

Summary
Stromal derived follicular dendritic cells (FDCs) are a major reservoir for antigen that is essential for formation of germinal centers, the site where memory and effector B cells differentiate. A long-standing question is how FDCs retain antigen in its native form for extended periods and how they display it to specific B cells. Here we found that FDC acquired complement-coated immune complexes (IC) from non-cognate B cells via complement receptors 1 and 2 (CD35 and CD21 respectively) and rapidly internalized them by an actin-dependent pathway. IC were retained intact within a non-degradative cycling compartment and were displayed periodically on the cell surface where they were accessible to antigen-specific B cells. This would explain how antigens are protected from damage and retained over long periods of time, while remaining accessible for B cells.
Introduction
Follicular dendritic cells (FDC) are centrally located within B cell follicles of secondary lymphoid tissues, including the spleen and lymph nodes (LN), where they are the major source of B cell attractant (CXCL-13)(Cyster et al., 2000; Tew et al., 1990). They are also a source of survival factors such as B cell activating factor (BAFF) and cytokines such as IL-6 and IL-10 that modulate the differentiation of B cells and T follicular helper cells within an active germinal center (GC) (Garin et al., 2010; Wu et al., 2009). FDC are stromal-derived and are identified by their extensive dendritic morphology and cell surface markers such as CD21, CD35, FDC-M1 (Mfge8), FDC M2 (complement C4), BP-3, complement C3 and FcγR (Kinoshita et al., 1991; Kranich et al., 2008; Taylor et al., 2002; Roozendaal and Carroll, 2007; Qin et al., 2000). In a recent elegant study, Aguzzi and colleagues identified the source of FDC as platelet-derived growth factor receptor beta positive perivascular cells that are located throughout the host and this would explain their capacity to develop at ectopic sites (Krautler et al., 2012). B cell surface lymphotoxin α and β and TNFα signal FDC precursors to develop into mature FDC (Alimzhanov et al., 1997; Endres et al., 1999; Fu et al., 1997; Pasparakis et al., 1996; Gonzalez et al., 1998).
Over 40 years ago, FDC were recognized to retain antigen within B cell follicles for extensive periods where it is required for maintenance of GC (Hanna and Szakal, 1968; Nossal et al., 1968; Mandel et al., 1980). Within GC, activated B cells that undergo somatic hypermutation and class switch recombination require antigen for survival signals, to enhance affinity maturation and for the formation of memory and effector B cells (Kelsoe, 1996; MacLennan, 1994). Although, affinity maturation can occur in the absence of GC in lymphotoxin-deficient mice, elimination of FDC by ablation or blockade of lymphotoxin signaling, antigen or complement receptor CD21 and CD35 results in a rapid elimination of GC (Fischer et al., 1998; Matsumoto et al., 1996; Wang et al., 2011; Gommerman et al., 2002). In mice complement receptor 1 (CD35) and complement receptor 2 (CD21) are both encoded by the Cr2 locus, since both are co-expressed on FDC and B cells CD21 and CD35 was referred to as Cr2.
Antigen acquisition from FDC in vivo by cognate B cells was recently visualized using multi-photon intravital imaging (Suzuki et al., 2009). How antigens are retained in a native state and made readily accessible to cognate B cells over long periods has remained an enigma. Based on electron microscopy studies, it was proposed that immune complex (IC) is retained on the surface of FDC in two forms, i.e. filiform and beaded structures termed “immune complex bodies or “ICCOSOMES”. Early in a GC response, it is held that the latter are released and taken-up by B cells for presentation to T cells but this model doesn't explain how antigens are sequestered by FDC without degradation (Burton et al., 1991; Kosco et al., 1988; Szakal et al., 1988).
Recent studies have identified a novel pathway by which LN resident subcapsular sinus macrophages (SSM) capture lymph-borne IC and shuttle them to non-cognate B cells in the underlying follicles (Phan et al., 2009; Phan et al., 2007). Both the initial capture of IC from the lymph by SSM and the uptake by non-cognate B cells is dependent on complement receptors (Cr), i.e. CD11b (Cr3) and CD21 (Cr2) and CD35 (Cr1), respectively. For example, using bone marrow chimeras in which WT mice are reconstituted with Cr2-deficient bone marrow, Phan et al show that substantially less IC is taken-up by the Cr2-deficient B cells relative to control WT chimeras and overall deposition of IC on FDC is reduced in the Cr2-deficient chimeras (Phan et al., 2009; Phan et al., 2007). Therefore, while other pathways such as conduits are capable of delivering antigen to FDC, non-cognate B cells represent one major pathway(Bajenoff and Germain, 2009; Roozendaal et al., 2009).
To study the cell biology of antigen acquisition and retention in living cells, we used a combination of flow cytometry and in vivo and ex vivo imaging of FDC. Using multi-photon intravital imaging, direct transfer of complement-coated IC from non-cognate B cells to FDC was observed. Unexpectedly, we found that FDC rapidly internalize intact IC into a non-degradative, cycling compartment. Notably, internalized IC undergo several rounds of surface recycling. The cycling of IC bound to FDC complement receptors helps explain the directional transfer of IC from non-cognate B cells to FDC; and it explains how IC are efficiently displayed on the FDC surface for extensive periods in order to be accessible to cognate B cells.
Results
FDC acquire immune complexes from non-cognate B cells
In WT mice, FDC become decorated with IC within 8 hours post-immunization suggesting that non-cognate B cells deliver and handoff the IC to the FDC. However, actual transfer of IC from the B cell to the FDC has not been visualized in vivo so it is possible that an intermediate cell type is involved or that the IC are released and later acquired by the FDC. Moreover, as both the capture by B cells and transfer to FDC is dependent in large part on Cr2 recognition of the complement C3d-coated IC, this raises a question about the directional transfer of IC, i.e. from B cell to FDC (Phan et al., 2009; Roozendaal et al., 2009). Thus, how are the IC removed from the B cell by the FDC when IC is bound by Cr2 on both cell types? To address this question, we first wanted to test in vivo whether non-cognate B cells directly transfer IC to FDC. As a model system, mice received 5 × 106 enriched GFP+ non-cognate B cells and were passively immunized with rabbit anti-phycoerythrin. Twenty-four hours later, anesthetized mice were injected in the footpad with 8 μg of phycoerythrin (PE) and the popliteal LN was imaged using multi-photon intravital microscopy (MP-IVM). As reported by Cyster et al. non-cognate B cells were identified bearing clusters of immune complexes (PE-IC) (Figure 1A; Movie S1) (Phan et al., 2009; Phan et al., 2007). Tracking of individual donor B cells within LN follicles, we visualized direct transfer of a cluster of PE-IC from the B cell to the FDC (Figure 1A; Movie S1). Thus, within minutes of contact between the B cell and FDC the complexes were efficiently transferred to FDC.
Figure 1.

(see also Figure S1, S2 and Movie S1, S2, S3). Acquisition of IC by FDC. (A) (left panel) Snap shot taken from multiphoton-intravital imaging of the popliteal LN in a mouse passively immunized with anti-PE antibody and adoptively transferred with WT fluorescently labeled B cells (green). LN was imaged from 60 min to 120 min following PE (red) injection into the footpad showing B cells transfer PE-IC onto FDC (blue). (right panel) Series of higher magnification images over 5 min showing transfer of PE-IC (red) bound to a B cell (green) to an FDC (blue) (arrowheads). (B) Live-cell imaging of cultured FDC labeled with lipophilic DiO dye (green) and incubated with PE-IC bound non-cognate B cells. Images were generated every 60 seconds. Images show transfer of C3-opsonized PE-IC clustered on the B cell to the FDC and subsequent fragmentation on the FDC surface. (right panel) Schematic representation of transfer of PE-IC clusters from B cells onto FDC and their fragmentation upon contact with FDC. (C) (Left panel) Spinning disk confocal image of PE-IC (red) within FDC (green). (right panel) Representative Z-stack series projection through 30 μm at approximate 0.4 μm intervals identifies PE-IC (grey and arrowheads) within FDC (outlined).
To examine transfer of antigen from the B cell to the FDC in more detail, we established an ex vivo model based on our in vivo observation of non-cognate B cell transfer of PE-IC directly to the FDC. Therefore, rabbit IgG anti-PE was mixed with PE antigen to form PE-IC and then fresh mouse serum was added to the complexes to activate and bind complement C3. On culture with non-cognate B cells the complement-coated PE-IC formed clusters on the B cell surface similar to those observed in vivo (Figure S1A). Inspection of the patches of gold-labeled PE-IC on non-cognate B cells loaded in vitro by electron microscopy revealed that the patches consist of multiple complexes (Figure S1B). Moreover, staining of the loaded B cells with anti-PE and analysis by flow cytometry confirmed that the majority of the complexes were retained on the outside of the B cell (Figure S1C). Therefore, for the experiments using ex vivo cultures of FDC, non-cognate B cells were loaded with complexes in vitro.
To follow transfer of PE-IC from the B cells to the FDC in living cells, non-cognate B cells loaded with complement coated PE-IC, were mixed with FDC in ex vivo cultures and analyzed by live cell fluorescence spinning disk confocal microscopy. FDC were harvested according to the procedures reported by Tew and colleagues and cultured on collagen-coated plates for 5-10 days where they regained their dendritic morphology(El Shikh et al., 2006). Isolated cells were >90% mature FDC based on morphology, cell surface markers (Cr2, FDC-M1, FcγRIIb) and binding of IC. Real time images demonstrate that on contact between donor B cells with FDC, the clustered PE-IC was rapidly transferred (Figure 1B; Movie S2). The large cluster of PE-IC on the B cell seemed to dissociate into much smaller particles upon transfer to the FDC (Figure 1B; Movie S2). In the absence of FDC, approximately 90% of non-cognate B cells retained PE-IC after 4 hours of incubation, showing that transfer from the B cells to FDC is specific. Consistent with these observations, the frequency of PE-IC positive B cells decreased in a time-dependent manner when incubated with FDC (Figure S2A) while FDC acquired PE-IC in a reciprocal manner (Figure S2B). Efficient uptake of PE-IC by FDCs was dependent on their expression of Cr2 but not FcγRIIb as predicted from results in vivo (Figure S2B) (Roozendaal et al., 2009; Fang et al., 1998). Thus, PE-IC-bearing donor B cells rapidly transfer the complexes to FDC in real time after cell contact and the efficiency of transfer is dependent on FDC expression of Cr2.
FDC rapidly internalize immune complexes
Antigen was tracked on and within FDC in the ex vivo model, using live-cell fluorescence spinning disk confocal microscopy. Analysis of optical sections (spaced approximately <0.4 μm) identified PE-IC both on the FDC surface and distributed within the FDC during the first 30 min post-delivery (Figure 1C; Movie S3). The relatively rapid internalization of PE-IC by FDC following contact with B cells suggests a possible mechanism to explain the unidirectional movement of complexes from the FDC to the B cell. For example, a mechanism by which Cr2 is periodically internalized could provide a one-way transfer of complementcoated complexes from the loaded non-cognate B cell to the FDC. To test whether uptake and internalization of PE-IC requires a dynamic actin cytoskeleton, FDC were pretreated for 30 min with 5 μM cytochalasin D (Cyt D) which blocks actin polymerization (Casella et al., 1981).Treatment of cells with 5 μM Cyt D for 30 min blocks cellular processes dependent on actin-polymerization such as phagocytosis in a reversible manner (May et al., 1998). To confirm that 5 μM Cyt D was not toxic in our ex vivo model, FDC were pre-treated with Cyt D prior to culture with fluorescent-labeled transferrin ligand (Tf). Comparison of the treated with untreated FDC demonstrated the actin-inhibitor did not impair uptake of Tf as expected (data not shown) (Boulant et al., 2011).
To circumvent any effects of the Cyt D on the non-cognate B cells loaded with PE-IC, Cyt D was removed from the media before culture for an additional 30 min with donor B cells. Subsequently, the FDC were washed, fixed and then stained with anti-FDC-M1 and Cr2 (Figure 2A). Confocal imaging of FDC cultured in media without Cyt D identified significant uptake of the PE-IC as expected based on total mean fluorescent intensity (MFI) (Figure 2B,C). By contrast, FDC pretreated with Cyt D acquired significantly less antigen from donor B cells, i.e. mean PE intensity 528 ± 56 vs. 338 ± 33, p< 0.002 (Figure 2B,C). Treatment with Cyt D did not completely block uptake, as FDC began to recover once Cyt D was removed from the media. The reduction in PE-IC uptake by FDC is unlikely due to effects of the Cyt D treatment on Cr2 expression because the amount of cell surface Cr2 was similar on FDC treated with either PBS or Cyt D (Figure 2D).
Figure 2.

Antigen uptake by FDC is actin dependent. (A) FDC were treated with 5 μM Cytochalasin D (Cyt D) or media for 30 min, washed and then incubated with C3-opsonized PE-IC-loaded non-cognate B cells for another 30 min. (B) Representative confocal images of fixed cells show PE-IC (red) uptake by FDC from B cells is diminished in Cyt D treated FDC. FDC were identified by FDC-M1 (green) and Cr2 (blue) staining. (C) Quantification of confocal images by Volocity software showing PE mean fluorescent intensity per cell (PE intensity/number of voxels) for each group. (D) Cr2 MFI is not altered by Cyt D treatment. Cumulative data of FDC culture from 3 mice over 2 independent experiments (minimum of 15 cells per group). Each symbol represents a single cell; horizontal bar is mean value. Statistics: p-values calculated by Student's t test; ns = not significant.
The results suggest a pathway by which capture of PE-IC by FDC requires actin-dependent internalization of the receptor-ligand complex that effectively removes the complex from the B cell surface. Thus, directional transfer of immune complexes on cell contact may be mediated by the active internalization of the FDC Cr2 protein.
Internalized IC return to FDC cell surface
The finding that PE-IC is internalized upon transfer suggests they may be returned to the cell surface to be acquired by cognate B cells as identified in vivo (Cyster et al., 2000; Garin et al., 2010; Suzuki et al., 2009; Tew et al., 1990; Wang et al., 2011). To test this possibility two separate approaches were used, i.e. imaging of the cell surface IC using antibody staining following a mild acid wash, and flow cytometry to detect acquisition of surface antigen by cognate B cells following a mild acid wash. FDC were loaded with PE-IC, then washed with a mild acid solution to remove surface bound IC and then either fixed and stained with anti-PE or cultured in media to allow recovery (Figure 3A). Staining of acid-washed and fixed FDC with anti-PE confirmed removal of detectable surface antigen (Figure 3A). By contrast, acid-washed cells allowed to recover in media for 30 min before fixation and antibody staining revealed PE on the cell surface (Figure 3A). Staining of the recovered cells with antibody to complement C3 identified co-localization with the PE-IC complex supporting that the IC are returned to the cell surface intact (Figure 3A). Quantification of these results show negligible staining for surface PE-IC on acid washed and fixed FDC relative to FDC allowed to recover prior to staining with anti-PE antibody (Figure 3B, left panel). Removal of surface bound C3 by the acid treatment was less efficient than PE-IC (Figure 3B right panel). Thus, based on imaging, endocytosed PE-IC are returned to the cell surface where it is accessible to antibody binding. Furthermore, PE-IC co-localize with C3 suggesting that the IC are retained intact during cycling.
Figure 3.

Antigen acquired by FDC resurfaces. (A) Schematic diagram of FDC loading and acid wash procedure and recovery. PE-IC appearing on the surface of the FDC after acid wash must come from inside the cell. FDC were loaded with C3-opsonized PE-IC, then either fixed, acid washed and fixed or acid washed, recovered for 30 min and then fixed. Cells were stained with anti-PE and anti-C3d antibody. (B) Quantification of the results shows efficient stripping of C3-opsonized PE-IC by acid wash and recovery of PE-IC on the surface after 30 min. C3d remains fixed on PE-IC during resurfacing. (C) Schematic representation of the experiment. FDC were loaded with TEL (turkey egg lysozyme)-PE-IC or PE-IC by non-cognate B cells in culture. FDC were acid washed to remove cell surface antigen and fixed with 1 % PFA or left in media alone to recover. MD4 Ig transgenic B cells (specific for TEL) were mixed with FDC for 2 or 4 hr in culture, then harvested, stained with B220 and analyzed by flow cytometry. (D) Results indicate efficient uptake of TEL-PE-IC by cognate MD4 B cells when cultured with live FDC but negligible uptake of PE-IC (without TEL) or TEL-PE-IC from FDC after acid wash and fixation. (E) MD4 B cells were cultured for an additional 20 hours before staining with anti-CD86 to assay for B cell activation. Results indicate cognate B cells express CD86 when exposed to specific antigen on FDC but negligible expression when cultured with FDC loaded with non-specific PE-IC. Statistics: p-values calculated by Student's t test; ns = not significant.
As a second approach to evaluate whether endocytosed PE-IC return to the cell surface and to determine if antigen is functionally intact, FDC, loaded with PE-IC (control) or turkey egg lysozyme (TEL) coupled to PE (TEL-PE-IC) and acid washed, as described above, were cultured with cognate B cells and uptake of antigen was assayed by flow cytometry (Figure 3C). FDC were incubated with MD4 B cells specific for hen and turkey lysozyme for 2 or 4 hr to allow for their acquisition of antigen(Shokat and Goodnow, 1995). After exposure to acid-stripped FDC, the B cells were harvested and prepared for analysis by flow cytometry. As expected, the TEL specific-MD4 B cells cultured with FDC loaded with PE-IC were negative for uptake of antigen, demonstrating that antigen uptake is specific (Figure 3D). Notably, cognate B cells incubated 2 or 4 hr with FDC loaded with TEL-PE-IC efficiently acquired PE relative to the control FDC. By contrast, FACS analysis of MD4 B cells cultured with acid-stripped FDC that were fixed with 1 % PFA showed negligible uptake of the labeled antigen (Figure 3D). Thus, based on flow cytometry analysis, endocytosed PE-IC return to the surface of FDC following acid stripping where it is accessible for acquisition by cognate B cells.
To determine if uptake of cognate antigen in the ex vivo model results in B cell activation, MD4 B cells were cultured as above for 2 hr with FDC loaded with PE-IC or FDC loaded with TEL-PE-IC. B cells were harvested and assayed by flow cytometry after overnight culture. As expected, culture of MD4 B cells with FDC loaded with non-specific antigen (PE-IC) resulted in negligible activation. By contrast, MD4 B cells exposed to FDC loaded with specific antigen (TEL-PE-IC) were activated and expressed the B cell activation marker CD86 (Figure 3E). The combined results demonstrate that internalized antigen returns to the FDC surface where its acquisition by cognate B cells can lead to activation.
It is possible that cognate B cells pick up antigen from dying FDC in the ex vivo cultures and that cell surface display of antigen is not an active process. To address this question and to determine if the return of antigen to the FDC surface is actin-dependent, B1-8 B cells specific for NP hapten; 4-hydroxy-3-nitrophenyl acetyl, were cultured with FDC loaded with NP-PE-IC(Lam and Rajewsky, 1999). To block actin-dependent cycling, FDC were first treated with 1 or 5 μM Cyt D for 30 min and then washed to remove Cyt D before mixing with cognate B cells (Figure 4A). Cyt D is known to interfere with B cell receptor internalization so it was important to remove the actin inhibitor before adding cognate B cells. The B1-8 NP-specific B cells were distinguished from non-transgenic B cells carried over from the knock-in B1-8 mice based on the IgMa allo-marker of the immunoglobulin heavy chain (IgH) knock-in allele (Figure 4B). Flow cytometry analysis of NP-specific B cells cultured with antigen-loaded FDC identified capture of a similar range of antigen as described above with the TEL-PE and MD4 B cells (Figure 4B,C). While 1 μM Cyt D had limited effect on uptake of cognate antigen, treatment of the FDC with 5 μM Cyt D resulted in a significant reduction in uptake by the IgMa+ B1-8 B cells relative to the media alone control, i.e. 12 ± 1 % vs. 21 ± 0.3 %, respectively (Figure 4C). As expected, the IgMb+ non-cognate B cells take up negligible amounts of NP-PE-IC (Figure 4B). It is noted that the effects of 5 μM Cyt D treatment on the FDC probably represent an underestimate as the FDC begin to recover from the actin-inhibitor during culture with the cognate B cells.
Figure 4.

FDC display of antigen for acquisition by cognate B cells is actin dependent. (A) Non-cognate B cells bearing clusters of NP haptenated PE-IC were incubated with FDC culture for 2 hr. Non-cognate B cells were removed by washing and pulsed FDC were treated with media alone, 1 or 5 μM Cyt D for 30 min. Cyt D was removed and the FDC were incubated with a mix of WT (IgMb) and NP-specific B1-8 (IgMa) B cells for 30 min before harvest and analysis by flow-cytometry. (B) Gating scheme for NP-PE-IC bound by B1-8 B cells (B220+, IgMa+). Histogram shows uptake is specific for B1-8 (IgMa) B cells but not WT (IgMb) as expected. (C) Percent PE positive B1-8 B cells (left panel) and the MFI of NP-PE-IC bound by B1-8 B cells (right panel). Results indicate that display of specific antigen on FDC is actin dependent. Results shown are from 3 separate experiments with 3 mice each. (D) FDC were loaded with NP-PE-IC as described above and cultured for 60 min before removal of the FDC. Subsequently, B1-8 B cells were mixed with FDC supernatant or directly with FDC in fresh media for 30 min. (E) B1-8 cells were harvested and analyzed by flow cytometry as in B and C. Results show efficient acquisition of antigen by B1-8 B cells requires intact FDC. Results represent 3 separate FDC cultures derived from 2 sets of mice. Statistics: p-values calculated by Student's t test; ns = not significant.
An alternative possibility for B cell acquisition of antigen in the ex vivo cultures is that antigen is released by viable FDC into the media before it is taken-up by cognate B cells. To test this possibility, NP-specific B cells were cultured for 30 min either with media taken from a 2 hr culture of FDC loaded with NP-PE-IC or with the FDC taken from the culture (Figure 4D). NP-specific B cells efficiently acquired antigen when mixed directly with FDC as expected; however, substantially less antigen was taken-up when the cognate B cells were incubated with the FDC culture media (Figure 4E). Importantly, acquisition of antigen by cognate B cells is dependent on presence of loaded FDC as supernatant from FDC culture alone is not an efficient source. The combined results support a dynamic process by which internalized IC is returned to the FDC surface by a process that is actin-dependent.
Immune complexes periodically cycle to FDC surface
Our results with the ex vivo cultures show that FDC capture C3-coated PE-IC from non-cognate B cells and rapidly internalize them into endosomal vesicles some of which return to the cell surface intact. A process in which IC cycle continuously from inside to outside of the FDC would provide a novel pathway to retain antigen for extended periods without degradation. To examine whether PE-IC are included within a cycling pathway, FDC were loaded with C3-coated PE-IC as described above and stained sequentially with donkey anti-rabbit IgG labeled in three different colors, i.e. DyLight 488 (green), DyLight 405 (blue) or DyLight 649 (red) (Figure 5A). After final staining, FDC were washed and fixed for confocal imaging. To confirm that the secondary donkey antibody stably binds to rabbit Ig IC, non-cognate B cells loaded with PE-IC were stained with either donkey anti-Ig labeled in DyLight 405 or 649 or both, then B cells were cultured at 37°C for 60 min Subsequently, the cell supernatants were added to B cells bearing PE-IC to determine if the donkey antibody label could transfer between PE-IC. Analysis of the stained cells by flow cytometry demonstrates negligible antibody transfer (Figure S3A,B).
Figure 5.
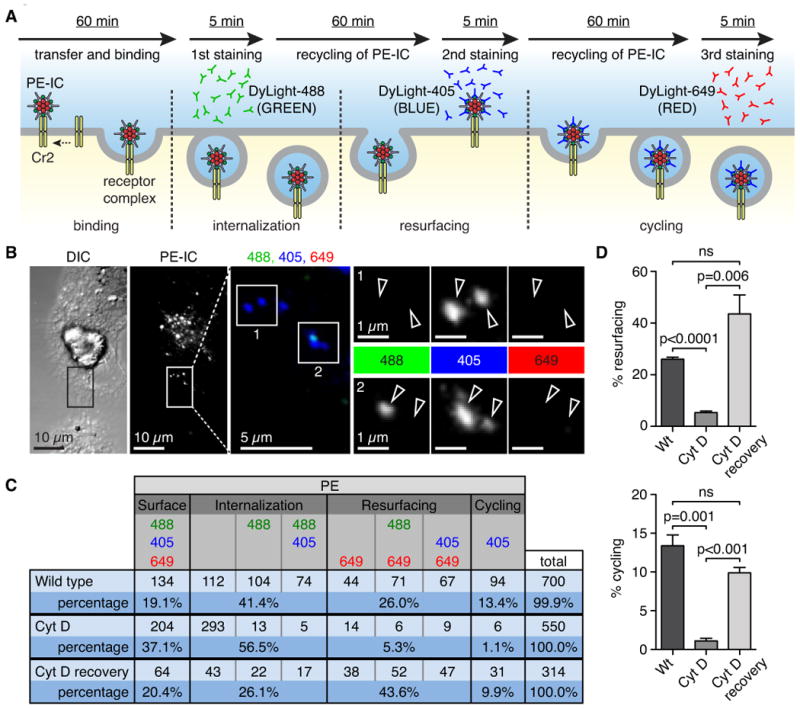
(see also Figure S3). Cycling of IC is actin dependent. (A) Schematic representation of the experimental set-up for antibody cell surface staining: C3-opsonized PE-IC is transferred to FDC for 60 min before removal of B cells. FDC are incubated with DyLight488 labeled donkey anti-rabbit IgG (green) for 5 minutes to detect surface PE-IC. FDC are washed, IC are allowed to cycle and the process is repeated with two additional labeled donkey anti-rabbit IgGs, i.e. DyLight405 (blue) and DyLight649 (red) respectively. This results in several color combinations that indicate cycling or resurfacing. (B) Representative confocal image of FDC with cycling PE-IC. PE channel (grey value) was used to identify objects and then objects were scored for green (DyLight488, first staining), blue (DyLight405, second staining) or red (DyLight649, third staining). If the PE-IC object was blue only it was scored as cycling (Inset 1). If the object was blue and green it was scored as internalization (Inset 2). (C) Table of all possible color combinations, their interpretation and the raw cumulative data. This setup will result in an underestimation of the actual number of cycling PE-IC. (D) Bar graph showing percent PE-IC resurfacing (top panel). Bar graph showing percent PE-IC cycling (bottom panel). Statistics: p-values calculated by Student's t test; ns = not significant; data are represented as mean +/- SEM.
Sequential staining of FDC with the three secondary antibodies results in four ‘states’ of the PE-IC based on the different color combinations: surface; internalization; resurfacing; or cycling (Figure 5B,C). For example, a high magnification image of a PE+ particle visualized in each channel of fluorescence identifies no fluorescence in the green or red channels but positive for blue (Figure 5B-1). Thus, the particle is scored as cycling as it was unavailable for the first antibody, stained with the second (blue) but inside again for the third antibody. In a second example, the PE+ particle was identified as internalized because it was positive for the first two antibodies (green and blue) but not the third (red) (Figure 5B-2). Overall, 700 PE+ particles were analyzed in each separate color channel and the results were quantitated for the resurfacing and cycling (Figure 5C). For example, 94 (13.4%) PE+ particles were identified as “cycling”, while 182 (40%) PE+ particles were surfacing (Figure 5C). As expected, fixation of cells after initial loading with PE-IC blocked cycling of PE-IC (results not shown)
To test if the cycling of antigen to the cell surface was actin-dependent, FDC were first loaded with PE-IC and then subsequently treated with 5 μM Cyt D to block actin polymerization prior to antibody staining. As a control, Cyt D was washed out (Cyt D recovery) before staining. Results demonstrate that treatment of loaded FDC with Cyt D before exposure to donkey anti-rabbit Ig significantly impairs resurfacing of PE-IC relative to the recovery control, i.e. 5.3% vs. 43.6% (p=0.006) and cycling, i.e. 1.1% vs. 9.9% (p<0.001) of antigen (Figure 5C,D).
Thus, rabbit IgG-PE complexes transferred from non-cognate B cells are not degraded appreciably over time but a fraction remains intact and periodically cycles to the cell surface via an actin-dependent mechanism.
Long-term retention of immune complexes within cycling endosomes
To compare results from ex vivo cultures with capture of antigen by FDC in vivo, mice previously treated with rabbit anti-PE were injected sub-cutaneous with PE antigen (Figure 6A). LNs were harvested at days 5 or 10 for analysis by immunohistochemistry (IHC) and extraction of FDC for culture ex vivo and characterization. IHC analysis of LN cryo-sections of immunized mice at both time points confirmed deposits of PE-IC on FDC as expected (Figure 6B and not shown). To prepare ex vivo cultures, FDC harvested at day 5 from immunized mice were cultured on collagen-coated slides for an additional 6 days. Slides were stained for Cr2 and analyzed by confocal microscopy to confirm retention of PE at day 11 post-immunization. Quantitation of individual FDC isolated from the immunized mice relative to non-immune controls, identifies a significant MFI of PE+ staining per cell as expected (Figure 6C). Notably, the pattern of PE retained by ex vivo cultured FDC is similar to that of the IHC images taken at day 10 post-immunization in that both show retention of PE antigen (Figure 6B,C). Thus, PE-IC acquired by FDC in vivo are retained by the FDC for at least 6 additional days in culture.
Figure 6.

Antigen acquired by FDC in vivo is periodically cycled to the cell surface. (A) Schematic representation, mice were passively immunized with rabbit anti-PE on day -1, injected with PE on day 0, then irradiated on day 4 or 9 and FDC isolated on day 5 or 10. Alternatively, draining popliteal LNs (pLN) were harvested and cryo-preserved on day 10. (B) Sections of pLN show PE (red) localizing with Cr2 (blue) that marks the FDC within the B cell (B220, green) follicle at day 10 post-immunization. (C) Representative confocal images of FDC harvested at day 5 post-immunization with PE (red) and cultured in vitro until day 11. Cells were stained with antibody to Cr2 (blue). Quantification of images shows mean fluorescent intensity of PE per cell for FDC isolated from immunized and PBS control mice. Each symbol represents a single cell. (D) Representative confocal images of FDC isolated from immune mice at day 10 and cultured until day 16. FDC were either fixed, acid washed and fixed or acid washed, recovered for 30 min and then fixed. Cells were stained with anti-PE and anti-C3d antibody. (E) Quantification shows efficient acid wash and recovery of PE-IC (upper panel) and C3 (lower panel) on the surface after 30 min. C3d remains fixed on PE-IC during resurfacing. Results indicate C3-PE-IC recycle at day 16 after loading in vivo. Statistics: p-values calculated by Student's t test; ns = not significant.
To determine if PE-IC loaded on FDC in vivo continued to undergo cycling for an extended period, FDC were harvested at day 10 post immunization. Cells were allowed to recover in culture for an additional 6 days before acid wash and staining cell surface IC with anti-PE. Image analysis confirmed removal of surface PE-IC by acid wash as expected (Figure 6D,E). By contrast, when FDC were allowed to recover after acid wash and then stained, PE was detected on the surface (Figure 6D,E). C3 staining was also detected on the surface of the recovered FDC, indicating that the complement-coated PE-IC are intact and cycle 16 days after loading (Figure 6D,E). Overall, these results validate the findings with cultures of FDC loaded with IC in vitro and provide an explanation for how GC B cells may sample antigens in vivo over an extended period.
Discussion
While it is well established that FDC retain antigen over extended periods and that this retention is important for both primary and secondary B cell responses, how antigen is initially taken-up and sequestered by FDC without degradation; and how it is subsequently made available to B cells has remained unexplained (Cyster et al., 2000; Garin et al., 2010; Suzuki et al., 2009; Tew et al., 1990; Wang et al., 2011). In the current study, we resolve this problem by identifying a set of steps by which non-cognate B cells, loaded with IC either in vivo or in vitro, deliver antigen in the form of large aggregates to FDC; where it is stripped from the B cells and rapidly internalized as smaller particles within a cycling compartment. Thus, we propose a cell-to-cell transfer followed by a cycling pathway in a non-degradative compartment that provides an efficient mechanism for FDC capture, internalization, long-term retention and presentation of native antigen to cognate B cells.
Earlier studies identified the novel pathway by which C3-coated IC are trapped by sub-capsular sinus macrophages and shuttled to non-cognate B cells in the underlying B cell follicles. Subsequently, the B cells transport the IC to the FDC. The pathway was shown to be dependent on B cell expression of Cr2 as substantially less antigen was deposited on FDC in chimeric mice bearing Cr2-deficient B cells(Phan et al., 2007; Roozendaal et al., 2009). Moreover, disruption of the pathway led to a reduction in affinity maturation of GC B cells (Phan et al., 2009). Our observation using MP-IVM in anesthetized mice immunized with PE demonstrate direct transfer of the complement-coated PE-IC from the B cells to FDC. Although the overall efficiency of the process cannot be determined from the present study, it does demonstrate that non-cognate B cells hand-off antigen complexes directly. It seems most probable that other cell types such as resident dendritic cells may also participate in delivery of IC to FDC as proposed for transport of particulate antigens such as influenza virus (Gonzalez et al., 2010).
FDC are the only cell type known to store antigen intact and accessible to cognate B cells for extended periods (Garin et al., 2010). Macrophage and dendritic cells efficiently phagocytose IC through immunoglobulin Fc receptors but the type of receptor determines the antigen fate (Nimmerjahn and Ravetch, 2006). For example, uptake of IC by activating FcγR, i.e. FcγRI, FcγRI III and FcγR IV, results in internalization and degradation. By contrast, uptake of IC via the inhibitory FcγRIIB results in a transient return of the complexes to the cell surface for possible engagement by B cells (Bergtold et al., 2005). However, unlike the pathway we describe for FDC, IC internalized by inhibitory FcγR are eventually degraded within 48 hours based on studies in vitro. Murine FDC and B cells also express FcγRIIB but its expression is not required for uptake either in vivo or in our in vitro model (Phan et al., 2007; Roozendaal and Carroll, 2007). However, given the findings of Bergtold et al, it is possible that the Fc inhibitory receptor may be important in the cycling of the IC once they are internalized via Cr2 and this will be an important question to address for future studies (Bergtold et al., 2005).
Earlier results, based on electron microscopy studies, proposed that FDC retained and periodically released antigen to B cells in the form of immune complex coated bodies or ICCOSOMES, where it can be directly acquired by cognate B cells (Burton et al., 1991; Szakal et al., 1988; Cyster et al., 2000; Garin et al., 2010; Suzuki et al., 2009; Tew et al., 1990; Wang et al., 2011). Our results are consistent with the earlier findings of antigen exposure on the FDC cell surface but add that the presentation of antigen is dynamic. Thus, we identify an active process involving cycles of surface presentation. The timing of these events is such that snapshots obtained by EM are unable to resolve how antigen is temporally displayed. Moreover, our results that uptake of antigen from FDC culture supernatant alone or disruption of cycling endosomes by actin inhibition or fixation leads to inefficient acquisition by cognate B cells support a direct contact model; however, we can not rule out a role for release of immune complexes as an alternative source of antigen. It is possible that multiple pathways exist in which FDC provide antigens to B cells. For example, in an inflammatory environment where there are ongoing GC, the activation status of the FDC may influence the display of antigen.
Our finding of periodic cycling of intact IC by FDC suggests a mechanism to explain the observed retention of antigen over long periods. Another important factor in long-term retention of antigen is the intrinsic turnover of FDC. It is unclear whether FDC actively divide and retain antigen in daughter cells or if they are periodically replaced by precursor cells such as those recently described as platelet-derived growth factor receptor beta- positive perivascular cells (Krautler et al., 2012). Given the relative dense network of FDC within the follicles, it is possible that following uptake and internalization, IC are relayed to neighboring FDC and this pathway could help explain the highly efficient retention. In future studies, it will be important to track the long-term fate of FDC and how the immune status alters the local retention of antigen.
In addition to their known role within secondary lymphatic tissues, FDC are found in tertiary lymphatic tissues that form at sites of chronic inflammation or infection where they are thought to be important in maintaining B cell follicles and germinal centers (Krautler et al., 2012). As these ectopic sites form outside of normal lymphatic tissues it is not clear whether antigen is delivered to the FDC by one of the known routes, i.e. non-cognate B cells and FRC conduits or possibly dendritic cells (Phan et al., 2009; Bajenoff and Germain, 2009; Roozendaal et al., 2009; Bergtold et al., 2005; Gonzalez et al., 2010; Qi et al., 2006). Moreover, as the ectopic sites are often transient relative to known lymphoid structures, it will be important to learn if the fate of IC is regulated differently. For example, are IC retained primarily in a non-degradative, cycling endosomal compartment or are IC more actively shuttled to the lysosomes limiting the period of antigen display?
Overall, the combined results resolve a long standing question in humoral immunity; and identify a pathway by which FDC take up complement-coated IC from non-cognate B cells via Cr2 and internalize them into a cycling endocytic pathway. For the most part, antigen is retained intact within a non-degradative compartment that periodically cycles to the cell surface exposing it to cognate B cells.
Methods
Mice
C57BL/B6 background CD45.1, CD45.2, MD4, B1-8, Cr2-deficient and EGFP mice were purchased from Jackson Laboratories and maintained in specific pathogen-free facilities at Boston Children's Hospital Program in Cellular and Molecular Medicine (PCMM), Harvard Medical School. Institutional Animal Care and Use Committees (IACUC) at Harvard Medical School and PCMM approved animal experimental protocols.
Immune complex generation
B-Phycoerythrin (PE) (Anaspec) was used as a model Ag. IC were generated by mixing 10 μg of PE, 5 μg of rabbit anti-PE IgG (Rockland) and 10 μl freshly isolated C57BL/B6 serum (as a source of complement) in GVB++ buffer (Complement Tech) for 30 min at 37°C. 1 × 107 splenocytes from a C57BL/B6 mouse were then incubated with the immune complex mix for 30 min at 37 °C to generate IC bound B cells.
FDC isolation and ex vivo culture
FDC isolation and culture procedures were modified from that described (El Shikh et al., 2006). In short, mice were irradiated with 1200 rads using a 137 Cs irradiator located at the Immune Disease Institute at Harvard Medical School, one day prior to FDC isolation. LNs (brachial, axillary, inguinal, popliteal, cervical, lumbar, sacral and mesenteric) were harvested from irradiated mice and digested with 0.26U Liberase DH and 0.2 mg/ml DNase I (Roche). FDC were enriched using MACS separation column (Miltenyi Biotec) following manufacturer's protocol, using 0.7 μg FDC-M1 antibody (in-house) and 5 μg/ml biotinylated anti-rat kappa antibody (clone MRK-1, BD Biosciences). Positively selected FDC were re-suspended in FDC media (DMEM supplemented with 10 % FBS, 20 mM HEPES buffer, 0.2 mM MEM non-essential amino acids, 2 mM L-glutamine and 1μg/ml gentamicin) and plated on collagen-coated (Rat tail derived collagen; Roche) glass coverslips (#1.5, 25mm diameter, Warner Instruments) at a density of 5 × 105 FDC per coverslip. 5-7 day cultured FDC were used for different assays.
B cell isolation
Splenocytes from C57BL/B6, B1-8 and MD4 mice were enriched for B cells by negative selection using biotinylated anti-CD43 (S7 clone; BD Biosciences) followed by streptavidin micro beads (Miltenyi Biotech). Cells were separated using MACS columns (Miltenyi Biotech) following manufacturer's protocol. Purity of the cells used for assays was greater than 90% as assessed by flow cytometry.
FDC and B cell assays
For different assays involving IC-bound B cells and FDC, 2 × 106 B cells per 5 × 105 cultured FDC were used. In experiments where the role of actin was analyzed, FDC were either left untreated or treated with Cytochalasin D (Sigma-Aldrich.
Flow cytometry
B cells were washed from FDC cultures after the assay and stained in a 96-well U-bottom plate. Anti- B220-PerCP and IgMb-FITC antibodies were purchased from Biolegend. FDC-M1-Alexa 633 was made in-house. FACSCanto II (BD Bioscience) was used to acquire the samples and FlowJo software (Tristar) and Prism (Graphpad Software) were used for analysis of the acquired data.
Live cell labeling
B cells were labeled with the CellTracker dye CMFDA (invitrogen) at final concentration of 2.5 μM for 15 min, followed by extensive washing and used for adoptive transfer. For spinning disk epifluorescent or confocal microscopy, B cells and FDC were labeled with DiI or DiO Vybrant Cell-labeling solution (Molecular Probes).
In vivo imaging
Mice were passively immunized with 1 mg of rabbit anti-PE IgG, and pretreated by intravenous injection with fluorescent anti-Cr2 to label the FDC in vivo. Purified, fluorescently labeled B cells were adoptively transferred as described. 24 hr later, 10 μg of PE was injected subcutaneously into the footpad. The popliteal LN was surgically exposed and MP-IVM was performed as described(Gonzalez et al., 2010).
Cultured cell time-lapse imaging and confocal images
Cultured FDC attached to glass cover slides were mounted in a chamber, which is filled with PE-IC bound B cells in FDC media and then installed in a heated stage plate (20/20 Technology) connected to a chamber insert (20/20 Technology), and placed in an environment chamber (Okolab), where temperature humidity and CO2 levels were controlled during imaging. Epifluorescence microscopy was performed using a Mariana™ system (Intelligent Imaging Innovations) with Axiovert 200M microscope (Carl Zeiss MicroImaging) equipped with 63 × oil, 100 × oil objectives (Plan Apochromat, 1.4 NA, Carl Zeiss MicroImaging), coolSnap HQ II camera (Photometrics). Xenon lamp (DG-4, Sutter Instruments), with WF HQE and Misc. filter set (485/20, 560/25 and 650/13, Chroma Technology Corp) was used as a light source. Time-lapse movies were acquired in one optical section with between 15 sec – 1min intervals, with multiple positions.
The microscope is connected with a spinning disk confocal head (CSU22, Yokogawa electric), cascade camera (Photometrics) and piezo-driven stage (Application Scientific Instrumentation) for sample axial position control. Three lasers (491 nm, 561 nm, 660 nm) coupled to spinning head through an acousticoptical tunable filter (AOTF) were used as a light source. Images were analyzed and 3D view rendered by Slidebook 4.2 or 5 (Intelligent Imaging Innovations).
Immunohistochemistry
LNs were dissected out and embedded in optimal cutting temperature compound (TissueTek). Cryosections were prepared at 10 μm thickness and fixed in 1% paraformaldehyde solution. Fixed samples were pre-incubated with anti-FcR (2.4G2, house produced) and 2% bovine serum albumin for blocking nonspecific and Fc mediated binding. FDC M1 Alexa 488(house labeled), donkey anti-rabbit DyLight 405, donkey anti-rabbit DyLight 488, donkey anti-rabbit DyLight 649 (Biolegend) were used for staining ex vivo cultures of FDC. Images were acquired with a FluoView FV1000 confocal microscope (Olympus) with a 20× lens (NA: 0.7) and processed with FluoView software (Olympus). Data was analyzed with Volocity software (Perkin-Elmer).
Electron microscopy
Isolated FDC were grown on collagen-coated ACLAR cover slips. Cells were pre-incubated with anti FcR (2.4G2) and then labeled with anti-Cr2 (clone 8C12, in-house), followed by rabbit anti-rat IgG and protein A gold (Cell Microscopy Center, Department of Cell Biology, University Medical Center Utrecht, The Netherlands). For detection of IC, cells were pre-treated as above and then labeled with protein A gold. Popliteal LNs were harvested and sections were prepared as described (Gonzalez et al., 2010). Samples were analyzed on a Tecnai G2 Spirit Bio TWIN electron microscope (FEI Company) at the Harvard Medical School EM facility.
Supplementary Material
Highlights.
Visualizing direct immune complex transfer from non-cognate B cells to FDC in vivo
Uptake of immune complexes from non-cognate B cells by FDC is actin-dependent
Immune complexes cycling to the FDC surface are displayed for B cell acquisition
In vivo, FDC retain immune complexes in a cycling compartment for at least 16 days
Acknowledgments
The authors thank all members of the Carroll lab and Patrick Reeves of the Kirchhausen lab for suggestions and help with experiments. We acknowledge the valuable technical assistance with live cell and confocal imaging from E. Marino and H. Leung, respectively, and help by M. Coughlin for preparation of EM sections. Supported by US National Institutes of Health (RO1 AI039246-16; R37 AI054636; PO1 AI078897-04) to M.C.C. and GM-075252 and U54 AI057159 (New England Regional Center of Excellence in Biodefense and Emerging Infectious Diseases) to T.K.; and Glaxo-Smith Kline (GSK-P-11-02) to M.C.C. and GSK post-doctoral fellowship to PC; Marie Curie International Outgoing Fellowship for Career Development (220044) to S.F.G. The authors declare no competing financial interests.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature Cited
- Alimzhanov MB, Kuprash DV, Kosco-Vilbois MH, Luz A, Turetskaya RL, Tarakhovsky A, Rajewsky K, Nedospasov SA, Pfeffer K. Abnormal development of secondary lymphoid tissues in lymphotoxin beta-deficient mice. Proc Natl Acad Sci USA. 1997;94:9302–9307. doi: 10.1073/pnas.94.17.9302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajenoff M, Germain RN. B cell follicle development remodels the conduit system and allows soluble antigen delivery to follicular dendritic cells. Blood. 2009;114:4989–4997. doi: 10.1182/blood-2009-06-229567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergtold A, Desai DD, Gavhane A, Clynes R. Cell surface recycling of internalized antigen permits dendritic cell priming of B cells. Immunity. 2005;23:503–514. doi: 10.1016/j.immuni.2005.09.013. [DOI] [PubMed] [Google Scholar]
- Boulant S, Kural C, Zeeh JC, Ubelmann F, Kirchhausen T. Actin dynamics counteract membrane tension during clathrin-mediated endocytosis. Nat Cell Biol. 2011;13:1124–1131. doi: 10.1038/ncb2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton GF, Kosco MH, Szakal AK, Tew JG. Iccosomes and the secondary antibody response. Immunology. 1991;73:271–276. [PMC free article] [PubMed] [Google Scholar]
- Casella JF, Flanagan MD, Lin S. Cytochalasin D inhibits actin polymerization and induces depolymerization of actin filaments formed during platelet shape change. Nature. 1981;293:302–305. doi: 10.1038/293302a0. [DOI] [PubMed] [Google Scholar]
- Cyster JG, Ansel KM, Reif K, Ekland EH, Hyman PL, Tang HL, Luther SA, Ngo VN. Follicular stromal cells and lymphocyte homing to follicles. Immunol Rev. 2000;176:181–193. doi: 10.1034/j.1600-065x.2000.00618.x. [DOI] [PubMed] [Google Scholar]
- El Shikh ME, El Sayed R, Szakal AK, Tew JG. Follicular dendritic cell (FDC)-FcgammaRIIB engagement via immune complexes induces the activated FDC phenotype associated with secondary follicle development. Eur J Immunol. 2006;36:2715–2724. doi: 10.1002/eji.200636122. [DOI] [PubMed] [Google Scholar]
- Endres R, Alimzhanov MB, Plitz T, Futterer A, Kosco-Vilbois MH, Nedospasov SA, Rajewsky K, Pfeffer K. Mature follicular dendritic cell networks depend on expression of lymphotoxin beta receptor by radioresistant stromal cells and of lymphotoxin beta and tumor necrosis factor by B cells. The Journal of experimental medicine. 1999;189:159–168. doi: 10.1084/jem.189.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y, Xu C, Fu YX, Holers VM, Molina H. Expression of complement receptors 1 and 2 on follicular dendritic cells is necessary for the generation of a strong antigen-specific IgG response. Journal of immunology. 1998;160:5273–5279. [PubMed] [Google Scholar]
- Fischer MB, Goerg S, Shen L, Prodeus AP, Goodnow CC, Kelsoe G, Carroll MC. Dependence of germinal center B cells on expression of CD21/CD35 for survival. Science. 1998;280:582–585. doi: 10.1126/science.280.5363.582. [DOI] [PubMed] [Google Scholar]
- Fu YX, Molina H, Matsumoto M, Huang G, Min J, Chaplin DD. Lymphotoxin-alpha (LTalpha) supports development of splenic follicular structure that is required for IgG responses. J Exp Med. 1997;185:2111–2120. doi: 10.1084/jem.185.12.2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garin A, Meyer-Hermann M, Contie M, Figge MT, Buatois V, Gunzer M, Toellner KM, Elson G, Kosco-Vilbois MH. Toll-like receptor 4 signaling by follicular dendritic cells is pivotal for germinal center onset and affinity maturation. Immunity. 2010;33:84–95. doi: 10.1016/j.immuni.2010.07.005. [DOI] [PubMed] [Google Scholar]
- Gommerman JL, Mackay F, Donskoy E, Meier W, Martin P, Browning JL. Manipulation of lymphoid microenvironments in nonhuman primates by an inhibitor of the lymphotoxin pathway. J Clin Invest. 2002;110:1359–1369. doi: 10.1172/JCI15975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez M, Mackay F, Browning JL, Kosco-Vilbois MH, Noelle RJ. The sequential role of lymphotoxin and B cells in the development of splenic follicles. The Journal of experimental medicine. 1998;187:997–1007. doi: 10.1084/jem.187.7.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez SF, Lukacs-Kornek V, Kuligowski MP, Pitcher LA, Degn SE, Kim YA, Cloninger MJ, Martinez-Pomares L, Gordon S, Turley SJ, Carroll MC. Capture of influenza by medullary dendritic cells via SIGN-R1 is essential for humoral immunity in draining lymph nodes. Nat Immunol. 2010;11:427–434. doi: 10.1038/ni.1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna MG, Jr, Szakal AK. Localization of 1251-labeled antigen in germinal centers of mouse spleen: histologic and ultrastructural autoradiographic studies of the secondary immune reaction. Journal of immunology. 1968;101:949–962. [PubMed] [Google Scholar]
- Kelsoe G. Life and death in germinal centers. Immunity. 1996;4:107–111. doi: 10.1016/s1074-7613(00)80675-5. [DOI] [PubMed] [Google Scholar]
- Kinoshita T, Fujita T, Tsunoda R. Expression of complement receptors CR1 and CR2 on murine follicular dendritic cells and B lymphocytes. Vol. 2. Amsterdam: Elsevier; 1991. [Google Scholar]
- Kosco MH, Monfalcone AP, Szakal AK, Tew JG. Germinal center B cells present antigen obtained in vivo to T cells in vitro and stimulate mixed lymphocyte reactions. Adv Exp Med Biol. 1988;237:883–888. doi: 10.1007/978-1-4684-5535-9_132. [DOI] [PubMed] [Google Scholar]
- Kranich J, Krautler NJ, Heinen E, Polymenidou M, Bridel C, Schildknecht A, Huber C, Kosco-Vilbois MH, Zinkernagel R, Miele G, Aguzzi A. Follicular dendritic cells control engulfment of apoptotic bodies by secreting Mfge8. J Exp Med. 2008;205:1293–1302. doi: 10.1084/jem.20071019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krautler N, Kana V, Kranich J, Tian Y, Perera D, Lemm D, Schwarz P, Armulik A, Browning J, Tallquist M, et al. Follicular dendritic cells emerge from ubiquitous pervascular precursors. Cell. 2012 doi: 10.1016/j.cell.2012.05.032. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam KP, Rajewsky K. B cell antigen receptor specificity and surface density together determine B-1 versus B-2 cell development. The Journal of experimental medicine. 1999;190:471–477. doi: 10.1084/jem.190.4.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLennan I. Germinal Centers. Ann Rev Immunol. 1994;12:117–139. doi: 10.1146/annurev.iy.12.040194.001001. [DOI] [PubMed] [Google Scholar]
- Mandel TE, Phipps RP, Abbot A, Tew JG. The follicular dendritic cell: long term antigen retention during immunity. Immunol Rev. 1980;53:29–59. doi: 10.1111/j.1600-065x.1980.tb01039.x. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Lo SF, Carruthers CJL, Min J, Mariathasan S, Huang G, Pias DR, Martin SM, Geha RS, Nahn MH, Chaplin DD. Affinity maturation without germinal centres in lymphotoxin-alpha-deficient mice. Nature. 1996;382:462–466. doi: 10.1038/382462a0. [DOI] [PubMed] [Google Scholar]
- May JA, Ratan H, Glenn JR, Losche W, Spangenberg P, Heptinstall S. GPIIb-IIIa antagonists cause rapid disaggregation of platelets pre-treated with cytochalasin D. Evidence that the stability of platelet aggregates depends on normal cytoskeletal assembly. Platelets. 1998;9:227–232. doi: 10.1080/09537109876744. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn F, Ravetch JV. Fcgamma receptors: old friends and new family members. Immunity. 2006;24:19–28. doi: 10.1016/j.immuni.2005.11.010. [DOI] [PubMed] [Google Scholar]
- Nossal GJ, Abbot A, Mitchell J, Lummus Z. Antigens in immunity. XV. Ultrastructural features of antigen capture in primary and secondary lymphoid follicles. J Exp Med. 1968;127:277–290. doi: 10.1084/jem.127.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasparakis M, Alexopoulou L, Episkopou V, Kollias G. Immune and inflammatory responses in TNF alpha-deficient mice: a critical requirement for TNF alpha in the formation of primary B cell follicles, follicular dendritic cell networks and germinal centers, and in the maturation of the humoral immune response. The Journal of experimental medicine. 1996;184:1397–1411. doi: 10.1084/jem.184.4.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan TG, Green JA, Gray EE, Xu Y, Cyster JG. Immune complex relay by subcapsular sinus macrophages and noncognate B cells drives antibody affinity maturation. Nat Immunol. 2009;10:786–793. doi: 10.1038/ni.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan TG, Grigorova I, Okada T, Cyster JG. Subcapsular encounter and complement-dependent transport of immune complexes by lymph node B cells. Nat Immunol. 2007;8:992–1000. doi: 10.1038/ni1494. [DOI] [PubMed] [Google Scholar]
- Qi H, Egen JG, Huang AY, Germain RN. Extrafollicular activation of lymph node B cells by antigen-bearing dendritic cells. Science. 2006;312:1672–1676. doi: 10.1126/science.1125703. [DOI] [PubMed] [Google Scholar]
- Qin D, Wu J, Vora KA, Ravetch JV, Szakal AK, Manser T, Tew JG. Fc gamma receptor IIB on follicular dendritic cells regulates the B cell recall response. J Immunol. 2000;164:6268–6275. doi: 10.4049/jimmunol.164.12.6268. [DOI] [PubMed] [Google Scholar]
- Roozendaal R, Carroll MC. Complement receptors CD21 and CD35 in humoral immunity. Immunol Rev. 2007;219:157–166. doi: 10.1111/j.1600-065X.2007.00556.x. [DOI] [PubMed] [Google Scholar]
- Roozendaal R, Mempel TR, Pitcher LA, Gonzalez SF, Verschoor A, Mebius RE, von Andrian UH, Carroll MC. Conduits mediate transport of low-molecular-weight antigen to lymph node follicles. Immunity. 2009;30:264–276. doi: 10.1016/j.immuni.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shokat K, Goodnow C. Antigen-induced B-cell death and elimination during germinal centre immune response. Nature. 1995;375:334–338. doi: 10.1038/375334a0. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Grigorova I, Phan TG, Kelly LM, Cyster JG. Visualizing B cell capture of cognate antigen from follicular dendritic cells. J Exp Med. 2009;206:1485–1493. doi: 10.1084/jem.20090209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szakal AK, Kosco MH, Tew JG. A novel in vivo follicular dendritic cell-dependent iccosome-mediated mechanism for delivery of antigen to antigen-processing cells. J Immunol. 1988;140:341–353. [PubMed] [Google Scholar]
- Taylor PR, Pickering MC, Kosco-Vilbois MH, Walport MJ, Botto M, Gordon S, Martinez-Pomares L. The follicular dendritic cell restricted epitope, FDC-M2, is complement C4; localization of immune complexes in mouse tissues. European journal of immunology. 2002;32:1888–1896. doi: 10.1002/1521-4141(200207)32:7<1883::AID-IMMU1888>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Tew JG, Kosco MH, Burton GF, Szakal AK. Follicular dendritic cells as accessory cells. Immunol Rev. 1990;117:185–211. doi: 10.1111/j.1600-065x.1990.tb00573.x. [DOI] [PubMed] [Google Scholar]
- Wang X, Cho B, Suzuki K, Xu Y, Green JA, An J, Cyster JG. Follicular dendritic cells help establish follicle identity and promote B cell retention in germinal centers. The Journal of experimental medicine. 2011;208:2497–2510. doi: 10.1084/jem.20111449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, El Shikh ME, El Sayed RM, Best AM, Szakal AK, Tew JG. IL-6 produced by immune complex-activated follicular dendritic cells promotes germinal center reactions, IgG responses and somatic hypermutation. Int Immunol. 2009;21:745–756. doi: 10.1093/intimm/dxp041. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.