

Abstract
Schizophrenia is a severe mental disorder affecting ~1% of the world population, with heritability of up to 80%. To identify new common genetic risk factors, we performed a genome-wide association study (GWAS) in the Han Chinese population. The discovery sample set consisted of 3,750 patients and 6,468 healthy controls (1,578 cases and 1,592 controls from the Northern Han; 1,238 cases and 2,856 controls from the Central Han; 934 cases and 2,020 controls from the Southern Han); and we followed up the top association signals in an additional independent cohort of 4,383 cases and 4,539 controls from the Han Chinese. Meta-analysis identified genome-wide significant association of common SNPs with schizophrenia on chromosome 8p12 (rs16887244, P=1.27×10−10) and 1q24.2 (rs10489202, P=9.50×10−9). Our findings provide new insights into the pathogenesis of schizophrenia.
Schizophrenia is a severe neuropsychiatric disorder, characterized by psychotic features (delusions and hallucinations), disorganization, dysfunction in normal affective responses, and altered cognitive functioning1. Recent genome-wide studies in European ancestry populations have revealed both common low risk SNPs and rare high-penetrance copy number variants predisposing to schizophrenia2-8. However much of the heritability in schizophrenia remains unaccounted for and the pathways or biological mechanisms that underlie susceptibility are still largely unknown.
To search for common variants associated with schizophrenia, we performed a genome-wide association study (GWAS) of 3,750 patients with schizophrenia and 6,468 healthy controls genotyped using Affymetrix SNP 6.0 genechips in Han Chinese from three geographic locations (Northern, Central and Southern). In total, 546,561 SNPs were used for statistical analysis after quality control (QC) in initial studies. Logistic regression was used to test the additive effects of minor allele dosage for each SNP. Potential population stratification was adjusted for using eigenvectors from principal component analysis (PCA) (Online Methods).
We carried out a combined analysis of 3 independent genome-wide cohorts as the discovery stage, and the most significant associated SNPs were followed up in additional samples. The initial studies included, i) a genome-wide association study of 1,578 schizophrenia patients and 1,592 healthy controls from the Northern Han population (Beijing and Shandong Province); ii) a genome-wide analysis of 1,238 cases and 2,856 controls from the Central Han population (Shanghai and Anhui Province); iii) a genome-wide scan of 934 cases and 2,020 controls recruited from the Southern Han population (Guangdong and Guangxi Provinces). Sample descriptions and characteristics can be found in Online Methods and Supplementary Table 1. A meta-analysis (Online Methods) was carried out based on the PCA-adjusted association results of the three cohorts (Supplementary Figure 1). For all the reported SNPs, the p values of the meta-analysis are identical for fixed- and random-effects models of meta-analysis, and no heterogeneity between the groups was found (Supplementary Table 2). Though no SNP showed genome-wide significance (P<5.0×10−8), four SNPs in the chromosome 8p12 (rs1488935, Pgwas-meta=2.81×10−6) and 1q24.2 (rs10489202, Pgwas-meta=2.65×10−6; rs1060041, Pgwas-meta=4.11×10−6; rs11586522, Pgwas-meta=1.53×10−6) regions showed strong association (Pgwas-meta<5.0×10−6, Table 1, Supplementary Figure 2, Supplementary Table 2). We followed up the top SNPs in these regions (3 SNPs for 1q24.2 and 2 SNPs for 8p12) in an independent cohort of 4,383 schizophrenia cases and 4,539 healthy controls, which, together with the discovery sample set, we defined as BIOX. We successfully replicated the association findings on 8p12 and 1q24.2, and the combined results of the initial associations and replications showed genome-wide significance (8p12: rs16887244, PBIOX-meta = 1.27×10 −10, G allele odds ratio (OR)=0.84, 95% confidence interval (CI)=0.79-0.88; rs1488935, PBIOX-meta = 5.06×10−9, T allele OR=0.85, 95% CI=0.81-0.90; & 1q24.2: rs10489202, PBIOX-meta =9.50×10−9, A allele OR=1.23, 95% CI=1.15-1.32; Table 1). To search for additional associations of common variants, we also carried out imputation analysis in the selected regions (Figure 1).
Table 1. GWAS, replication study and meta-analysis results for selected SNPs.
| CHR | SNP | BP | Frequencya | BIOX GWAS |
BIOX FOLLOW-UP (4383 cases, 4539 controls) |
BIOX GWAS +FOLLOW-UP |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NORTHERN HAN (1578 cases, 1592 controls) |
CENTRAL HAN (1238 cases, 2856 controls) |
SOUTHERN HAN (934 cases, 2020 controls) |
INITIAL META | ||||||||||||
|
| |||||||||||||||
| Alleleb | OR (95% CI) |
P* | OR (95% CI) |
P* | OR (95% CI) |
P* | Pgwas-meta | Allele | OR (95% CI) |
Prep | PBIOX-meta | ||||
| 1 | rs10489202 | 166169703 | 0.141 | A | 1.273 (1.084-1.494) |
0.003 | 1.309 (1.122-1.528) |
6.19×10−4 | 1.180 (0.953-1.461) |
0.130 | 2.65×10−6 | A | 1.189 (1.094-1.293) |
4.76×10−5 | 9.50×10−9 |
| 1 | rs1060041 | 166240600 | 0.139 | T | 1.272 (1.083-1.493) |
0.003 | 1.324 (1.133-1.547) |
4.06×10−4 | 1.136 (0.916-1.410) |
0.245 | 4.11×10−6 | T | 1.113 (1.025-1.211) |
0.012 | 5.31×10−7 |
| 1 | rs11586522 | 166357655 | 0.160 | T | 1.277 (1.099-1.483) |
0.001 | 1.267 (1.101-1.459) |
9.77×10−4 | 1.181 (0.956-1.46) |
0.123 | 1.53×10−6 | T | 1.040 (0.957-1.129) |
0.362 | 1.17 ×10−4 |
| 8 | rs16887244 | 38150502 | 0.317 | G | 0.852 (0.747-0.972) |
0.017 | 0.809 (0.718-0.912) |
5.16×10−4 | 0.876 (0.745-1.029) |
0.107 | 9.27×10−6 | G | 0.833 (0.781-0.888) |
2.23×10−8 | 1.27×10−10 |
| 8 | rs1488935 | 38252950 | 0.316 | T | 0.842 (0.737-0.962) |
0.011 | 0.795 (0.703-0.898) |
2.30×10−4 | 0.871 (0.742-1.023) |
0.093 | 2.81×10−6 | T | 0.871 (0.816-0.929) |
3.42×10−5 | 5.06×10−9 |
Frequency is the average minor allelic frequency of controls in BIOX data
minor allele
These P values were adjusted by PCA methods in each GWAS of geographical sample groups.
Figure 1.
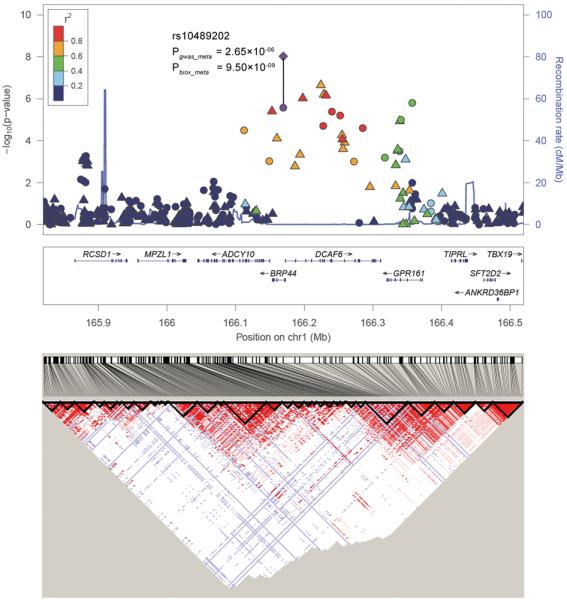
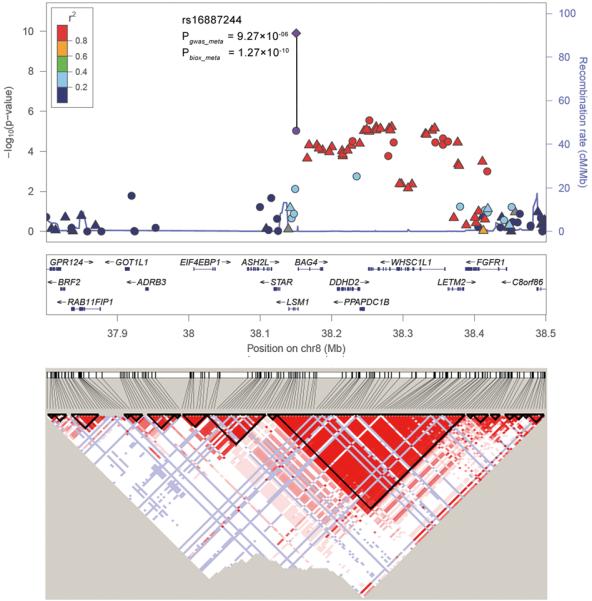
Regional plots of the 2 schizophrenia loci (8p12 and 1q24.2). Results (−log10 P) are shown for SNPs for the region flanking 350 kb on either side of the marker SNPs. Pgwas_meta is for the combined results of the three initial datasets, represented by the circle (for genotyped SNPs) and the triangle (for imputed SNPs), and Pbiox_meta is for the combined results of the initial and follow-up dataset (BIOX), represented by the diamond. The marker SNP is shown in purple and the r2 values of the rest of the SNPs are indicated by different colors. The genes within the relevant region are annotated and shown as arrows. The D′ LD block structure of the related surrounding region according to the CHB+JPT HapMap data are provided at the bottom.
We further validated our top association signals by obtaining results from a GWAS of the SGENE-plus project (aims to identify genetic variants associating with schizophrenia, and study their impact on phenotype and their interactions with environmental factors contributing to the pathogenesis of the disease); this included 3,830 cases and 14,724 controls of European ancestry. Both SNPs rs16887244 (PSGENE-plus=0.026, G allele OR=0.92, 95% CI=0.85-0.99) and rs1488935 (PSGENE-plus=0.027, T allele OR=0.92, 95% CI=0.85-0.99) showed nominal association with schizophrenia in the European ancestry population, and the direction of the effect was consistent with our findings (Supplementary Table 2 and 3). The effect size of rs10489202 (A allele OR=1.01, 95% CI=0.94-1.09) was small in the SGENE-plus data (Supplementary Table 3). A statistical power comparison between the two datasets was also carried out (Supplementary Table 4).
On 8p12, both SNPs rs16887244 and rs1488935 demonstrated genome-wide significance in the combined analysis (Table 1, Figure 1a). Controlling for rs16887244, conditional logistic regression analysis revealed that there were no additional association signals (Supplementary Table 5). SNP rs16887244 is located in intron 1 of LSM1 (MIM: 607281); rs1488935 is located in intron 23 of WHSC1L1 (MIM: 607083), a gene which is related to the Wolf-Hirschhorn syndrome 1 candidate-like1 gene. We used data from two published expression quantitative trait loci (eQTL) datasets (derived from lymphoblastoid cell lines9,10 and the brain11) to determine whether rs16887244 is associated with the expression of the genes nearby. In the expression data of the lymphoblastoid cell lines (Supplementary Table 6), rs16887244 is nominally associated with the expression of several genes (ASH2L, LSM1, BAG4, DDHD2, and PPAPDC1B; P < 0.05), and in the expression data of the brain (Supplementary Table 7), it is nominally associated with genes ASH2L, DDHD2, PPAPDC1B and LETM2 (P < 0.05).
Notably, rs1488935 is located ~135kb upstream of FGFR1 (MIM: 136350), the fibroblast growth factor receptor 1 gene. Hippocampal FGFR1 mRNA expression has been reported to be upregulated in schizophrenia and major depression patients12 and transgenic mice expressing a dominant negative mutant [FGFR1(TK-)] from the catecholaminergic, neuron-specific tyrosine hydroxylase (TH) gene promoter have been found to exhibit a schizophrenia-like syndrome13. Terwisscha et al. in a review found several lines of evidence, including functional plausibility, positional and functional genetic studies, knockout mouse models, effects of using FGF in animals and humans, and associations between FGFs and environmental risk factors of schizophrenia, supporting a role for FGFs in schizophrenia14. What is more O’Donovan et al. have also reported another fibroblast growth factor receptor gene FGFR2 associated with schizophrenia (P=0.0009)15.
On 1q24.2, rs10489202 is located in intron 1 of BRP44 gene, the brain protein 44 gene (Figure 1b), rs1060041 is a coding-synonymous SNP in DCAF6 (MIM: 610494) and rs11586522 is located in intron 2 region of GPR161 (MIM: 612250). Controlling for the most significant signal rs10489202, logistic regression analysis indicated that no additional signal was observed in the genomic region around rs10489202 (Supplementary Table 5). In the expression data of the lymphoblastoid cell lines (Supplementary Table 6), rs10489202 is nominally associated with the expression of several genes (MPZL1, DCAF6, and TIPRL; P<0.05), however, in the expression data of the brain, we observed no association (Supplementary Table 7). Interestingly, rs10489202 is also ~140kb downstream of MPZL1 (MIM: 604376), the myelin protein zero like 1 or protein zero-related gene. Tkachev et al.16 found that MPZL1/PZR was significantly upregulated in schizophrenia patients compared with healthy controls (1.29-fold, P=0.0263). Common SNPs in MPZL1/PZR gene were recently reported to be associated with schizophrenia in 523 case-parent triad family of Han Chinese (P=0.0017)17.
Population stratification is a major concern in GWAS that uses case-control design. It has been demonstrated using a set of GWAS datasets that there is a “north-south” population structure in China18. The PCA analysis of all our initial study samples also revealed such a structure (Supplementary Figure 3 and 4).We therefore stratified our samples into Northern, Central and Southern groups according to their geographical regions, and carried out GWAS on individual groups, then obtained the combined association using meta-analysis.
The MHC region on chromosome 6p21.3-22.1 has previously been associated with schizophrenia in GWASs of European ancestry populations4-6, and these findings have been replicated in a large sample of Han Chinese population19. In our initial study, no common SNP in these regions met the selection criteria (Pgwas-meta<5.0×10−6). However, 149 SNPs out of 1,786 (8.34%, Pgwas-meta<0.05) showed nominal association signals with schizophrenia in the MHC region (Supplementary Table 8), and the most significant signal was observed at rs2394514 (Pgwas-meta=1.16×10−5), which is located at 6p22.1. We found no significant association at rs131940534,5 and rs69325906 from 6p22.1, and rs31312966 from 6p21.32, which was previously reported as genome-wide significant associations (P<5.0×10−8) with schizophrenia in European ancestry populations (derived from the NHGRI GWAS Catalog20, full SNPs are listed in the Supplementary Table 9). We compared the allele frequencies of these SNPs between HapMap CEU and HCB samples (Supplementary Table 10). The allele frequencies for most of the SNPs obviously differ between the European and Chinese populations. In addition, we measured linkage disequilibrium (LD), r2 values of these SNPs with the 149 SNPs (in the MHC region with Pgwas-meta < 0.05 in our study) in HapMap CEU and HCB samples (Supplementary Table 11 and 12). SNP rs7749823 which is in LD with rs13194053 (r2=0.644) and rs6932590 (r2=0.623) in the European populations shows association with schizophrenia in our study (Pgwas-meta=4.18×10−5, A allele OR=1.64, Risk allele frequency (RAF)=0.97). Interestingly, haplotype analysis of the HapMap CEU data shows the frequency of haplotype TTA of rs13194053-rs6932590-rs7749823 is 0.808. As both T alleles of rs13194053 and rs6932590 are risk alleles for schizophrenia in the European population studies, our association signal with the A allele of rs7749823 of the three marker haplotype in the Han Chinese population is consistent with the European findings.
We also examined other known schizophrenia susceptibility loci (NRGN, 11q24.2; and TCF4, 18q21.2) which were reported as genome-wide significant in the published studies of schizophrenia (Supplementary Table 9). However, none of the SNPs within the loci in our dataset show association with schizophrenia. However, genetic heterogeneity obviously exists for schizophrenia risk variants across ethnicities (Supplementary Table 10). This reinforces the need for genome-wide association studies of schizophrenia to be carried out in diverse populations.
In summary we have identified common genetic variants in 8p12 and 1q24.2 to be associated with schizophrenia in the Han Chinese population. Several promising candidate genes are implicated in these two regions. This makes it difficult to decide precisely which genes contain the causative variants in these two chromosomal regions. Nevertheless identification of these novel common genetic risk variants that predispose to schizophrenia is an encouraging first step in a process that has the potential to be translated into improved methods for the prediction/treatment of the disease.
Online Methods
Subjects
All schizophrenia patients analyzed in the BIOX study were interviewed by 2 independent psychiatrists, diagnosed according to DSM-IV criteria and had a 2-year history of schizophrenia. All cases met 2 criteria: preoccupation with one or more delusions and frequent auditory hallucinations; however, none of the following symptoms were prominent: disorganized speech, disorganized or catatonic behavior, or flat or inappropriate affect. The schizophrenia cases in the initial and replication stages of our study were recruited using the same diagnostic criteria.
Healthy controls were randomly selected from Chinese Han volunteers who were requested to reply to a written invitation to evaluate their medical history. Practice lists of controls were screened for potentially suitable volunteers by excluding subjects with major mental illness.
The discovery phase (BIOX GWAS) includes three datasets: the Northern Han sample set of 1,578 cases and 1,592 controls were recruited from Beijing and Shandong Province; the Central Han sample set of 1,238 cases and 2,856 controls were recruited from Shanghai and Anhui Province; and the Southern Han sample set of 934 cases and 2,020 controls were recruited from Guangdong and Guangxi Provinces. Sample descriptions can be found in the Supplementary Table 1.
The replication stage (BIOX FOLLOW-UP) included 4,383 schizophrenia cases and 4,539 healthy controls recruited from Shanghai. All subjects in the replication stage were unrelated and born in Shanghai and their parents had to be local residents of Shanghai as well. This replication sample is expected to have minimal population stratification given that Shanghai used to have the strictest resident-registration system in China before the 1980s. Sample descriptions and characteristics can be found in the Supplementary Table 1.
The SGENE-plus samples included 513 patients and 471 controls from Denmark/Copenhagen; 93 schizophrenia patients and 88 unrelated controls from England; 182 schizophrenia patients and 197 controls from Finland; 1048 patients and 971 controls from Germany; 531 cases and 11,615 controls from Iceland; 84 patients and 89 controls from Italy; 693 patients and 629 controls from The Netherlands; 658 schizophrenia cases and 661 controls from Scotland. All case samples were diagnosed according to ICD-10 or DSM-IV criteria. These samples are the same as the genome-wide typed samples used in a previous Nature publication6 with two exceptions: slight changes in the Icelandic schizophrenia patients used (~10% difference), and two newly genome-wide typed groups from Netherlands and Denmark.
All participants provided written informed consent. Approval was received for our study from the local Ethics Committee of Human Genetic Resources. Other details of sample description were listed in the Supplementary Sample Information.
DNA extraction
EDTA anti-coagulated venous blood samples were collected from all participants. Genomic DNA was extracted from peripheral blood lymphocytes by standard procedures using Flexi Gene DNA kits (Fuji), diluted to working concentrations of 50 ng/μl for SNP chip genotyping, and 10~20 ng/μl for replication genotyping.
GWAS genotyping and quality control
The genome-wide scan was performed using the Affymetrix Genome-Wide Human SNP Array 6.0. Quality control (QC) filtering of the GWAS data was performed by excluding arrays with a Contrast QC < 0.4 from further data analysis. Genotype data were generated using the birdseed algorithm21. For sample filtering, arrays that generated genotypes at < 95% of loci were excluded. For SNP filtering (after sample filtering), SNPs with call rates < 95% in either cases or controls were removed in each geographical group. SNPs with minor allele frequency (MAF) < 1% or significant deviation from Hardy Weinberg Equilibrium (HWE; P ≤ 1×10−3) in controls were also excluded. SNPs passing QC in the Northern, Central and Southern Han groups were used for further analysis. After QC, there were 546,561 SNPs remaining for the combined initial study.
SNP selection criteria and replication genotyping
We selected all SNPs with Pinitial-meta<5.0×10−6 for the replication study, according to a previous GWAS22. To ensure that there were at least 2 SNPs included for each region, we added the 2nd most significant SNP among the 8p12 findings for replication. Genotyping for the replication study was performed using the Ligation Detection Reaction (LDR) method23,24, with technical support from Shanghai Biowing Applied Biotechnology Company.
Analysis of population substructure
Population substructure was evaluated using principal components analysis as implemented in the software EIGENSTRAT25,26. 20 principal components (PCs), some of which will reflect ancestry differences among subjects, were generated for each subject. Logistic regression was used to determine whether there was a significant difference in PC scores between cases and controls; significant PCs were used as covariates in the association analysis to correct for population stratification. We identified 6 significant PCs (for the Northern data set), 8 significant PCs (for the Central data set), and 4 significant PCs (for the Southern data set).
Imputation analysis of untyped SNPs
We selected the two new identified potential susceptibility loci (1q24.2, rs10489202 ± 350kb and 8p12, rs16887244 ± 350kb) for imputation. Hapmap SNPs in the 2 regions were imputed using MACH 1.0. Phased haplotypes for 90 CHB+JPT subjects (180 haplotypes) were used as the reference for imputing genotypes. Any SNP imputed with information content r2<0.3 was excluded from association analysis because of lack of power. The criteria for SNP QC filtering were the same as the genotyped ones.
Statistical methods
Association of single SNPs with schizophrenia was tested by logistic regression using PLINK27, separately for the Northern, Central and Southern data sets, correcting for principal component scores that had statistically significant differences between cases and controls. The Hardy-Weinberg-Equilibrium analysis was performed using PLINK, and Haploview28 was used for the genome wide P value plot. The Q-Q plot was created using the R package. The regional plots were generated using LocusZoom (see URLs). In the replication study, allelic association analysis was conducted using SHEsis29. A meta-analysis using the random-effect model was carried out based on the PCA-adjusted association results of the three cohorts in the initial GWAS, the inflation factor λ value was 0.97. The meta-analysis was conducted using PLINK. Heterogeneity across the data sets was evaluated using Cochran’s Q test. The meta-analysis was carried out using the Mantel-Haenzel method with a random-effects model30. Conditional logistic regression was used to test for independent effects of an individual SNP22,31. Based on the sample size of the BIOX and SGENE-plus datasets, we compared the statistical power between BIOX and SGENE-plus (Supplementary Table 4; alpha=5×10−8)5,32.
Expression quantitative trait loci (eQTL) analysis
Expression profiles were analyzed within two eQTL datasets (lymphoblastoid cell lines and brain). We downloaded the expression data sets from NCBI GEO (http://www.ncbi.nlm.nih.gov/geo/). The first dataset, published by Stranger and colleagues9,10, consists of gene expression profiles generated using RNA extracted from lymphoblastoid cell lines of all 210 unrelated HapMap individuals from four populations (CEU: 60 Utah residents with ancestry from northern and western Europe; CHB: 45 Han Chinese in Beijing; JPT: 45 Japanese in Tokyo; YRI: 60 Yoruba in Ibadan, Nigeria). Expression analysis was performed using Sentrix Human-6 Expression BeadChip (Illumina, San Diego, California, United States). The SNP genotypes from phase II HapMap were used in the analysis. The second eQTL dataset11 consists of gene expression profiles for frozen tissue samples obtained from four brain regions (cerebellum, pons, frontal and temporal cortices) of 143 neurologically normal subjects of European ancestry. Expression analysis was performed using Illumina HumanRef-8 Expression BeadChips, and SNP genotyping was performed using Infinium HumanHap550 BeadChips. The expression data were normalized and log transformed as described in the original reports9-11. The eQTLs were tested by linear regression of normalized expression levels on SNP genotypes (coded as the number of minor alleles at each SNP: 0, 1 or 2). For the lymphoblastoid cell lines dataset, the analyses were conducted for each population and the combined dataset. For the brain dataset, the analysis of each tissue region was performed separately.
Supplementary Material
Acknowledgements
We are deeply grateful to all the participants as well as to the doctors working on this project. The authors also thank editors and anonymous reviewers for their valuable comments on the manuscript. This work was supported by the 863 Program (2006AA02A407, 2009AA022701), the Natural Science Foundation of China (31000553), the 973 Program (2010CB529600), the Funding Awarded to Authors for National Excellence of Doctoral Dissertation Research (201026), the Program for New Century Excellent Talents in University (NCET-09-0550), the Shanghai Municipal Health Bureau program (2008095), the Shanghai Changning Health Bureau program (2008406002), the Shanghai Municipal Commission of Science and Technology Program (09DJ1400601), and National Key Technology R&D Program (2006BAI05A09), and the Shanghai Leading Academic Discipline Project (B205).
Footnotes
Competing Financial Interests
The authors declare no competing financial interests.
URLs.
LocusZoom, http://csg.sph.umich.edu/locuszoom/
EIGENSTRAT, http://genepath.med.harvard.edu/~reich/Software.htm
PLINK, http://pngu.mgh.harvard.edu/~purcell/plink/
Haploview program, http://www.broad.mit.edu/mpg/haploview
The International HapMap project, http://www.hapmap.org
SHEsis, http://analysis.bio-x.cn/myAnalysis.php
MACH, http://www.sph.umich.edu/csg/abecasis/MACH/index.html
NCBI GEO, http://www.ncbi.nlm.nih.gov/geo/
References
- 1.Craddock N, O’donovan MC, Owen MJ. The genetics of schizophrenia and bipolar disorder: dissecting psychosis. Journal of Medical Genetics. 2005;42:193–204. doi: 10.1136/jmg.2005.030718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McCarthy SE, et al. Microduplications of 16p11. 2 are associated with schizophrenia. Nature genetics. 2009;41:1223–1227. doi: 10.1038/ng.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O’Donovan MC, et al. Identification of loci associated with schizophrenia by genome-wide association and follow-up. Nature genetics. 2008;40:1053–1055. doi: 10.1038/ng.201. [DOI] [PubMed] [Google Scholar]
- 4.Purcell SM, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shi J, et al. Common variants on chromosome 6p22. 1 are associated with schizophrenia. Nature. 2009;460:753–757. doi: 10.1038/nature08192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stefansson H, et al. Common variants conferring risk of schizophrenia. Nature. 2009;460:744–747. doi: 10.1038/nature08186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vacic V, et al. Duplications of the neuropeptide receptor gene VIPR2 confer significant risk for schizophrenia. Nature. 2011;471:499–503. doi: 10.1038/nature09884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu B, et al. Strong association of de novo copy number mutations with sporadic schizophrenia. Nature genetics. 2008;40:880–885. doi: 10.1038/ng.162. [DOI] [PubMed] [Google Scholar]
- 9.Stranger BE, et al. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science. 2007;315:848–853. doi: 10.1126/science.1136678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stranger BE, et al. Population genomics of human gene expression. Nature genetics. 2007;39:1217–1224. doi: 10.1038/ng2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gibbs JR, et al. Abundant quantitative trait loci exist for DNA methylation and gene expression in human brain. PLoS Genet. 2010;6:e1000952. doi: 10.1371/journal.pgen.1000952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gaughran F, Payne J, Sedgwick PM, Cotter D, Berry M. Hippocampal FGF-2 and FGFR1 mRNA expression in major depression, schizophrenia and bipolar disorder. Brain research bulletin. 2006;70:221–227. doi: 10.1016/j.brainresbull.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 13.Klejbor I, et al. Fibroblast growth factor receptor signaling affects development and function of dopamine neurons – inhibition results in a schizophrenia - like syndrome in transgenic mice. Journal of neurochemistry. 2006;97:1243–1258. doi: 10.1111/j.1471-4159.2006.03754.x. [DOI] [PubMed] [Google Scholar]
- 14.Terwisscha SAF, Bakker SC, Kahn RS. Fibroblast Growth Factors in Schizophrenia. Schizophrenia bulletin. 2010;36:1157–1166. doi: 10.1093/schbul/sbp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O’Donovan MC, et al. Analysis of 10 independent samples provides evidence for association between schizophrenia and a SNP flanking fibroblast growth factor receptor 2. Molecular psychiatry. 2008;14:30–36. doi: 10.1038/mp.2008.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tkachev D, et al. Oligodendrocyte dysfunction in schizophrenia and bipolar disorder. The Lancet. 2003;362:798–805. doi: 10.1016/S0140-6736(03)14289-4. [DOI] [PubMed] [Google Scholar]
- 17.He G, et al. MPZL1/PZR, a novel candidate predisposing schizophrenia in Han Chinese. Molecular psychiatry. 2006;11:748–751. doi: 10.1038/sj.mp.4001841. [DOI] [PubMed] [Google Scholar]
- 18.Chen J, et al. Genetic structure of the Han Chinese population revealed by genome-wide SNP variation. The American Journal of Human Genetics. 2009;85:775–785. doi: 10.1016/j.ajhg.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li T, et al. Common Variants in Major Histocompatibility Complex Region and TCF4 Gene Are Significantly Associated with Schizophrenia in Han Chinese. Biological psychiatry. 2010;68:671–673. doi: 10.1016/j.biopsych.2010.06.014. [DOI] [PubMed] [Google Scholar]
- 20.Hindorff LA, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proceedings of the National Academy of Sciences. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Korn JM, et al. Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs. Nature genetics. 2008;40:1253–1260. doi: 10.1038/ng.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen ZJ, et al. Genome-wide association study identifies susceptibility loci for polycystic ovary syndrome on chromosome 2p16. 3, 2p21 and 9q33. Nature Genetics. 2011;43:55–59. doi: 10.1038/ng.732. [DOI] [PubMed] [Google Scholar]
- 23.Thomas G, et al. Capillary and microelectrophoretic separations of ligase detection reaction products produced from low - abundant point mutations in genomic DNA. Electrophoresis. 2004;25:1668–1677. doi: 10.1002/elps.200405886. [DOI] [PubMed] [Google Scholar]
- 24.Yi P, et al. PCR/LDR/capillary electrophoresis for detection of single - nucleotide differences between fetal and maternal DNA in maternal plasma. Prenatal diagnosis. 2009;29:217–222. doi: 10.1002/pd.2072. [DOI] [PubMed] [Google Scholar]
- 25.Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PLoS Genet. 2006;2:e190. doi: 10.1371/journal.pgen.0020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Price AL, et al. Principal components analysis corrects for stratification in genome-wide association studies. Nature genetics. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 27.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. The American Journal of Human Genetics. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 29.Yong Y, Lin HE. SHEsis, a powerful software platform for analyses of linkage disequilibrium, haplotype construction, and genetic association at polymorphism loci. Cell research. 2005;15:97–98. doi: 10.1038/sj.cr.7290272. [DOI] [PubMed] [Google Scholar]
- 30.Mantel N, Haenszel W. Statistical aspects of the analysis of data from retrospective studies. J Natl Cancer Inst. 1959;22:719–748. [PubMed] [Google Scholar]
- 31.Petukhova L, et al. Genome-wide association study in alopecia areata implicates both innate and adaptive immunity. Nature. 2010;466:113–117. doi: 10.1038/nature09114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Menashe I, Rosenberg PS, Chen BE. PGA: power calculator for case-control genetic association analyses. BMC genetics. 2008;9:36. doi: 10.1186/1471-2156-9-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.