

Abstract
Probes for use in time-resolved fluorescence competitive binding assays at melanocortin receptors based on the parental ligands MSH(4), MSH(7), and NDP-α-MSH were prepared by solid phase synthesis methods, purified, and characterized. The saturation binding of these probes was studied using HEK-293 cells engineered to overexpress the human melanocortin 4 receptor (hMC4R) as well as the human cholecystokinin 2 receptor (hCCK2R). The ratios of non-specific binding to total binding approached unity at high concentrations for each probe. At low probe concentrations, receptor-mediated binding and uptake was discernable, and so probe concentrations were kept as low as possible in determining Kd values. The Eu-DTPA-PEGO-MSH(4) probe exhibited low specific binding relative to non-specific binding, even at low nanomolar concentrations, and was deemed unsuitable for use in competition binding assays. The Eu-DTPA-PEGO probes based on MSH(7) and NDP-α-MSH exhibited Kd values of 27±3.9 nM and 4.2±0.48 nM, respectively, for binding with hMC4R. These probes were employed in competitive binding assays to characterize the interactions of hMC4R with monovalent and divalent MSH(4), MSH(7), and NDP-α-MSH constructs derived from squalene. Results from assays with both probes reflected only statistical enhancements, suggesting improper ligand spacing on the squalene scaffold for the divalent constructs. The Ki values from competitive binding assays that employed the MSH(7)-based probe were generally lower than the Ki values obtained when the probe based on NDP-α-MSH was employed, which is consistent with the greater potency of the latter probe. The probe based on MSH(7) was also competed with monovalent, divalent, and trivalent MSH(4) constructs that previously demonstrated multivalent binding in competitive binding assays against a variant of the probe based on NDP-α-MSH. Results from these assays confirm multivalent binding, but suggest a more modest increase in avidity for these MSH(4) constructs than was previously reported.
Keywords: competition binding assays, fluorescent probes, melanocortin 4 receptor, saturation binding assays, time-resolved fluorescence
1. Introduction
The affinity of a molecule for binding to a receptor is often quantified by a competitive binding assay against a labeled ligand of known potency. For example, labeled forms of Ac-Ser-Tyr-Ser-Nle-Glu-His-DPhe-Arg-Trp-Gly-Lys-Pro-Val-NH2 [NDP-α-MSH], a superpotent ligand that binds to melanocortin receptors,1,2 have been used for this purpose.3,4 In such assays, one often assumes thermodynamic control, i.e., that the respective on-rates and off-rates of both the competing and competed ligands are similar so that all bound and unbound states are in equilibrium. If this is not the case, details of how the assay is carried out (order and timing of reagent addition, timing of measurements taken) can affect the outcome. Determination of on-rates and off-rates for binding of molecules to living cells is difficult since ligands and labeled probes may be taken up by the cells and because receptors may cycle to and from the cell surface. In the absence of knowledge of ligand and probe on-rates and off-rates, a close match between the affinities of the competed probe and the competing ligand in a competitive binding assay would seem prudent–but is it necessary? This issue was examined experimentally as reported herein.
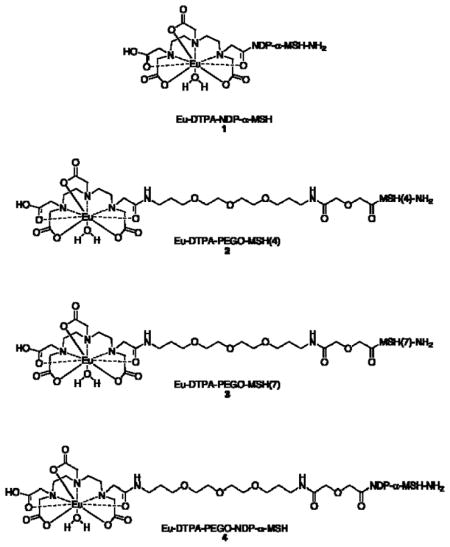
We have investigated the binding of multivalent molecules, including several derived from the weak ligand Ac-His-DPhe-Arg-Trp-NH2 [MSH(4)]5–7, to human melanocortin 4 receptors (hMC4R).8 MSH(4) was selected because synergistic effects are generally more easily detected for multivalent constructs of low-affinity ligands.9–13 Probe 1 is based on NDP-α-MSH and was used to determine the values for many of Ki our multivalent constructs. However, we became concerned that competitions between superpotent probes such as 1 and multivalent constructs based on much weaker ligands such as MSH(4) were inherently unbalanced, and that perhaps the measured avidity of a competing multivalent construct depended on the affinity of the competed fluorescent probe. In a preliminary study, we prepared the Eu-DTPA-PEGO-MSH(4) probe 2 and tested it in saturation and competitive binding assays.14 The Kd of 2, determined by saturation binding to HEK-293 cells overexpressing hMC4R, was 9.1 μM, compared with a reported Kd for 1 of 8.3 nM.15 We report herein syntheses of the structurally related probes Eu-DTPA-PEGO-MSH(7) 3 and Eu-DTPA-PEGO-NDP-α-MSH 4, studies of the saturation binding of 2–4 with hMC4R, and the use of 3 and 4 in competitive binding assays involving monovalent and divalent MSH(4), MSH(7), and NDP-α-MSH constructs derived from squalene16 and monovalent, divalent, and trivalent MSH(4) constructs that previously exhibited multivalent binding in competitive binding assays against a variant of probe 4.17
2. Materials and methods
2.1. Chemical synthesis
2.1.1. General experimental
Dichloromethane (DCM), diethyl ether, and tetrahydrofuran (THF) were dried by passage through activated alumina. Other solvents and commercial reagents were used as supplied. For moisture sensitive reactions, glassware was flame-dried under argon. Analytical thin-layer chromatography (TLC) was carried out on pre-coated silica gel 60 F-254 plates with visualization by UV exposure, by exposure to I2 vapor, or by staining with 10% phosphomolybdic acid solution in ethanol or with 5% H2SO4 in ethanol and heat. Gravity column chromatography was accomplished using silica gel 60 (63–210 μm). NMR spectra were recorded at 300 MHz or 500 MHz for 1H NMR and at 75 MHz or 125 MHz for 13C NMR. Chemical shifts (δ) are expressed in ppm and are internally referenced (7.24 ppm for CDCl3 and 3.31 ppm for CD3OD for 1H NMR and 77.0 ppm for CDCl3 and 49.15 ppm for CD3OD for 13C NMR). Preparative HPLC was performed on a 19 × 256 mm Waters X-Bridge Preparative C18 column. The mobile phase was 10–90% acetonitrile and water containing 0.1% trifluoroacetic acid (TFA) within 50 min. The flow rate was 15 mL/min. The dual UV detector system operated at 230 and 280 nm. ESI experiments were performed on an ESI Bruker Apex Qh 9.4 T FT-ICR instrument using standard ESI conditions. The samples were dissolved in acetonitrile/water 1:1 containing 0.1% formic acid in a concentration range of 1–30 μM.
2.1.2. Solid phase synthesis
For the synthesis of probes 2–4, resin-bound MSH(4), MSH(7), and NDP-α-MSH were synthesized manually using an N -Fmoc/t-Bu solid-phase peptide synthesis strategy and standard DIC/HOBt activation on Rink amide Tentagel S resin (approximately 0.24 mmol of active sites per gram). For the synthesis of ligands 5–7, 16, and 17, resin-bound MSH(4), MSH(7), and NDP-α-MSH were synthesized manually using an N -Fmoc/t-Bu solid-phase peptide synthesis strategy and standard DIC/Cl-HOBt activation on Rink amide resin (approximately 0.68 mmol of active sites per gram) as follows. Resin (1.0 g) in a syringe (polypropylene reaction tube equipped with a polypropylene frit) was allowed to swell in THF for 1 h. THF was removed, and 20% piperidine in DMF (15 mL) was added to deprotect the Fmoc functionality. After 2 min, the DMF/piperidine solution was removed, 20% piperidine in DMF (15 mL) was again added, and the mixture was shaken for 18 min. The DMF/piperidine solution was removed and the resin washed with DMF (3 × 15 mL), DCM (3 × 15 mL), DMF (3 × 15 mL), 0.5 M HOBt in DMF (1 × 15 mL), 0.5 M HOBt in DMF (1 × 15 mL) plus a drop of 0.01 M bromophenol blue in DMF, DMF (2 × 15 mL), and DCM (1 × 15 mL), in that order. For Rink amide resin, a mixture of the appropriate Fmoc-amino acid (3 equivalents), Cl-HOBt (3 equivalents), and DIC (6 equivalents) in DMF (15 mL) was allowed to react for 2 min, was then added to the resin, and the mixture shaken for 1 h, during which time the blue color disappeared. The resin was then washed with DMF (3 × 15 mL), DCM (3 × 15 mL), and DMF (3 × 15 mL). Free NH2 groups were capped by addition of a 1:1 mixture of acetic anhydride and pyridine (6 mL). After the mixture was shaken for 20 min, the resin was washed with DMF (3 × 15 mL), DCM (3 × 15 mL), and DMF (3 × 15 mL). The absence of free amine groups was confirmed by the Kaiser test. The above cycle of procedures was repeated for coupling of the other amino acids in the sequence, producing the resin-bound peptide derivatives. Further details for production of probes 2–4 and azides 5–7 are given in sections 2.1.2.1. and 2.1.2.2.
2.1.2.1. Synthesis and characterization of probes 2–4
Attachment of the PEGO linker was performed using DIC/HOBt activation (3 equiv Fmoc-PEGO18, 3 equiv of HOBt, and 3 equiv of DIC). Next, the DTPA chelator was attached to the N-terminus of the resin–bound construct as follows. After Fmoc removal, the resin was washed with DMSO. DTPA dianhydride19 (10 equiv) and HOBt (30 equiv) were dissolved in dry DMSO (1 mL) at 50 °C and then stirred for 20 min at rt. This mixture was injected into the syringe reactor which was shaken overnight, then the resin washed with DMSO, THF, 20% aqueous THF, THF, 5% DIEA in THF (5 min), THF, DMF, THF, and DCM. A cleavage mixture consisting of trifluoroacetic acid, water, 1,2-ethanedithiol, and thioanisole (91:3:3:3, 10 mL/g of resin) was injected into the syringe reactor containing the resin and the mixture shaken for 4 h at rt. The solution was then filtered off and the resin washed twice with TFA. Filtrates were collected, concentrated under a stream of nitrogen, and the product was precipitated by addition of cold ether to the residue. The peptide pellet was washed three times with cold ether, dried, dissolved in 1.0 M acetic acid, and lyophilized. The lyophilized DTPA-PEGO-MSH(4), DTPA-PEGO-MSH(7), and DTPA-PEGO-NDP-α-MSH constructs were purified by preparative HPLC and characterized by FT-ICR MS.
The metal-free precursors were dissolved in 0.1 M ammonium acetate, the pH was adjusted to 8 with aqueous 0.1 M NH4OH, and 3 equiv of EuCl3•6H2O in water were added. The reaction mixture was stirred at rt overnight. The excess EuCl3 and ammonium salts were removed using a Sep-Pak® C18 reverse-phase column with repetitive washing (20 mL of HPLC grade water). The final products 2–4 were eluted using 50% aqueous acetonitrile (4 mL), concentrated, lyophilized, and characterized by analytical HPLC and FT-ICR MS. Data appear in Table 1.
Table 1.
MS and HPLC characterization of compounds 2–7.
| Compound | Formula [M] | Calculated Masses [Ion] | Masses Found (error) | tR (min) | Yields (%) |
|---|---|---|---|---|---|
| 2a | C60H85N16O19151Eu | 1486.5532 [M+1]+ | 1486.5526 (0.4 ppm) | 10.5 | 20 |
| C60H85N16O19153Eu | 1488.5546 [M+1]+ | 1488.5549 (0.2 ppm) | |||
| 3a | C74H108N19O25151Eu | 1815.7119 [M+1]+ | 1815.7113 (0.3 ppm) | 12.9 | 9 |
| C74H108N19O25153Eu | 1817.7133 [M+1]+ | 1817.7139 (0.3 ppm) | |||
| 4a | C104H155N26O33151Eu | 817.0254 [M+3]3+ | 817.0248 (0.7 ppm) | 12.6 | 11 |
| C104H155N26O33153Eu | 817.6925 [M+3]3+ | 817.6926 (0.1 ppm) | |||
| 5b | C38H50N14O5 | 783.4161 [M+1]+ | 783.4158 (0.4 ppm) | 13.6 | 39–45 |
| 6b | C52H73N17O11 | 556.7910 [M+2]2+ | 556.7909 (0.3 ppm) | 15.4 | 42–50 |
| 7b | C82H118N24O19 | 872.4575 [M+2]2+ | 872.4567 (0.9 ppm) | 14.6 | 50–60 |
Analyzed on a 3 × 150 mm 3.5 Å Waters C18 X-Bridge column, flow rate 0.3 mL/min, linear gradient from 10–60% B in A over 45 min, where A is 0.1% TEAA in water (pH 6) and B is 90% acetonitrile and 10% A, detection at 220 and 280 nm. Purity >95%. Characterized by FT-ICR MS.
Analyzed on a 3 × 150 mm 3.5 Å Waters C18 X-Bridge column, flow rate 0.3 mL/min, linear gradient from 10–90% B in A over 45 min, where A is 0.1% TFA in water and B is 0.1% TFA in acetonitrile, detection at 220 and 280 nm. Purity >95%. Characterized by ESI MS.
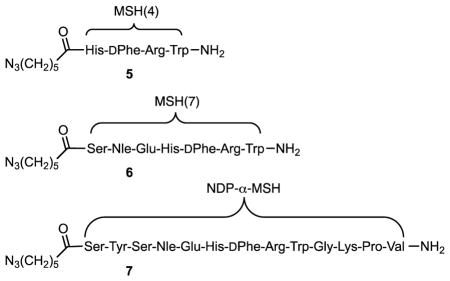
2.1.2.2. Synthesis and characterization of azides 5–7
Azide 5 was prepared from the corresponding resin-bound tetrapeptide and 6-azidohexanoic acid as previously described.20 Azides 6 and 7 were prepared in a manner similar to 5. Cleavage from the resin and deprotection were achieved using a cleavage mixture of trifluoroacetic acid, triisopropylsilane, thioanisole, and water (91:3:3:3). The mixture of cleavage cocktail and resin was shaken overnight, the solution was separated from the resin, volatiles were evaporated, the residue triturated with ether, and the crude product separated by centrifugation. Following purification by preparative HPLC, compounds 5–7 were characterized by ESI MS. Data appear in Table 1.
2.1.3. Scaffold synthesis and characterization (Scheme 1)
Scheme 1.

Synthesis of monovalent constructs 12a–15a and divalent constructs 12b–15b.a
aCompounds 8, 9, and 10 were obtained as complex mixtures of stereoisomers and regioisomers. Compounds 12a–15a and 12b–15b, derived from 9 and 10, respectively, are likewise assumed to be the related mixtures of stereoisomers and regioisomers.
2.1.3.1. Synthesis and characterization of 2,6,10,15,19,23-hexamethyltetracosane-3,7,11,14,18,22-hexaol (8)
In a three-necked flask a solution of borane-tetrahydrofuran complex in THF (1 M, 132 mL, 132 mmol) was deoxygenated with argon gas. The flask was immersed in an ice bath, and 2-methyl-2-butene (2 M, 132 mL, 264 mmol) also deoxygenated with argon gas was added to the borane solution dropwise with stirring at 0 °C. The resulting disiamyl borane was maintained at 0 °C for 3 h prior to use. A solution of squalene (3.0 gm, 7.3 mmol) in THF was then added dropwise at 0 °C. The mixture was stirred at 0 °C for 5 h, kept at 4 °C for 3 d, then 3 N NaOH (80 mL) and 30% H2O2 (80 mL) were added. The reaction mixture was stirred at rt for 12 h and was then diluted with EtOAc (500 mL), washed with water (2 × 200 mL), brine (2 × 100 mL), dried (Na2SO4), filtered, and concentrated in vacuo to leave a viscous oil. The oil was subjected to gravity column chromatography on silica gel 60 eluted with chloroform-methanol (95:5), giving 1.94 g (51%) of 8 as a white, foamy solid, mp 42–46 °C, Rf 0.3 (silica gel 60, 1:9 MeOH:CHCl3). IR (neat) cm−1 3350 (br), 2956, 1464, 1376. NMR spectra are complex since the product is a mixture of regioisomers and stereoisomers. The 1H NMR and 13C NMR spectra appear in the Supplementary Data that accompanies this paper. Significant lines from the 1H NMR spectrum are listed here. 1H NMR (500 MHz, CDCl3) δ 0.85–0.97 (approximately 24H, overlapping methyl doublets), 1.10–1.21 (approximately 6H, methyl singlets), 1.20–1.80 (approximately 26H, m), 3.30–3.50 (approximately 6H, m); HRMS (ESI) calcd for C30H63O6 (M+H)+ 519.4619, found 519.4619.
2.1.3.2. Synthesis and characterization of alkynes 9 and 10
To a suspension of NaH (113 mg, 4.74 mmol) in DMF (10 mL) was added a solution of 8 (821 mg, 1.58 mmol) in DMF (5 mL) and the mixture was stirred at rt for 15 min. Tetrabutylammonium iodide (294 mg, 0.8 mmol) and a solution of 1-bromo-5-hexyne21 (1.27 g, 7.90 mmol) in DMF (3 mL) were added to the reaction mixture and stirring was continued for 24 h. The mixture was then diluted with ether (150 mL), washed with water (3 × 50 mL), brine (20 mL), dried (Na2SO4), filtered, and concentrated in vacuo to give a pale yellow oil. The oil was subjected to gravity column chromatography on silica gel 60 eluted with 2% MeOH/CHCl3, giving 220 mg (21%) of bisalkyne 10 as a viscous gum, Rf 0.7 (silica gel 60, 1:9 MeOH/CHCl3). Further elution of the column with 5% MeOH/CHCl3 gave 410 mg (43%) of monoalkyne 9, also as a viscous gum, Rf 0.4 (silica gel 60, 1:9 MeOH/CHCl3). NMR spectra are complex since the products are mixtures of regioisomers and stereoisomers. The 1H NMR and 13C NMR spectra appear in the Supplementary Data that accompanies this paper. Significant lines from the 1H NMR spectra are listed here. Spectral data for 9: IR (neat) cm−1 3350 (br), 2955, 2117, 1462, 1378; 1H NMR (500 MHz, CDCl3) δ 0.80–1.00 (approximately 24H, overlapping methyl doublets), 1.10–1.22 (approximately 6H, methyl singlets), 1.10–1.80 (approximately 29H, m), 1.96 (1H, t, J = 2.5 Hz), 2.22 (2H, td, J = 7.0 Hz, 2.5 Hz), 3.30–3.50 (approximately 8H, m); HRMS (ESI) calcd for C36H71O6 (M+H)+ 599.5245, found 599.5241. Spectral data for 10: IR (neat) cm−1 3350 (br), 2955, 2117, 1462, 1376; 1H NMR (500 MHz, CDCl3) δ 0.80–1.00 (approximately 24H, overlapping methyl doublets), 1.10–1.22 (approximately 6H, methyl singlets), 1.10–1.90 (approximately 32H, m), 1.92–1.95 (2H, m), 2.16–2.22 (4H, m), 2.90–3.60 (approximately 10H, m); HRMS (ESI) calcd for C42H79O6 (M+H)+ 679.5871, found 679.5862.
2.1.4. Multimer synthesis and characterization (Scheme 1)
2.1.4.1. Synthesis and characterization of 12a
To a mixture of alkyne 9 (30 mg, 50 μmol), TBTA (3 mg, 6 μmol), and tetrakis(acetonitrile)copper(I) hexafluorophosphate (2 mg, 6 μmol) in dry methanol (1 mL) was added serinamide azide 1120 (18 mg, 75 μmol). The reaction mixture was subjected to microwave irradiation (Biotage Initiator, 100 °C) and the reaction monitored by TLC (20% MeOH/CHCl3). After 4 h, volatiles were evaporated and the residue loaded onto a silica gel 60 column. Elution with 5% MeOH/CHCl3 afforded 22 mg (26 μmol, 52%) of 12a as an gummy solid, Rf 0.3 (silica gel 60, 1:9 MeOH/CHCl3). IR (neat) cm−1 3350 (br), 2933, 1677, 1546, 1461, 1377. NMR spectra are complex since the product is a mixture of regioisomers and stereoisomers. The 1H NMR and 13C NMR spectra appear in the Supplementary Data that accompanies this paper. Significant lines from the 1H and 13C NMR spectra are listed here. 1H NMR (500 MHz, CD3OD) δ 0.85–0.94 (approximately 24H, overlapping methyl doublets), 1.12–1.19 (approximately 6H, methyl singlets), 1.20–1.80 (approximately 29H, m), 1.92 (2H, pentet, J = 7 Hz), 2.29 (2H, t, J = 7 Hz), 2.72 (2H, t, J = 7 Hz), 2.90–3.50 (approximately 7H, m), 3.73–3.81 (2H, m), 4.36 (2H, t, J = 7 Hz), 4.39–4.44 (1H, m), 7.74 (1H, s); 13C NMR (125 MHz, CD3OD) δ 15.9, 17.8, 18.1, 19.5, 19.6, 19.7, 26.1, 26.2, 26.3, 27.1, 27.5, 27.6, 29.4, 29.7, 30.9, 31.1, 32.8, 34.9, 36.6, 36.8, 39.4, 40.3, 51.2, 52.4, 56.5, 63.2, 73.4, 77.7, 78.0, 78.3, 79.1, 79.4, 79.6, 123.3, 175.2. 176.0; HRMS (ESI) calcd for C45H88N5O9 (M+H)+ 842.6577, obsd 842.6582.
2.1.4.2. Synthesis and characterization of 12b
To a mixture of alkyne 10 (25 mg, 36 μmol), TBTA (3 mg, 6 μmol), and tetrakis(acetonitrile)copper(I) hexafluorophosphate (2 mg, 6 μmol) in dry methanol (1 mL) was added serinamide azide 1120 (22 mg, 90 μmol). The reaction mixture was subjected to microwave irradiation (Biotage Initiator, 100 °C) and the reaction monitored by TLC (20% MeOH/CHCl3). After 4 h, volatiles were evaporated and the residue loaded onto a silica gel 60 column. Elution with 10% MeOH/CHCl3 afforded 38 mg (32 μmol, 90%) of 12b as a gummy solid, Rf 0.5 (silica gel 60, 2:8 MeOH/CHCl3). IR (neat) cm−1 3335 (br), 2936, 1658, 1546, 1461, 1374. NMR spectra are complex since the product is a mixture of regioisomers and stereoisomers. The 1H NMR and 13C NMR spectra appear in the Supplementary Data that accompanies this paper. Significant lines from the 1H and 13C NMR spectra are listed here. 1H NMR (500 MHz, CD3OD) δ 0.85–0.94 (approximately 24H, overlapping methyl doublets), 1.12–1.19 (approximately 6H, methyl singlets), 1.20–1.80 (approximately 35H, m), 1.91 (4H, pentet, J = 7 Hz), 2.29 (4H, t, J = 7 Hz), 2.72 (4H, t, J = 7 Hz), 2.80–3.50 (approximately 10H, m), 3.73–3.80 (4H, m), 4.36 (4H, t, J = 7 Hz), 4.41 (2H, t J = 5 Hz), 7.75 (2H, s); 13C NMR (125 MHz, CD3OD) δ 16.0, 18.1, 18.8, 19.0, 19.6, 26.1, 26.3, 27.2, 27.6, 30.9, 31.1, 36.6, 51.2, 56.6, 63.3, 73.4, 77.7, 123.3, 175.2, 176.0; HRMS (ESI) calcd for C60H114N10O12 (M+2H)2+ 583.4303, obsd 583.4305.
2.1.4.3. Synthesis and characterization of 13a
To a mixture of alkyne 9 (20 mg, 33 μmol), TBTA (3 mg, 6 μmol), and tetrakis(acetonitrile)copper(I) hexafluorophosphate (2 mg, 6 μmol) in dry methanol (1 mL) was added MSH(4) azide 5 (29 mg, 36 μmol). The reaction mixture was subjected to microwave irradiation (Biotage Initiator, 100 °C) for 4 h. The reaction mixture was diluted with water (25 mL) and extracted (3 × 30 mL) with a solution of dithizone (3 mg) in CHCl3 (150 mL). The aqueous phase was washed with CHCl3 (2 × 20 mL) and lyophilized to give a white solid. Purification by HPLC (stationary phase 19 × 256 mm X-Bridge Preparative C18 column, mobile phase 10–90% acetonitrile and water containing 0.1% TFA within 50 min, flow rate 15 mL/min, UV detection at 230 nm) yielded 8 mg (5 μmol, 18%) of 13a, tR 21.6–24.9 min; analytical HPLC (stationary phase 3 × 150 mm 3.5 Å Waters C18 X-Bridge column, flow rate 0.3 mL/min, linear gradient from 10–90% B in A over 45 min, where A is 0.1% TFA in water and B is 0.1% TFA in acetonitrile, detection at 220 and 280 nm), tR 20.2 min (broad); HRMS (ESI) calcd for C74H122N14O11 (M+2H)2+ 691.4703, obsd 691.4699.
2.1.4.4. Synthesis and characterization of 13b
To a mixture of alkyne 10 (15 mg, 22 μmol), TBTA (3 mg, 6 μmol), and tetrakis(acetonitrile)copper(I) hexafluorophosphate (2 mg, 6 μmol) in dry methanol (1 mL) was added MSH(4) azide 5 (36 mg, 46 μmol). The reaction mixture was subjected to microwave irradiation (Biotage Initiator, 100 °C) for 4 h. The reaction mixture was diluted with water (25 mL) and extracted (3 × 30 mL) with a solution of dithizone (3 mg) in CHCl3 (150 mL). The aqueous phase was washed with CHCl3 (2 × 20 mL) and lyophilized to give a white solid. Purification by HPLC (stationary phase 19 × 256 mm X-Bridge Preparative C18 column, mobile phase 10–90% acetonitrile and water containing 0.1% TFA within 50 min, flow rate 15 mL/min, UV detection at 230 nm) yielded 26 mg (11 μmol, 53%) of 13b, tR 18.2 - 22.6 min; analytical HPLC (stationary phase 3 × 150 mm 3.5 Å Waters C18 X-Bridge column, flow rate 0.3 mL/min, linear gradient from 10–90% B in A over 45 min, where A is 0.1% TFA in water and B is 0.1% TFA in acetonitrile, detection at 220 and 280 nm), tR 18.6 min (broad); HRMS (ESI) calcd for C118H181N28O16 (M+3H)3+ 748.8064, obsd 748.8063.
2.1.4.5. Synthesis and characterization of 14a
To a mixture of alkyne 9 (10 mg, 17 μmol), TBTA (3 mg, 6 μmol), and tetrakis(acetonitrile)copper(I) hexafluorophosphate (2 mg, 6 μmol) in dry methanol (1 mL) was added MSH(7) azide 6 (20 mg, 18 μmol). The reaction mixture was subjected to microwave irradiation (Biotage Initiator, 100 °C) for 4 h. The reaction mixture was diluted with water (25 mL) and extracted (3 × 30 mL) with a solution of dithizone (3 mg) in CHCl3 (150 mL). The aqueous phase was washed with CHCl3 (2 × 20 mL) and lyophilized to give a white solid. Purification by HPLC (stationary phase 19 × 256 mm X-Bridge Preparative C18 column, mobile phase 10–90% acetonitrile and water containing 0.1% TFA within 50 min, flow rate 15 mL/min, UV detection at 230 nm) yielded 7 mg (4 μmol, 24%) of 14a, tR 25.2–27.9 min; analytical HPLC (stationary phase 3 × 150 mm 3.5 Å Waters C18 X-Bridge column, flow rate 0.3 mL/min, linear gradient from 10–90% B in A over 45 min, where A is 0.1% TFA in water and B is 0.1% TFA in acetonitrile, detection at 220 and 280 nm), tR 20.2 min (broad); HRMS (ESI) calcd for C88H146N17O17 (M+3H)3+ 571.0355, obsd 571.0365.
2.1.4.6. Synthesis and characterization of 14b
To a mixture of alkyne 10 (7.5 mg, 11 μmol), TBTA (3 mg, 6 μmol), and tetrakis(acetonitrile)copper(I) hexafluorophosphate (2 mg, 6 μmol) in dry methanol (1 mL) was added MSH(7) azide 6 (26 mg, 23 μmol). The reaction mixture was subjected to microwave irradiation (Biotage Initiator, 100 °C) for 4 h. The reaction mixture was diluted with water (25 mL) and extracted (3 × 30 mL) with a solution of dithizone (3 mg) in CHCl3 (150 mL). The aqueous phase was then washed with CHCl3 (2 × 20 mL) and lyophilized to give a white solid. Purification by HPLC (stationary phase 19 × 256 mm X-Bridge Preparative C18 column, mobile phase 10–90% acetonitrile and water containing 0.1% TFA within 50 min, flow rate 15 mL/min, UV detection at 230 nm) yielded 13 mg (5 μmol, 40%) of 14b, tR 23.1–25.9 min; analytical HPLC (stationary phase 3 ± 150 mm 3.5 Å Waters C18 X-Bridge column, flow rate 0.3 mL/min, linear gradient from 10–90% B in A over 45 min, where A is 0.1% TFA in water and B is 0.1% TFA in acetonitrile, detection at 220 and 280 nm), tR 19.4 min (broad); HRMS (ESI) calcd for C146H228N34O28 (M+4H)4+ 726.4360, obsd 726.4353.
2.1.4.7. Synthesis and characterization of 15a
To a mixture of alkyne 9 (5 mg, 8 μmol), TBTA (3 mg, 6 μmol), and tetrakis(acetonitrile)copper(I) hexafluorophosphate (2 mg, 6 μmol) in dry methanol (1 mL) was added NDP- -MSH azide 7 (16 mg, 9 μmol). The reaction mixture was subjected to microwave irradiation (Biotage Initiator, 100 °C) for 4 h. The reaction mixture was diluted with water (25 mL) and extracted (3 × 30 mL) with a solution of dithizone (3 mg) in CHCl3 (150 mL). The aqueous phase was then washed with CHCl3 (2 × 20 mL) and lyophilized to give a white solid. Purification by HPLC (stationary phase 19 × 256 mm X-Bridge Preparative C18 column, mobile phase 10–90% acetonitrile and water containing 0.1% TFA within 50 min, flow rate 15 mL/min, UV detection at 230 nm) yielded 11 mg (5 μmol, 58%) of 15a, tR 23.7–26.2 min; analytical HPLC (stationary phase 3 × 150 mm 3.5 Å Waters C18X-Bridge column, flow rate 0.3 mL/min, linear gradient from 10–90% B in A over 45 min, where A is 0.1% TFA in water and B is 0.1% TFA in acetonitrile, detection at 220 and 280 nm), tR 19.5 min (broad); HRMS (ESI) calcd for C118H191N24O25 (M+3H)3+ 781.4799, obsd 781.4789.
2.1.4.8. Synthesis and characterization of 15b
To a mixture of alkyne 10 (3.7 mg, 5 μmol), TBTA (3 mg, 6 μmol), and tetrakis(acetonitrile)copper(I) hexafluorophosphate (2 mg, 6 μmol) in dry methanol (1 mL) was added NDP- -MSH azide 7 (20 mg, 11 μmol). The reaction mixture was subjected to microwave irradiation (Biotage Initiator, 100 °C) for 4 h. The reaction mixture was diluted with water (25 mL) and extracted (3 × 30 mL) with a solution of dithizone (3 mg) in CHCl3 (150 mL). The aqueous phase was then washed with CHCl3 (2 × 20 mL) and lyophilized to give a white solid. Purification by HPLC (stationary phase 19 × 256 mm X-Bridge Preparative C18 column, mobile phase 10–90% acetonitrile and water containing 0.1% TFA within 50 min, flow rate 15 mL/min, UV detection at 230 nm) yielded 13 mg (3 μmol, 62%) of 15b, tR 22.1–24.9 min; analytical HPLC (stationary phase 3 × 150 mm 3.5 Å Waters C18 X-Bridge column, flow rate 0.3 mL/min, linear gradient from 10–90% B in A over 45 min, where A is 0.1% TFA in water and B is 0.1% TFA in acetonitrile, detection at 220 and 280 nm), tR 17.9 min (broad); HRMS (ESI) calcd for C206H317N48O44 (M+3H)3+ 1389.1342, obsd 1389.1344.
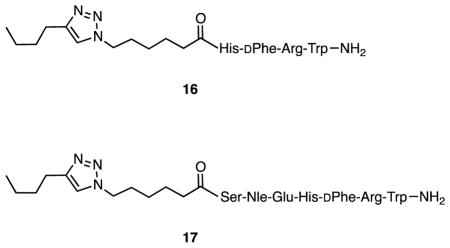
2.1.4.9. Synthesis and characterization of 16 and 17
Triazole 16 was prepared from the corresponding resin-bound tetrapeptide as previously described.20 Triazole 17 was prepared in a similar manner from the corresponding resin-bound heptapeptide. Following attachment of the N-terminal 6-(4-butyl-1H-1,2,3-triazol-1-yl)hexanoic acid residue, cleavage and deprotection were achieved using a 91:3:3:3 mixture of trifluoroacetic acid, triisopropylsilane, thioanisole, and water (10 mL). The mixture of cleavage cocktail and resin was shaken overnight, the solution was separated from the resin, volatiles were evaporated, the residue triturated with ether, and the crude product separated by centrifugation. Purification of 17 was accomplished by reversed phase chromatography using 19 × 256 mm X-Bridge Preparative C18 column. The mobile phase was 10–90% acetonitrile and water containing 0.1% TFA within 50 min; the flow rate was 15 mL/min and the UV detector system operated at 230 nm. HRMS (ESI) calcd for C58H85N17O11 (M + 2H)2+ 597.8301, obsd 597.8295.
2.1.4.10. Synthesis and characterization of 18–20
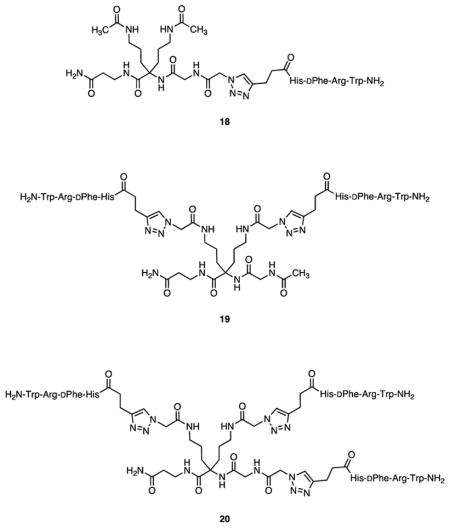
The preparation and characterization of triazoles 18–20 was previously described.17
2.2. Biological studies
2.2.1. Formulation of Solutions
Stock solutions of probes 2–4, squalene-derived constructs 12a–15a and 12b–15b, and control compounds 16,20 17, and 7 were made up in water. Concentrations were initially based on measured weights of solutes and were confirmed by UV analysis.
2.2.2. Binding Assays
2.2.2.1. Saturation Binding Assays
Quantitative receptor-binding assays were carried out following previously described methods.3,4,14,15 HEK-293 cells engineered to express both hMC4R and hCCK2R were used to assess ligand binding.15 Cells were grown in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum. Cells were seeded in Costar, tissue culture treated, black frame with transparent well, 96-well plates (part #3603) at a density of 2 × 104 cells per well and were incubated at 37 °C to reach 80–90% confluence. Ligands were diluted in binding buffer (1L DMEM, 5.97 g Hepes, 0.2% BSA, 1 mM 1,10-phenanthroline, 0.5 mg/L leupeptin, 200 mg/L bacitracin, pH adjusted to 7.4 using 2 N NaOH). On the day of an experiment, serial dilutions of Eu-labeled probes 2–4 (100 μL total volume) were made in different well plates in preparation for addition to plates containing cells. Plates containing cells were removed from the incubator and media was removed. Binding buffer (50 μL/well) was added to the first four rows of a plate and 25 μM NDP-α-MSH in binding buffer (50 μL/well) was added to the next four rows to determine non-specific binding. Fifty μL of the solutions of pre-diluted probes 2–4 were then added to the cell-containing plates and the plates were incubated at 37 °C. After 1 h the media was removed and the plates were washed (3 × 175 μL/well) using wash buffer (1L DMEM, 1 g BSA, 20 μM EDTA, 0.01% Tween20). Enhancement solution (PerkinElmer 1244–105) was added (105 μL/well) and the plates were incubated for 30 min at 37 °C before fluorescence was measured using a VICTOR™ X4 2030 Multilabel Reader (PerkinElmer) employing the standard Eu time-resolved fluorescence (TRF) measurement settings (340 nm excitation, 400 μs delay, and emission collection for 400 μs at 615 nm).
2.2.2.2. Competition Binding Assays
Serial dilutions of compounds to be tested were made in different well plates in binding buffer (1L DMEM, 5.97 g Hepes, 0.2% BSA, 1 mM 1,10-phenanthroline, 0.5 mg/L leupeptin, 200 mg/L bacitracin, pH adjusted to 7.4 using 2 N NaOH, 50 μL total volume) before adding to plates containing cells. Stock solutions of Eu-labeled probes were diluted with binding buffer to a concentration of 40 nM for 3 and 10 nM for 4. Plates containing cells were removed from the incubator, media was removed and replaced with binding buffer containing the Eu-labeled probe (50 μL/well), the solutions of compounds to be tested (concentration range generally 10−4–10−11 M) were added, and the plates incubated at 37 °C. After 1 h the media was removed and the plates were washed (3 × 175 μL/well) using wash buffer (1L DMEM, 1 g BSA, 20 μM EDTA, 0.01% Tween20). Enhancement solution (PerkinElmer 1244–105) was added (105 μL/well) and the plates were incubated for 30 min at 37 °C before fluorescence was measured using using a VICTOR™ X4 2030 Multilabel Reader (PerkinElmer) employing the standard Eu time-resolved fluorescence (TRF) measurement settings (340 nm excitation, 400 μs delay, and emission collection for 400 μs at 615 nm).
2.2.2.3. Data Analysis
Binding data were analyzed using GraphPad Prism software. Saturation binding data were analyzed using nonlinear regression analysis and fitted to classic one site total binding and nonspecific binding equations. Results from saturation binding experiments are depicted in Figure 2 and given in Table 2. The values of Kd given represent averages of four independent saturation binding experiments, each consisting of four data points per concentration tested. Competitive binding data were analyzed using nonlinear regression analysis and fitted to a classic one site binding competition equation. Each IC50 value was generated from individual competitive binding assays and converted to a Ki value using the equation Ki = IC50/(1 + ([ligand]/Kd)) where [ligand] refers to the concentration of the probe used as the labeled competed ligand. For probe 3, [ligand] = 20 nM and Kd = 27 nM. For probe 4, [ligand] = 5 nM and Kd = 4.2 nM. Results from competition binding experiments are given in Table 3. The values of Ki given represent averages of four independent competition binding experiments, each consisting of four data points per concentration tested.
Figure 2.

Saturation binding curves for Eu-DTPA-PEGO probes 2–4. Total binding (●). Non-specific binding (■). Specific binding (▲). The graphs in column 3 are truncated expansions of the graphs in column 2 and include a specific binding curve.
Table 2.
Binding constants for Eu-DTPA-PEGO probes 2–4 with hMC4R.
The value given is the average of four independent binding experiments, each done in quadruplicate.
Includes data up to [2] = 400 nM.
Includes data up to [3] = 150 nM.
Includes data up to [4] = 30 nM.
Table 3.
Competitive binding of Eu-DTPA-PEGO probes 3 and 4 against monovalent constructs 12a–15a, bivalent constructs 12b–15b, monovalent, divalent, and trivalent compounds 18–20, and control compounds 16, 17, and 7 with hMC4R.
| Compounds | Probe 3 Kia,b (nM) |
Probe 4 Kia,c (nM) |
|---|---|---|
| 12a | ncbd | ncbd |
| 12b | ncbd | ncbd |
| 16 | 350±28 | 990±40 |
| 13a | 600±16 | 2200±150 |
| 13b | 150±38 | 810±56 |
| 17 | 1.8±0.2 | 18±1.0 |
| 14a | 2.4±0.8 | 33±0.6 |
| 14b | 1.7±0.2 | 15±1.5 |
| 7 | 3.1±0.4 | 0.14±0.01 |
| 15a | 6.0±0.2 | 15±1.8 |
| 15b | 1.5±0.9 | 3.4±1.4 |
| 18 | 330±52 | 3900±600e |
| 19 | 37±10 | 250±58e |
| 20 | 4.3±0.4 | 11±1.2e |
The Ki was calculated using the equation Ki = IC50/(1+([ligand]/Kd)), where [ligand] refers to the concentration of the probe used as the labeled competed ligand.
Here [ligand] = 20 nM, the concentration of probe 3.
Here [ligand] = 5 nM, the concentration of probe 4.
ncb = no competitive binding.
Calculated from EC50 values taken from reference 17, wherein a probe similar to 4 was employed.
3. Results
3.1. Chemistry
Fully protected resin-bound precursors to probes 2–4 and azides 5–7 were prepared by solid phase synthesis on Rink amide Tentagel S resin (for probe synthesis) or on Rink amide resin (for ligand synthesis). For the preparation of probes 2–4, Fmoc-PEGO18 was coupled to the N-terminus of the resin-bound peptides. Following removal of the Fmoc, DTPA,19 activated as the HOBt ester, was attached to the N-terminus of the PEGO spacer. Simultaneous side chain deprotection and cleavage of the peptide from the resin produced the metal-free precursors to 2–4 which were purified by preparative HPLC and characterized by FT-ICR MS. Following complexation of Eu3+, the Eu-DTPA-PEGO probes 2–4 were purified by reversed phase chromatography and characterized by analytical HPLC and FT-ICR MS (Table 1).
For the preparation of azides 5–7, 6-azidohexanoic acid22 was coupled to the N-terminus of the resin-bound peptides. Simultaneous side chain deprotection and cleavage of the peptide from the resin produced compounds 5–7 which were purified by reversed phase preparative HPLC and characterized by ESI MS (Table 1). Yields ranged from 39–60%.
Reaction of squalene with excess disiamylborane, followed by oxidation, produced hexaol 8 in 51% yield as a white foam (Scheme 1). Analysis by 13C NMR (Figure 1) confirmed that hydroboration had been regioselective and had yielded a mixture of diastereomeric secondary alcohols as depicted in 8.16,20 Treatment of 8 with three equivalents of sodium hydride and five equivalents of 6-bromo-1-hexyne21 in DMF gave a mixture of alkylation products from which monoalkynes 9 and dialkynes 10 were isolated in 21% and 43% yields, respectively, by silica gel column chromatography. It is assumed that 9 and 10 are mixtures of regioisomers due to random alkylation at oxygen atoms along the carbon backbone.
Figure 1.

Top: A portion of the DEPT 135 13C NMR spectrum of 8 in CD3OD. Bottom: A portion of the 13C NMR spectrum of 8 in CD3OD. The lines near 73 ppm are due to tertiary carbons bearing OH groups and the lines between 76–79 ppm are due to secondary carbons bearing OH groups. The spectral complexity occurs because 8 is a mixture of regioisomers and stereoisomers.
Copper(1)-catalyzed azide-alkyne cycloaddition (CuAAC) reactions23 of 9 and 10 with excesses of the serine amide-derived azide 1120 gave the corresponding triazole-containing products 12a and 12b in 52% and 90% yields, respectively, following chromatographic purification (Scheme 1). These compounds were characterized by NMR and ESI MS. CuAAC reactions of 9 and 10 with excesses of azide 5 gave the corresponding triazoles 13a and 13b. Copper ions were removed from the crude product mixtures by complexation with dithizone and removal of the complex by extraction with CHCl3.24 The water-soluble products 13a and 13b were purified by preparative reversed phase HPLC, recovered by lyophilization in 18% and 53% yields, respectively, and characterized by ESI MS. In a similar manner, CuAAC reactions of 9 and 10 with excesses of azides 6 and 7 gave the corresponding triazole-containing products 14a, 14b, 15a, and 15b in 24%, 40%, 58%, and 62% yields, respectively, after workup and purification (Scheme 1). These compounds were characterized by ESI MS. The synthesis and characterization of compound 16 was previously reported.20 Compound 17 was prepared in a similar manner by N-terminal acylation of the resin-bound heptapeptide with 6-(4-butyl-1H-1,2,3-triazol-1-yl)hexanoic acid.20 The synthesis and characterization of compounds 18–20 were previously reported.17
3.2. Bioassays
HEK-293 cells overexpressing both hMC4R8,25 and hCCK2R26 were used to measure the affinities of probes 2–4 for binding to hMC4R by means of saturation binding assays (Figure 2).15 Kd values for probes 2–4 are given in Table 2. Europium-based time-resolved fluorescence competitive binding assays using Eu-DTPA-PEGO-MSH(7) (3) and Eu-DTPA-PEGO-NDP-α-MSH-NH2 (4) as the competed probes were employed to study the binding of monovalent and bivalent serinamide, MSH(4), MSH(7), and NDP-α-MSH constructs 12a–15a and 12b–15b. Competitive binding assays using probe 3 as the competed probe were employed to study the binding of monovalent, divalent, and trivalent MSH(4) constructs 18–20.17 Ki values for these compounds and for the control compounds 16, 17, and 7 are given in Table 3.
4. Discussion
Previously we described the preparation and testing of scaffolds derived from squalene,14,16 solanesol,20 and sucrose27 with one or more sidechains bearing MSH(4), a ligand with a low micromolar affinity for binding to hMC4R. MSH(4) was chosen because synergistic effects are generally more easily detected for multivalent constructs of low-affinity ligands.9–13 All of the monovalent and multivalent MSH(4) constructs derived from squalene, solanesol, and sucrose exhibited competitive binding to hMC4R that was comparable to the parental ligand. The competed probe used in these studies was Eu-DTPA-NDP-α-MSH (1), which is based on the superpotent ligand NDP-α-MSH. The off-rate of NDP-α-MSH is approximately eight hours.28 In addition, evidence suggests that receptor-bound 1 is taken up by the cells. 29 For these reasons, it was clear that competition binding assays involving 1 and hMC4R on living cells do not take place under thermodynamic control. This fact raised the possibility that the competition for binding to hMC4R between 1 and any variant of MSH(4) was biased in favor of 1, to the detriment of multivalent binding.
To determine what effects might result from the use of probes with a lesser affinity for hMC4R, the set of Eu-DTPA-PEGO probes 2–4 was prepared by standard solid phase methods. Saturation binding assays for 2–4 were carried out using HEK-293 cells that stably overexpress both hMC4R (6 × 105 receptors per cell) and hCCK2R (1 × 106 receptors per cell).15 These cells were employed so that results obtained here will be comparable with results from studies with multimeric constructs that bear ligands targeted to hCCK2R and with multimeric constructs that bear mixtures of ligands targeted to both receptors.30 In all cases, non-specific binding was determined by blocking binding to hMC4R with a high concentration of NDP-α-MSH. Although probe 2 exhibited 20% as much non-specific binding as did probes 3 and 4 (see Figure 2), the difference between total binding and non-specific binding for 2 was small, even at very low concentrations of the probe. Thus, the measured Kd has a large associated error (93 ± 100 nM), and plans to use probe 2 in competition binding assays were abandoned.31
As with probe 2, graphs of total and non-specific binding for probes 3 and 4 appear to be linear and parallel at high concentrations (see Figure 2), consistent with some form of receptor-independent binding and/or uptake as opposed to receptor saturation. However, at sufficiently low concentrations, the differences between total and non-specific binding for probes 3 and 4 were significant, and permitted the calculation of reasonable Kd values (see Table 2) so that use of these probes in competition binding assays was deemed possible.
For competition assays, a range of ligand potencies attached to monovalent, bivalent, and trivalent constructs was desired. Azides 5–7 were prepared by standard solid phase methods. Scaffolds 9 and 10 were prepared from squalene via hexaol 8 as depicted in Scheme 1.16 Attachment of azides 5–7 to 9 and 10 by means of microwave-assisted copper(I)-catalyzed azide-alkyne cycloaddition (CuAAC)23 produced monovalent and bivalent constructs 13a and 13b incorporating MSH(4), 14a and 14b incorporating MSH(7), and 15a and 15b incorporating NDP-α-MSH. Control compounds 12a and 12b were prepared by CuAAC reaction of serine amide-derived azide 1120 with 9 and 10. In addition, MSH(4)-containing compounds 18–20, which were recently shown to exhibit enhanced avidity with increased ligand number when competed against an NDP-α-MSH-based probe,17 were here competed against MSH(7)-based probe 3. Compounds 16,20 17, and 7 were used as reference compounds in place of the parental ligands.
Results of the competition binding assays are given in Table 3. The serine amide-containing control compounds 12a and 12b were both ineffective at blocking probes 3 and 4 from binding to hMC4R over the range of concentrations tested. For the squalene-derived monovalent and divalent constructs, the trends in Ki values are consistent for all three ligands tested, MSH(4) (compare 16, 13a, and 13b), MSH(7) (compare 17, 14a, and 14b), and NDP-α-MSH (compare 7, 15a, and 15b). Squalene-derived monovalent compounds exhibit a higher Ki value than the control, often a factor of two, presumably due to compromised ligand binding, while the corresponding divalent compound exhibits a lower Ki value, often a factor of two that can be ascribed to statistical doubling of the number of ligands. In all but one case, the Ki value obtained using probe 4 was higher than the corresponding Ki value obtained using probe 3, a result that is consistent with the stronger binding of 4 versus 3 to hMC4R. In view of past results with monovalent and multivalent compounds derived from squalene,14,16 solanesol,20 sucrose,27 and compounds 18–20,17 the results herein suggest that the MSH(4) ligands in compounds 13b–15b are spaced too far apart for manifestation of multivalent binding.
Enhanced avidity interpretable as multivalent binding was observed when compounds 18–20 were competed against probe 3, although the lowering of the Ki values with each additional ligand (approximately nine-fold at each step) was attenuated when compared with Ki values calculated from published EC50 data for competition binding assays that employed a Eu-DTPA-PEGO-NDP-α-MSH probe similar to 4 (approximately 16-fold and 22-fold, see Table 3).17
Although different sets of data were produced, similar conclusions were reached from competition binding experiments that employed probes 3 and 4, regardless of the potency of the ligand being competed. Probe 3 requires less time and effort to prepare, and so may find favor over probe 4 in future competition binding studies. However, confirmation of observed binding behaviors by the use of both probes would seem a prudent practice.
Supplementary Material
Acknowledgments
The authors thank Renata Patek for laboratory assistance. This work was supported by grants R33 CA 95944, RO1 CA 97360, RO1 CA 123547, and P30 CA 23074 from the National Cancer Institute.
ABBREVIATIONS
- BSA
bovine serum albumin
- Cl-HOBt
6-chloro-1-hydroxybenzotriazole
- CuAAC
copper(I)-catalyzed azide-alkyne cycloaddition
- DCM
dichloromethane
- DIC
diisopropyl carbodiimide
- DIEA
diisopropylethylamine
- DMEM
Dulbecco’s Modified Eagle Medium
- DMF
N,N-dimethylformamide
- DMSO
dimethyl sulfoxide
- DTPA
diethylenetriaminepentaacetic acid
- IC50
half maximal inhibitory concentration
- ESI MS
electrospray ionization mass spectrometry
- Fmoc
9-fluorenylmethyoxycarbonyl
- Fmoc-PEGO
1-(9H-fluoren-9-yl)-3,19-dioxo-2,8,11,14,21-pentaoxa-4,18-diazatricosan-23-oic acid
- FT-ICR MS
Fourier transform ion cyclotron resonance mass spectrometry
- hMC4R
human melanocortin 4 receptor
- HOBt
1-hydroxybenzotriazole
- HRMS
high resolution mass spectroscopy
- MEM
Minimum Essential Medium
- MSH(4)
His-DPhe-Arg-Trp
- MSH(7)
Ser-Nle-Glu-His-DPhe-Arg-Trp
- NDP-α-MSH
Ac-Ser-Tyr-Ser-Nle-Glu-His-DPhe-Arg-Trp-Gly-Lys-Pro-Val-NH2
- PEGO
19-amino-5-oxo-3,10,13,16-tetraoxa-6-azanonadecan-1-oic acid
- TBTA
tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine
- TEAA
triethylammonium acetate
- TFA
trifluoroacetic acid
- THF
tetrahydrofuran
- TLC
thin-layer chromatography
- TRF
time-resolved fluorescence
Footnotes
Supplementary data (copies of the HPLC chromatograms for compounds 2–7, 13a–15a, and 13b–15b, HRMS data for compounds 2–4, and the 1H and 13C NMR spectra of compounds 8–10, 12a, and 12b) associated with this article can be found, in the online version, at doi:
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Sawyer TK, Sanfilippo PJ, Hruby VJ, Engel MH, Heward CB, Burnett JB, Hadley ME. Proc Natl Acad Sci USA. 1980;77:5754. doi: 10.1073/pnas.77.10.5754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hadley ME, Anderson B, Heward CB, Sawyer TK, Hruby VJ. Science. 1981;213:1025. doi: 10.1126/science.6973820. [DOI] [PubMed] [Google Scholar]
- 3.Handl HL, Vagner J, Yamamura HI, Hruby VJ, Gillies RJ. Anal Biochem. 2004;330:242. doi: 10.1016/j.ab.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 4.De Silva CR, Vagner J, Lynch RM, Gillies RJ, Hruby VJ. Anal Biochem. 2010;398:15. doi: 10.1016/j.ab.2009.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hruby VJ, Wilkes BC, Hadley ME, Al-Obeidi F, Sawyer TK, Staples DJ, de Vaux AE, Dym O, de Lauro Castrucci AM, Hintz MF, Riehm JP, Rao KR. J Med Chem. 1987;30:2126. doi: 10.1021/jm00394a033. [DOI] [PubMed] [Google Scholar]
- 6.Castrucci AML, Hadley ME, Sawyer TK, Wilkes BC, Al-Obiedi F, Staples DJ, de Vaux AE, Dym O, Hintz MF, Riehm JP, Rao KR, Hruby VJ. Gen Comp Endocrinol. 1989;73:157. doi: 10.1016/0016-6480(89)90066-x. [DOI] [PubMed] [Google Scholar]
- 7.Haskell-Luevano C, Hendrata S, North C, Sawyer TK, Hadley ME, Hruby VJ, Dickinson C, Gantz I. J Med Chem. 1997;40:2133. doi: 10.1021/jm960840h. [DOI] [PubMed] [Google Scholar]
- 8.The hMC4R vector was originally received from Dr. Ira Gantz; see Gantz I, Miwa H, Konda Y, Shimoto Y, Tashiro T, Watson SJ, DelValle J, Yamada T. J Biol Chem. 1993;268:15174.
- 9.Mammen M, Choi SK, Whitesides GM. Angew Chem Int Ed. 1998;37:2754. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 10.Kiessling LL, Gestwicki JE, Strong LE. Angew Chem Int Ed. 2006;45:2348. doi: 10.1002/anie.200502794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krishnamurthy VM, Estroff LA, Whitesides GM. In: Fragment-based Approaches in Drug Discovery. Jahnke W, Erlanson DA, editors. Wiley-VCH; Weinheim: 2006. pp. 11–53. [Google Scholar]
- 12.Carlson CB, Mowery P, Owen RM, Dykhuizen EC, Kiessling LL. ACS Chemical Biology. 2007;2:119. doi: 10.1021/cb6003788. [DOI] [PubMed] [Google Scholar]
- 13.Choi S-K. Synthetic Multivalent Molecules. Wiley-Interscience; Hoboken, NJ: 2004. [Google Scholar]
- 14.Xu L, Vagner J, Alleti R, Rao V, Jagadish B, Morse DL, Hruby VJ, Gillies RJ, Mash EA. Bioorg Med Chem Lett. 2010;20:2489. doi: 10.1016/j.bmcl.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu L, Vagner J, Josan JS, Lynch RM, Morse DL, Baggett B, Han H, Mash EA, Hruby VJ, Gillies RJ. Mol Cancer Ther. 2009;8:2356. doi: 10.1158/1535-7163.MCT-08-1183. Cells with a passage number less than 50 were used in assays. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jagadish B, Sankaranarayanan R, Xu L, Richards R, Vagner J, Hruby VJ, Gillies RJ, Mash EA. Bioorg Med Chem Lett. 2007;17:3310. doi: 10.1016/j.bmcl.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brabez N, Lynch RM, Xu L, Gillies RJ, Chassaing G, Lavielle S, Hruby VJ. J Med Chem. 2011;54:7375. doi: 10.1021/jm2009937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jolimaitre P, Poirier C, Richard A, Blanpain A, Delord B, Roux D, Bourel-Bonnet L. Eur J Med Chem. 2007;42:114. doi: 10.1016/j.ejmech.2006.08.011. 1-(9H-fluoren-9-yl)-3,19-dioxo-2,8,11,14,21-pentaoxa-4,18-diazatricosan-23-oic acid (Fmoc-PEGO) is commercially available from Novabiochem as O-(N-Fmoc-3-aminopropyl)-O′-(N-diglycolyl-3-aminopropyl)diethyleneglycol. [DOI] [PubMed] [Google Scholar]
- 19.Diethylenetriamine pentaacetic acid dianhydride is commercially available.
- 20.Alleti R, Rao V, Xu L, Gillies RJ, Mash EA. J Org Chem. 2010;75:5895. doi: 10.1021/jo101043m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sharma S, Oehlschlager AC. J Org Chem. 1989;54:5064. [Google Scholar]
- 22.Grandjean C, Boutonnier A, Guerreiro C, Fournier JM, Mulard LA. J Org Chem. 2005;70:7123. doi: 10.1021/jo0505472. [DOI] [PubMed] [Google Scholar]
- 23.Hein JE, Fokin VV. Chem Soc Rev. 2010;39:1302. doi: 10.1039/b904091a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Caplin S. Tissue Culture Association Manual. 1976;2:439. [Google Scholar]
- 25.Yang X, Wang Z, Dong W, Ling L, Yang H, Chen R. J Prot Chem. 2003;22:335. doi: 10.1023/a:1025386022852. [DOI] [PubMed] [Google Scholar]
- 26.Noble F, Wank SA, Crawley JN, Bradwejn J, Seroogy KB, Hamon M, Roques BP. Pharmacol Rev. 1999;51:745. [PubMed] [Google Scholar]
- 27.Rao V, Alleti R, Xu L, Tafreshi NK, Morse DL, Gillies RJ, Mash EA. Bioorg Med Chem. 2011;19:6474–6482. doi: 10.1016/j.bmc.2011.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haskell-Luevano C, Miwa H, Dickinson C, Hadley ME, Hruby VJ, Yamada T, Gantz I. J Med Chem. 1996;39:432. doi: 10.1021/jm950407s. [DOI] [PubMed] [Google Scholar]
- 29.Xu L. Unpublished work [Google Scholar]
- 30.Josan JS, Handl HL, Sankaranarayanan R, Xu L, Lynch RM, Vagner J, Mash EA, Hruby VJ, Gillies RJ. Bioconjugate Chem. 2011;22:1270–1278. doi: 10.1021/bc1004284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.In a prior study (see reference 14), a Kd of 9.1 ± 1.4 μM was measured for probe 2, and much less non-specific binding relative to total binding was observed. The study employed HEK-293 cells that transiently overexpressed only hMC4R (6 × 105 receptors per cell), a large excess of MSH(4) instead of NDP-α-MSH as the blocking agent in the determination of non-specific binding, and a different brand of 96-well plates. It is unclear which factors, if any, account for the different outcome for probe 2 in the present study.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.