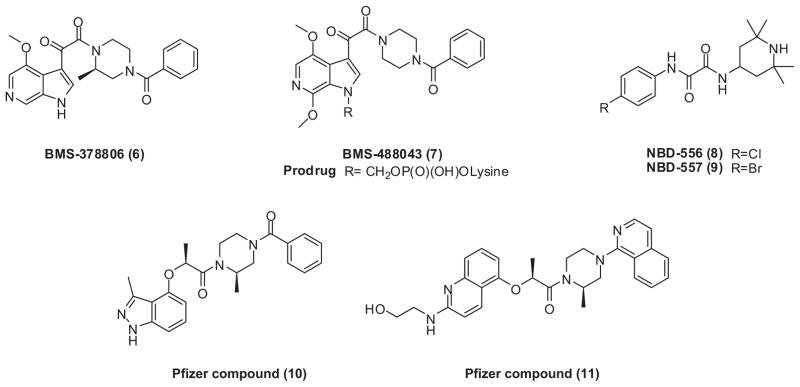
Selected structures of CD4-gp120 binding inhibitors.
The New York Blood Center identified similar structures, NBD-556 (8) and NBD-557 (9) (Fig. 2), which also contain an oxalamide moiety. These two compounds showed micromolar potency against HIV-1.47 Compounds 10 and 11 (Fig. 2) disclosed by Pfizer, Inc. in patents (WO-2005016344 and WO-2005121094) also share similar structures with BMS analogs and have nanomolar anti-HIV activity.46
Zintevir (AR177, Aronex Pharmaceuticals), a 17-base G-quartet phosphorothioate nucleotide, was first reported as a HIV integrase inhibitor.48 However, later research discovered that it functions by blocking CD4-gp120 binding at sub-micromolar concentration.49 However, an acute toxicity study in mice showed that AR177 caused male-specific mortality, and changes in serum chemistry, hematology, and histology at doses of 250 and 600 mg/kg, with renal necrosis and histologic vacuolization being prominent in several organs.50 In comparison, AR177 did not cause significant hemodynamic toxicity in cynomolgus monkeys, although lower doses of 20 and 50 mg/kg were administered.51 Overall, the toxicity of AR177 is considered species specific. Unfortunately, zintevir was discontinued in Phase II clinical trials with no further indicated reasons.52
3. Down-Regulation of CD4 Expression
CADA analogs (cyclotriazadisulfonamide) (12) (Fig. 2) have been identified as potent anti-HIV compounds with novel mechanism of action.53 Binding studies with HIV-1 revealed that CADA did not directly interact with the CD4 receptor and/or viral envelope glycoproteins. Further investigation discovered that CADA analogs function by a specific CD4 down-modulating potency.54 Analysis of CD4 mRNA levels suggested that CADA regulation is not involved at the transcriptional level but most probably interacts at a (post)translational level.55 Vermeire et al. further demonstrated that the antiviral potency of the CADA analogs depended primarily on the down-regulation of CD4 receptor expression. Removal of CADA compounds from the cell culture medium resulted in complete restoration of CD4 expression. This category of compounds showed anti-HIV entry activity at micromolar or sub-micromolar concentrations.56,57 Most recently, this same group reported that they have successfully integrated a dansyl fluorophore into the chemical structure of some CADA compounds, and showed the feasibility of tracking a receptor and its down-modulator simultaneously.58 These fluorescent CADA analogs can now be applied in further exploration on receptor modulation.

B. Co-Receptor Binding Inhibitors
Chemokine receptors belong to the seven transmembrane G protein coupled receptor family. They are involved in the progression of many diseases, including rheumatoid arthritis, transplant rejection, asthma, cancer, HIV, and others. CCR5 and CXCR4 are the main chemokine receptors involved in the HIV entry process.59 Based on their chemokine receptor usage, HIV isolates may be divided into R5, X4, and R5/X4 strains.60 R5 isolates are the predominant virus strains, which are preferentially transmitted between HIV-infected patients.61 However, R5 variants will eventually evolve into X4 isolates or R5/X4 isolates in approximately 50% of HIV-1 positive individuals, resulting in increased viral replication rate, faster disease progression, and the onset of AIDS.62
During the HIV entry process, the CD4-gp120 complex must further bind to chemokine co-receptors in order to enter the target cells.63 The third variable region, V3, of gp120 is the major region implicated in co-receptor interactions. It is an approximately 35-residue-long, frequently glycosylated, highly variable, disulfide-bonded structure that has a major influence on HIV-1 tropism.64 The amino-acid sequences of V-3 can determine the usage of CCR5 or CXCR4.65 Besides V3, a relatively well-conserved structure located partly on the bridging sheet is also important for binding to both CCR5 and CXCR4 co-receptors.64 Epidemiology data shows that individuals homozygous for δ32 CCR5 allele (no expression of CCR5 on cell surface) are highly resistant to HIV infection66,67 and apparently healthy. This fact highlights the merits of targeting co-receptor binding to inhibit HIV entry.
1. CCR5 Inhibitors
The CCR5 co-receptor binding site on HIV gp120 is concealed by V1, V2, and V3. The conformational change caused by the binding of gp120 with CD4 exposes this CCR5 binding site. When a CCR5 inhibitor is present, binding of the CCR5 N-terminus and ECL2 (second extracellular loop) with the V3 stem of gp120 is blocked, resulting in inhibition of viral entry.56 The strategies to block CCR5 co-receptor binding include modified non-agonistic CCR5 chemokine natural ligands, CCR5 antibodies and CCR5 small molecule antagonists.
RANTES (13) was discovered as the natural β-chemokines ligand, which can compete with gp120 to bind the CCR5 co-receptor,68 and thus, inhibit HIV infection. However, it also triggers CCR5-mediated down-stream signaling pathways, which may lead to undesired side effects. Modification of RANTES at the N-terminus led to the discovery of AOP-RANTES and NNY-RANTES, which are nanomolar level inhibitors of HIV R5 variants.69 C1,C5-RANTES has two Ser to Cys substitutions at positions 1 and 5 of the wild type. It was the first RANTES derivative without agonistic functions while maintaining high potency towards HIV.70 Currently, PSC-RANTES (14) is under development as a potential microbicide71 (Fig. 3).
Figure 3.
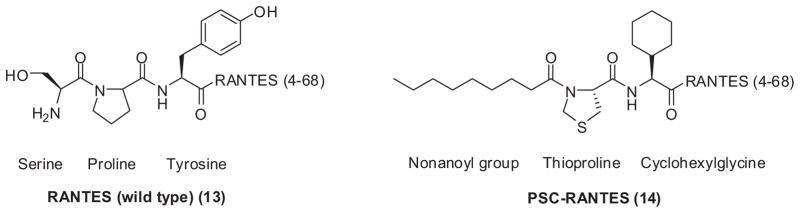
Structures of modified chemokine derivatives as HIV inhibitors.
PRO 140 (Progenics Pharmaceuticals) is another promising large molecule CCR5 co-receptor binding inhibitor. It is a murine anti-CCR5 monoclonal antibody that binds a complex epitope spanning multiple extracellular domains on CCR5.72 It does not prevent CC-chemokine signaling, but still blocks the interactions of gp120 to CCR5. PRO 140 demonstrates a potent and genetic-subtype-independent anti-HIV-1 activity and is currently in Phase II clinical trials.
Because large molecules are often difficult to administer, their clinical application may be limited. Therefore, the search for orally bioavailable small molecule inhibitors remains of great interest. This search has led to the discovery of many small molecule CCR5 antagonists, which are very promising for development as therapeutic antiviral drugs. These small molecule CCR5 antagonists share a common binding site formed by the transmembrane (TM) domains of CCR5. Although all antagonists sit in the same big hydrophobic pocket, they occupy different subcavities. The residues involved in ligand interaction are quite different for each of the antagonists.73 The putative binding pocket was defined by Kondru et al.,73 and the key residues that line the binding pocket are identified as Trp86, Tyr108, Phe109, Thr195, Ile198, Trp248, Tyr251, Glu283 (interacts with all antagonists), and Met287.
TAK-799 (Takeda Chemical Industries) (15) (Fig. 4) is the first reported nonpeptide small molecule that antagonizes the binding of RANTES to CCR5-expressing Chinese hamster ovary cells at nanomolar concentrations (1.6–3.7 nM).74 TAK-799 selectively inhibits CCR5 within the chemokine receptor family by binding inside a cavity formed by the transmembrane helices 1, 2, 3, and 7 of the CCR5 receptor.75 Although it showed no cytotoxicity to the host cells, lack of oral bioavailability limits its further development. Clinical trials of an injectable quaternary ammonium salt TAK-799 were discontinued due to local reactions at injection sites. Modification of TAK-799 by replacing the quaternary ammonium moiety with a polar sulfoxide moiety led to the discovery of TAK-652 (16)76 (Fig. 4). TAK-652 showed increased anti-HIV-1 activity against different clinical HIV subtypes with a mean IC50 value of 0.25 nM. More importantly, it has oral bioavailability and is well absorbed after oral administration in different preclinical animal studies. TAK-652 is currently in Phase I/II clinical trials. Additional high throughput screening against CCR5 in the same company led to the discovery of a different compound skeleton exemplified by 17 (Fig. 4), which exhibits nanomolar anti-HIV-1 activity. However, 17 was rapidly degraded in metabolic stability studies. Continuing modification resulted in the identification of TAK-220 (18)77 (Fig. 4), an orally bioavailable, metabolically stable piperidine-4-carboxamide derivative, which is also in clinical studies.
Figure 4.

Structures of TAK analogs.
Maraviroc (MVC, UK-427,857, Pfizer, Inc.) (19) (Fig. 5) is another selective CCR5 antagonist with potent antiviral activity against all CCR5-tropic HIV-1 viruses at low nanomolar concentrations (mean 90% inhibitory concentration of 2.0 nM).78 It was obtained by medicinal chemistry optimization of a high throughput screening lead, the imidazopyridine derivative UK-107,543 (20) (Fig. 5).78 The initial modification of 20 by introducing an amide bond into the structure (21) (Fig. 5) solved the high lipophilicity problem of the lead.79 Later study revealed that only the S enantiomer of 21 showed good potency. However, this type of piperidine-based CCR5 antagonist is also a potent CYP 2D6 inhibitor, which may create problems with drug–drug interactions in drug combinational use. Further development led to the discovery of tropane-based compound 22 (Fig. 5), which does not inhibit CYP 2D6 liabilities, but retains high potency towards CCR5.80 Unfortunately, 22 is also a potent inhibitor of the hERG ion channel. This undesired side effect was successfully conquered by replacing the benzimidazole group with a triazole moiety and replacing the cyclobutyl group with a difluorocyclohexane moiety, which resulted in the identification of maraviroc (19).81 Mechanism of action study revealed that 19 binds to the cavity formed by the transmembrane helices 2, 3, 6, and 7 of CCR5 receptor, which is different with the binding site of TAK-799.82 Radioligand binding competition assays demonstrated that 19 blocks the binding of gp120 to CCR5, resulting in the prevention of membrane fusion events necessary for viral entry. Compound 19 inhibits some downstream CCR5 signaling events following its inhibition of several natural chemokine ligands such as MIP-1α, MIP-1β, and RANTES. However, it does not trigger release of intracellular calcium or CCR5 internalization, indicating that it is devoid of CCR5 agonist activity. Thus, 19 is a functional antagonist of CCR5.78 In clinical trials, it was administered orally with twice-daily dosing.83 In August 2007, 19 was approved by the US FDA as a CCR5 antagonist HIV entry inhibitor under the brand name Selzentry.
Figure 5.
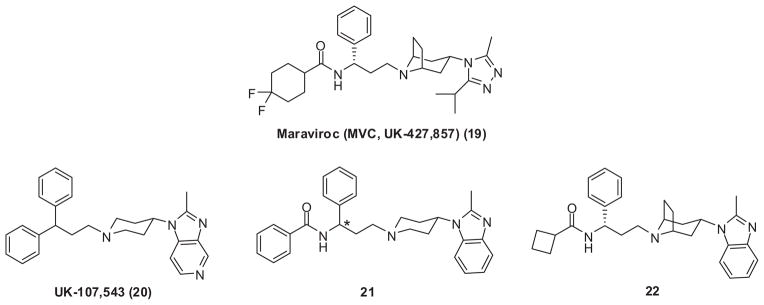
Lead optimization and identification of Maraviroc.
Vicriviroc (SCH-D, SCH-417,690, Schering-Plough) (23) (Fig. 6), a piperzine-based CCR5 antagonist, is also currently in Phase III clinical study. It is a second-generation compound with improved anti-HIV activity and pharmacokinetic profile compared with the first-generation ancriviroc (SCH-C, SCH-351,125, Schering-Plough) (24) (Fig. 6), a piperidino-piperidine derivative.84,85 Both compounds are orally bioavailable and interact within the cavity formed by CCR5 transmembrane regions, resulting in inhibition of HIV gp120 binding.86 Compound 24 was discovered first by modification of a high throughput screening lead. However, later study revealed that high doses of 24 led to CNS side effects and prolongation of the corrected QT cardiac interval (QTc interval: the time between depolarization and repolarization of the heart muscle adjusted for heart rate; the longer the QTc interval, the slower the heart rate).85 The clinical studies of 24 were then discontinued. Within the piperazino-piperidine series, replacing the N-oxide of 25 by 4,6-dimethylpyrimidine carboxamide as in 26 (Fig. 6) resulted in better CV profiles.84 Further optimization of 26 led to the discovery of vicriviroc (23), which has potent, broad-spectrum antiviral activity against diverse HIV-1 subtypes and synergistic anti-HIV activity in combinational use with drugs from other classes. Another promising candidate in the piperazino-piperidine family is AD101 (SCH-350,581) (27) (Fig. 6), which also shows potent anti-HIV-1 activity by blocking gp120/CD4 complex and CCR5 interaction. AD101 is currently in preclinical study.87
Figure 6.

Structures of Vicriviroc and Ancriviroc analogs.
INCB9471 (Incyte Corporation) (28) (Fig. 7) is another promising candidate in Phase II clinical trials. It also belongs to the piperazino-piperidine family. The similar structures of INCB9471 and vicriviroc suggest that they might share the same mechanism of action. INCB9471 is an orally bioavailable CCR5 antagonist with very potent antiviral activity against a wide range of HIV-1 strains, including drug resistant isolates.88
Figure 7.

Structures of INCB9471and Aplaviroc.
Aplaviroc (GW873140, formerly ONO4128, AK602, GlaxoSmithKline) (29) (Fig. 7) failed in Phase II clinical trials in October 2005, due to unexpected serious hepatotoxicity. It belongs to the spirodiketopiperazine family and exhibits potent antiviral activity with IC50 values around 0.1–0.6 nM.89 Aplaviroc exerts its antiviral function by interacting with CCR5 ECL2 rather than binding to the CCR5 transmembrane cavity, which is distinct from other CCR5 antagonists.89,90
Other classes of compounds have also been reported to be CCR5 antagonists. GlaxoSmithKline filed a patent (WO2004054974) describing some newly discovered anti-HIV-1 CCR5 antagonists. Among these compounds, GSK 163929 (30) (Fig. 8) showed nanomolar level inhibition activity towards HIV replication and successfully completed preclinical studies.91 NSC651016 (31) (Fig. 8) inhibits chemokine binding of CCR5, CXCR4, CCR1, and CCR3, but does not interact with CXCR2 or CCR2b. It inhibits a wide range of HIV-1 variants and HIV-2 isolates, and could be developed as a topical microbicide.92 Shikonin (32) (Fig. 8), a major component of purple gromwell (zicao, dried root of Lithospermum erythrorhizon) is a CCR5 chemokine antagonist with sub-micromolar level antiviral activity.93 Merck reported the discovery of several compounds (33–37) (Fig. 9) with potent CCR5 binding affinity. However, the anti-HIV activities of these compounds are only moderate.94–97
Figure 8.
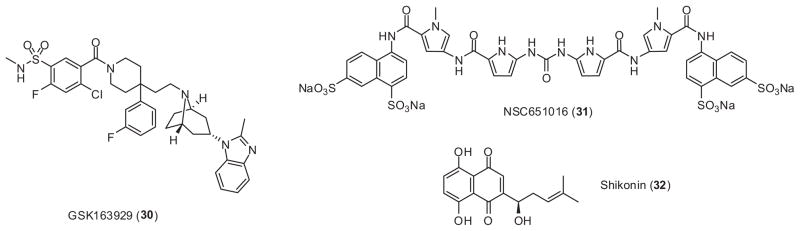
Selected structures of promising CCR5 antagonists as anti-HIV agents.
Figure 9.

Merck analogs showing potent CCR5 binding affinity.
2. CXCR4 Inhibitors
Although the deletion of CCR5 has little impact on an individual’s health, CXCR4 is involved in a number of physiological processes. In a mice knockout study, knocking out either CXCR4 or its only known natural ligand SDF-1 (stromal cell-derived factor) resulted in abnormal cerebral development and was embryonically lethal.98,99 Whether CXCR4/SDF-1 is vital only in embryonic development needs to be further determined. Deletion of SDF-1 also caused defects in B-cell lymphopoiesis and bone marrow myelopoiesis in mice.100 These findings make CXCR4 antagonists less feasible. However, although R5 variants are dominant in initial HIV transmission, a switch in co-receptor use occurs after transmission, and the virus gains ability to use both R5 and X4 during AIDS progression.62 Therefore, CXCR4 antagonists are still necessary. The ideal CXCR4 antagonist should block HIV entry without affecting CXCR4 down-stream signaling pathways or causing CXCR4 internalization.
The initial strategy for developing CXCR4 antagonists also focused on mimics of the CXCR4 natural ligand SDF-1. T-22 is a synthetic 18 amino-acid peptide with highly polar cationic character.101 Its downsized analogs T134 and T-140 contain fourteen amino-acid residues.102,103 Both compounds inhibit replication of X4 isolates by specifically binding to the CXCR4 co-receptor. Further SAR study concluded that four amino-acid residues, Arg2, NaI3 [L-3-(2-naphthyl)alanine3], Tyr5, and Arg14, are essential for T-140 antiviral potency (IC50 ~ 60 nM)104 which led to the identification of FC131 [cyclo(-L-NaI1-L-Gly2-D-Tyr3-L-Arg4-L-Arg5-)] with antiviral IC50 around 38 nM.105
ALX40-4C (N-α-acetyl-nona-D-arginine amide) is the first co-receptor inhibitor to be tested in humans prior to the elucidation of its mechanism of action as a CXCR4 antagonist.106 Unfortunately, development of this compound was stopped due to negligible reduction of viral load in clinical studies.
Norvatis Ltd. reported the discovery of a nine-residue hybrid peptoid/peptide oligomer, CGP64222, which specifically blocks the CXCR4 co-receptor binding.107 It shows anti-HIV activity at a nanomolar concentration.108 Mechanism of action study further revealed that CGP64222 also inhibits Tat/TAR RNA interaction as a second mechanism of action to inhibit HIV replication. (Recruitment of Tat protein to the transcription machinery after its binding to the RNA response element TAR is one of the first steps in HIV gene expression.)
In addition to large molecule polypeptides, several small molecule CXCR4 antagonists are also under development. AMD3100 (AnorMED, Inc., now Genzyme Corporation) (38) (Fig. 10) is a low molecular weight bicyclam analog with potent anti-X4 HIV variant activity (IC50 ~ 1.4 nM).109 Its development was terminated when abnormal cardiac activity was observed in Phase II clinical studies. The second generation cyclam analog AMD3465 (39) (Fig. 10) has only one cyclam unit; the other is replaced by N-pyridinylmethylene.110 However, although the antiviral activity of 39 is slightly better than that of 38, it still lacks oral bioavailability. The mechanism of action of cyclam analogs is related to the multiple positive charges associated with the basic nitrogen atoms,111 which interact with negatively charged acidic residues in CXCR4. Studies showed that residues D171, D262, and E288 are important in CXCR4 binding by 38.111 The third generation AMD070 (AMD11070) (40) (Fig. 10) is structurally different from the prior analogs and is also orally bioavailable.112 It is highly specific for the CXCR4 receptor. Compounds 38 and 40 have similar binding sites111 and exhibits similar antiviral activity against a broad range of X4 and R5X4 isolates, but are not active against R5 strains. Compound 40 is currently in Phase II clinical trials.
Figure 10.

Structures of AMD analogs.
KRH-1636 (Kureha Chemical Industries) (41) is an arginine-based CXCR4 antagonist with good anti-HIV-1 activity (IC50 ~ 42 nM).113 It was obtained from optimization of a high throughput screening lead. It can be absorbed after intraduodenal administration to rats, suggesting that it may be orally bioavailable.113 Further modification led to the discovery of KRH-2731 · 5HCl (structure has not been released), which is a 41-analog with improved antiviral potency (IC50 = 1.0 nM) against X4 and R5X4 HIV isolates.114 KRH-2731 · 5HCl is orally bioavailable. Mechanism of action study revealed that it binds to ECL2 and ECL3 (second and third extracellular loops) of the CXCR4 receptor.65

C. Fusion Inhibitors
As previously mentioned, the binding of CD4/gp120 complex with a chemokine co-receptor leads to a conformational rearrangement in gp120 that enables gp41 to reorient parallel to viral and cellular membranes and expose a highly hydrophobic fusion-peptide (FP) region of trimeric gp41, which will later insert into the cell membrane. FP is located at the N-terminus of gp41, adjacent to two heptad repeats (HRs). During the fusion process, HR1 and HR2 form a thermostable, six-helix bundle structure.23,26 The change in free energy associated with this critical step provides the force needed for formation of the fusion pore, through which the viral capsid is released into the target cell.26
1. Large Molecule Fusion Inhibitors
As early as the 1990s, researchers realized that synthetic peptides based on the HR1 and HR2 sequences of gp41 have anti-HIV properties.115,116 The mechanism of action of this oligopeptide class is believed to be competitive binding of the synthetic peptides with the corresponding HR regions, which is also called the “dominant-negative inhibition model”.17
Until now, fusion inhibitors have been the most successful class of HIV entry inhibitors. Enfuvirtide (T20, Fuzeon, Trimeris/Roche) (42) was approved for use in combination with other ARTs in treatment-experienced patients in March 2003 by the US FDA.117,118 It is a 36-amino acid synthetic peptide that mimics the HR2 region (residues 127–162 in C-terminal) of gp41.118 By competitively binding to the HR1 region of gp41, 42 prevents the formation of the six-helix bundle structure that is critical as the energy source for the fusion process.119 Compound 42 shows good antiviral potency and long-term safety in clinical use. A synergistic effect has also been observed in its combinational use with other ARTs. However, it is administered by subcutaneous injection of 90 mg twice daily, which is highly inconvenient. Moreover, inhibitor-naive primary HIV-1 isolates exhibited wide ranging susceptibilities to 42, and the compound has a relatively low genetic barrier for resistance.120 Thus, enfuvirtide is considered only a second-line antiretroviral agent.

The second-generation fusion inhibitor tifuvirtide (T-1249) shows 10-fold more potent antiviral activity compared with 42.121 It preserves antiviral efficacy to 42-resistant HIV-1 isolates as well as HIV-2 and SIV variants,121 and is a 39-amino acid peptide mimicking a different region of HR2.122 However, its clinical development was halted due to formulation challenges.123
TRI-999 and TRI-1144 (Trimeris)124 are additional oligopeptide fusion inhibitors, which show better pharmacokinetic profiles. Using rational design, researchers in Trimeris further designed a series of oligomeric HR2 peptides with increased helical structure that self-associate into stable oligomeric structures, resulting in exceptionally high HR1/HR2 bundle stability.125 The engineered peptides (e.g., T-2635, T-267221, T-267227) are found to be as much as 3,600-fold more active than 42 and T-1249 against HR2-resistant viruses, and are shown to have 100-fold improved pharmacokinetics in cynomolgus monkeys. More importantly, viruses appear to require more mutations to decrease sensitivity to the engineered peptides, and the impact of these mutations on activity is significantly less than that observed for 42.125 Taken together, these newly engineered peptides are attractive for further clinical development.
5-Helix is a chimeric gp41 containing three N- and two C-terminal helices connected by a linker, which showed nanomolar activity against diverse HIV-1 isolates by targeting the HR2 region of gp41.126
2. Small Molecule Fusion Inhibitors—Recent Progress With Triterpene Derivatives as HIV Entry Inhibitors
Until now, most reported fusion inhibitors have been large polypeptides, which lack oral bioavailability and are relatively costly. The azo dye ADS-J1 (43) was originally defined as a fusion inhibitor;127 however, recent study discovered that it acts before the gp41-dependent fusion step.128 Small molecules that can be easily purified during manufacture and are more likely to be orally administrated hold better potential to be developed into clinical drugs.

a. Discovery of RPR103611 and IC9564
Certain triterpene derivatives represent promising small molecule fusion inhibitors (exact binding site is currently unknown, but it has been demonstrated that this compound class functions at a post-binding, envelope-dependent step involved in fusion of the virus to the cell membrane, as discussed below). Betulinic acid (BA, 3β-hydroxy-lup-20(29)-en-28-oic acid, 44) and platanic acid (45), both of which are pentacyclic triterpenes extracted from Syzigium claviflorum, were first reported in 1994 to suppress HIV-1IIIB replication in vitro, with IC50 values of 1.4 and 6.5 μM, respectively.129 Because of its promising anti-HIV activity and ready availability from various natural sources, including white birch trees, BA was selected as a lead in an anti-HIV drug discovery program.

C-28 ω-aminoalkanoic acid derivatives of BA were then synthesized and evaluated for antiviral activity by Soler et al.130 Initially, the chain length between the C-28 amide bond moiety and the terminal carboxylic acid group (46a–l, m = 1–12) (Fig. 11) was explored, and incremental chain lengthening significantly influenced the anti-HIV-1 potency of the derivatives. Compounds with amide side chains between amino-octanoic acid and amino-dodecanoic acid (46g–k, m = 7–11) showed increased antiviral potency, with amino-undecanoic acid (46j, m = 10) being optimal. Condensation of these C-28 ω-aminoalkanoic acid derivatives with a second aminoalkanoic acid produced compounds with two amide moieties at different positions of the C-28 side chain. It was then discovered that small peptide amide derivatives of the parent octanoic acid analog 46g were more potent than both the parent compound and 46j. The presence of a small lipophilic space between the two amide moieties increased the anti-HIV-1 activity of 46g by around 10-fold, and substituents on this second aminoalkanoic acid moiety also modulated the antiviral potency. This effort led to the discovery of RPR103611 (47) (Fig. 11), a statine derivative, which inhibits the infectivity of several HIV-1 strains in a 10 nM concentration range.130,131
Figure 11.
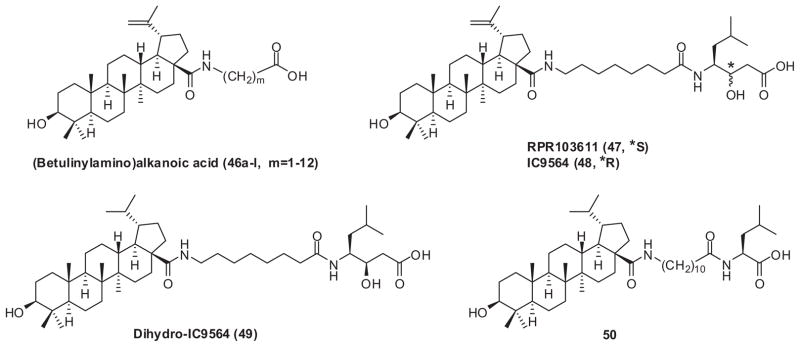
C-28 modified BA as HIV entry inhibitors.
Research in the authors’ laboratories on BA derivatives as HIV entry inhibitors focused on the modifications in the isopropylene and C-28 side chains. The study showed that IC9564 (48) (Fig. 11), a stereoisomer of RPR103611, exhibits equally potent anti-HIV-1 activity.132 This same study also revealed that dihydro-IC9564 (49) (Fig. 11), with a saturated isopropyl rather than isopropenyl group, and 50, a L-leucine derivative of (betulinylamino)-undecyclic acid (Fig. 11) also were equipotent against HIV-1 virus compared with RPR103611 and IC9564.132 Compound 50 is synthesized more easily than RPR103611 and IC9564.
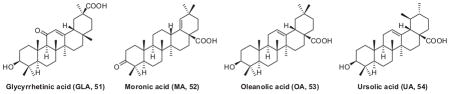
Other triterpene skeletons, including glycyrrhetinic acid (GLA, 51), moronic acid (MA, 52), oleanolic acid (OA, 53), and ursolic acid (UA, 54) as well as their derivatives modified at a position corresponding to C-28 of BA, were also synthesized and evaluated.133 However, none of these analogs were better than RPR103611 and IC9564, indicating that the BA triterpenoid skeleton itself, in addition to the C-28 side chain moiety, is also a pharmacophore for antiviral potency.
b. Mechanism of action study of RPR103611 and IC9564
RPR103611 does not inhibit RT, integrase, or protease. A time-of-addition assay indicated that postponing the addition time of RPR103611 for 2 h cancelled its inhibitory potency on HIV-1 replication, suggesting that this compound functions at an early stage of the virus life cycle.131 A virus binding assay using recombinant soluble CD4 ruled out the possibilities that attachment of virus to target cells or CD4-gp120 binding were inhibited. RPR103611 does blockage syncytium formation, which indicates that it functions at a post-binding step necessary for virus-membrane fusion. Thus, this compound is the first nonpeptidic small molecule found to act as a fusion inhibitor.131
A sequence analysis of RPR103611-resistant mutants indicated that a single amino acid change, I84S, in the HIV-1 gp41 region is sufficient to confer drug resistance.134 The I84 residue is highly conserved and is only rarely replaced by another hydrophobic residue (e.g., phenylalanine). However, I84 is also found in gp41 of some naturally RPR103611-resistant HIV-1 strains, such as NDK or ELI.134 Thus, although the drug-resistant mutants occur on gp41, it would be imprudent to conclude that gp41 is the direct target for RPR103611. It is possible that the mutation of I84 to a polar amino acid may alter the gp41 conformation so that the stability of the gp120-gp41 complex is affected.134 The mutation could also modify access of a compound to a target on gp120. Indeed, a later study from the same group reported that the antiviral efficacy of RPR103611 depends on both the sequence of the gp41 loop region (I84, L91) and the stability of the gp120-gp41 complex.135 Virus with a decreased affinity between gp120 and gp41 is more susceptible to RPR103611.135
A mechanism of action study on IC9564 by Chen et al. revealed that mutations in gp120 are responsible for resistance to the compound.136 No mutations on gp41 were found to significantly affect the antiviral activity of the compound.136 However, this discovery does not rule out the possibility that IC9564 and RPR103611 share the same mechanism of action, since IC9564-resistant viral strains carry mutations of G237R and R252K, which locate in the inner domain of gp120 core. The inner domain of gp120 is believed to interact with gp41, indicating that these mutations on gp120 may also affect the affinity between gp120 and gp41.136 In this particular study, no mutations were found in the regions of gp120 that are well defined for CD4 binding or chemokine co-receptor binding. In continuing studies, a conformational change in gp120 induced by IC9564 was observed by using conformational monoclonal antibody.137,138 It was discovered that IC9564 induces a nonproductive gp120 conformation that is not able to trigger a conformational rearrangement in gp41 for membrane fusion.138 This discovery supports the previous speculation that triterpenes in this class function as nonpeptidic small molecule fusion inhibitors.
In their most recent publication, Dr. Chen’s group reported that IC9564 can increase the antiviral activity of TAK-779 by more than 100-fold.139 Mutations near the tip of the V3 loop were found among IC9564 escaping viruses. Moreover, IC9564 can compete with the binding of V3 loop monoclonal antibodies and with the binding of CD4-gp120 complex to chemokine receptors.139 These results suggested that IC9564 may also function by targeting the V3 loop of gp120, a domain involved in chemokine receptor binding.139,140 The exact binding target of triterpene derivatives is still under investigation.
c. Current status of triterpene derivatives as potent HIV-1 inhibitors
Interestingly, the anti-HIV-1 targets of triterpene analogs can vary depending on the side chain modification positions. C-28 modified BA analogs are potent HIV entry inhibitors, while C-3 modified BA derivatives function by blocking virus maturation.16 Bevirimat (DSB, PA-457, Panacos Pharmaceuticals) (55), which is a C-3 substituted dimethylsuccinyl BA analog, represents the first HIV-1 maturation inhibitor. It is currently in Phase IIb clinical trials and shows good oral bioavailability, safety and pharmacokinetic profiles.141–145 It is encouraging that triterpene analogs can be safely and efficiently used in humans.
Recent progress in this research area has included the development of bi-functional BA analogs, which contain both the C-3 and C-28 side chains, such as A12-2 (56), with IC50 value of 2.6 nM.146 This category of compounds preserves the anti-HIV-1 entry and anti-HIV-1 maturation activities of the parent compounds, and shows a synergistic effect on antiviral potency.

3. CONCLUSION
With increasing knowledge of the HIV entry process, various targets involved in different stages of viral entry have emerged. Inhibitors of these novel targets hold great potential to supplement current HAART, especially to treat individuals harboring multidrug-resistant virus. The success of enfuvirtide and maraviroc validates the clinical application of viral entry inhibitors as a new class of antiretroviral drugs. Many more HIV entry inhibitors, including attachment inhibitors, co-receptor binding inhibitors and fusion inhibitors, are currently in clinical trials, and some have already advanced to Phase III clinical studies.
However, along with great success in the development of HIV entry inhibitors, some problems also arise. It is well known that the envelop glycoprotein is among the most variable HIV proteins with diverse genotypes. Thus, the susceptibility of treatment-naive viral strains to different entry inhibitors targeting viral envelop glycoprotein may vary significantly. In addition, co-receptor binding inhibitors are phenotype specific, including CCR5 antagonists and CXCR4 antagonists. Thus, HIV patients must be analyzed for which viral isolates (R5, X4, or R5/X4) they harbor for appropriate selection of treatment with CCR5 or CXCR4 antagonist. Moreover, because CCR5 and CXCR4 co-receptors are actually a host cellular target rather than a viral enzyme or glycoprotein target, the long-term efficacy and safety of drug use must also be closely monitored. The recent FDA approval of maraviroc as the first CCR5 antagonist HIV entry inhibitor, however, suggests that this class of entry inhibitors may be safely used in HIV-infected individuals. This approval, together with the approval of the first fusion inhibitor enfuvirtide, further confirms that HIV entry inhibitors represent significant complements to AIDS therapy. With the emergence of HIV virus resistant to different current ARTs, the development of new drugs with novel targets is still the focus of much research.
Acknowledgments
Contract grant sponsor: National Institute of Allergies and Infectious Diseases; Contract grant numbers: AI-077417, AI-33066.
This investigation was supported by Grants AI-077417 and AI-33066 from the National Institute of Allergies and Infectious Diseases awarded to K. H. Lee.
Biographies
Keduo Qian is a Postdoctoral Research Associate in the Natural Products Research Laboratories, School of Pharmacy, University of North Carolina at Chapel Hill (UNC-CH). She received her B.S. in pharmacy in 2004 from Peking University, Health Science Center, China and Ph.D. in pharmaceutical sciences in 2008 under the direction of Dr. Kuo-Hsiung Lee from UNC-CH. She is currently engaged in the further studies on anti-HIV triterpenes for discovering potential clinical trials candidates.
Susan L. Morris-Natschke is Research Associate Professor, Division of Medicinal Chemistry and Natural Products, School of Pharmacy, University of North Carolina at Chapel Hill (UNC-CH). She received her B.S. degree in 1975 in chemistry from the University for Maryland and Ph.D. in 1982 in organic chemistry from UNC-CH under the direction of Dr. Ernest L. Eliel. Her research interests have included the design and synthesis of antiviral and anticancer phospholipids, leading to a position as Scientific Advisor for Kucera Pharmaceutical Company, Winston-Salem, NC. She is also highly involved in natural products chemistry and scientific writing and editing.
Kuo-Hsiung Lee is Kenan Professor of Medicinal Chemistry and Director of the Natural Products Research Laboratories at the University of North Carolina at Chapel Hill (UNC-CH), and Academician of Academia Sinica, Taiwan. He received his B.S. degree from Kaohsiung Medical College, Taiwan (1961); M.S. in Pharmaceutical Chemistry from Kyoto University, Japan (1965); and Ph.D. in Medicinal Chemistry from the University of Minnesota, Minneapolis (1968). He has published over 625 articles, served on 18 journal editorial advisory boards, and obtained over 65 patents dealing with bioactive natural products and received numerous awards, including the Lifu Academic Award for Chinese Medicine (1994), the Outstanding Achievement Award, University of Minnesota (1999), Taiwanese-American Foundation Achievement Award in Science and Engineering (2003), and Kitasato Microbial Chemistry Medal (2005). As a world-recognized leader in medicinal and natural products chemistry, he and his associates have discovered over 3,000 bioactive natural products and their synthetic derivatives/analogs from medicinal plants by combining modern chemistry and biology. These lead structures come from all natural product classes and serve as templates for new generation drug design involved in new drug discovery to potentially treat cancer, AIDS, or other diseases. A specific example is the successful discovery of Bevirimat (dimethyl succinyl betulinic acid), a synthetic analog of the natural triterpene betulinic acid, and the first in a new class of HIV drugs called Maturation Inhibitors. It is currently in an anti-AIDS Phase IIb clinical trial in the US.
APPENDIX
Pharmaceutical Companies
Aronex Pharmaceuticals, 8707 Technology Forest Place, The Woodlands, TX 77381-1191
Bristol-Myers Squibb Company, P.O. Box 4000, Princeton, NJ 08543-4000 (US)
Cellegy Pharmaceuticals Inc., 2085 B Quaker Pointe Rd., Quakertown PA 18951
Genzyme Corporation, 500 Kendall Street, Cambridge, MA 02142
GlaxoSmithKline US Headquarters, 1500 Spring Garden St, Philadelphia, PA 19130
Incyte Corporation, Experimental Station, Route 141 & Henry Clay Road, Building E336, Wilmington, DE 19880
Indevus Pharmaceuticals, Inc. - NJ, 7 Clarke Drive, Cranbury, NJ 08512
Kureha Chemical Industries, 3-3-2, Nihonbashi-Hamacho, Chuo-ku, Tokyo 103-855
Novartis, Ltd., Pharmaceuticals, Pharma Research, CH-4002 Basle, Switzerland
Panacos Pharmaceuticals, Gaithersburg, Maryland Research & Development Facility, 209 Perry Parkway, Suite 7, Gaithersburg, Maryland 20877
Pfizer Research and Development, Ramsgate Road, Sandwich, Kent CT13 9NJ, UK
Progenics Pharmaceuticals, Inc., 777 Old Saw Mill River Road, Tarrytown, NY 10591
Schering-Plough Corporation, World Headquarters, 2000 Galloping Hill Road, Kenilworth, NJ 07033-0530
Tanox Inc., 10555 Stella Link, Houston, TX 77025
Footnotes
See Appendix for location of pharmaceutical companies.
References
- 1.UNAIDS. AIDS epidemic update. Dec, 2007. Report nr UNAIDS/07.27E/JC1322E. [Google Scholar]
- 2.Girard MP, Osmanov SK, Kieny MP. A review of vaccine research and development: The human immunodeficiency virus (HIV) Vaccine. 2006;24(19):4062–4081. doi: 10.1016/j.vaccine.2006.02.031. [DOI] [PubMed] [Google Scholar]
- 3.Barbaro G, Scozzafava A, Mastrolorenzo A, Supuran CT. Highly active antiretroviral therapy: Current state of the art, new agents and their pharmacological interactions useful for improving therapeutic outcome. Curr Pharm Des. 2005;11(14):1805–1843. doi: 10.2174/1381612053764869. [DOI] [PubMed] [Google Scholar]
- 4.Ho DD, Neumann AU, Perelson AS, Chen W, Leonard JM, Markowitz M. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature. 1995;373(6510):123–126. doi: 10.1038/373123a0. [DOI] [PubMed] [Google Scholar]
- 5.Hammer SM, Katzenstein DA, Hughes MD, Gundacker H, Schooley RT, Haubrich RH, Henry WK, Lederman MM, Phair JP, Niu M, Hirsch MS, Merigan TC. A trial comparing nucleoside monotherapy with combination therapy in HIV-infected adults with CD4 cell counts from 200 to 500 per cubic millimeter. AIDS Clinical Trials Group Study 175 Study Team. N Engl J Med. 1996;335(15):1081–1090. doi: 10.1056/NEJM199610103351501. [DOI] [PubMed] [Google Scholar]
- 6.Gulick RM, Meibohm A, Havlir D, Eron JJ, Mosley A, Chodakewitz JA, Isaacs R, Gonzalez C, McMahon D, Richman DD, Robertson M, Mellors JW. Six-year follow-up of HIV-1-infected adults in a clinical trial of antiretroviral therapy with indinavir, zidovudine, and lamivudine. AIDS. 2003;17(16):2345–2349. doi: 10.1097/00002030-200311070-00009. [DOI] [PubMed] [Google Scholar]
- 7.Chun TW, Carruth L, Finzi D, Shen X, DiGiuseppe JA, Taylor H, Hermankova M, Chadwick K, Margolick J, Quinn TC, Kuo YH, Brookmeyer R, Zeiger MA, Barditch-Crovo P, Siliciano RF. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature. 1997;387(6629):183–188. doi: 10.1038/387183a0. [DOI] [PubMed] [Google Scholar]
- 8.Finzi D, Hermankova M, Pierson T, Carruth LM, Buck C, Chaisson RE, Quinn TC, Chadwick K, Margolick J, Brookmeyer R, Gallant J, Markowitz M, Ho DD, Richman DD, Siliciano RF. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science. 1997;278(5341):1295–1300. doi: 10.1126/science.278.5341.1295. [DOI] [PubMed] [Google Scholar]
- 9.Wong JK, Hezareh M, Gunthard HF, Havlir DV, Ignacio CC, Spina CA, Richman DD. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science. 1997;278(5341):1291–1295. doi: 10.1126/science.278.5341.1291. [DOI] [PubMed] [Google Scholar]
- 10.Piscitelli SC, Flexner C, Minor JR, Polis MA, Masur H. Drug interactions in patients infected with human immunodeficiency virus. Clin Infect Dis. 1996;23(4):685–693. doi: 10.1093/clinids/23.4.685. [DOI] [PubMed] [Google Scholar]
- 11.Louie M, Markowitz M. Goals and milestones during treatment of HIV-1 infection with antiretroviral therapy: A pathogenesis-based perspective. Antiviral Res. 2002;55(1):15–25. doi: 10.1016/s0166-3542(02)00022-0. [DOI] [PubMed] [Google Scholar]
- 12.Boden D, Hurley A, Zhang L, Cao Y, Guo Y, Jones E, Tsay J, Ip J, Farthing C, Limoli K, Parkin N, Markowitz M. HIV-1 drug resistance in newly infected individuals. JAMA. 1999;282(12):1135–1141. doi: 10.1001/jama.282.12.1135. [DOI] [PubMed] [Google Scholar]
- 13.Wegner SA, Brodine SK, Mascola JR, Tasker SA, Shaffer RA, Starkey MJ, Barile A, Martin GJ, Aronson N, Emmons WW, Stephan K, Bloor S, Vingerhoets J, Hertogs K, Larder B. Prevalence of genotypic and phenotypic resistance to anti-retroviral drugs in a cohort of therapy-naive HIV-1 infected US military personnel. AIDS. 2000;14(8):1009–1015. doi: 10.1097/00002030-200005260-00013. [DOI] [PubMed] [Google Scholar]
- 14.Hammer SM, Pedneault L. Antiretroviral resistance testing comes of age. Antivir Ther. 2000;5(1):23–26. [PubMed] [Google Scholar]
- 15.Pereira CF, Paridaen JT. Anti-HIV drug development—An overview. Curr Pharm Des. 2004;10(32):4005–4037. doi: 10.2174/1381612043382459. [DOI] [PubMed] [Google Scholar]
- 16.Yu D, Wild CT, Martin DE, Morris-Natschke SL, Chen CH, Allaway GP, Lee KH. The discovery of a class of novel HIV-1 maturation inhibitors and their potential in the therapy of HIV. Expert Opin Investig Drugs. 2005;14(6):681–693. doi: 10.1517/13543784.14.6.681. [DOI] [PubMed] [Google Scholar]
- 17.Poveda E, Briz V, Soriano V. Enfuvirtide, the first fusion inhibitor to treat HIV infection. AIDS Rev. 2005;7(3):139–147. [PubMed] [Google Scholar]
- 18.Roux KH, Taylor KA. AIDS virus envelope spike structure. Curr Opin Struct Biol. 2007;17(2):244–252. doi: 10.1016/j.sbi.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 19.Lu M, Blacklow SC, Kim PS. A trimeric structural domain of the HIV-1 transmembrane glycoprotein. Nat Struct Biol. 1995;2(12):1075–1082. doi: 10.1038/nsb1295-1075. [DOI] [PubMed] [Google Scholar]
- 20.Kwong PD, Wyatt R, Robinson J, Sweet RW, Sodroski J, Hendrickson WA. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature. 1998;393(6686):648–659. doi: 10.1038/31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Speck RF, Wehrly K, Platt EJ, Atchison RE, Charo IF, Kabat D, Chesebro B, Goldsmith MA. Selective employment of chemokine receptors as human immunodeficiency virus type 1 coreceptors determined by individual amino acids within the envelope V3 loop. J Virol. 1997;71(9):7136–7139. doi: 10.1128/jvi.71.9.7136-7139.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tomaras GD, Greenberg ML. Mechanisms for HIV-1 entry: Current strategies to interfere with this step. Curr Infect Dis Rep. 2001;3(1):93–99. doi: 10.1007/s11908-001-0064-x. [DOI] [PubMed] [Google Scholar]
- 23.Chen CH, Matthews TJ, McDanal CB, Bolognesi DP, Greenberg ML. A molecular clasp in the human immunodeficiency virus (HIV) type 1 TM protein determines the anti-HIV activity of gp41 derivatives: Implication for viral fusion. J Virol. 1995;69(6):3771–3777. doi: 10.1128/jvi.69.6.3771-3777.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chan DC, Fass D, Berger JM, Kim PS. Core structure of gp41 from the HIV envelope glycoprotein. Cell. 1997;89(2):263–273. doi: 10.1016/s0092-8674(00)80205-6. [DOI] [PubMed] [Google Scholar]
- 25.Weissenhorn W, Dessen A, Harrison SC, Skehel JJ, Wiley DC. Atomic structure of the ectodomain from HIV-1 gp41. Nature. 1997;387(6631):426–430. doi: 10.1038/387426a0. [DOI] [PubMed] [Google Scholar]
- 26.Melikyan GB, Markosyan RM, Hemmati H, Delmedico MK, Lambert DM, Cohen FS. Evidence that the transition of HIV-1 gp41 into a six-helix bundle, not the bundle configuration, induces membrane fusion. J Cell Biol. 2000;151(2):413–423. doi: 10.1083/jcb.151.2.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mondor I, Ugolini S, Sattentau QJ. Human immunodeficiency virus type 1 attachment to HeLa CD4 cells is CD4 independent and gp120 dependent and requires cell surface heparans. J Virol. 1998;72(5):3623–3634. doi: 10.1128/jvi.72.5.3623-3634.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moulard M, Lortat-Jacob H, Mondor I, Roca G, Wyatt R, Sodroski J, Zhao L, Olson W, Kwong PD, Sattentau QJ. Selective interactions of polyanions with basic surfaces on human immunodeficiency virus type 1 gp120. J Virol. 2000;74(4):1948–1960. doi: 10.1128/jvi.74.4.1948-1960.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Callahan LN, Phelan M, Mallinson M, Norcross MA. Dextran sulfate blocks antibody binding to the principal neutralizing domain of human immunodeficiency virus type 1 without interfering with gp120-CD4 interactions. J Virol. 1991;65(3):1543–1550. doi: 10.1128/jvi.65.3.1543-1550.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boyd MR, Gustafson KR, McMahon JB, Shoemaker RH, O’Keefe BR, Mori T, Gulakowski RJ, Wu L, Rivera MI, Laurencot CM, Currens MJ, Cardellina JH, II, Buckheit RW, Jr, Nara PL, Pannell LK, Sowder RC, II, Henderson LE. Discovery of cyanovirin-N, a novel human immunodeficiency virus-inactivating protein that binds viral surface envelope glycoprotein gp120: Potential applications to microbicide development. Antimicrob Agents Chemother. 1997;41(7):1521–1530. doi: 10.1128/aac.41.7.1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dey B, Lerner DL, Lusso P, Boyd MR, Elder JH, Berger EA. Multiple antiviral activities of cyanovirin-N: Blocking of human immunodeficiency virus type 1 gp120 interaction with CD4 and coreceptor and inhibition of diverse enveloped viruses. J Virol. 2000;74(10):4562–4569. doi: 10.1128/jvi.74.10.4562-4569.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Botos I, O’Keefe BR, Shenoy SR, Cartner LK, Ratner DM, Seeberger PH, Boyd MR, Wlodawer A. Structures of the complexes of a potent anti-HIV protein cyanovirin-N and high mannose oligosaccharides. J Biol Chem. 2002;277(37):34336–34342. doi: 10.1074/jbc.M205909200. [DOI] [PubMed] [Google Scholar]
- 33.Rusconi S, Moonis M, Merrill DP, Pallai PV, Neidhardt EA, Singh SK, Willis KJ, Osburne MS, Profy AT, Jenson JC, Hirsch MS. Naphthalene sulfonate polymers with CD4-blocking and anti-human immunodeficiency virus type 1 activities. Antimicrob Agents Chemother. 1996;40(1):234–236. doi: 10.1128/aac.40.1.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Van Damme L, Wright A, Depraetere K, Rosenstein I, Vandersmissen V, Poulter L, McKinlay M, Van Dyck E, Weber J, Profy A, Laga M, Kitchen V. A phase I study of a novel potential intravaginal microbicide, PRO 2000, in healthy sexually inactive women. Sex Transm Infect. 2000;76(2):126–130. doi: 10.1136/sti.76.2.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smita J, Soma D, Beverly B, Albert P, JoAnn K, Fang G, Missy C, Lydia ST, Anjali P, Arun R, Sanjay M, Steven JR. Phase I safety study of 0. 5% PRO 2000 vaginal Gel among HIV un-infected women in Pune, India. AIDS Res Ther. 2006;3:4. doi: 10.1186/1742-6405-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang L, Chen CH. Molecular targets of anti-HIV-1 triterpenes. Curr Drug Targets Infect Disord. 2002;2(1):33–36. doi: 10.2174/1568005024605936. [DOI] [PubMed] [Google Scholar]
- 37.Allaway GP, Davis-Bruno KL, Beaudry GA, Garcia EB, Wong EL, Ryder AM, Hasel KW, Gauduin MC, Koup RA, McDougal JS, et al. Expression and characterization of CD4-IgG2, a novel heterotetramer that neutralizes primary HIV type 1 isolates. AIDS Res Hum Retroviruses. 1995;11(5):533–539. doi: 10.1089/aid.1995.11.533. [DOI] [PubMed] [Google Scholar]
- 38.Jacobson JM, Israel RJ, Lowy I, Ostrow NA, Vassilatos LS, Barish M, Tran DN, Sullivan BM, Ketas TJ, O’Neill TJ, Nagashima KA, Huang W, Petropoulos CJ, Moore JP, Maddon PJ, Olson WC. Treatment of advanced human immunodeficiency virus type 1 disease with the viral entry inhibitor PRO 542. Antimicrob Agents Chemother. 2004;48(2):423–429. doi: 10.1128/AAC.48.2.423-429.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jacobson JM, Lowy I, Fletcher CV, O’Neill TJ, Tran DN, Ketas TJ, Trkola A, Klotman ME, Maddon PJ, Olson WC, Israel RJ. Single-dose safety, pharmacology, and antiviral activity of the human immunodeficiency virus (HIV) type 1 entry inhibitor PRO 542 in HIV-infected adults. J Infect Dis. 2000;182(1):326–329. doi: 10.1086/315698. [DOI] [PubMed] [Google Scholar]
- 40.Moore JP, Sattentau QJ, Klasse PJ, Burkly LC. A monoclonal antibody to CD4 domain 2 blocks soluble CD4-induced conformational changes in the envelope glycoproteins of human immunodeficiency virus type 1 (HIV-1) and HIV-1 infection of CD4+ cells. J Virol. 1992;66(8):4784–4793. doi: 10.1128/jvi.66.8.4784-4793.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kuritzkes DR, Jacobson J, Powderly WG, Godofsky E, DeJesus E, Haas F, Reimann KA, Larson JL, Yarbough PO, Curt V, Shanahan WR., Jr Antiretroviral activity of the anti-CD4 monoclonal antibody TNX-355 in patients infected with HIV type 1. J Infect Dis. 2004;189(2):286–291. doi: 10.1086/380802. [DOI] [PubMed] [Google Scholar]
- 42.Lin PF, Blair W, Wang T, Spicer T, Guo Q, Zhou N, Gong YF, Wang HG, Rose R, Yamanaka G, Robinson B, Li CB, Fridell R, Deminie C, Demers G, Yang Z, Zadjura L, Meanwell N, Colonno R. A small molecule HIV-1 inhibitor that targets the HIV-1 envelope and inhibits CD4 receptor binding. Proc Natl Acad Sci USA. 2003;100(19):11013–11018. doi: 10.1073/pnas.1832214100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guo Q, Ho HT, Dicker I, Fan L, Zhou N, Friborg J, Wang T, McAuliffe BV, Wang HG, Rose RE, Fang H, Scarnati HT, Langley DR, Meanwell NA, Abraham R, Colonno RJ, Lin PF. Biochemical and genetic characterizations of a novel human immunodeficiency virus type 1 inhibitor that blocks gp120-CD4 interactions. J Virol. 2003;77(19):10528–10536. doi: 10.1128/JVI.77.19.10528-10536.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ho HT, Fan L, Nowicka-Sans B, McAuliffe B, Li CB, Yamanaka G, Zhou N, Fang H, Dicker I, Dalterio R, Gong YF, Wang T, Yin Z, Ueda Y, Matiskella J, Kadow J, Clapham P, Robinson J, Colonno R, Lin PF. Envelope conformational changes induced by human immunodeficiency virus type 1 attachment inhibitors prevent CD4 binding and downstream entry events. J Virol. 2006;80(8):4017–4025. doi: 10.1128/JVI.80.8.4017-4025.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Si Z, Madani N, Cox JM, Chruma JJ, Klein JC, Schon A, Phan N, Wang L, Biorn AC, Cocklin S, Chaiken I, Freire E, Smith AB, III, Sodroski JG. Small-molecule inhibitors of HIV-1 entry block receptor-induced conformational changes in theviral envelope glycoproteins. Proc Natl Acad Sci USA. 2004;101(14):5036–5041. doi: 10.1073/pnas.0307953101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kadow J, Wang HG, Lin PF. Small-molecule HIV-1 gp120 inhibitors to prevent HIV-1 entry: An emerging opportunity for drug development. Curr Opin Investig Drugs. 2006;7(8):721–726. [PubMed] [Google Scholar]
- 47.Zhao Q, Ma L, Jiang S, Lu H, Liu S, He Y, Strick N, Neamati N, Debnath AK. Identification of N-phenyl-N′-(2,2,6,6-tetramethyl-piperidin-4-yl)-oxalamides as a new class of HIV-1 entry inhibitors that prevent gp120 binding to CD4. Virology. 2005;339(2):213–225. doi: 10.1016/j.virol.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 48.Ojwang JO, Buckheit RW, Pommier Y, Mazumder A, De Vreese K, Este JA, Reymen D, Pallansch LA, Lackman-Smith C, Wallace TL, et al. T30177, an oligonucleotide stabilized by an intramolecular guanosine octet, is a potent inhibitor of laboratory strains and clinical isolates of human immunodeficiency virus type 1. Antimicrob Agents Chemother. 1995;39(11):2426–2435. doi: 10.1128/aac.39.11.2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Este JA, Cabrera C, Schols D, Cherepanov P, Gutierrez A, Witvrouw M, Pannecouque C, Debyser Z, Rando RF, Clotet B, Desmyter J, De Clercq E. Human immunodeficiency virus glycoprotein gp120 as the primary target for the antiviral action of AR177 (Zintevir) Mol Pharmacol. 1998;53(2):340–345. doi: 10.1124/mol.53.2.340. [DOI] [PubMed] [Google Scholar]
- 50.Wallace TL, Gamba-Vitalo C, Loveday KS, Cossum PA. Acute, multiple-dose, and genetic toxicology of AR177, an anti-HIV oligonucleotide. Toxicol Sci. 2000;53(1):63–70. doi: 10.1093/toxsci/53.1.63. [DOI] [PubMed] [Google Scholar]
- 51.Wallace TL, Bazemore SA, Kornbrust DJ, Cossum PA. Single-dose hemodynamic toxicity and pharmacokinetics of a partial phosphorothioate anti-HIV oligonucleotide (AR177) after intravenous infusion to cynomolgus monkeys. J Pharmacol Exp Ther. 1996;278(3):1306–1312. [PubMed] [Google Scholar]
- 52.Weber J, Piontkivska H, Quinones-Mateu ME. HIV type 1 tropism and inhibitors of viral entry: Clinical implications. AIDS Rev. 2006;8(2):60–77. [PubMed] [Google Scholar]
- 53.Vermeire K, Zhang Y, Princen K, Hatse S, Samala MF, Dey K, Choi HJ, Ahn Y, Sodoma A, Snoeck R, Andrei G, De Clercq E, Bell TW, Schols D. CADA inhibits human immunodeficiency virus and human herpesvirus 7 replication by down-modulation of the cellular CD4 receptor. Virology. 2002;302(2):342–353. doi: 10.1006/viro.2002.1624. [DOI] [PubMed] [Google Scholar]
- 54.Vermeire K, Schols D. Specific CD4 down-modulating compounds with potent anti-HIV activity. J Leukoc Biol. 2003;74(5):667–675. doi: 10.1189/jlb.0403177. [DOI] [PubMed] [Google Scholar]
- 55.Vermeire K, Bell TW, Choi HJ, Jin Q, Samala MF, Sodoma A, De Clercq E, Schols D. The Anti-HIV potency of cyclotriazadisulfonamide analogs is directly correlated with their ability to down-modulate the CD4 receptor. Mol Pharmacol. 2003;63(1):203–210. doi: 10.1124/mol.63.1.203. [DOI] [PubMed] [Google Scholar]
- 56.Vermeire K, Schols D, Bell TW. CD4 down-modulating compounds with potent anti-HIV activity. Curr Pharm Des. 2004;10(15):1795–1803. doi: 10.2174/1381612043384547. [DOI] [PubMed] [Google Scholar]
- 57.Bell TW, Anugu S, Bailey P, Catalano VJ, Dey K, Drew MG, Duffy NH, Jin Q, Samala MF, Sodoma A, Welch WH, Schols D, Vermeire K. Synthesis and structure-activity relationship studies of CD4 down-modulating cyclotriazadisulfonamide (CADA) analogues. J Med Chem. 2006;49(4):1291–1312. doi: 10.1021/jm0582524. [DOI] [PubMed] [Google Scholar]
- 58.Vermeire K, Lisco A, Grivel JC, Scarbrough E, Dey K, Duffy N, Margolis L, Bell TW, Schols D. Design and cellular kinetics of dansyl-labeled CADA derivatives with anti-HIV and CD4 receptor down-modulating activity. Biochem Pharmacol. 2007;74(4):566–578. doi: 10.1016/j.bcp.2007.05.018. [DOI] [PubMed] [Google Scholar]
- 59.Berger EA, Murphy PM, Farber JM. Chemokine receptors as HIV-1 coreceptors: Roles in viral entry, tropism, and disease. Annu Rev Immunol. 1999;17:657–700. doi: 10.1146/annurev.immunol.17.1.657. [DOI] [PubMed] [Google Scholar]
- 60.Berger EA, Doms RW, Fenyo EM, Korber BT, Littman DR, Moore JP, Sattentau QJ, Schuitemaker H, Sodroski J, Weiss RA. A new classification for HIV-1. Nature. 1998;391(6664):240. doi: 10.1038/34571. [DOI] [PubMed] [Google Scholar]
- 61.Zhu T, Mo H, Wang N, Nam DS, Cao Y, Koup RA, Ho DD. Genotypic and phenotypic characterization of HIV-1 patients with primary infection. Science. 1993;261(5125):1179–1181. doi: 10.1126/science.8356453. [DOI] [PubMed] [Google Scholar]
- 62.Moyle GJ, Wildfire A, Mandalia S, Mayer H, Goodrich J, Whitcomb J, Gazzard BG. Epidemiology and predictive factors for chemokine receptor use in HIV-1 infection. J Infect Dis. 2005;191(6):866–872. doi: 10.1086/428096. [DOI] [PubMed] [Google Scholar]
- 63.Cormier EG, Dragic T. The crown and stem of the V3 loop play distinct roles in human immunodeficiency virus type 1 envelope glycoprotein interactions with the CCR5 coreceptor. J Virol. 2002;76(17):8953–8957. doi: 10.1128/JVI.76.17.8953-8957.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hartley O, Klasse PJ, Sattentau QJ, Moore JP. V3: HIV’s switch-hitter. AIDS Res Hum Retroviruses. 2005;21(2):171–189. doi: 10.1089/aid.2005.21.171. [DOI] [PubMed] [Google Scholar]
- 65.Briz V, Poveda E, Soriano V. HIV entry inhibitors: Mechanisms of action and resistance pathways. J Antimicrob Chemother. 2006;57(4):619–627. doi: 10.1093/jac/dkl027. [DOI] [PubMed] [Google Scholar]
- 66.Liu R, Paxton WA, Choe S, Ceradini D, Martin SR, Horuk R, MacDonald ME, Stuhlmann H, Koup RA, Landau NR. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell. 1996;86(3):367–377. doi: 10.1016/s0092-8674(00)80110-5. [DOI] [PubMed] [Google Scholar]
- 67.Samson M, Libert F, Doranz BJ, Rucker J, Liesnard C, Farber CM, Saragosti S, Lapoumeroulie C, Cognaux J, Forceille C, Muyldermans G, Verhofstede C, Burtonboy G, Georges M, Imai T, Rana S, Yi Y, Smyth RJ, Collman RG, Doms RW, Vassart G, Parmentier M. Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature. 1996;382(6593):722–725. doi: 10.1038/382722a0. [DOI] [PubMed] [Google Scholar]
- 68.Cocchi F, DeVico AL, Garzino-Demo A, Arya SK, Gallo RC, Lusso P. Identification of RANTES, MIP-1 alpha, and MIP-1 beta as the major HIV-suppressive factors produced by CD8+ T cells. Science. 1995;270(5243):1811–1815. doi: 10.1126/science.270.5243.1811. [DOI] [PubMed] [Google Scholar]
- 69.Simmons G, Clapham PR, Picard L, Offord RE, Rosenkilde MM, Schwartz TW, Buser R, Wells TN, Proudfoot AE. Potent inhibition of HIV-1 infectivity in macrophages and lymphocytes by a novel CCR5 antagonist. Science. 1997;276(5310):276–279. doi: 10.1126/science.276.5310.276. [DOI] [PubMed] [Google Scholar]
- 70.Polo S, Nardese V, De Santis C, Arcelloni C, Paroni R, Sironi F, Verani A, Rizzi M, Bolognesi M, Lusso P. Enhancement of the HIV-1 inhibitory activity of RANTES by modification of the N-terminal region: Dissociation from CCR5 activation. Eur J Immunol. 2000;30(11):3190–3198. doi: 10.1002/1521-4141(200011)30:11<3190::AID-IMMU3190>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 71.Lederman MM, Veazey RS, Offord R, Mosier DE, Dufour J, Mefford M, Piatak M, Jr, Lifson JD, Salkowitz JR, Rodriguez B, Blauvelt A, Hartley O. Prevention of vaginal SHIV transmission in rhesus macaques through inhibition of CCR5. Science. 2004;306(5695):485–487. doi: 10.1126/science.1099288. [DOI] [PubMed] [Google Scholar]
- 72.Trkola A, Ketas TJ, Nagashima KA, Zhao L, Cilliers T, Morris L, Moore JP, Maddon PJ, Olson WC. Potent, broad-spectrum inhibition of human immunodeficiency virus type 1 by the CCR5 monoclonal antibody PRO 140. J Virol. 2001;75(2):579–588. doi: 10.1128/JVI.75.2.579-588.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kondru R, Zhang J, Ji C, Mirzadegan T, Rotstein D, Sankuratri S, Dioszegi M. Molecular interactions of CCR5 with major classes of small-molecule anti HIV CCR5 antagonists. Mol Pharmacol. 2007;73(3):789–800. doi: 10.1124/mol.107.042101. [DOI] [PubMed] [Google Scholar]
- 74.Baba M, Nishimura O, Kanzaki N, Okamoto M, Sawada H, Iizawa Y, Shiraishi M, Aramaki Y, Okonogi K, Ogawa Y, Meguro K, Fujino M. A small-molecule, nonpeptide CCR5 antagonist with highly potent and selective anti-HIV-1 activity. Proc Natl Acad Sci USA. 1999;96(10):5698–5703. doi: 10.1073/pnas.96.10.5698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dragic T, Trkola A, Thompson DA, Cormier EG, Kajumo FA, Maxwell E, Lin SW, Ying W, Smith SO, Sakmar TP, Moore JP. A binding pocket for a small molecule inhibitor of HIV-1 entry within the transmembrane helices of CCR5. Proc Natl Acad Sci USA. 2000;97(10):5639–5644. doi: 10.1073/pnas.090576697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Seto M, Aikawa K, Miyamoto N, Aramaki Y, Kanzaki N, Takashima K, Kuze Y, Iizawa Y, Baba M, Shiraishi M. Highly potent and orally active CCR5 antagonists as anti-HIV-1 agents: Synthesis and biological activities of 1-benzazocine derivatives containing a sulfoxide moiety. J Med Chem. 2006;49(6):2037–2048. doi: 10.1021/jm0509703. [DOI] [PubMed] [Google Scholar]
- 77.Imamura S, Ichikawa T, Nishikawa Y, Kanzaki N, Takashima K, Niwa S, Iizawa Y, Baba M, Sugihara Y. Discovery of a piperidine-4-carboxamide CCR5 antagonist (TAK-220) with highly potent Anti-HIV-1 activity. J Med Chem. 2006;49(9):2784–2793. doi: 10.1021/jm051034q. [DOI] [PubMed] [Google Scholar]
- 78.Dorr P, Westby M, Dobbs S, Griffin P, Irvine B, Macartney M, Mori J, Rickett G, Smith-Burchnell C, Napier C, Webster R, Armour D, Price D, Stammen B, Wood A, Perros M. Maraviroc (UK-427,857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob Agents Chemother. 2005;49(11):4721–4732. doi: 10.1128/AAC.49.11.4721-4732.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wood A, Armour D. The discovery of the CCR5 receptor antagonist, UK-427,857, a new agent for the treatment of HIV infection and AIDS. Prog Med Chem. 2005;43:239–271. doi: 10.1016/S0079-6468(05)43007-6. [DOI] [PubMed] [Google Scholar]
- 80.Armour D, de Groot MJ, Edwards M, Perros M, Price DA, Stammen BL, Wood A. The discovery of CCR5 receptor antagonists for the treatment of HIV infection: Hit-to-lead studies. Chem Med Chem. 2006;1(7):706–709. doi: 10.1002/cmdc.200600031. [DOI] [PubMed] [Google Scholar]
- 81.Price DA, Armour D, de Groot M, Leishman D, Napier C, Perros M, Stammen BL, Wood A. Overcoming HERG affinity in the discovery of the CCR5 antagonist maraviroc. Bioorg Med Chem Lett. 2006;16(17):4633–4637. doi: 10.1016/j.bmcl.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 82.Castonguay LA, Weng Y, Adolfsen W, Di Salvo J, Kilburn R, Caldwell CG, Daugherty BL, Finke PE, Hale JJ, Lynch CL, Mills SG, MacCoss M, Springer MS, DeMartino JA. Binding of 2-aryl-4-(piperidin-1-yl)butanamines and 1,3,4-trisubstituted pyrrolidines to human CCR5: A molecular modeling-guided mutagenesis study of the binding pocket. Biochemistry. 2003;42(6):1544–1550. doi: 10.1021/bi026639s. [DOI] [PubMed] [Google Scholar]
- 83.Carter NJ, Keating GM. Maraviroc. Drugs. 2007;67(15):2277–2288. doi: 10.2165/00003495-200767150-00010. discussion 2289–2290. [DOI] [PubMed] [Google Scholar]
- 84.Tagat JR, McCombie SW, Nazareno D, Labroli MA, Xiao Y, Steensma RW, Strizki JM, Baroudy BM, Cox K, Lachowicz J, Varty G, Watkins R. Piperazine-based CCR5 antagonists as HIV-1 inhibitors. IV. Discovery of 1-[(4,6-dimethyl-5-pyrimidinyl)carbonyl]-4-[4-[2-methoxy-1(R)-4-(trifluoromethyl)phenyl]ethyl-3(S)-methyl-1-piperaz inyl]-4-methylpiperidine (Sch-417690/Sch-D), a potent, highly selective, and orally bioavailable CCR5 antagonist. J Med Chem. 2004;47(10):2405–2408. doi: 10.1021/jm0304515. [DOI] [PubMed] [Google Scholar]
- 85.Strizki JM, Tremblay C, Xu S, Wojcik L, Wagner N, Gonsiorek W, Hipkin RW, Chou CC, Pugliese-Sivo C, Xiao Y, Tagat JR, Cox K, Priestley T, Sorota S, Huang W, Hirsch M, Reyes GR, Baroudy BM. Discovery and characterization of vicriviroc (SCH 417690), a CCR5 antagonist with potent activity against human immunodeficiency virus type 1. Antimicrob Agents Chemother. 2005;49(12):4911–4919. doi: 10.1128/AAC.49.12.4911-4919.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tsamis F, Gavrilov S, Kajumo F, Seibert C, Kuhmann S, Ketas T, Trkola A, Palani A, Clader JW, Tagat JR, McCombie S, Baroudy B, Moore JP, Sakmar TP, Dragic T. Analysis of the mechanism by which the small-molecule CCR5 antagonists SCH-351125 and SCH-350581 inhibit human immunodeficiency virus type 1 entry. J Virol. 2003;77(9):5201–5208. doi: 10.1128/JVI.77.9.5201-5208.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Seibert C, Ying W, Gavrilov S, Tsamis F, Kuhmann SE, Palani A, Tagat JR, Clader JW, McCombie SW, Baroudy BM, Smith SO, Dragic T, Moore JP, Sakmar TP. Interaction of small molecule inhibitors of HIV-1 entry with CCR5. Virology. 2006;349(1):41–54. doi: 10.1016/j.virol.2006.01.018. [DOI] [PubMed] [Google Scholar]
- 88.Cohenl C, Dejesus E, Mills A, Pierone G, Kumar P, Jr, Ruane P, Elion R, Fusco G, Levy R, Solomon K, Erickson-Viitanen S. Potent Antiretroviral Activity of the Once-daily CCR5 Antagonist INCB009471 over 14 Days of Monotherapy. 4th IAS Conference on HIV Pathogenesis, Treatment and Prevenation; 2007; July; Sydney, Australia. (# TUAB106) [Google Scholar]
- 89.Maeda K, Nakata H, Koh Y, Miyakawa T, Ogata H, Takaoka Y, Shibayama S, Sagawa K, Fukushima D, Moravek J, Koyanagi Y, Mitsuya H. Spirodiketopiperazine-based CCR5 inhibitor which preserves CC-chemokine/CCR5 interactions and exerts potent activity against R5 human immunodeficiency virus type 1 in vitro. J Virol. 2004;78(16):8654–8662. doi: 10.1128/JVI.78.16.8654-8662.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Watson C, Jenkinson S, Kazmierski W, Kenakin T. The CCR5 receptor-based mechanism of action of 873140, a potent allosteric noncompetitive HIV entry inhibitor. Mol Pharmacol. 2005;67(4):1268–1282. doi: 10.1124/mol.104.008565. [DOI] [PubMed] [Google Scholar]
- 91.Kazmierski WMAC, Bifulco N, Boros EE, Chauder BA, Chong PY, Duan M, Deanda F, Koble CS, McLean EW, Peckham JP, Perkins AC, Thompson JB, Vanderwall D. WO 2004054974
- 92.Howard OM, Korte T, Tarasova NI, Grimm M, Turpin JA, Rice WG, Michejda CJ, Blumenthal R, Oppenheim JJ. Small molecule inhibitor of HIV-1 cell fusion blocks chemokine receptor-mediated function. J Leukoc Biol. 1998;64(1):6–13. doi: 10.1002/jlb.64.1.6. [DOI] [PubMed] [Google Scholar]
- 93.Chen X, Yang L, Zhang N, Turpin JA, Buckheit RW, Osterling C, Oppenheim JJ, Howard OM. Shikonin, a component of chinese herbal medicine, inhibits chemokine receptor function and suppresses human immunodeficiency virus type 1. Antimicrob Agents Chemother. 2003;47(9):2810–2816. doi: 10.1128/AAC.47.9.2810-2816.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dorn CP, Finke PE, Oates B, Budhu RJ, Mills SG, MacCoss M, Malkowitz L, Springer MS, Daugherty BL, Gould SL, DeMartino JA, Siciliano SJ, Carella A, Carver G, Holmes K, Danzeisen R, Hazuda D, Kessler J, Lineberger J, Miller M, Schleif WA, Emini EA. Antagonists of the human CCR5 receptor as anti-HIV-1 agents. part 1: Discovery and initial structure-activity relationships for 1-amino-2-phenyl-4-(piperidin-1-yl)butanes. Bioorg Med Chem Lett. 2001;11(2):259–264. doi: 10.1016/s0960-894x(00)00637-5. [DOI] [PubMed] [Google Scholar]
- 95.Finke PE, Meurer LC, Oates B, Shah SK, Loebach JL, Mills SG, MacCoss M, Castonguay L, Malkowitz L, Springer MS, Gould SL, DeMartino JA. Antagonists of the human CCR5 receptor as anti-HIV-1 agents. Part 3: A proposed pharmacophore model for 1-[N-(methyl)-N-(phenylsulfonyl)amino]-2-(phenyl)-4-[4-(substituted)piperi din-1-yl]butanes. Bioorg Med Chem Lett. 2001;11(18):2469–2473. doi: 10.1016/s0960-894x(01)00491-7. [DOI] [PubMed] [Google Scholar]
- 96.Hale JJ, Budhu RJ, Holson EB, Finke PE, Oates B, Mills SG, MacCoss M, Gould SL, DeMartino JA, Springer MS, Siciliano S, Malkowitz L, Schleif WA, Hazuda D, Miller M, Kessler J, Danzeisen R, Holmes K, Lineberger J, Carella A, Carver G, Emini E. 1,3,4-Trisubstituted pyrrolidine CCR5 receptor antagonists. Part 2: Lead optimization affording selective, orally bioavailable compounds with potent anti-HIV activity. Bioorg Med Chem Lett. 2001;11(20):2741–2745. doi: 10.1016/s0960-894x(01)00545-5. [DOI] [PubMed] [Google Scholar]
- 97.Kim D, Wang L, Caldwell CG, Chen P, Finke PE, Oates B, MacCoss M, Mills SG, Malkowitz L, Gould SL, DeMartino JA, Springer MS, Hazuda D, Miller M, Kessler J, Danzeisen R, Carver G, Carella A, Holmes K, Lineberger J, Schleif WA, Emini EA. Discovery of human CCR5 antagonists containing hydantoins for the treatment of HIV-1 infection. Bioorg Med Chem Lett. 2001;11(24):3099–3102. doi: 10.1016/s0960-894x(01)00654-0. [DOI] [PubMed] [Google Scholar]
- 98.Tachibana K, Hirota S, Iizasa H, Yoshida H, Kawabata K, Kataoka Y, Kitamura Y, Matsushima K, Yoshida N, Nishikawa S, Kishimoto T, Nagasawa T. The chemokine receptor CXCR4 is essential for vascularization of the gastrointestinal tract. Nature. 1998;393(6685):591–594. doi: 10.1038/31261. [DOI] [PubMed] [Google Scholar]
- 99.Zou YR, Kottmann AH, Kuroda M, Taniuchi I, Littman DR. Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature. 1998;393(6685):595–599. doi: 10.1038/31269. [DOI] [PubMed] [Google Scholar]
- 100.Ma Q, Jones D, Borghesani PR, Segal RA, Nagasawa T, Kishimoto T, Bronson RT, Springer TA. Impaired B-lymphopoiesis, myelopoiesis, and derailed cerebellar neuron migration in CXCR4- and SDF-1-deficient mice. Proc Natl Acad Sci USA. 1998;95(16):9448–9453. doi: 10.1073/pnas.95.16.9448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tamamura H, Murakami T, Masuda M, Otaka A, Takada W, Ibuka T, Nakashima H, Waki M, Matsumoto A, Yamamoto N, et al. Structure-activity relationships of an anti-HIV peptide, T22. Biochem Biophys Res Commun. 1994;205(3):1729–1735. doi: 10.1006/bbrc.1994.2868. [DOI] [PubMed] [Google Scholar]
- 102.Tamamura H, Xu Y, Hattori T, Zhang X, Arakaki R, Kanbara K, Omagari A, Otaka A, Ibuka T, Yamamoto N, Nakashima H, Fujii N. A low-molecular-weight inhibitor against the chemokine receptor CXCR4: A strong anti-HIV peptide T140. Biochem Biophys Res Commun. 1998;253(3):877–882. doi: 10.1006/bbrc.1998.9871. [DOI] [PubMed] [Google Scholar]
- 103.Arakaki R, Tamamura H, Premanathan M, Kanbara K, Ramanan S, Mochizuki K, Baba M, Fujii N, Nakashima H. T134, a small-molecule CXCR4 inhibitor, has no cross-drug resistance with AMD3100, a CXCR4 antagonist with a different structure. J Virol. 1999;73(2):1719–1723. doi: 10.1128/jvi.73.2.1719-1723.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tamamura H, Omagari A, Oishi S, Kanamoto T, Yamamoto N, Peiper SC, Nakashima H, Otaka A, Fujii N. Pharmacophore identification of a specific CXCR4 inhibitor, T140, leads to development of effective anti-HIV agents with very high selectivity indexes. Bioorg Med Chem Lett. 2000;10(23):2633–2637. doi: 10.1016/s0960-894x(00)00535-7. [DOI] [PubMed] [Google Scholar]
- 105.Fujii N, Oishi S, Hiramatsu K, Araki T, Ueda S, Tamamura H, Otaka A, Kusano S, Terakubo S, Nakashima H, Broach JA, Trent JO, Wang ZX, Peiper SC. Molecular-size reduction of a potent CXCR4-chemokine antagonist using orthogonal combination of conformation- and sequence-based libraries. Angew Chem Int Ed Engl. 2003;42(28):3251–3253. doi: 10.1002/anie.200351024. [DOI] [PubMed] [Google Scholar]
- 106.Doranz BJ, Filion LG, Diaz-Mitoma F, Sitar DS, Sahai J, Baribaud F, Orsini MJ, Benovic JL, Cameron W, Doms RW. Safe use of the CXCR4 inhibitor ALX40-4C in humans. AIDS Res Hum Retroviruses. 2001;17(6):475–486. doi: 10.1089/08892220151126508. [DOI] [PubMed] [Google Scholar]
- 107.Daelemans D, Schols D, Witvrouw M, Pannecouque C, Hatse S, van Dooren S, Hamy F, Klimkait T, de Clercq E, VanDamme AM. A second target for the peptoid Tat/transactivation response element inhibitor CGP64222: Inhibition of human immunodeficiency virus replication by blocking CXC-chemokine receptor 4-mediated virus entry. Mol Pharmacol. 2000;57(1):116–124. [PubMed] [Google Scholar]
- 108.Hamy F, Felder ER, Heizmann G, Lazdins J, Aboul-ela F, Varani G, Karn J, Klimkait T. An inhibitor of the Tat/TAR RNA interaction that effectively suppresses HIV-1 replication. Proc Natl Acad Sci USA. 1997;94(8):3548–3553. doi: 10.1073/pnas.94.8.3548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Blanco J, Barretina J, Henson G, Bridger G, De Clercq E, Clotet B, Este JA. The CXCR4 antagonist AMD3100 efficiently inhibits cell-surface-expressed human immunodeficiency virus type 1 envelope-induced apoptosis. Antimicrob Agents Chemother. 2000;44(1):51–56. doi: 10.1128/aac.44.1.51-56.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hatse S, Princen K, De Clercq E, Rosenkilde MM, Schwartz TW, Hernandez-Abad PE, Skerlj RT, Bridger GJ, Schols D. AMD3465, a monomacrocyclic CXCR4 antagonist and potent HIVentry inhibitor. Biochem Pharmacol. 2005;70(5):752–761. doi: 10.1016/j.bcp.2005.05.035. [DOI] [PubMed] [Google Scholar]
- 111.Seibert C, Sakmar TP. Small-molecule antagonists of CCR5 and CXCR4: A promising new class of anti-HIV-1 drugs. Curr Pharm Des. 2004;10(17):2041–2062. doi: 10.2174/1381612043384312. [DOI] [PubMed] [Google Scholar]
- 112.Stone ND, Dunaway SB, Flexner C, Tierney C, Calandra GB, Becker S, Cao YJ, Wiggins IP, Conley J, MacFarland RT, Park JG, Lalama C, Snyder S, Kallungal B, Klingman KL, Hendrix CW. Multiple-dose escalation study of the safety, pharmacokinetics, and biologic activity of oral AMD070, a selective CXCR4 receptor inhibitor, in human subjects. Antimicrob Agents Chemother. 2007;51(7):2351–2358. doi: 10.1128/AAC.00013-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ichiyama K, Yokoyama-Kumakura S, Tanaka Y, Tanaka R, Hirose K, Bannai K, Edamatsu T, Yanaka M, Niitani Y, Miyano-Kurosaki N, Takaku H, Koyanagi Y, Yamamoto N. A duodenally absorbable CXC chemokine receptor 4 antagonist, KRH-1636, exhibits a potent and selective anti-HIV-1 activity. Proc Natl Acad Sci USA. 2003;100(7):4185–4190. doi: 10.1073/pnas.0630420100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Murakami T, Yoshida A, Tanaka R, Mitsuhashi S, Hirose K, Yanaka M, Yamamoto N, Tanaka Y. KRH-2731: An Orally Bioavailable CXCR4 Antagonist Is a Potent Inhibitor of HIV-1 Infection. 11th Conference on Retroviruses and Opportunistic Infections; 2004; San Francisco, CA. p. abstract #541. [Google Scholar]
- 115.Wild C, Oas T, McDanal C, Bolognesi D, Matthews T. A synthetic peptide inhibitor of human immunodeficiency virus replication: Correlation between solution structure and viral inhibition. Proc Natl Acad Sci USA. 1992;89(21):10537–10541. doi: 10.1073/pnas.89.21.10537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Jiang S, Lin K, Strick N, Neurath AR. HIV-1 inhibition by a peptide. Nature. 1993;365(6442):113. doi: 10.1038/365113a0. [DOI] [PubMed] [Google Scholar]
- 117.Robertson D. US FDA approves new class of HIV therapeutics. Nat Biotechnol. 2003;21(5):470–471. doi: 10.1038/nbt0503-470. [DOI] [PubMed] [Google Scholar]
- 118.Wild C, Greenwell T, Matthews T. A synthetic peptide from HIV-1 gp41 is a potent inhibitor of virus-mediated cell-cell fusion. AIDS Res Hum Retroviruses. 1993;9(11):1051–1053. doi: 10.1089/aid.1993.9.1051. [DOI] [PubMed] [Google Scholar]
- 119.Weiss CD. HIV-1 gp41: Mediator of fusion and target for inhibition. AIDS Rev. 2003;5(4):214–221. [PubMed] [Google Scholar]
- 120.Poveda E, Rodes B, Lebel-Binay S, Faudon JL, Jimenez V, Soriano V. Dynamics of enfuvirtide resistance in HIV-infected patients during and after long-term enfuvirtide salvage therapy. J Clin Virol. 2005;34(4):295–301. doi: 10.1016/j.jcv.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 121.Lalezari JP, Bellos NC, Sathasivam K, Richmond GJ, Cohen CJ, Myers RA, Jr, Henry DH, Raskino C, Melby T, Murchison H, Zhang Y, Spence R, Greenberg ML, Demasi RA, Miralles GD. T-1249 retains potent antiretroviral activity in patients who had experienced virological failure while on an enfuvirtide-containing treatment regimen. J Infect Dis. 2005;191(7):1155–1163. doi: 10.1086/427993. [DOI] [PubMed] [Google Scholar]
- 122.Markovic I, Clouse KA. Recent advances in understanding the molecular mechanisms of HIV-1 entry and fusion: Revisiting current targets and considering new options for therapeutic intervention. Curr HIV Res. 2004;2(3):223–234. doi: 10.2174/1570162043351327. [DOI] [PubMed] [Google Scholar]
- 123.Martin-Carbonero L. Discontinuation of the clinical development of fusion inhibitor T-1249. AIDS Rev. 2004;6(1):61. [PubMed] [Google Scholar]
- 124.Delmedico M, Bray B, Cammack N. Next generation HIV peptide fusion inhibitor candidates achieve potent, durable suppression of virus replication in vitro and improved pharmacokinetic properties. 13th Conference on Retroviruses and Opportunistic Infections; 2006; Denver. p. abstract #48. [Google Scholar]
- 125.Dwyer JJ, Wilson KL, Davison DK, Freel SA, Seedorff JE, Wring SA, Tvermoes NA, Matthews TJ, Greenberg ML, Delmedico MK. Design of helical, oligomeric HIV-1 fusion inhibitor peptides with potent activity against enfuvirtide-resistant virus. Proc Natl Acad Sci USA. 2007;104(31):12772–12777. doi: 10.1073/pnas.0701478104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ferrer M, Kapoor TM, Strassmaier T, Weissenhorn W, Skehel JJ, Oprian D, Schreiber SL, Wiley DC, Harrison SC. Selection of gp41-mediated HIV-1 cell entry inhibitors from biased combinatorial libraries of non-natural binding elements. Nat Struct Biol. 1999;6(10):953–960. doi: 10.1038/13324. [DOI] [PubMed] [Google Scholar]
- 127.Debnath AK, Radigan L, Jiang S. Structure-based identification of small molecule antiviral compounds targeted to the gp41 core structure of the human immunodeficiency virus type 1. J Med Chem. 1999;42(17):3203–3209. doi: 10.1021/jm990154t. [DOI] [PubMed] [Google Scholar]
- 128.Armand-Ugon M, Clotet-Codina I, Tintori C, Manetti F, Clotet B, Botta M, Este JA. The anti-HIV activity of ADS-J1 targets the HIV-1 gp120. Virology. 2005;343(1):141–149. doi: 10.1016/j.virol.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 129.Fujioka T, Kashiwada Y, Kilkuskie RE, Cosentino LM, Ballas LM, Jiang JB, Janzen WP, Chen IS, Lee KH. Anti-AIDS agents, 11. Betulinic acid and platanic acid as anti-HIV principles from Syzigium claviflorum, and the anti-HIV activity of structurally related triterpenoids. J Nat Prod. 1994;57(2):243–247. doi: 10.1021/np50104a008. [DOI] [PubMed] [Google Scholar]
- 130.Soler F, Poujade C, Evers M, Carry JC, Henin Y, Bousseau A, Huet T, Pauwels R, De Clercq E, Mayaux JF, Le Pecq JB, Dereu N. Betulinic acid derivatives: A new class of specific inhibitors of human immunodeficiency virus type 1 entry. J Med Chem. 1996;39(5):1069–1083. doi: 10.1021/jm950669u. [DOI] [PubMed] [Google Scholar]
- 131.Mayaux JF, Bousseau A, Pauwels R, Huet T, Henin Y, Dereu N, Evers M, Soler F, Poujade C, De Clercq E, et al. Triterpene derivatives that block entry of human immunodeficiency virus type 1 into cells. Proc Natl Acad Sci USA. 1994;91(9):3564–3568. doi: 10.1073/pnas.91.9.3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Sun IC, Chen CH, Kashiwada Y, Wu JH, Wang HK, Lee KH. Anti-AIDS agents 49. Synthesis, anti-HIV, and anti-fusion activities of IC9564 analogues based on betulinic acid. J Med Chem. 2002;45(19):4271–4275. doi: 10.1021/jm020069c. [DOI] [PubMed] [Google Scholar]
- 133.Yu D, Sakurai Y, Chen CH, Chang FR, Huang L, Kashiwada Y, Lee KH. Anti-AIDS agents 69. Moronic acid and other triterpene derivatives as novel potent anti-HIV agents. J Med Chem. 2006;49(18):5462–5469. doi: 10.1021/jm0601912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Labrosse B, Pleskoff O, Sol N, Jones C, Henin Y, Alizon M. Resistance to a drug blocking human immunodeficiency virus type 1 entry (RPR103611) is conferred by mutations in gp41. J Virol. 1997;71(11):8230–8236. doi: 10.1128/jvi.71.11.8230-8236.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Labrosse B, Treboute C, Alizon M. Sensitivity to a nonpeptidic compound (RPR103611) blocking human immunodeficiency virus type 1 Env-mediated fusion depends on sequence and accessibility of the gp41 loop region. J Virol. 2000;74(5):2142–2150. doi: 10.1128/jvi.74.5.2142-2150.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Holz-Smith SL, Sun IC, Jin L, Matthews TJ, Lee KH, Chen CH. Role of human immunodeficiency virus (HIV) type 1 envelope in the anti-HIV activity of the betulinic acid derivative IC9564. Antimicrob Agents Chemother. 2001;45(1):60–66. doi: 10.1128/AAC.45.1.60-66.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Yuan X, Huang L, Ho P, Labranche C, Chen CH. Conformation of gp120 determines the sensitivity of HIV-1 DH012 to the entry inhibitor IC9564. Virology. 2004;324(2):525–530. doi: 10.1016/j.virol.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 138.Huang L, Lai W, Ho P, Chen CH. Induction of a nonproductive conformational change in gp120 by a small molecule HIV type 1 entry inhibitor. AIDS Res Hum Retroviruses. 2007;23(1):28–32. doi: 10.1089/aid.2006.0137. [DOI] [PubMed] [Google Scholar]
- 139.Lai W, Huang L, Ho P, Li Z, Montefiori D, Chen CH. Betulinic acid derivatives that target gp120 and inhibit multiple genetic subtypes of human immunodeficiency virus type 1. Antimicrob Agents Chemother. 2008;52(1):128–136. doi: 10.1128/AAC.00737-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Huang L, Zhang L, Chen CH. Potential drug targets on the HIV-1 envelope glycoproteins, gp120 and gp41. Curr Pharm Des. 2003;9(18):1453–1462. doi: 10.2174/1381612033454720. [DOI] [PubMed] [Google Scholar]
- 141.Kashiwada Y, Hashimoto F, Cosentino LM, Chen CH, Garrett PE, Lee KH. Betulinic acid and dihydrobetulinic acid derivatives as potent anti-HIV agents. J Med Chem. 1996;39(5):1016–1017. doi: 10.1021/jm950922q. [DOI] [PubMed] [Google Scholar]
- 142.Kanamoto T, Kashiwada Y, Kanbara K, Gotoh K, Yoshimori M, Goto T, Sano K, Nakashima H. Anti-human immunodeficiency virus activity of YK-FH312 (a betulinic acid derivative), a novel compound blocking viral maturation. Antimicrob Agents Chemother. 2001;45(4):1225–1230. doi: 10.1128/AAC.45.4.1225-1230.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Li F, Goila-Gaur R, Salzwedel K, Kilgore NR, Reddick M, Matallana C, Castillo A, Zoumplis D, Martin DE, Orenstein JM, Allaway GP, Freed EO, Wild CT. PA-457: A potent HIV inhibitor that disrupts core condensation by targeting a late step in Gag processing. Proc Natl Acad Sci USA. 2003;100(23):13555–13560. doi: 10.1073/pnas.2234683100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Martin DE, Blum R, Wilton J, Doto J, Galbraith H, Burgess GL, Smith PC, Ballow C. Safety and pharmacokinetics of Bevirimat (PA-457), a novel inhibitor of human immunodeficiency virus maturation, in healthy volunteers. Antimicrob Agents Chemother. 2007;51(9):3063–3066. doi: 10.1128/AAC.01391-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Smith PF, Ogundele A, Forrest A, Wilton J, Salzwedel K, Doto J, Allaway GP, Martin DE. Phase I and II study of the safety, virologic effect, and pharmacokinetics/pharmacodynamics of single-dose 3-o-(3′,3′-dimethylsuccinyl)betulinic acid (bevirimat) against human immunodeficiency virus infection. Antimicrob Agents Chemother. 2007;51(10):3574–3581. doi: 10.1128/AAC.00152-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Huang L, Ho P, Lee KH, Chen CH. Synthesis and anti-HIV activity of bi-functional betulinic acid derivatives. Bioorg Med Chem. 2006;14(7):2279–2289. doi: 10.1016/j.bmc.2005.11.016. [DOI] [PubMed] [Google Scholar]