

Abstract
Biological signaling pathways interact with one another to form complex networks. Complexity arises from the large number of components, many with isoforms that have partially overlapping functions; from the connections among components; and from the spatial relationship between components. The origins of the complex behavior of signaling networks and analytical approaches to deal with the emergent complexity are discussed here.
Signaling in biological systems occurs at multiple levels. In its broad sense, one could use the term “signaling” to describe events ranging from interactions between single molecules to interactions between species in ecological systems. The aim here is to deal with complexity in signaling at a single level: intracellular signaling within a cell. We will outline how current and forthcoming tools in biochemistry, cell and molecular biology, and physiology, as well as theoretical analysis and simulation methods, may be used to study this complex system.
In a general sense, the adjective “complex” describes a system or component that by design or function or both is difficult to understand and verify. In the past decade, analysis of complex systems (the field of complexity) has emerged as a distinct facet of mathematical and physical sciences. Understanding of biological systems may be enhanced by analysis of their complex nature. In physical systems, complexity is determined by such factors as the number of components and the intricacy of the interfaces between them, the number and intricacy of conditional branches, the degree of nesting, and the types of data structures. Biological signaling networks possess many of these attributes, as well as dynamic assembly, translocation, degradation, and channeling of chemical reactions. All of these activities occur simultaneously, and each component participates in several different activities.
One approach to understanding complexity is to start with a conceptually simple view of signaling and add details that introduce new levels of complexity. As this process unfolds, it becomes clear where experimental data end and how progressively more difficult it becomes to understand the system as a whole in terms of the functional details of individual components.
A Signaling Wire
The simplest description of signaling may be in terms of elementary chemistry in a homogenous well-stirred cell where all molecules have equal access to each other. Here, the most upstream component of the signaling pathway interacts with an external source and transfers information about that interaction to an effector that is capable of eliciting a biological response. This scheme is illustrated in Fig. 1A. Bacterial two-component signal transduction (1) is one example of such a system. Some mammalian signaling pathways, such as the β-adrenergic receptor to the glycogen-phosphorylase pathway, can also be considered within this framework. The properties of such a simplified system are completely determined by the concentrations of each of the components and the reaction rates. Here the role of the many types of signaling components, including receptors, transducers, enzymes, and diffusible second messengers, is simply to give different signals a unique identity. Each pathway can be thought of as a wire carrying information. Because the well-stirred cell does not have wires that are spatially separated by insulators, the identity of the signals must be carried by distinct molecules so that the information can be processed in an orderly fashion.
Fig. 1.
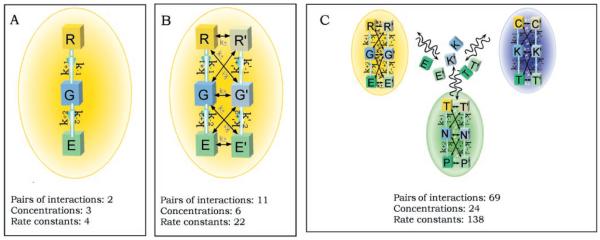
The increasing complexity of signaling pathways inside a cell. In each panel, k is the rate constant for the first pathway and k′ represents constants for the second pathway; plus and minus signs indicate forward and reverse, respectively; 1 and 2 indicate pathways 1 and 2. (A) A simple three-component pathway. The arrows indicate the direction of the signal flow. Each component interacts only with its adjacent component. This system represents a typical design of a transmembrane G protein signaling pathway, and the lettering for the components R (receptor), G (G protein), and E (effector) reflects this. (B) Two interactive signaling pathways in one compartment. Here, interactions are restricted to adjacent partners to represent real situations and limit the complexity of the system. (C) A complex system consisting of two interactive-pathways in each of three interacting compartments, colored yellow, blue, and green. Such a system could represent the first level of compartmentalization of the cell into membrane, cytoplasmic, and nuclear compartments. C, cytoplasmic components; K, kinase; T, transcription factors; N, nucleic acids; P, protein components in the nucleus. The communications between the compartments are carried out by the translocation of the signaling molecules. The number of interactions and the minimal number of parameters (concentration of reactants plus rate constants) for each system is given. The increasing complexity in terms of the number of parameters needed to specify the system can be readily seen.
Even in this highly oversimplified analysis a first order of complexity is evident: The vast array of signaling molecules and isoforms with apparently redundant signal transfer functions often have different kinetic properties. This makes the estimation of reaction rates and reactant concentrations a crucial issue in obtaining an accurate quantitative description of the system. Unfortunately, the measurements of these system parameters are often not available or possible with current technologies. Nevertheless, analysis of linear pathways provides valuable insights into system properties such as threshold stimuli required to trigger a response (2) and time courses for signal output. Mathematical analysis of enzyme function has long been an integral part of rigorous biochemistry, and the various models of regulation developed for enzymes (3) have been useful in analyzing signaling systems. These analyses provide mechanistic insights into the interactions between individual components, such as between ligands and receptors (4), as well as between intracellular components of the system (5). Often such mathematical analyses have served to discriminate between alternative reaction mechanisms (6).
Interactions Between Pathways
A simple three-component transmembrane signaling system is depicted in Fig. 1A. This organization is representative of many heterotrimeric GTP–binding protein (G-protein) signaling pathways. But the interaction between pathways necessitates a first elaboration of this simple scheme. Distinct pathways now become parts of an interacting signaling network. Each interaction between components in different pathways is a potential site of computation (7). Therefore, in a system consisting of two interactive pathways of n components, each one would, in principle, need to collect data of n2 interactions (one for every possible pair of interactions). Figure 1B describes a simplified situation where interactions occur only between two adjacent components. Such simplification often reflects the specificity in interactions between pathways. Even in such simple situations, the experimental challenges are considerable. In addition to specifying the concentrations of the reactants and rate constants for each step of each pathway, one needs an accurate estimate of how these values are affected by the presence of the interacting pathway. Intuitive approaches to the analysis of such networks are difficult. Nevertheless, such a system is amenable to quantitative analysis using reductionist chemical data from reconstituted test tube experiments. We have adapted GENE-SIS, a neural network simulator, to analyze a simplified network consisting of four different interacting signaling pathways. Such a network exhibits interesting emergent properties, including integration of signals across different time scales, generation of distinct outputs depending on the amplitude and duration of the input signals, and the presence of feedback loops that behave as bistable switches to process information flow through the network (8). Although this first glimpse of emergent complexity appears to be intriguing, rapidly accumulating experimental evidence suggests that several other considerations need to be taken into account in order to develop a minimally accurate picture of a living cell. Prime considerations among these are compartmentalization and regional organization of signaling components.
Compartments
Compartmentalization introduces several levels of complexity. First, many signaling components and their substrates are anchored in the plasma membrane. The plasma membrane provides a milieu for biochemical reactions that is quite distinct from the cytoplasm in its properties. The lipid environment enables a new class of reactions involving hydrophobic interactions. It also introduces a two-dimensional reaction environment, with alterations in component access, effective concentrations, and component orientation relative to the membrane. Organelle formation leads to a further expansion of the possible cellular microenvironments, each with different biochemical properties and signaling capabilities. Second, the separation of reactions in space allows the same molecules in the same cell to carry entirely different signals. In other words, we already have signaling “wires” distinguished by the identity of the molecules in the pathways. Compartmentalization duplicates these existing wires and separates them in space. This multiplies the number of signals they can carry.
These features cause trouble experimentally. Techniques for measuring rates and concentrations of reactants in their natural lipid or compartmentalized environments are often not available, and even when they are, the techniques require progressive refinement. The measurement of Ca2+ concentrations in organelles, for example, has required several generations of new probes to accurately estimate the Ca2+ concentrations in intracellular stores (9). Simulation studies have provided a useful framework for analyzing systems at this level. Studies of Ca2+ oscillations (10) and Ca2+ waves (11), for example, bring in testable hypotheses about the critical signaling interactions for information transfer within the cell. Even when there are a minimal number of components that move between compartments, the number of parameters needed to accurately describe the system becomes large, and experimental approaches to determining these parameters stretch the limits of current technologies. A three-compartment system with six translocatable components is shown in Fig. 1C, and the complexity of such a system is readily apparent.
Scaffolds and Reaction Channeling
In addition to subcellular compartmentalization, recent research has highlighted the role of molecular scaffolds that provide regional organization by assembling signaling components into functional complexes. The cytoskeleton is a dynamic framework on which the cell builds this regional organization. The most dramatic example of its dynamism is cell division. In the quiescent cell, it is both the substrate and the scaffold for signaling processes. A prime example of its dual role is the synapse. Here the cytoskeleton, in particular the pre- and postsynaptic structures, are the anchors for a wide array of synaptic signaling molecules. Consequently, modification of the synaptic cytoskeleton is a likely candidate for causing long-term changes in synaptic efficacy (12).
The term “scaffold” is also used for a new class of signaling proteins that do not have information transfer capability of their own but interact with multiple signaling proteins in a pathway. The scaffold provides an assembly line along which a series of enzymes process their substrates in a well-defined sequence and with an efficiency and specificity that are orders of magnitude higher than would be possible in freely diffusing systems. Scaffolds for the MAP kinase pathways are prototypical examples of such organization, and a number of other scaffold proteins have been identified (13). In vitro, this organization can result in reaction channeling, leading to dramatic increases in the efficiency of signal transfer as well as to enhanced specificity of signal flow, despite possible cross-reactivity with other pathways that are apparent in the test tube. A striking example of reaction channeling is the synthesis machinery for many antibiotics, which are composed of enzyme modules that are physically and chemically attached to each other (14). The substrate molecule proceeds stepwise down the chain of enzymes and is systematically extended and modified in a manner reminiscent of a factory assembly line. Efficiencies in signal transmission can be achieved by similar organization, and scaffolds are likely to play a role in achieving such efficiencies. A key experimental challenge is to accurately quantify these efficiencies.
Within the cell, signals in different compartments do not work in isolation. Compartments communicate with each other via translocating molecules. Translocation is often an integral part of the signal flow. Figure 2 shows four interacting pathways in the postsynaptic region of a neuron. These pathways include signaling components that can translocate from plasma membrane to cytoplasm and vice versa or from cytoplasm to nucleus. The major linear routes of signal flow are color-coded, and the cross-connections both positive (arrows) and negative (dots) are in black. The complexity of even such a minimal network is immediately obvious. However, most of these interactions can be identified, parameterized, and analyzed (8). Thus, the major hurdle is the development of methods to track, organize, and analyze the large number of parameters needed to specify such a system rather than the development of new methods of mathematical analysis.
Fig. 2.

Four major signaling pathways in the postsynaptic region of a neuron that combine to form a local signaling network. The major linear routes of signal flow are depicted by the thick arrows of four different colors: orange [phospholipase C (PLC) pathway], pink (Ras pathway), green (adenylyl cyclase pathway), and blue [Ca2+/calmodulin (CaM) pathway]. The interactions between different pathways are represented by black lines with arrow (representing activation) or a dot (representing inhibition). Although most major interactions in the network are shown, these connections are not meant to be all-inclusive; additional connections could exist. The three-colored background represents three different cell compartments: the plasma membrane (light yellow), cytosol (light blue), and nucleus (light green). Some of the signaling proteins that translocate between different compartments are shown in both compartments. Examples include MAP kinase, which when activated translocates from the cytoplasm to the nucleus to phosphorylate and activate transcription factors; and the transcription factor CREB, which upon phosphorylation by protein kinase A (PKA) translocates to the nucleus.
Although compartmentalization confines certain interactions between components, molecular trafficking between compartments raises the number of system parameters by at least another power. This qualitative shift in complexity (and the relative paucity of understanding of it) also marks the border between biochemistry and cell biology. One needs to consider transport as a whole new industry in the cellular economy. The movement of signals may be as simple as diffusion down concentration gradients (although the formation of those gradients may not always be simple) or as complex as the veritable rail network of the actin-tubulin cytoskeleton, which is traversed by cytoplasmic motors with precisely addressed proteins directed to their destinations by target sequences. The endoplasmic reticulum carries out the enormous job of sorting molecules between the nucleus, several organelles, the cell surface, and the outside and does a great deal of molecular assembly on the side. A comparison would be if the post office not only reliably supplied components from a dozen different sources but also assembled them en route and delivered a functioning computer to your door.
Although the role of the endoplasmic reticulum in the assembly of the cell is now well recognized, its role in signaling is just starting to be understood. Theoretically, compartmentalization and molecular trafficking using the endoplasmic reticulum introduce a qualitative difference in the kinds of analysis that could be done, even if all the data were at hand. Chemistry is now replaced by reaction-diffusion systems of complex geometry, and each cellular compartment has its own set of reactions that need to be independently analyzed first and then analyzed in a progressively interdependent manner, so that the effect of each compartment on all others is accounted for. And in the dual cell assembly and signaling role of the cytoskeleton and compartments, we can see the beginnings of a deeper level of analytical complexity: The system is self-modifying. This problem reaches its full expression in genetic regulation.
Regulation in the Nucleus
The core signaling system of the cell is, of course, the genetic machinery. We are already remote from our initial description of cellular signaling as a group of chemical reactions in a well-stirred test tube. Each of the previous levels of cellular signal flow has introduced new levels of complexity in experiment and analysis. At face value, the genetic machinery is based on the same set of components—enzymes, compartments, and tightly controlled signal trafficking—plus a gigabyte-sized program written into the DNA. Indeed, fairly accurate abstractions of some simple genetic systems can be made in terms of networks of genes, without dealing with the intricate details of the machinery involved (15). It is a major experimental challenge to understand all the biochemical reactions in the nucleus. These include protein-protein and protein-DNA interactions, mechanisms of transcription and splicing and processing of the transcripts and exporting of RNA, and how these processes are regulated intrinsically, as well as by signals from outside the nucleus. Although many of these questions are daunting, they may turn out to be more experimentally tractable than the spatial and organizational questions in cell biology described above. The defining feature, which makes the system as a whole extremely difficult to analyze, is that it is not a machine (however complex) drawn to a well-defined design, but a machine that can and does constantly rebuild itself within a range of variable parameters. For a systematic approach, what is needed is a relatively clear definition of the boundary of this variability. In principle, these boundaries are determined by an as-yet-unknown combination of intrinsic capability and external inputs. The balance between intrinsic capability and the response to external signals is likely to be a central issue in understanding gene expression. This is a difficult situation to analyze, and currently we are unsure of how to approach it. Nevertheless, it is the crux of one of the classic mysteries of biology: how the developing organism starts from a single cell, which divides and modifies itself into many different classes of cells and many specific shapes, yet produces a complete organism with little individual variation. A large body of emerging data indicates that early development occurs through signaling interactions that are genetically programmed, whereas at the later stages, the development of complex traits is dependent on external inputs as well. A quantitative description of this entire process would be a culmination and synthesis of much of biology.
Approaches to Analyze Complex Signaling Networks
A recurring theme in our discussion is the necessity for tightly coupling experiments and theory in particular computer simulations. There is simply too much essential detail in biological signaling for the unaided human mind to organize and understand. It appears that a paradigm shift from the qualitative to the quantitative is taking place in biology; that we are moving from a descriptive to a predictive science. Gene discovery and the consequent biochemical characterization of gene products has led to the accumulation of a treasure trove of quantitative properties of these gene products, many of which are components of signaling systems. What is now needed is a twofold effort to develop a signaling database and the tools to integrate these data. A systematic cataloging of proteins, then lipids, complex sugars, and other signaling molecules within the various organelles of a mammalian cell, including the locations, concentrations, and core kinetic properties, would in itself be a very large project requiring enormous resources. The analytical tools would rely on emerging databases, Internet access, and visualization and simulation techniques.
Key experimental tools for quantifying signaling at the level of compartments are already becoming available. The principal ones among these are likely to be a combination of genetically encoded fluorescent reporters and high-resolution imaging. Selective expression of these reporters in combination with high-resolution visualization techniques should allow the semi-quantitative estimation of molecular concentrations and interactions. Together with the knowledge available from a completely sequenced genome, these should enable systematic monitoring of many levels of signaling reactions in vivo and simultaneously keep track of changes in cellular structure. Likewise, the computational tools for handling this vast array of data are starting to take shape. Database and Web-based query systems on comparable scales already exist for protein structure and the genome projects (16). Advances in computer hardware have brought large-scale calculations and fast graphic visualization out of the domain of supercomputer centers onto reasonably priced machines in the laboratory. As is the case with genome databases, the main remaining issue is analysis. Simulation techniques for handling thousands of single-molecule signaling reactions taking place in the intricate cellular geometry will require (at least) a combination of finite element analysis and Monte Carlo methods. Although these techniques are well developed in engineering contexts, we are not aware of any applications that approach the scale and complexity of the geometry and interactions in the cell. In addition to the purely numerical issues, it is a significant challenge to develop user interfaces that will enable experimental biologists who are not expert computer programmers to use such complex computational programs with relative ease. Several efforts are under way to develop interfaces with databases and simulators that can meet these requirements (17).
Benefits of Understanding Complex Signaling Networks
The origins of many human diseases, including cancer, diabetes, and neural disorders, are in the functioning (and malfunctioning) of signaling components. Often malfunctioning of a single entity does not cause problems, but the combined effects of multiple malfunctioning complexes are substantial. An understanding of how individual components function within the context of the entire system under a variety of situations should be helpful in understanding why interactions between aberrant signaling pathways often result in pathophysiology. Understanding complex signaling networks may also provide a clear molecular view of the interactions of individuals with their environment.
References and Notes
- 1.Alex LA, Simon MI. Trends Genet. 1994;10:133. doi: 10.1016/0168-9525(94)90215-1. [DOI] [PubMed] [Google Scholar]
- 2.Huang CY, Ferrell JEJ. Proc. Natl. Acad. Sci. U.S.A. 1996;93:10078. doi: 10.1073/pnas.93.19.10078. [DOI] [PMC free article] [PubMed] [Google Scholar]; Ferrell JEJ, Machleder EM. Science. 1998;280:895. doi: 10.1126/science.280.5365.895. [DOI] [PubMed] [Google Scholar]; LaPorte DC, Walsh K, Koshland DEJ. J. Biol. Chem. 1984;259:14068. [PubMed] [Google Scholar]
- 3.Segel IH. Enzyme Kinetics–Behavior and Analysis of Rapid Equilibrium and Steady State Enzyme Systems. Wiley; New York: 1975. [Google Scholar]
- 4.Black JW, Gerskowitch VP, Leff P, Shankley NP. Br. J. Pharmacol. 1986;89:547. doi: 10.1111/j.1476-5381.1986.tb11155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Birnbaumer L, Swartz TL, Abramowitz J, Mintz PW, Iyengar R. J. Biol. Chem. 1980;255:3542. [PubMed] [Google Scholar]; Birnbaumer L, Bearer CF, Iyengar R. ibid. :3552. [PubMed] [Google Scholar]; Iyengar R, Abramowitz J, Bordelon-Riser M, Birnbaumer L. ibid. :3558. [PubMed] [Google Scholar]
- 6.Braun S, Levitzki A. Biochemistry. 1979;18:2134. doi: 10.1021/bi00577a045. [DOI] [PubMed] [Google Scholar]
- 7.Berridge MJ, Bray D. Nature. ibid. 1993;1995;361378:315, 419. doi: 10.1038/361315a0. [DOI] [PubMed] [Google Scholar]; Marder E. ibid. 1996;384:113. doi: 10.1038/384113a0. [DOI] [PubMed] [Google Scholar]
- 8.Bhalla US, Iyengar R. Science. 1999;283:381. doi: 10.1126/science.283.5400.381. [DOI] [PubMed] [Google Scholar]
- 9.Miyawaki A, et al. Nature. 1997;388:882. doi: 10.1038/42264. [DOI] [PubMed] [Google Scholar]
- 10.Tang Y, Othmer HG. Philos. Trans. R. Soc. London B Biol. Sci. 1995;349:179. doi: 10.1098/rstb.1995.0102. [DOI] [PubMed] [Google Scholar]; Meyer T, Stryer L. Proc. Natl. Acad. Sci. U.S.A. 1990;87:3841. doi: 10.1073/pnas.87.10.3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Amundson J, Clapham D. Curr. Opin. Neurobiol. 1993;3:375. doi: 10.1016/0959-4388(93)90131-h. [DOI] [PubMed] [Google Scholar]
- 12.Bliss TV, Collingridge GL. Nature. 1993;361:31. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]; Collingridge GL, Bliss TV. Trends Neurosci. 1995;18:54. [PubMed] [Google Scholar]
- 13.Whitmarsh AJ, Cavanagh J, Tournier C, Yasuda J, Davis RJ. Science. 1998;281:1671. doi: 10.1126/science.281.5383.1671. [DOI] [PubMed] [Google Scholar]; Schaeffer HJ, et al. ibid. :1668. [Google Scholar]; Faux MC, Scott JD. Cell. 1996;85:9. doi: 10.1016/s0092-8674(00)81075-2. [DOI] [PubMed] [Google Scholar]; Dong H, et al. Nature. 1997;386:279. doi: 10.1038/386279a0. [DOI] [PubMed] [Google Scholar]
- 14.Leadlay PF. Curr. Opin. Chem. Biol. 1997;1:162. doi: 10.1016/s1367-5931(97)80005-1. [DOI] [PubMed] [Google Scholar]
- 15.McAdams HH, Shapiro L. Science. 1995;269:650. doi: 10.1126/science.7624793. [DOI] [PubMed] [Google Scholar]
- 16. Protein structure query systems are available at www.pdb.bnl.gov/ and www.biosupplynet.com/cfdocs/btk/btk.cfm; the genome projects are listed at www.ncbi.nlm.nih.gov/Web/Genbank/index.html.
- 17.Bhalla US. In: The Book of GENESIS: Exploring Realistic Neural Models with GEneral NEural SImulation System. Bower JM, Beeman D, editors. Springer-Verlag; Berlin: 1998. [Google Scholar]; Destexhe A, Mainen ZF, Sejnowski TJ. J. Comput. Neurosci. 1994;1:195. doi: 10.1007/BF00961734. [DOI] [PubMed] [Google Scholar]; Anglister L, Stiles JR, Salpeter MM. Neuron. 1994;12:783. doi: 10.1016/0896-6273(94)90331-x. [DOI] [PubMed] [Google Scholar]; Stiles JR, Van Helden D, Bartol TM, Salpeter EE, Salpeter MM. Proc. Natl. Acad. Sci. U.S.A. 1996;93:5747. doi: 10.1073/pnas.93.12.5747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.G.W. is a Revson Fellow. Research in the Iyengar Laboratory is supported by NIH grant GM-54508