

Abstract
Rett Syndrome (RTT) is one of most prevalent female neurodevelopmental disorders. De novo mutations in X-linked MECP2 are mostly responsible for RTT. Since the identification of MeCP2 as the underlying cause of RTT, murine models have contributed to understanding the pathophysiology of RTT and function of MeCP2. Reprogramming is a procedure to produce induced pluripotent stem cells (iPSCs) by overexpression of four transcription factors. iPSCs obtain similar features as embryonic stem cells and are capable of self-renewing and differentiating into cells of all three layers. iPSCs have been utilized in modeling human diseases in vitro. Neurons differentiated from RTT-iPSCs showed the recapitulation of RTT phenotypes. Despite the early success, genetic and epigenetic instability upon reprogramming and ensuing maintenance of iPSCs raise concerns in using RTT-iPSCs as an accurate in vitro model. In this review, we update the current of iPSC-based RTT modeling, and concerns and challenges.
Rett syndrome (RTT;MIM 312750) is a severe neurodevelopmental disorder, affecting predominantly females. It was first described by Dr. Andreas Rett in 1966 [Rett, 1966]. It is the second most common cause of mental retardation in females affecting 1 in 10,000 [Chahrour and Zoghbi, 2007]. RTT patients show relatively normal development for the first 18 months, and then present with symptoms, including regression in speech and hand movements, postnatal microcephaly, hand dyspraxia, ataxia, abnormal breathing, growth retardations and autistic like symptoms [Chahrour and Zoghbi, 2007]. Over 90% of RTT cases result from de novo mutations in the X-linked gene encoding methyl CpG binding protein 2 (MECP2) [Amir et al., 1999; Chahrour and Zoghbi, 2007]. The MECP2 gene is located on the long arm of the X chromosome (Xq28). To date more than 100 mutations in the MECP2 gene have been described in RTT patients. In human, MECP2 seems essential for development. Hemizygotic MECP2 mutations in male lead to fatality and the prevalence of male RTT is extremely rare. Symptoms in male RTT patients are much more severe than heterozygotic females, exhibiting severe encephalopathy with death at birth or X-linked recessive mental retardation [Evans et al., 2006; Renieri et al., 2003]. In a subset of patients, RTT results from mutations in either another X-linked gene, cyclin-dependent kinase-like 5 (CDKL5) or in forkhead box G1 (FOXG1) which is located at 14q13 on chromosome 14 [Chahrour and Zoghbi, 2007].
Following the discovery of the genes responsible for RTT, investigation of how mutations in MECP2, CDKL5 or FOXG1 cause the observed phenotypes has been actively pursued, using murine models and in vitro cell culture based models [Ricceri et al., 2008; Weng et al., 2011]. Although MeCP2 seems to be essential for human development, homozygous female MeCP2 null or hemizygotic male mice are born normally, but they show the motor phenotypes observed in human patients and eventually die within 2 – 3 months. Heterozygous Mecp2 mutant female mice, an equivalent of female RTT patients, develop symptoms within 10 – 12 months. Murine model can give us insight into the function of MeCP2 in specific cell types. Phenotypes in mouse deleted of MeCP2 in neurons are similar to complete MeCP2 null mouse, suggesting that abnormal functions in MeCP2 in neurons may be determinant of RTT [Chen et al., 2001]. Recently, the manifestation of RTT symptoms in mice with MeCP2 knocked out in either excitatory or inhibitory neurons further corroborates that well coordinated expression of MeCP2 in specific neurons is critical in the normal function of neurons [Chao et al., 2010; Chao et al., 2007]. In addition, MeCP2 seems to have an essential function in non-neuronal cell types in the brain, such as astrocytes and microglia [Derecki et al., 2012; Lioy et al., 2011]. In vitro neuronal culture models have also facilitated understanding the molecular mechanism of MeCP2 function in terms of their gene expression patterns and chromatin structure [Adkins and Georgel, 2011].
Reprogramming is a procedure to convert differentiated somatic cells to a pluripotent state. Four transcription factors (Oct4, Sox2, Klf4 and Myc) are generally used to derive so-called “induced pluripotent stem cells” (iPSCs) [Park et al., 2008b; Takahashi and Yamanaka, 2006; Yu et al., 2009]. iPSCs exhibit many of the characteristics of embryonic stem cells (ESCs), and are capable of both self-renewal and differentiation into cells representative of the three germ layers. Because iPSCs maintain the same genetic composition of donors, iPSC or iPSC-derivatives are ideal for investigating the contribution of phenotypes in a given genotype. Here, we will review the recent advancements in reprogramming and its application in disease modeling, especially neuronal diseases, focusing on RTT.
Factor-Based Reprogramming
It has been more than 50 years since the demonstration of nuclear transfer to generate viable adult offspring in Xenopus [Gurdon, 1962]. This was a turning point as it demonstrated that the vertebrate genome was not fixed, but was plastic and amenable to cell fate changes. Mammalian cells exhibited similar epigenetic flexibility and a number of healthy animals have been cloned using nuclear transfer technologies. In the 1980s Harold Weintraub's group demonstrated that cellular fate could be changed by the expression of a transcription factor/ master regulator MyoD [Davis et al., 1987]. Then in 2006, the Yamanaka group demonstrated that the expression of four transcription factors (Oct4, Sox2, Klf4 and Myc) was sufficient to reprogram a somatic cell such as a fibroblast to a pluripotent state – induced pluripotency [Takahashi and Yamanaka, 2006]. These remarkable findings have been recognized and resulted in the presentation of the Nobel prize for the contribution of Gurdon and Yamanaka [Surani, 2012].
Reprogramming of human somatic cells using similar factors succeeded in generating human iPSCs [Park et al., 2008b; Takahashi et al., 2007; Yu et al., 2009]. iPSCs are pluripotent and can differentiate in any cell types in a body. The ultimate biomedical use of iPSCs will be in cell replacement therapy using iPSC derived cell types as autologous donor cells. Because utility of pluripotent stem cells in cell therapy still has biosafety issues, the immediate impact of iPSCs will be in the arena of in vitro disease modeling [Dimos et al., 2008; Park et al., 2008a]. To date there have been over 200 papers reporting the development of disease specific iPSCs and the recapitulation of disease phenotypes to a certain extent (for an in depth overview see [Siller et al., 2013]). More importantly, in the field of neuronal disease, iPSC technology has been used to successfully model neurodevelopmental and neurodegenerative disorders, such as Rett syndrome, Fragile X syndrome, Schizophrenia, Alzheimer's diseases (AD), Parkinson's diseases (PD), spinal muscular dystrophy (SMA), amyotrophic lateral sclerosis (ALS) and Familial dysautomia (FD) [Siller et al., 2013].
Investigation of RTT using iPSC
After successful reports of deriving human iPSCs, the first RTT-iPSCs were generated by the Ellis group [Hotta et al., 2009]. Since then, multiple laboratories have derived iPSCs from RTT patients with MECP2 mutations and studied neuronal phenotypes in detail (Table 1). Neurons from RTT-iPSCs have recapitulated phenotypes observed in both murine models and patients. In vitro phenotypes include, reduced soma/ nuclear size, lower expression of neuronal markers, and reduced dendrite spine density [Ananiev et al., 2011; Cheung et al., 2011; Kim et al., 2011b; Marchetto et al., 2010]. RTT-iPSC derived neurons also displayed a reduction in the transient rise of intracellular calcium levels typical of active synapse as well as a decrease in the frequency/amplitude of spontaneous excitatory and inhibitory postsynaptic currents [Marchetto et al., 2010]. These in vitro recapitulations of RTT phenotypes using patients specific RTT-iPSCs provide a strong proof of principle of the utility of iPSC in studying RTT. The Ellis group further elaborated the neurophysiological features of neurons differentiated from Mecp2 mutant mouse iPSCs [Farra et al., 2012]. They found that MeCP2-deficient neurons have fewer action potentials, decreased action potential amplitude, diminished peak inward currents and higher input resistance [Farra et al., 2012]. Comparative analysis of neurons derived from murine RTT-iPSC and patient's RTT-iPSC alike would potentially undercover the neurophysiological difference between human and mouse that cause similar but distinct phenotypes in RTT patients and Mecp2 null murine model.
Table 1.
Reports on derivation and characterization of iPSCs from RTT patients.
| Lab | Mutations | In vitro difference in phenotype | Therapeutics | Reference |
|---|---|---|---|---|
| The Ellis lab | R306C missense mutation in MeCP2 | N/A | N/A | [Hotta et al., 2009] |
| The Muotri lab | T148M, R306C, Q244X, or 1155del32 in MeCP2 | Fewer synapses Reduced spine density Smaller soma size Altered calcium transient Electrophysical defects | IGF1 Gentamicin | [Marchetto et al., 2010] |
| The Ellis lab | Deletion in exons 3 and 4 of MeCP2 | Reduction in soma size | N/A | [Cheung et al., 2011] |
| The Park lab | T158M, E235fs, Q244X, R306C, or X487W in MeCP2 | Reduction in TuJ positive cells | N/A | [Kim et al., 2011b] |
| The Chang lab | T158, V247X, R306C, or R294X in MeCP2 | Reduction in neuron size | N/A | [Ananiev et al., 2011] |
| The Broccoli lab | R59X, or L220P in MeCP2 | Aberrant spine structure | N/A | [Ricciardi et al., 2012] |
Mutations in CDKL5 have been found in patients showing phenotypes overlapping with those of MeCP2 mutant RTT patients [Weaving et al., 2004]. CDKL5 was found to regulate spinal density and dendritic structures of neurons in MeCP2 pathway [Mari et al., 2005]. Neurons differentiated from iPSCs of CDKL5 patients exhibited similar morphological phenotypes as observed in murine models, including the reduced number of synaptic contacts [Amenduni et al., 2011; Ricciardi et al., 2012]. iPSCs from patients with FOXG1 have yet to be reported. The comparison of phenotypes among iPSCs with MeCP2, CDKL5 and FOXG1 mutations could potentially uncover the molecular mechanism of RTT, and would facilitate the discovery of therapeutics for RTT.
RTT caused by MeCP2 mutations is a monogenic disease. However, each patient displays variability in symptoms and disease progression [Chahrour and Zoghbi, 2007; Schanen et al., 2004]. Difference in symptoms appears to result from the location of mutations in MECP2 gene. MeCP2 has three major functional domains; methyl CpG binding domain (MBD), transcription repression domain (TRD), and unique C-terminal domain (CTD) [Chahrour and Zoghbi, 2007]. In short, the overall severity of the phenotype is a reflection of the way mutations affect the function of MeCP2, the more deleterious to the function the more severe the observed phenotype. Thus, the non sense mutations causing the loss of functional C-terminal region of MeCP2 causes severe microcephaly (e.g. R270X, Q244X), while the point mutations in MBD (R133C) or TRD (R306C) domains results in mild symptoms. We have isolated RTT-iPSCs from patients with different MeCP2 mutations ([Kim et al., 2011b], Table 1). The analysis of in vitro phenotypes of these iPSCs would probe the function of each domain in RTT and potentially enable the development of mutation specific therapy.
Another major factor responsible for the manifestation of different phenotypes in female RTT patients is the skewing in X chromosome inactivation status. Female cells within the inner cell mass of blastocyst of early embryonic development have two active X chromosomes, one of which undergo random X chromosome inactivation (XCI) upon gastrulation [Hysolli et al., 2012]. Thus, adult female cells are mosaic in terms of their X chromosome status. Since MeCP2 is on X chromosome, the skewing of activation status of X chromosome with mutant MeCP2 determines the severity of symptoms [Archer et al., 2007; Young and Zoghbi, 2004]. When more cells are present that have an active X chromosome with mutant MeCP2, patients exhibit a severe phenotype. This unique phenomenon of XCI affects allele specific expression of genes on X chromosome in iPSCs derived from female cells. Like epiblast cells that have undergone XCI, human pluripotent stem cells, including ESCs and iPSCs, maintain only one active X chromosome unless kept in physiological oxygen concentration and with HDAC inhibitors in culture medium [Diaz Perez et al., 2012; Lengner et al., 2010]. As a consequence, iPSC clones from female RTT patients that are genotypically equivalent, have differential expression of genes on X chromosome, including MeCP2. We, and others have isolated iPSC clones from RTT patients that only express either wild type or mutant MeCP2 (Table 1). Comparison of wild type and mutant iPSCs from same patients overcomes the issue of deriving control cell lines in studying diseases using iPSCs because these are isogenically controlled. In vitro studies to date using neurons from RTT-iPSCs have made use of these isogenically matched wild type and mutant clones (Table 1). Interestingly, the Muotri group and our group were able to isolate RTT-iPSC clones that biallelically expressed both wild type and mutant MeCP2 genes. However, it is not yet fully defined whether that clones showing biallelic expression of MeCP2 have two fully active X chromosomes, or have undergone X chromosome erosion and thus obtained a partial activation of the prior inactive X chromosome [Mekhoubad et al., 2012].
Considerations in using iPSC technology in modeling RTT
Despite numerous successes in studying disease phenotypes in vitro, multifaceted considerations should be made in using iPSCs in disease modeling. Major issues arise from reprogramming per se and those of in vitro differentiation. Since the initial report, reprogramming technology has both evolved rapidly and improved the efficiency of reprogramming and quality of iPSCs [Sohn et al., 2012]. Issues associated with retroviral or lentiviral vectors can potentially affect the expression of genes adjacent to integration sites. Although most retroviral or lentiviral genes are silenced in iPSCs, there are incidences of re-expression of the viral genes in iPSC or iPSC-derivatives that could be detrimental [Okita et al., 2007]. Thus, non-integrating methods, such as episomal vectors, modified mRNA and proteins, have been actively pursued and demonstrated production of high quality iPSCs albeit at a lower efficiency in reprogramming. Currently, reprogramming by Sendai virus is considered efficient and results in high quality iPSCs [Fusaki et al., 2009]. In applying iPSC for cellular disease models, we do not deem derivation of iPSCs as a hurdle. However, extensive analysis of genomic and epigenomic status of iPSCs has revealed that iPSCs may acquire reprogramming-specific epigenetic marks and genomic footprints [Gore et al., 2011; Hussein et al., 2011; Lister et al., 2011]. Because it is not predictable how reprogramming-mediated epigenomic and genomic changes affect the in vitro phenotypes of the given iPSCs, as a consequence further investigation is required to establish ways to maintain the integrity of genome in iPSCs. Below, we discuss further the findings on genetic and epigenetic change in iPSCs.
Early passage iPSCs display a de novo Copy Number Variations (CNV) arising during reprogramming [Hussein et al., 2011]. Although some of the CNVs are present in parental fibroblasts, changes in number and type of CNVs in iPSCs appear to be dynamic during passaging, and became stabilized at later passage iPSCs. iPSC cells that have obtained detrimental CNVs seem to vanish during passaging, suggesting the selective pressure on proliferation of iPSCs. How and when during reprogramming the de novo CNV arise are critical questions to be answered in order to obtain high quality iPSCs for disease modeling and cell therapy.
Pluripotent ESCs have unique histone modification and DNA methylation status. Genes defining differentiated or developmental states show active (H3K4me3) as well as inactive (H3K27me3) histone marks. Meanwhile, pluripotent genes are marked by only active histone marks [Bernstein et al., 2006]. DNA methylation on CpG islands determines the expression of the given genes. Reprogramming procedures using the four factors reset the epigenomic status of the somatic cell to that in pluripotent cells. Despite controversy, the comparison of histone marks in iPSCs with ESCs seems to support that reprogramming faithfully convert somatic histone marks to pluripotent histone marks [Chin et al., 2010; Guenther et al., 2010]. However, genome-wide DNA methylation analysis showed that iPSCs are close to ESCs in DNA methylation status, but acquire reprogramming specific differentially methylated regions as well as retain some epigenetic memory of the cellular origin [Doi et al., 2009; Lister et al., 2011]. The DNA methylation marks appear to be permanent and are maintained during differentiation, thus affecting the differentiation potential of iPSCs [Kim et al., 2011a; Lister et al., 2011]. This imposes a potential challenge in using iPSCs for in vitro disease models. Extensive epigenetic or gene expression analysis perhaps will be a prerequisite to select the most relevant iPSC clones for further use [Bock et al., 2011].
As briefly mentioned above, modeling of X-linked diseases using female iPSCs poses a particular challenge because of the unstable X chromosome status in current culture condition used in maintaining human pluripotent stem cells. Unless derived under physiological hypoxic conditions, one of X chromosomes in female human ESCs undergoes XCI [Lengner et al., 2010]. Female iPSCs display a similar X chromosome status and retain an inactive X chromosome after cellular reprogramming. This also provides the potential in developing isogenic controls that express either wild type or mutant gene on X chromosome as exemplified in MeCP2. However, the inactive X chromosome is not epigenetically stable and undergo co-called erosion of inactivation that produce a quasi-activation state [Mekhoubad et al., 2012]. Change in expression of genes on X chromosome with multiple passages would raise the concern in making in vitro disease modeling using female iPSCs. There have been efforts in maintaining two active X chromosomes in human ESCs or iPSC using chemicals or culture conditions as previously described [Diaz Perez et al., 2012]. Alternative approaches have been employed in maintaining two active X chromosomes, such as by overexpressing NANOG or OCT 4 and KL4 in combination with a small molecule cocktail have succeeded [Hanna et al., 2010]. However, there has been no investigation to stabilize the inactive state of X chromosome in human female pluripotent stem cells to date. The utilization of iPSCs for disease modeling will only increase in the future, and consequently there will be a need for more investment in studying change in X chromosome status in iPSCs.
The grand assumption in modeling diseases in vitro is that what one observes as an in vitro difference is a consequence of the phenotype of the iPSC-derivatives with the given mutations. In addition, it has to be taken into account that human pluripotent stem cells, either ESCs or iPSCs, display a large range of clonal variation in their proliferation and differentiation potential. An early study on 17 human ESCs found marked difference of differentiation potential of ESCs [Osafune et al., 2008]. iPSCs derived from same parental somatic cells also show difference in differentiation potential [Hu et al., 2010]. Lineage scoring based on gene expression and DNA methylation profiling may identify the best candidate iPSC clones for differentiation analysis [Bock et al., 2011]. However, the selective picking of clones only suitable for the experimental purposes may lessen the significance of the findings in in vitro disease modeling. Improving differentiation potential of iPSC or overriding the observed clonal variation with potent small molecules will potentially result in more reliable in vitro outcome [Chambers et al., 2009; Chetty et al., 2013; Kim et al., 2010].
Future directions
Reprogramming technology now provides an unprecedented approach to study RTT and indeed other diseases. The basic stream to follow for modeling any disease, such as RTT, would be the derivation of validated patient specific iPSCs and subsequent differentiation to the cell types implicated in disease, in the case of RTT, neurons. Next the researcher would probe for an in vitro phenotype, and this approach has already proved successful (see Table 1). Brain disorders, however, are complicated diseases that encompass a myriad of interactions of neurons with other neurons and non-neuronal cells. In the case of RTT, glial cells, such as astrocytes and microglia, were shown to be critical in RTT [Derecki et al., 2012; Lioy et al., 2011]. Investigation of interaction of neurons and glial cells in human brain is challenging, but RTT-iPSC by reprogramming technology enables differentiation of neurons and glial cells, and investigating the functional interaction. In addition to cell-to-cell interaction, we need to consider a higher-level neuronal circuitry in brain as a whole. Advanced brain tissue engineering that positions the in vitro differentiated neurons and glial cells to their physiological niche as a functional unit may allow studying RTT in 3 dimensions. In solid organs, decellurization of an organ to produce matrix scaffold and ensuing recellurization have given promising results in cellular replacement in lung, heart and liver [Song and Ott, 2011]. However, brain is composed of soft tissue and would be a challenging organ for whole cell replacement. Investigation of murine models transplanted with neurons and/ or non-neuronal cells derived from human RTT-iPSCs will be another important approach to study RTT physio-pathogenesis [Espuny-Camacho et al., 2013; Maroof et al., 2013].
When the exact genetic mutations are known for a disease, such as in RTT with a variety of MECP2 mutations, introducing the given mutations in a standardized ESC or iPSC would facilitate investigating the effect of mutations in one isogenic background [Soldner et al., 2011]. A number of approaches have been developed to precisely edit the genomes of both ESCs and iPSCs, these include Zinc finger nucleases (ZFNs) and the transcription activator-like effector nucleases (TALENs) [Joung and Sander, 2013]. In brief, the target sequences are recognized by either ZF or TALE motifs and then cut by a linked nuclease, this in turn promotes homologous recombination. Although these approaches are effective, there have been concerns in producing off-target mutagenesis [Mussolino et al., 2011; Pattanayak et al., 2011]. Recently, clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated (Cas) system that was identified as a bacterial adaptive immune defense has been applied in genome editing [Jinek et al., 2012; Mali et al., 2013]. CRISPR system use guide RNA to recognize target sequence, and CRISPR-associated nuclease causes specific double strand breaks in a DNA target to increase homologous recombination efficiency. Although the frequency and causes of off-target activity are to be determined, CRISPR allows the multiplex genome editing [Mali et al., 2013]. Because a series of mutations in MeCP2 are already known from clinical genetic studies, a standardized ESC or iPSC lines engineered with known mutations within MeCP2 would provide a great resource for RTT disease modeling.
Success of in vitro disease modeling absolutely depends on the faithful differentiation of pluripotent cells to the cell types that are afflicted. Function of MeCP2 in neurons seems to be foremost essential in RTT. To this end sophisticated deletions of Mecp2 in different type of neurons using conditional null allele models have been developed. These in turn have demonstrated that Mecp2 is essential in different types of neurons within the brain, including forebrain excitatory neurons, inhibitory neurons, hypothalamic neurons, basolateral amygdala neurons, and aminergic neurons [Adachi et al., 2009; Chao et al., 2010; Fyffe et al., 2008; Samaco et al., 2009]. In addition, it has been demonstrated that non-neuronal cell types such as astrocytes and microglia are equally important players in RTT [Derecki et al., 2012; Lioy et al., 2011]. Pluripotency of iPSCs now allow the research community to assess the function of MeCP2 in each of these neuronal and non-neuronal cell types. However, we still need to be cautious, because in vitro differentiation methodologies are not yet fully defined to produce these functionally distinct neuronal subtypes, astrocytes and microglia. Recent success in differentiating cortical excitatory and inhibitor neurons from human ESC and iPSCs is encouraging in studying RTT, because these are major neuronal cell types affected in RTT [Espuny-Camacho et al., 2013; Maroof et al., 2013]. The future will require the development of robust methodologies to produce many other neuronal subtypes. This will be an important weapon in the arsenal in order to study disease mechanism of many diseases including schizophrenia, autism spectrum disorders and RTT. Therefore, as methods are honed and refined to produce more specific neuronal subtypes and other functionally relevant cell types from iPSCs, we will acquire a more comprehensive understanding RTT.
Here, we have given an overview of RTT and reprogramming as a novel research tool to facilitate a better understanding of RTT. In order to channel knowledge and resources in a focused manner, the RTT research community has compiled a database which will enable the study of RTT genotype-phenotype correlations [Grillo et al., 2012]. Armed with the information from the database and combined with in vitro data from RTT-iPSC derivatives, this will provide a critical resource. This gained knowledge will aid the development of diagnostics as well as personalized medicine to improve current drug regimes and to identify potential new therapeutics. As in RTT, reprogramming technologies in combination with robust methodologies to faithfully direct iPSCs to the required cell types, will provide an unique opportunity for disease modeling and pathogenesis research for many other diseases.
Figure 1. Application of reprogramming in RTT studies.
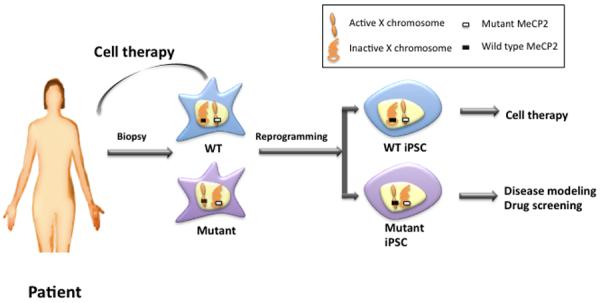
Somatic cells are obtained from RTT patients. Female RTT patient's cells are mosaic in expression of genes in X chromosome due to random XCI during early embryogenesis. When reprogrammed, iPSCs in general maintain one active X chromosome. Thus, RTT-iPSC clones expressing either wild type or mutant MeCP2 are produced from RTT patients. Mutant RTT-iPSCs can be differentiated into relevant cell types and used for in vitro disease modeling in comparison of wild type iPSC. Wild type iPSC can be used in cell therapy as autologous cells.
Figure 2. Challenges in studying RTT using iPSCs.
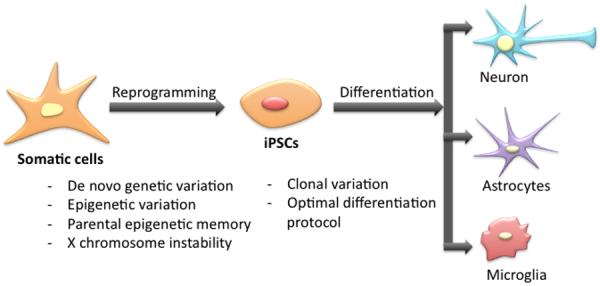
Reprogramming can cause de novo genetic or epigenetic variations in iPSCs. In female iPSCs, maintaining X chromosome in stable is challenging. iPSCs display clonal variation. Developing optimal differentiation potential to produce RTT relevant cell types are critical in successful modeling.
Acknowledgements
Gareth J Sullivan was supported by the Research Council of Norway and Helse Sør Øst. In-Hyun Park was partly supported by NIH (GM0099130-01), CSCRF (12-SCB-YALE-11), Charles Hood Foundation and by CTSA Grant UL1 RR025750 from the National Center for Advancing Translational Science (NCATS), a component of the National Institutes of Health (NIH), and NIH roadmap for Medical Research. Its contents are solely the responsibility of the authors and do not necessarily represent the official view of NIH.
References
- Adachi M, Autry AE, Covington HE, 3rd, Monteggia LM. MeCP2-mediated transcription repression in the basolateral amygdala may underlie heightened anxiety in a mouse model of Rett syndrome. J Neurosci. 2009;29:4218–27. doi: 10.1523/JNEUROSCI.4225-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adkins NL, Georgel PT. MeCP2: structure and function. Biochem Cell Biol. 2011;89:1–11. doi: 10.1139/O10-112. [DOI] [PubMed] [Google Scholar]
- Amenduni M, De Filippis R, Cheung AY, Disciglio V, Epistolato MC, Ariani F, Mari F, Mencarelli MA, Hayek Y, Renieri A, Ellis J, Meloni I. iPS cells to model CDKL5-related disorders. Eur J Hum Genet. 2011;19:1246–55. doi: 10.1038/ejhg.2011.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–8. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- Ananiev G, Williams EC, Li H, Chang Q. Isogenic pairs of wild type and mutant induced pluripotent stem cell (iPSC) lines from Rett syndrome patients as in vitro disease model. PLoS One. 2011;6:e25255. doi: 10.1371/journal.pone.0025255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer H, Evans J, Leonard H, Colvin L, Ravine D, Christodoulou J, Williamson S, Charman T, Bailey ME, Sampson J, de Klerk N, Clarke A. Correlation between clinical severity in patients with Rett syndrome with a p.R168X or p.T158M MECP2 mutation, and the direction and degree of skewing of X-chromosome inactivation. J Med Genet. 2007;44:148–52. doi: 10.1136/jmg.2006.045260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, Jaenisch R, Wagschal A, Feil R, Schreiber SL, Lander ES. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–26. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- Bock C, Kiskinis E, Verstappen G, Gu H, Boulting G, Smith ZD, Ziller M, Croft GF, Amoroso MW, Oakley DH, Gnirke A, Eggan K, Meissner A. Reference Maps of human ES and iPS cell variation enable high-throughput characterization of pluripotent cell lines. Cell. 2011;144:439–52. doi: 10.1016/j.cell.2010.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahrour M, Zoghbi HY. The story of Rett syndrome: from clinic to neurobiology. Neuron. 2007;56:422–37. doi: 10.1016/j.neuron.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, Studer L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol. 2009;27:275–80. doi: 10.1038/nbt.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao HT, Chen H, Samaco RC, Xue M, Chahrour M, Yoo J, Neul JL, Gong S, Lu HC, Heintz N, Ekker M, Rubenstein JL, Noebels JL, Rosenmund C, Zoghbi HY. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature. 2010;468:263–9. doi: 10.1038/nature09582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao HT, Zoghbi HY, Rosenmund C. MeCP2 controls excitatory synaptic strength by regulating glutamatergic synapse number. Neuron. 2007;56:58–65. doi: 10.1016/j.neuron.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen RZ, Akbarian S, Tudor M, Jaenisch R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet. 2001;27:327–31. doi: 10.1038/85906. [DOI] [PubMed] [Google Scholar]
- Chetty S, Pagliuca FW, Honore C, Kweudjeu A, Rezania A, Melton DA. A simple tool to improve pluripotent stem cell differentiation. Nat Methods. 2013 doi: 10.1038/nmeth.2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung AY, Horvath LM, Grafodatskaya D, Pasceri P, Weksberg R, Hotta A, Carrel L, Ellis J. Isolation of MECP2-null Rett Syndrome patient hiPS cells and isogenic controls through X-chromosome inactivation. Hum Mol Genet. 2011;20:2103–15. doi: 10.1093/hmg/ddr093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin MH, Pellegrini M, Plath K, Lowry WE. Molecular analyses of human induced pluripotent stem cells and embryonic stem cells. Cell Stem Cell. 2010;7:263–9. doi: 10.1016/j.stem.2010.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RL, Weintraub H, Lassar AB. Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell. 1987;51:987–1000. doi: 10.1016/0092-8674(87)90585-x. [DOI] [PubMed] [Google Scholar]
- Derecki NC, Cronk JC, Lu Z, Xu E, Abbott SB, Guyenet PG, Kipnis J. Wild-type microglia arrest pathology in a mouse model of Rett syndrome. Nature. 2012;484:105–9. doi: 10.1038/nature10907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz Perez SV, Kim R, Li Z, Marquez VE, Patel S, Plath K, Clark AT. Derivation of new human embryonic stem cell lines reveals rapid epigenetic progression in vitro that can be prevented by chemical modification of chromatin. Hum Mol Genet. 2012;21:751–64. doi: 10.1093/hmg/ddr506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimos JT, Rodolfa KT, Niakan KK, Weisenthal LM, Mitsumoto H, Chung W, Croft GF, Saphier G, Leibel R, Goland R, Wichterle H, Henderson CE, Eggan K. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science. 2008;321:1218–21. doi: 10.1126/science.1158799. [DOI] [PubMed] [Google Scholar]
- Doi A, Park IH, Wen B, Murakami P, Aryee MJ, Irizarry R, Herb B, Ladd-Acosta C, Rho J, Loewer S, Miller J, Schlaeger T, Daley GQ, Feinberg AP. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat Genet. 2009;1:1. doi: 10.1038/ng.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espuny-Camacho I, Michelsen KA, Gall D, Linaro D, Hasche A, Bonnefont J, Bali C, Orduz D, Bilheu A, Herpoel A, Lambert N, Gaspard N, Peron S, Schiffmann SN, Giugliano M, Gaillard A, Vanderhaeghen P. Pyramidal neurons derived from human pluripotent stem cells integrate efficiently into mouse brain circuits in vivo. Neuron. 2013;77:440–56. doi: 10.1016/j.neuron.2012.12.011. [DOI] [PubMed] [Google Scholar]
- Evans JC, Archer HL, Whatley SD, Clarke A. Germline mosaicism for a MECP2 mutation in a man with two Rett daughters. Clin Genet. 2006;70:336–8. doi: 10.1111/j.1399-0004.2006.00691.x. [DOI] [PubMed] [Google Scholar]
- Farra N, Zhang WB, Pasceri P, Eubanks JH, Salter MW, Ellis J. Rett syndrome induced pluripotent stem cell-derived neurons reveal novel neurophysiological alterations. Mol Psychiatry. 2012;17:1261–71. doi: 10.1038/mp.2011.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusaki N, Ban H, Nishiyama A, Saeki K, Hasegawa M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc Jpn Acad Ser B Phys Biol Sci. 2009;85:348–62. doi: 10.2183/pjab.85.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fyffe SL, Neul JL, Samaco RC, Chao HT, Ben-Shachar S, Moretti P, McGill BE, Goulding EH, Sullivan E, Tecott LH, Zoghbi HY. Deletion of Mecp2 in Sim1-expressing neurons reveals a critical role for MeCP2 in feeding behavior, aggression, and the response to stress. Neuron. 2008;59:947–58. doi: 10.1016/j.neuron.2008.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gore A, Li Z, Fung HL, Young JE, Agarwal S, Antosiewicz-Bourget J, Canto I, Giorgetti A, Israel MA, Kiskinis E, Lee JH, Loh YH, Manos PD, Montserrat N, Panopoulos AD, Ruiz S, Wilbert ML, Yu J, Kirkness EF, Izpisua Belmonte JC, Rossi DJ, Thomson JA, Eggan K, Daley GQ, Goldstein LS, Zhang K. Somatic coding mutations in human induced pluripotent stem cells. Nature. 2011;471:63–7. doi: 10.1038/nature09805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillo E, Villard L, Clarke A, Ben Zeev B, Pineda M, Bahi-Buisson N, Hryniewiecka-Jaworska A, Bienvenu T, Armstrong J, Roche-Martinez A, Mari F, Veneselli E, Russo S, Vignoli A, Pini G, Djuric M, Bisgaard AM, Mejaski Bosnjak V, Polgar N, Cogliati F, Ravn K, Pintaudi M, Melegh B, Craiu D, Djukic A, Renieri A. Rett networked database: an integrated clinical and genetic network of Rett syndrome databases. Hum Mutat. 2012;33:1031–6. doi: 10.1002/humu.22072. [DOI] [PubMed] [Google Scholar]
- Guenther MG, Frampton GM, Soldner F, Hockemeyer D, Mitalipova M, Jaenisch R, Young RA. Chromatin structure and gene expression programs of human embryonic and induced pluripotent stem cells. Cell Stem Cell. 2010;7:249–57. doi: 10.1016/j.stem.2010.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurdon JB. Adult frogs derived from the nuclei of single somatic cells. Dev Biol. 1962;4:256–73. doi: 10.1016/0012-1606(62)90043-x. [DOI] [PubMed] [Google Scholar]
- Hanna J, Cheng AW, Saha K, Kim J, Lengner CJ, Soldner F, Cassady JP, Muffat J, Carey BW, Jaenisch R. Human embryonic stem cells with biological and epigenetic characteristics similar to those of mouse ESCs. Proc Natl Acad Sci U S A. 2010;107:9222–7. doi: 10.1073/pnas.1004584107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotta A, Cheung AY, Farra N, Vijayaragavan K, Seguin CA, Draper JS, Pasceri P, Maksakova IA, Mager DL, Rossant J, Bhatia M, Ellis J. Isolation of human iPS cells using EOS lentiviral vectors to select for pluripotency. Nat Methods. 2009;6:370–6. doi: 10.1038/nmeth.1325. [DOI] [PubMed] [Google Scholar]
- Hu BY, Weick JP, Yu J, Ma LX, Zhang XQ, Thomson JA, Zhang SC. Neural differentiation of human induced pluripotent stem cells follows developmental principles but with variable potency. Proc Natl Acad Sci U S A. 2010;107:4335–40. doi: 10.1073/pnas.0910012107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussein SM, Batada NN, Vuoristo S, Ching RW, Autio R, Narva E, Ng S, Sourour M, Hamalainen R, Olsson C, Lundin K, Mikkola M, Trokovic R, Peitz M, Brustle O, Bazett-Jones DP, Alitalo K, Lahesmaa R, Nagy A, Otonkoski T. Copy number variation and selection during reprogramming to pluripotency. Nature. 2011;471:58–62. doi: 10.1038/nature09871. [DOI] [PubMed] [Google Scholar]
- Hysolli E, Jung YW, Tanaka Y, Kim KY, Park IH. The lesser known story of X chromosome reactivation: a closer look into the reprogramming of the inactive X chromosome. Cell Cycle. 2012;11:229–35. doi: 10.4161/cc.11.2.18998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–21. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joung JK, Sander JD. TALENs: a widely applicable technology for targeted genome editing. Nat Rev Mol Cell Biol. 2013;14:49–55. doi: 10.1038/nrm3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DS, Lee JS, Leem JW, Huh YJ, Kim JY, Kim HS, Park IH, Daley GQ, Hwang DY, Kim DW. Robust enhancement of neural differentiation from human ES and iPS cells regardless of their innate difference in differentiation propensity. Stem Cell Rev. 2010;6:270–81. doi: 10.1007/s12015-010-9138-1. [DOI] [PubMed] [Google Scholar]
- Kim K, Zhao R, Doi A, Ng K, Unternaehrer J, Cahan P, Huo H, Loh YH, Aryee MJ, Lensch MW, Li H, Collins JJ, Feinberg AP, Daley GQ. Donor cell type can influence the epigenome and differentiation potential of human induced pluripotent stem cells. Nat Biotechnol. 2011a;29:1117–9. doi: 10.1038/nbt.2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KY, Hysolli E, Park IH. Neuronal maturation defect in induced pluripotent stem cells from patients with Rett syndrome. Proc Natl Acad Sci U S A. 2011b;108:14169–74. doi: 10.1073/pnas.1018979108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengner CJ, Gimelbrant AA, Erwin JA, Cheng AW, Guenther MG, Welstead GG, Alagappan R, Frampton GM, Xu P, Muffat J, Santagata S, Powers D, Barrett CB, Young RA, Lee JT, Jaenisch R, Mitalipova M. Derivation of pre-X inactivation human embryonic stem cells under physiological oxygen concentrations. Cell. 2010;141:872–83. doi: 10.1016/j.cell.2010.04.010. [DOI] [PubMed] [Google Scholar]
- Lioy DT, Garg SK, Monaghan CE, Raber J, Foust KD, Kaspar BK, Hirrlinger PG, Kirchhoff F, Bissonnette JM, Ballas N, Mandel G. A role for glia in the progression of Rett's syndrome. Nature. 2011;29 doi: 10.1038/nature10214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, Pelizzola M, Kida YS, Hawkins RD, Nery JR, Hon G, Antosiewicz-Bourget J, O'Malley R, Castanon R, Klugman S, Downes M, Yu R, Stewart R, Ren B, Thomson JA, Evans RM, Ecker JR. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature. 2011;471:68–73. doi: 10.1038/nature09798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–6. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchetto MC, Carromeu C, Acab A, Yu D, Yeo GW, Mu Y, Chen G, Gage FH, Muotri AR. A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell. 2010;143:527–39. doi: 10.1016/j.cell.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mari F, Azimonti S, Bertani I, Bolognese F, Colombo E, Caselli R, Scala E, Longo I, Grosso S, Pescucci C, Ariani F, Hayek G, Balestri P, Bergo A, Badaracco G, Zappella M, Broccoli V, Renieri A, Kilstrup-Nielsen C, Landsberger N. CDKL5 belongs to the same molecular pathway of MeCP2 and it is responsible for the early-onset seizure variant of Rett syndrome. Hum Mol Genet. 2005;14:1935–46. doi: 10.1093/hmg/ddi198. [DOI] [PubMed] [Google Scholar]
- Maroof AM, Keros S, Tyson JA, Ying SW, Ganat YM, Merkle FT, Liu B, Goulburn A, Stanley EG, Elefanty AG, Widmer HR, Eggan K, Goldstein PA, Anderson SA, Studer L. Directed differentiation and functional maturation of cortical interneurons from human embryonic stem cells. Cell Stem Cell. 2013;12:559–72. doi: 10.1016/j.stem.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mekhoubad S, Bock C, de Boer AS, Kiskinis E, Meissner A, Eggan K. Erosion of dosage compensation impacts human iPSC disease modeling. Cell Stem Cell. 2012;10:595–609. doi: 10.1016/j.stem.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mussolino C, Morbitzer R, Lutge F, Dannemann N, Lahaye T, Cathomen T. A novel TALE nuclease scaffold enables high genome editing activity in combination with low toxicity. Nucleic Acids Res. 2011;39:9283–93. doi: 10.1093/nar/gkr597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature. 2007;448:313–7. doi: 10.1038/nature05934. [DOI] [PubMed] [Google Scholar]
- Osafune K, Caron L, Borowiak M, Martinez RJ, Fitz-Gerald CS, Sato Y, Cowan CA, Chien KR, Melton DA. Marked differences in differentiation propensity among human embryonic stem cell lines. Nat Biotechnol. 2008;26:313–5. doi: 10.1038/nbt1383. [DOI] [PubMed] [Google Scholar]
- Park IH, Arora N, Huo H, Maherali N, Ahfeldt T, Shimamura A, Lensch MW, Cowan C, Hochedlinger K, Daley GQ. Disease-specific induced pluripotent stem cells. Cell. 2008a;134:877–86. doi: 10.1016/j.cell.2008.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park IH, Zhao R, West JA, Yabuuchi A, Huo H, Ince TA, Lerou PH, Lensch MW, Daley GQ. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2008b;451:141–6. doi: 10.1038/nature06534. [DOI] [PubMed] [Google Scholar]
- Pattanayak V, Ramirez CL, Joung JK, Liu DR. Revealing off-target cleavage specificities of zinc-finger nucleases by in vitro selection. Nat Methods. 2011;8:765–70. doi: 10.1038/nmeth.1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renieri A, Meloni I, Longo I, Ariani F, Mari F, Pescucci C, Cambi F. Rett syndrome: the complex nature of a monogenic disease. J Mol Med (Berl) 2003;81:346–54. doi: 10.1007/s00109-003-0444-9. [DOI] [PubMed] [Google Scholar]
- Rett A. [On a unusual brain atrophy syndrome in hyperammonemia in childhood] Wien Med Wochenschr. 1966;116:723–6. [PubMed] [Google Scholar]
- Ricceri L, De Filippis B, Laviola G. Mouse models of Rett syndrome: from behavioural phenotyping to preclinical evaluation of new therapeutic approaches. Behav Pharmacol. 2008;19:501–17. doi: 10.1097/FBP.0b013e32830c3645. [DOI] [PubMed] [Google Scholar]
- Ricciardi S, Ungaro F, Hambrock M, Rademacher N, Stefanelli G, Brambilla D, Sessa A, Magagnotti C, Bachi A, Giarda E, Verpelli C, Kilstrup-Nielsen C, Sala C, Kalscheuer VM, Broccoli V. CDKL5 ensures excitatory synapse stability by reinforcing NGL-1-PSD95 interaction in the postsynaptic compartment and is impaired in patient iPSC-derived neurons. Nat Cell Biol. 2012;14:911–23. doi: 10.1038/ncb2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samaco RC, Mandel-Brehm C, Chao HT, Ward CS, Fyffe-Maricich SL, Ren J, Hyland K, Thaller C, Maricich SM, Humphreys P, Greer JJ, Percy A, Glaze DG, Zoghbi HY, Neul JL. Loss of MeCP2 in aminergic neurons causes cell-autonomous defects in neurotransmitter synthesis and specific behavioral abnormalities. Proc Natl Acad Sci U S A. 2009;106:21966–71. doi: 10.1073/pnas.0912257106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schanen C, Houwink EJ, Dorrani N, Lane J, Everett R, Feng A, Cantor RM, Percy A. Phenotypic manifestations of MECP2 mutations in classical and atypical Rett syndrome. Am J Med Genet A. 2004;126A:129–40. doi: 10.1002/ajmg.a.20571. [DOI] [PubMed] [Google Scholar]
- Siller R, Greenhough S, Park IH, Sullivan GJ. Modelling human disease with pluripotent stem cells. Curr Gene Ther. 2013;13:99–110. doi: 10.2174/1566523211313020004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn YD, Han JW, Yoon YS. Generation of induced pluripotent stem cells from somatic cells. Prog Mol Biol Transl Sci. 2012;111:1–26. doi: 10.1016/B978-0-12-398459-3.00001-0. [DOI] [PubMed] [Google Scholar]
- Soldner F, Laganiere J, Cheng AW, Hockemeyer D, Gao Q, Alagappan R, Khurana V, Golbe LI, Myers RH, Lindquist S, Zhang L, Guschin D, Fong LK, Vu BJ, Meng X, Urnov FD, Rebar EJ, Gregory PD, Zhang HS, Jaenisch R. Generation of isogenic pluripotent stem cells differing exclusively at two early onset Parkinson point mutations. Cell. 2011;146:318–31. doi: 10.1016/j.cell.2011.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song JJ, Ott HC. Organ engineering based on decellularized matrix scaffolds. Trends Mol Med. 2011;17:424–32. doi: 10.1016/j.molmed.2011.03.005. [DOI] [PubMed] [Google Scholar]
- Surani MA. Cellular reprogramming in pursuit of immortality. Cell Stem Cell. 2012;11:748–50. doi: 10.1016/j.stem.2012.11.014. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–72. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–76. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Weaving LS, Christodoulou J, Williamson SL, Friend KL, McKenzie OL, Archer H, Evans J, Clarke A, Pelka GJ, Tam PP, Watson C, Lahooti H, Ellaway CJ, Bennetts B, Leonard H, Gecz J. Mutations of CDKL5 cause a severe neurodevelopmental disorder with infantile spasms and mental retardation. Am J Hum Genet. 2004;75:1079–93. doi: 10.1086/426462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng SM, Bailey ME, Cobb SR. Rett syndrome: from bed to bench. Pediatr Neonatol. 2011;52:309–16. doi: 10.1016/j.pedneo.2011.08.002. [DOI] [PubMed] [Google Scholar]
- Young JI, Zoghbi HY. X-chromosome inactivation patterns are unbalanced and affect the phenotypic outcome in a mouse model of rett syndrome. Am J Hum Genet. 2004;74:511–20. doi: 10.1086/382228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Hu K, Smuga-Otto K, Tian S, Stewart R, Slukvin II, Thomson JA. Human induced pluripotent stem cells free of vector and transgene sequences. Science. 2009;324:797–801. doi: 10.1126/science.1172482. [DOI] [PMC free article] [PubMed] [Google Scholar]