

Abstract
Pseudouridine is the most abundant modified nucleotide in ribosomal RNA throughout eukaryotes and archaea but its role is not known. Here we produced mouse embryonic fibroblast cells expressing only catalytically inactive dyskerin, the pseudouridine synthase that converts uridine to pseudouridine in ribosomal RNA. The mutant dyskerin protein, D125A, was extremely unstable but cells were able to divide and grow very slowly. Abnormalities in ribosome RNA synthesis were apparent but mature cytoplasmic RNAs lacking pseudouridine were produced and were very unstable. We conclude that pseudouridine is required to stabilize the secondary structure of ribosomal RNA that is essential for its function.
Structured summary of protein interactions∷
fibrillarin and Dkc1 colocalize by fluorescence microscopy (View interaction)
Keywords: Pseudouridine, Dyskerin, Ribosome biogenesis, snoRNA
1. Introduction
Post transcriptional modification of specific nucleotides in ribosomal RNA (rRNA) and spliceosomal RNA (snRNA) is carried out by small nucleolar ribonucleoproteins (snoRNPs) [1]. The two most frequent modifications are methylation, and pseudouridylation. Pseudouridylation is carried out by H/ACA snoRNPs, which consist of an H/ACA snoRNA and 4 proteins. The pseudouridine synthase is Cbf5p, also known as Nap57 and dyskerin.
snoRNA guided pseudouridylation, the structure of H/ACA snoRNAs and the sequence of their protein components have been highly conserved in evolution suggesting that they play an important functional role. Nevertheless the precise functional importance of pseudouridine is not clear. In yeast, temperature sensitive mutants in Cbf5p show slow growth and poor rRNA synthesis at the permissive temperature. Mutation of an aspartic acid residue D95 in yeast Cbf5p (equivalent to D125 in human and mouse dyskerin) abolishes pseudouridine formation and inhibits 18S rRNA synthesis. This aspartic acid residue is the only conserved residue in 4 classes of pseudouridine synthases and is crucial for activity in all enzymes tested to date. Ribosomes from the yeast Cbf5p D95A mutant have a decreased affinity for tRNA binding and decreased translational fidelity, suggesting pseudouridine in rRNA may be important in translation [2]. By deleting individual guide RNAs in yeast it was shown that some specific modified nucleotides can affect translational fidelity and ribosome biogenesis [3,4].
Information on the effects of perturbed pseudouridylation in vertebrates is scant. Mutations in the DKC1 gene encoding dyskerin cause the X-linked form of the severe bone marrow failure syndrome dyskeratosis congenita (DC). Most of the features of this disease are due to altered telomere maintenance [5]. Dyskerin and the other H/ACA snoRNP proteins are also required, in vertebrates, for the assembly and function of telomerase. Although no pseudouridylation defects or alterations in ribosome biogenesis or function have been shown in patients [6], mouse models with mutations in Dkc1 have decreased levels of pseudouridine in rRNA [7,8] and a variety of other defects including changes in translational fidelity [2,9] and perturbed ribosome biogenesis [7,8]. In one case it was shown that in Dkc1 mutant mouse cells, pseudouridylation at a specific site was decreased and that rRNA in the cells, and in cells from a DC patient, had altered electrophoretic mobility under conditions of incomplete denaturation, suggesting changes in the stability or the secondary structure of rRNA [10]. However, a causative relationship between the pseudouridylation status of rRNA and the downstream effects observed in mice or in mouse cells has not been established.
Here we have specifically ablated the pseudouridine synthase activity of dyskerin in mouse embryo fibroblasts by replacing the endogenous Dkc1 gene with a transgene that expresses dyskerin with a mutation that alters the catalytic aspartic acid residue, D125. Cells expressing this protein have no detectable pseudouridine in their rRNA, grow very slowly, have altered rRNA processing and unstable mature rRNA.
2. Materials and methods
2.1. Production of MEF cells expressing mutant Dkc1 cDNAs
Dkc1 mutant (Dkc1D125A) or WT cDNAs were cloned into MSCV-IRES-GFP vector using standard procedures. Production of viral particles and transduction into p53−/−, Dkc1flox MEF cells [11,12] was carried out using standard protocols. A plasmid expressing the Cre-ERT2 gene, that produces a tamoxifen inducible Cre [13] was introduced using puromycin selection. 24 h after infection, 500 nM tamoxifen was added to the medium to induce the deletion of the endogenous Dkc1 gene. 72 h after infection GFP positive single cells were sorted into 96 well plates. Growing cells were expanded via increasing sized wells to eventually be transferred into 10 cm dishes.
2.2. Growth rate of MEF cells
2 × 104 MEF cells were plated into a 24-well plate at Day 0, then OD490 was measured from day 1 to day 5 by using CellTiter 96 Aqueous One Solution Cell Proliferations Assay (Promega, Madison, WI).
2.3. RNA isolation
Total RNA was extracted from MEF cells by using TRIzol Reagent (Invitrogen, Carlsbad, CA). A rotor-stator homogenizer was used to thoroughly disrupt and homogenize mouse tissues. Aliquots of RNA were stored at −80 °C for further use.
2.4. Real-time RT/PCR
Real-time RT/PCR was carried out using Power SYBR-Green PCR Master Mix (Applied Biosystems, Carlsbad, California) and Superscript II MMLV transcriptase and RNase inhibitors (Invitrogen) and a 7900HT Real-time PCR system equipped with SDS software (Applied Biosystems).
2.5. Senescence associated β-galactosidase (SA-β-gal) staining
Senescent cells were detected by using a β-galactosidase Staining Kit (Cell Signaling Technology, Danvers, MA), Briefly, MEF cells were plated in a 6-well plate 24 h before analysis. After washed twice in PBS, the cells were fixed for 10 min in 4% formaldehyde and 0.2% glutaraldehyde in PBS and washed 3 times in PBS for 5 min each. The staining reaction was performed with 2 ml staining solution with 1 mg/ml X-Gal at 37 °C for overnight in the dark.
2.6. Analysis of apoptosis of MEF cells
Apoptotic cells were detected by using a PE-Annexin V and SYTOX Green double staining method from manufacture's protocol (Invitrogen). Briefly, 5 × 105 MEF cells were plated in a 10-cm dish the day before the analysis. After harvesting, cells were washed in 1 × Annexin binding buffer and suspended in binding buffer at a final concentration of about 1 × 106 ml. 5 μl PE Annexin V antibody and 1 μl of the 1 μM SYTOX Green stain working solution were added to each 100 μl cell suspension. After 30 min incubation at room temperature, 400 μl of the Annexin binding buffer were added and the cells analyzed by flow cytometry.
2.7. Pulse chase analysis of rRNA processing
MEF cells were preincubated for 45 min in methionine-free medium and then incubated for 30 min in medium containing l-[methyl-3H]methionine (50 μCi/ml). The cells were then chased in non-radioactive fresh medium for various times. Total RNA was separated on 1.25% agarose formaldehyde gel and transfered to nylon membrane. The membranes were sprayed with EN3-HANCE Spray (Applied Biosystems) and exposed to X-ray films at −80 °C.
2.8. Analysis of pseudouridylation in 28S and 18S rRNA
MEF cells were cultured in phosphate-free DMEM medium for 1 h and then labeled for 3 h with [32P] orthophosphate (0.1 mCi/ml). Total RNA was extracted by using TRIzol and electrophoresed through a 1% agarose formaldehyde gel. The 28S and 18S rRNA were purified by electroelution, phenol/chloroform extracted, ethanol precipitated, and digested with RNase T2 (Sigma) in 50 mM ammonium acetate, pH 4.5/0.05% SDS/1 mM EDTA at 37 °C. Digested RNA (20000 cpm each) was analyzed by two-dimensional cellulose TLC (EM Science, Gibbstown, NJ) by using isobutyric acid/NH4OH/H2O (577:38:385, by volume) in the first dimension and 2-propanol/HCl/H2O (70:15:15, by volume) in the second dimension. The TLC plates were exposed to X-ray films at −80 °C.
2.9. Western blot analysis
Total protein from cells and mouse tissues was prepared by using RIPA lysis buffer (1×TBS, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 0.004% sodium azide and 1×protease inhibitor cocktail). Protein concentration was measured by using the Bio-Rad protein assay (Bio-Rad, Hercules, CA). Relative quantitation was carried out using Quantity One software (Bio Rad).
2.10. Antibodies
The sources of antibodies were as follows: anti-Flag (Sigma, F7425), anti-Fibrillarin (Abcam, ab4566), anti-dyskerin was as previously described [8], anti-β-Actin was used as total protein loading control (Abcam, ab20272).
2.11. Immunofluorescence
Immunofluorescence for Flag and Fibrillarin was performed by using a rabbit anti-Flag and a mouse anti-Fibrillarin with a standard technique. The cells were examined at 1000′ magnification using a fluorescence microscope (Nikon, Melville, NY, USA). FITC, Alexa 568 and DAPI images were overlapped by using the advanced software.
3. Results and discussion
3.1. Isolation of Dkc1D125A MEF cells
FLAG-tagged mutant Dkc1 cDNA, cloned into the MSCV-IRES-GFP vector, was introduced into p53−/− MEF cells in which the Dkc1 gene contained lox elements in intron 11 and the 3′UTR (Dkc1floxed – Fig. 1A [12]. The endogenous Dkc1 gene was then deleted using tamoxifen inducible Cre recombinase. Finally cells were selected by flow cytometry for those that expressed GFP from the integrated viral DNA and were sorted as single cells into 96 well plates (Fig. 1). Cells that could be successfully expanded into 10 cm plates were selected for further experiments. Clones that grew were screened by PCR for those that contained the Dkc1D125A gene and did not contain the endogenous gene. At this stage we obtained 16 clones containing WT dyskerin-FLAG from 192 tested and 2/1248 clones containing D125A-dyskerin-FLAG. Single cell cloning was necessary because cells expressing no dyskerin may not be viable. In addition the cloning allowed us to select for cells whatever their growth properties. Without cloning, non-targeted cells would likely outgrow those carrying the mutation. In fact the D125A clones grew very slowly from the time of sorting with a doubling time of approximately 1 week.
Fig. 1.
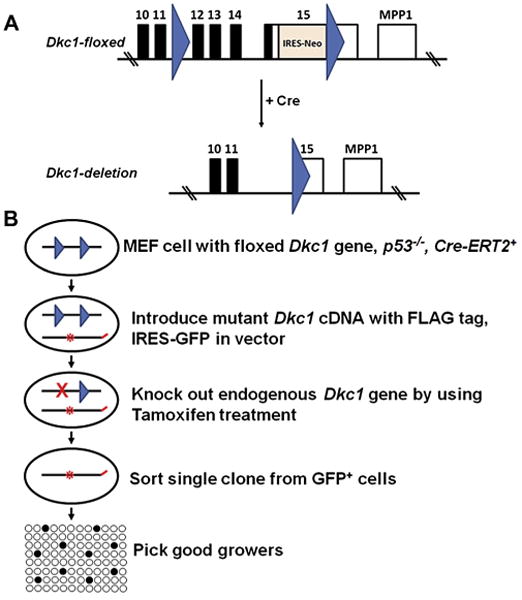
Scheme for isolation of Dkc1D125A MEF cells. (A) Scheme for removing endogenous Dkc1 gene by using Cre-Loxp system. Dkc1-floxed MEF cells were prepared from Dkc1-floxed mice [12]. (B) Scheme for isolation of MEF cells expressing Flag-tagged dyskerin but with no endogenous dyskerin protein. MSCV-IRES-GFP vectors containing the FLAG-tagged WT or mutant Dkc1 cDNA were introduced into p53−/− MEF cells in which the Dkc1 gene contained Loxp sites as shown above. The MEF cells also contained the tamoxifen inducible Cre gene Cre-ERT2. The endogenous Dkc1 gene was then by treatment with tamoxifen. Finally cells were selected by flow cytometry for those that expressed GFP and were sorted as single cells into 96 well plates. Clones that grew were selected by PCR for those that contained the Dkc1D125A gene and did not contain the endogenous wild type gene.
3.2. Dkc1D125A cells grow very slowly and the D125A mutant dyskerin is extremely unstable
Both of the Dkc1D125A clones grew very slowly with a doubling time of approximately 1 week while clones containing the WT cDNA doubled in 2 days (WT-High – Fig. 2A). The proportion of cells in the G2/M phase of the cell cycle was increased in both D125A cell lines, suggesting that a G2/M cell cycle checkpoint may be responsible for the slow cell growth (Fig. 2B). G2/M arrest has been observed in transformed dyskerin depleted cells [14], in yeast cells with mutated Cbf5 [15] and in fibroblasts from dyskeratosis congenita patients [16]. D125A cells also contained a significant number of cells in super G2, representing cells with more than 4n genomic content. To determine if the slow growth was due to increased senescence or apoptosis we stained early passage cells for β-galactosidase (Fig. 2C) and Annexin V (Fig. 2D). D125 cells did not differ from WT cells in these assays leading us to conclude the slow growth is due to the delayed progression through the cell cycle.
Fig. 2.
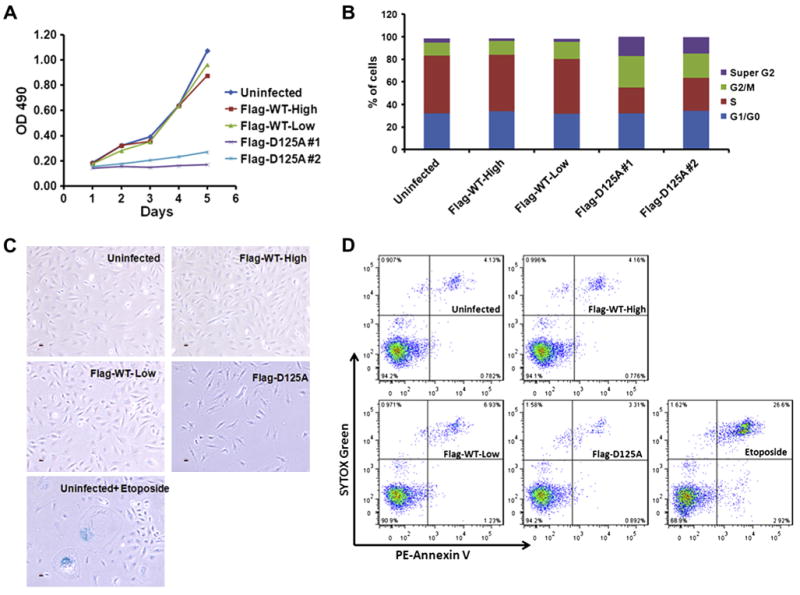
Dkc1D125A cells grow slowly and show cell cycle arrest. (A) Growth curve of MEF cells. 2 × 104 MEF cells were plated into a 24-well plate at Day 0, then OD490 was measured from day 1 to day 5 by using CellTiter 96 Aqueous One Solution Cell Proliferations Assay. (B) Cell cycle analysis of MEF cells. 5 × 105 MEF cells were plated into a 10-CM dish the day before the analysis. After harvesting, cells were fixed with cold 70% ethanol and then stained with freshly made propidium iodide/Triton X-100 staining solution (0.1% Triton X-100, 0.2 mg/ml Rnase A, 20mg/ml propidium iodide) at 37 °C for 30 min before analysis by flow cytometry. (C) Analysis of senescence β-Galactosidase staining (blue). Etoposide treatment at 100 μM for 24 h is used as a positive control. (D) Analysis of apoptosis in MEF cells. Etoposide treatment at 100 μM for 24 h is used as positive control.
A striking feature of both D125A cell lines was that the expression of dyskerin was very low at the protein level, 4–6% of the level in control cell lines, but the cells contained greatly increased amounts of Dkc1 mRNA (Fig. 3A and C). The dramatically decreased D125A protein could be partially rescued by proteasome inhibitor (MG132) treatment (Fig. 3B). Together, these results strongly suggest the D125A protein is very unstable. The extreme instability of the mouse D125A protein might explain the inefficient production of the D125A clones if we assume that a certain minimum amount of dyskerin is required for cell viability and that cells overexpressing the transduced gene sufficiently to produce this amount of dyskerin are rare. The intracellular localization of dyskerin in the D125A cells did not differ from that in cells expressing WT dyskerin, being restricted to nucleoli and Cajal bodies (Fig. 3D).
Fig. 3.
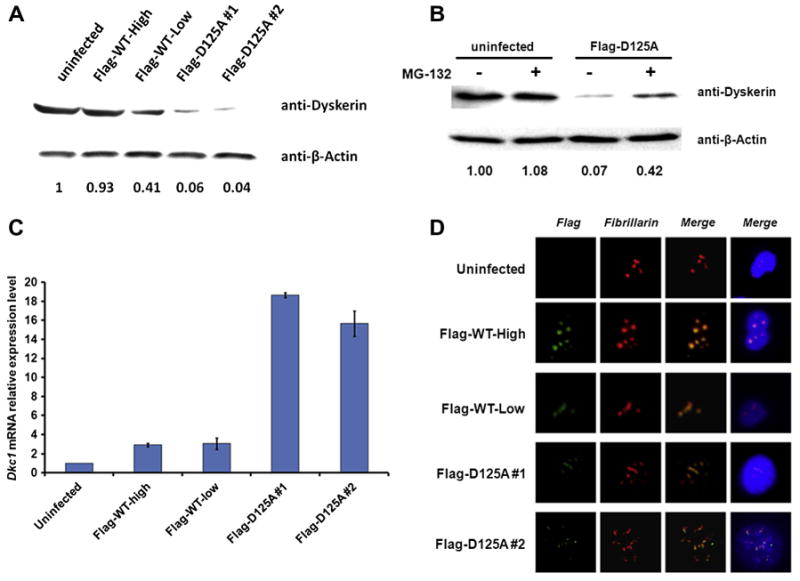
The D125A mutant dyskerin is extremely unstable. (A) Levels of dyskerin protein in MEF cells. 20 μg of total protein for each sample was used for Western blot with antibodies to dyskerin and β-actin. Relative amounts are shown. (B) Effect of proteasome inhibitor (MG132) treatment on MEF cells. 10 μM MG132 was added into culture medium 10 h before harvesting the cells for Western blot. Relative amounts are shown. (C) Total RNA was extracted from growing MEF cells. Dkc1 mRNA expression levels were measured by real time RT/PCR using Power SYBR-Green PCR Master Mix (Applied Biosystems) and Superscript II MMLV transcriptase and RNase inhibitors (Invitrogen) and a 7900HT Real-time PCR system equipped with SDS software (Applied Biosystems). GAPDH was used as an internal control. (D) Flag Tagged WT and D125 dyskerin show normal sub-cellular localization. Immunofluorescence was performed with a standard paraformaldehyde technique (fixed in PBS buffered 4% paraformaldehyde for 10 min, permeabilised with 0.5% Triton-PBS for 15 min, blocked with 30% normal goat serum for 1 h). Primary antibodies, Flag (Green) and Fibrillarin (Red), were used at 1/500 in 1.5% normal goat serum for 2 h. After washing with PBS, cells were incubated with a secondary goat anti-rabbit IgG conjugated with FITC and/or Alexa 568 at 1/1000 in 1.5% normal goat serum for 45 min. All blocking and incubation steps were carried out at room temperature. DNA was counterstained with DAPI (blue). In each case dyskerin colocalizes with fibrillarin in nucleoli and Cajal bodies.
We do not know if the growth and cell cycle changes in D125A cells are due to the specific D125A mutation, to the low amounts of unstable protein, or to a combination of both. To try and solve this problem we attempted to isolate a clone with low levels of wild type dyskerin. Despite intensive efforts we were unable to isolate a clone with amounts of dyskerin as low as that in the D125A cells. The clones with the lowest dyskerin levels that we produced, an example being WT-Low, had about half of the amount of dyskerin, in the steady state, as untreated MEF cells. They did not grow appreciably slower than WT cells (Fig. 2A).
3.3. Perturbed RNA processing and unstable rRNA lacking pseudouridine
Yeast and bacterial enzymes with mutations in the highly conserved aspartic acid residue equivalent to the mouse D125 show no pseudouridylation activity [17,18]. To confirm that the D125A cells were similarly affected by the mutation we did not detect any pseudouridine in mature 18S or 28S rRNA extracted and purified from D125A cells after labeling with 32P-labeled phosphate (Fig. 4A). We investigated pre-rRNA processing in these cells using pulse-chase labeling with tritiated methyl methionine (Fig. 4B–F). This provides a sensitive assay for rRNA processing because cellular pools of methyl-methionine are low, facilitating rapid wash-out during the chase. Uninfected wild type MEF cells, MEF cells ectopically expressing FLAG-tagged WT dyskerin (Flag-WT-High) and MEF cells expressing D125A mutated dyskerin were labeled for 30′ with 3H-methyl-methionine and then chased with unlabelled methionine. The results show that in D125A cell lines (clone 1, Fig. 4B and C; clone 2, Fig. 4D and E) there is a small delay in the appearance of the mature rRNA species and differences in the molecular weights of the transient precursor molecules, reflecting differences in rRNA processing. In particular, species migrating slower than the 32S and 41S precursor molecules are much more abundant in the D125A cells than in WT cells and the abundance of the 30S species decreases more slowly in the mutant cells. Examination of the WT tracks reveals that the same bands are present but at a much lower intensity (Fig. 4B), suggesting some cleavage steps in rRNA processing, but not others, are inhibited by lack of pseudouridine in the template. The alternative is that low dyskerin levels and low snoRNA levels inhibit cleavage of rRNA precursors by a different mechanism. We believe this is unlikely because the only known cleavage events in which H/ACA snoRNPs are essential, are those catalyzed by snR30/U17 snoRNP and leading to mature 18S RNA [19]. In D125A cells 18S and 28S rRNAs are delayed in production to about the same extent, suggesting no specific defect in the snoRNP mediated cleavage event.
Fig. 4.
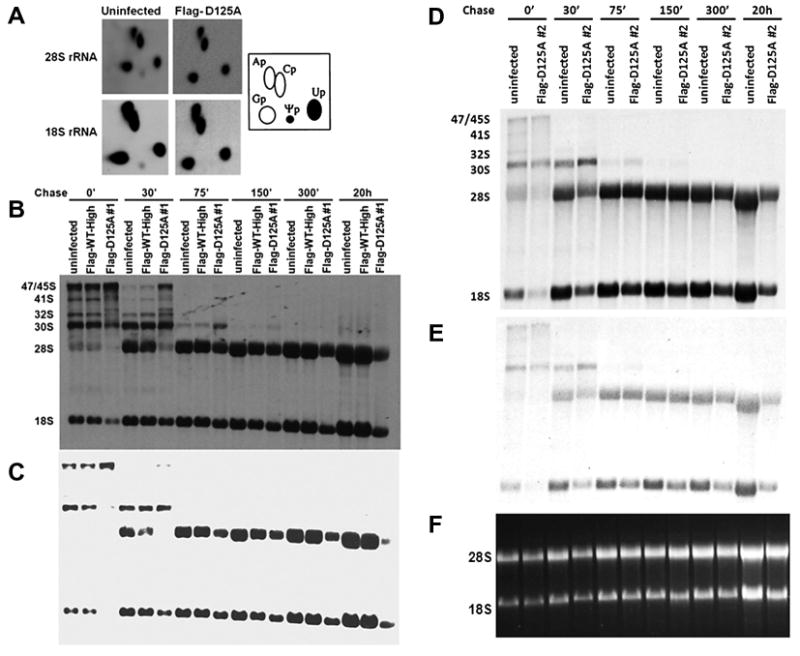
Dkc1D125A cells show rRNA processing abnormalities. (A) Pseudouridine in 18S and 28S rRNA from uninfected and Dkc1D125A MEF cells. MEF cells were cultured in phosphate-free DMEM medium for 1 h and then labeled for 3 h with [32P] orthophosphate (0.1 mCi/ml). Total RNA was electrophoresed through a 1% agarose formaldehyde gel. The 28S and 18S rRNA were purified by electroelution and digested with RNase T2 in 50 mM ammonium acetate, pH 4.5/0.05% SDS/1 mM EDTA at 37 °C. Digested RNA (20000 cpm each) was analyzed by two-dimensional cellulose TLC by using isobutyric acid/NH4OH/H2O (577:38:385) in the first dimension and 2-propanol/HCl/H2O (70:15:15) in the second dimension. The TLC plates were exposed to a tritium screen for 3 days and scanned with phosphorimager (GE Healthcare, Pittsburgh, USA). The positions of the labeled ribonucleotides are indicated. (B) Pulse chase labeling experiments of rRNA isolated from uninfected, Flag-WT-High and Dkc1D125A clone 1 MEF cells. 2 × 106 MEF cells were cultured in methionine free DMEM medium for 45 min and labeled with l-[methyl-3H]methionine for 30′ and then chased in non-radioactive medium for the times shown. The RNA (equal cpm per lane) was separated on a 1.25% formaldehyde agarose gel, transferred to a nylon filter and exposed to X-ray film for 2 weeks. (C) The gel shown in B was exposed for 3 days. (D) Pulse chase labeling experiments of rRNA isolated from uninfected, and Dkc1D125A clone 2 MEF cells. The exposure time was 2 weeks. (E) The gel shown in D was exposed for 3 days. (F) Ethidium bromide staining of the 28S and 18S RNA of D and E.
The most dramatic difference, though, is in the mature rRNAs (Fig. 4C and E). After 30′ of chase the mature 18S and particularly 28S rRNAs in both the D125A cell lines are barely labeled while the mature rRNAs in the WT cells are strongly labeled. As the chase continues the amount of labeled mature rRNA in the WT cells continues to increase up to 20 h of chase. In the D125A cells, however, the label in the mature rRNA species increases up to 150′ but then begins to decrease, with little labeled 18S or 28S rRNA remaining after 20 h of chase. The overall levels of mature rRNAs showed no significant difference (Fig. 4F). This indicates that mature rRNA in D125A cells is unstable compared with that in wild type cells. Our previous results showed that ribosomal RNAs deficient in pseudouridine had altered secondary structure, evident by altered mobility in agarose gels after partial denaturation [10]. These results, together with the finding of rRNA instability presented here, suggest that pseudouridine is important for stabilizing the secondary structure of rRNA that is essential for its function.
3.4. Decreased levels of H/ACA snoRNA but not C/D snoRNA or snRNA in Dkc1D125A cells
We were interested to know if there may be any affect of the D125A mutation on the stability of spliceosomal snRNAs, which contain pseudouridine residues that are produced by dyskerin in H/ACA snoRNA complexes. Fig. 5 shows that while H/ACA snoRNAs are depleted in mutant cells there is no major effect on the abundance of C/D snoRNAs or on snRNAs. Whether lack of pseudouridine residues in snRNA affects their function remains to be seen.
Fig. 5.
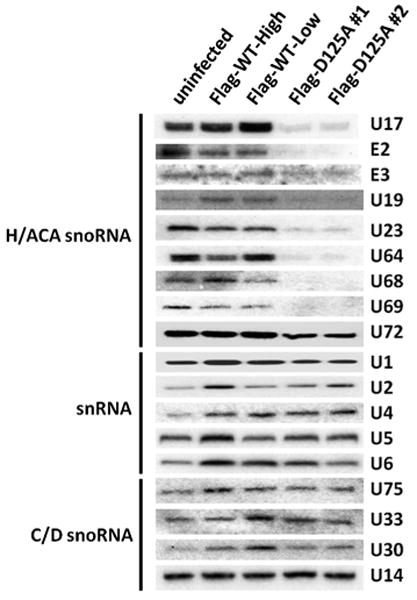
The Dkc1D125A cells show decreased level of box H/ACA snoRNA. Decreased levels of specific H/ACA RNAs in Dkc1D125A cells as assessed by Northern blot analysis. 5 μg of total RNA from MEF cells was loaded in each lane and hybridized with 32P labeled mouse H/ACA snoRNA, C/D box snoRNA and snRNA probes as Indicated.
In conclusion MEF cells with no pseudouridylation activity in H/ACA snoRNPs can produce mature ribosomes. The ribosomes are very unstable and the cells show very slow cell growth and cell division. The results are compatible with a role for pseudouridine in stabilizing the functional secondary structure of ribosomal RNA.
Acknowledgments
We would like to thank the NCI and NIH and for financial support through grants to PJM (CA106995) and MB (CA105312). We also would like to thank the America Society of Hematology (ASH) for financial support through a grant to B-W G (ASH Scholar Award).
References
- 1.Watkins NJ, Bohnsack MT. The box C/D and H/ACA snoRNPs: key players in the modification, processing and the dynamic folding of ribosomal RNA. Wiley Interdiscip Rev RNA. 2011;3:397–414. doi: 10.1002/wrna.117. [DOI] [PubMed] [Google Scholar]
- 2.Jack K, et al. RRNA pseudouridylation defects affect ribosomal ligand binding and translational fidelity from yeast to human cells. Mol Cell. 2011;44:660–666. doi: 10.1016/j.molcel.2011.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baxter-Roshek JL, Petrov AN, Dinman JD. Optimization of ribosome structure and function by rRNA base modification. PLoS ONE. 2007;2:e174. doi: 10.1371/journal.pone.0000174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baudin-Baillieu A, Fabret C, Liang XH, Piekna-Przybylska D, Fournier MJ, Rousset JP. Nucleotide modifications in three functionally important regions of the Saccharomyces cerevisiae ribosome affect translation accuracy. Nucleic Acids Res. 2009;37:7665–7677. doi: 10.1093/nar/gkp816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Walne AJ, Marrone A, Dokal I. Dyskeratosis congenita: a disorder of defective telomere maintenance? Int J Hematol. 2005;82:184–189. doi: 10.1532/IJH97.05067. [DOI] [PubMed] [Google Scholar]
- 6.Wong JM, Collins K. Telomerase RNA level limits telomere maintenance in X-linked dyskeratosis congenita. Genes Dev. 2006;20:2848–2858. doi: 10.1101/gad.1476206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ruggero D, Grisendi S, Piazza F, Rego E, Mari F, Rao PH, Cordon-Cardo C, Pandolfi PP. Dyskeratosis congenita and cancer in mice deficient in ribosomal RNA modification. Science. 2003;299:259–262. doi: 10.1126/science.1079447. [DOI] [PubMed] [Google Scholar]
- 8.Mochizuki Y, He J, Kulkarni S, Bessler M, Mason PJ. Mouse dyskerin mutations affect accumulation of telomerase RNA and small nucleolar RNA, telomerase activity, and ribosomal RNA processing. Proc Natl Acad Sci USA. 2004;101:10756–10761. doi: 10.1073/pnas.0402560101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yoon A, Peng G, Brandenburger Y, Zollo O, Xu W, Rego E, Ruggero D. Impaired control of IRES-mediated translation in X-linked dyskeratosis congenita. Science. 2006;312:902–906. doi: 10.1126/science.1123835. [DOI] [PubMed] [Google Scholar]
- 10.Gu BW, Zhao C, Fan JM, Dai Q, Bessler M, Mason PJ. Anomalous electrophoretic migration of newly synthesized ribosomal RNAs and their precursors from cells with DKC1 mutations. FEBS Lett. 2009;583:3086–3090. doi: 10.1016/j.febslet.2009.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ge J, Rudnick DA, He J, Crimmins DL, Ladenson JH, Bessler M, Mason PJ. Dyskerin ablation in mouse liver inhibits rRNA processing and cell division. Mol Cell Biol. 2010;30:413–422. doi: 10.1128/MCB.01128-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.He J, Navarrete S, Jasinski M, Vulliamy T, Dokal I, Bessler M, Mason PJ. Targeted disruption of Dkc1, the gene mutated in X-linked dyskeratosis congenita, causes embryonic lethality in mice. Oncogene. 2002;21:7740–7744. doi: 10.1038/sj.onc.1205969. [DOI] [PubMed] [Google Scholar]
- 13.Imai T, Jiang M, Chambon P, Metzger D. Impaired adipogenesis and lipolysis in the mouse upon selective ablation of the retinoid X receptor alpha mediated by a tamoxifen-inducible chimeric Cre recombinase (Cre-ERT2) in adipocytes. Proc Natl Acad Sci USA. 2001;98:224–228. doi: 10.1073/pnas.011528898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alawi F, Lin P. Dyskerin is required for tumor cell growth through mechanisms that are independent of its role in telomerase and only partially related to its function in precursor rRNA processing. Mol Carcinog. 2011;50:334–345. doi: 10.1002/mc.20715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiang W, Middleton K, Yoon HJ, Fouquet C, Carbon J. An essential yeast protein, CBF5p, binds in vitro to centromeres and microtubules. Mol Cell Biol. 1993;13:4884–4893. doi: 10.1128/mcb.13.8.4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dokal I, Bungey J, Williamson P, Oscier D, Hows J, Luzzatto L. Dyskeratosis congenita fibroblasts are abnormal and have unbalanced chromosomal rearrangements. Blood. 1992;80:3090–3096. [PubMed] [Google Scholar]
- 17.Huang L, Pookanjanatavip M, Gu X, Santi DV. A conserved aspartate of tRNA pseudouridine synthase is essential for activity and a probable nucleophilic catalyst. Biochemistry. 1998;37:344–351. doi: 10.1021/bi971874+. [DOI] [PubMed] [Google Scholar]
- 18.Zebarjadian Y, King T, Fournier MJ, Clarke L, Carbon J. Point mutations in yeast CBF5 can abolish in vivo pseudouridylation of rRNA. Mol Cell Biol. 1999;19:7461–7472. doi: 10.1128/mcb.19.11.7461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Atzorn V, Fragapane P, Kiss T. U17/snR30 is a ubiquitous snoRNA with two conserved sequence motifs essential for 18S rRNA production. Mol Cell Biol. 2004;24:1769–1778. doi: 10.1128/MCB.24.4.1769-1778.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]