

Abstract
Nitric oxide (NO) has been implicated in the pathogenesis of septic shock. However, the role of NO synthase 3 (NOS3) during sepsis remains incompletely understood. Here, we examined impact of NOS3 deficiency on systemic inflammation and myocardial dysfunction during peritonitis-induced polymicrobial sepsis. Severe polymicrobial sepsis was induced by colon ascendens stent peritonitis (CASP) in wild-type (WT) and NOS3-deficient (NOS3KO) mice. NOS3KO mice exhibited shorter survival time than did WT mice after CASP. NOS3 deficiency worsened systemic inflammation assessed by the expression of inflammatory cytokines in the lung, liver, and heart. CASP markedly increased the number of leukocyte infiltrating the liver and heart in NOS3KO but not in WT mice. The exaggerated systemic inflammation in septic NOS3KO mice was associated with more marked myocardial dysfunction than in WT mice 22h after CASP. The detrimental effects of NOS3-deficiency on myocardial function after CASP appear to be caused by impaired Ca2+ handling of cardiomyocytes. The impaired Ca2+ handling of cardiomyocytes isolated from NOS3KO mice subjected to CASP was associated with depressed mitochondrial ATP production, a determinant of the Ca2+ cycling capacity of sarcoplasmic reticulum (SR) Ca2+-ATPase. The NOS3-deficiency-induced impairment of the ability of mitochondria to produce ATP after CASP was at least in part attributable to reduction in mitochondrial respiratory chain complex I activity. These observations suggest that NOS3 protects against systemic inflammation and myocardial dysfunction after peritonitis-induced polymicrobial sepsis in mice.
Keywords: septic shock, inflammation, cardiomyocyte function, calcium handling, mitochondrial function
INTRODUCTION
Septic shock is a complex syndrome that claims over 200,000 lives per year in the United States (1). Although cytokines and nitric oxide (NO) have been implicated in the pathogenesis of septic shock, the underlying mechanisms of organ dysfunction in sepsis remain incompletely understood.
NO is synthesized by a family of enzymes referred as nitric oxide synthases (NOSs). There are three known isoforms (NOS1, NOS2, and NOS3), that are obligate homodimers that catalyze NADPH-dependent oxidation of L-arginine to NO and L-citrulline. NOS1 (neuronal NOS) is constitutively expressed in neuronal cells and cardiomyocytes. NOS2 (inducible NOS) was first identified in macrophages but has since been detected in a wide variety of cells, typically after exposure to lipopolysaccharide and/or cytokines. NOS3 (endothelial NOS) is constitutively expressed in the endothelial cells, endocardial cells, and cardiomyocytes (2).
High levels of NO produced by NOS2 contribute to the systemic hypotension and organ dysfunction associated with sepsis (3). However, despite the prominent role of NOS2 in cardiovascular dysfunction of sepsis, a clinical trial using NOS inhibitors that are not isoform-specific was associated with increased mortality in septic patients (4). Although these observations indirectly suggest that NOS1 and/or NOS3 may have salutary effects on cardiovascular function in sepsis, role of NOS3 in sepsis remains controversial. Studies showed that NOS3 has no effects, pro-inflammatory, or anti-inflammatory effects in sepsis. For example, Shesely and colleagues reported that NOS3 deficiency does not affect mortality after endotoxin challenge (5). Recent studies showed that endotoxin-induced systemic inflammation and hypotension were attenuated in NOS3KO mice compared with WT mice (6), suggesting a pro-inflammatory role of NOS3 in sepsis. In contrast, NOS3-overexpression in vascular endothelial cells attenuated endotoxin-induced lung injury and mortality in mice (7). While these studies were performed using endotoxin challenge models of sepsis, it has been suggested that a bolus injection of endotoxin is unlikely to reflect the pathophysiology of clinical sepsis (8). Using a clinically-relevant polymicrobial sepsis model, we recently reported that cardiomyocyte-specific overexpression of NOS3 prevents myocardial dysfunction and death after sepsis (9). Nonetheless, role of endogenous levels of NOS3 in septic shock remains to be defined.
The goal of the current study was to elucidate the impact of NOS3 on systemic inflammation and myocardial dysfunction during a clinically-relevant polymicrobial sepsis. Based on our previous study, we hypothesized that NOS3 has anti-inflammatory effects and protects myocardial function during sepsis. To address this hypothesis, we examined the impact of NOS3 deficiency on systemic inflammation and myocardial dysfunction in vivo and in cardiomycytes isolated from mice subjected to peritonitis-induced polymicrobial sepsis. Here, we report that NOS3 protects against systemic inflammation and myocardial dysfunction during polymicrobial sepsis.
MATERIALS AND METHODS
Animals
After approval by the Massachusetts General Hospital Subcommittee on Research Animal Care, all the animal experiments were performed in accordance with the guidelines of the National Institutes of Health. Male WT (C57BL/6) and NOS3KO (Strain Name: B6.129P2-Nos3tm1Unc/J) mice (5) backcrossed onto C57BL/6 background more than 10 generations (2–6 months old, 20–30g) were purchased from the Jackson Laboratory (Bar Harbor, ME) and given access to food and water ad libitum in our animal facility until the time of experiments.
Sepsis model
Colon ascendens stent peritonitis (CASP) operation was performed to induce polymicrobial sepsis as described previously (9, 10). Briefly, under anesthesia (80 mg/kg ketamine and 12 mg/kg xylazine ip) and after disinfection of the abdomen, a 14 gauge stent (14G Angiocath, Becton Dickinson) was placed approximately 10 mm from the ileocecal valve through the anti-mesenteric wall into the lumen of the ascending colon and then fixed with 7–0 stitches. To ensure proper intraluminal positioning of the stent, stool was milked from the cecum into the ascending colon toward the stent until a small drop of stool appeared. Fluid resuscitation of mice was performed by flushing 1.0 ml of sterile saline into the peritoneal cavity before closure of the abdominal walls. Sham operation was performed in the same manner except that only a stitch was made without placing a stent.
Survival analysis
Survival after surgery was assessed in age-matched male WT mice (n = 20) and NOS3KO mice (n = 18). In accordance with the institutional animal care guidelines, a sepsis score developed by Zantl and colleagues (10) was used with some modification to quantitate severity of sepsis, that consists of assessment of 5 clinical parameters (Table 1). A score (0, 1, 2, or 3) was assigned to each item by an investigator blinded to the genotype of mice and type of surgery. Lethal outcome was assumed when mice showed a sepsis score of more than 9 points on a scale ranging from 0 to 15. Mice scoring 10 or more points were euthanized. We chose 9 points based on our pilot studies in which mice that were assigned a sepsis score of 9 or higher showed more than 90% mortality.
Table 1.
Sepsis score
| Parameter | Criteria | scores |
|---|---|---|
|
| ||
| Appearance | ▪normal, smooth fur | 0 |
| ▪roughened fur | 1 | |
| ▪wet fur | 2 | |
| ▪mucous eyes | 3 | |
|
| ||
| Breathing pattern | ▪normal | 0 |
| ▪fast | 1 | |
| ▪slow | 2 | |
| ▪weak and intermittent | 3 | |
|
| ||
| Weight change | ▪−5% | 0 |
| ▪−15% | 1 | |
| ▪−20 % | 2 | |
| ▪> −20% | 3 | |
|
| ||
| Behavior | ▪normal, agile, prying | 0 |
| ▪slow movements, sitting position | 1 | |
| ▪dull, slouched, tottering movements | 2 | |
| ▪lateral position | 3 | |
|
| ||
| Provoked reaction | ▪escape reaction when cage is opened | 0 |
| ▪flight when approached by hand | 1 | |
| ▪flight when touched | 2 | |
| ▪no flight reaction at all | 3 | |
Measurements of gene expression
Total RNA was extracted from heart, lung, and liver tissues of WT and NOS3KO mice at baseline (healthy control) and at 10h (heart, lung, and liver) and 22h (heart) after CASP using the illustra RNA spin Mini kit (GE healthcare) and cDNA was synthesized using MMLV-RT (Promega). TNF-α, IL-1β, IL-6, ICAM-1, NOS1, NOS2 and 18S ribosomal RNA transcript levels were measured by real-time PCR using a Realplex 2 system (Eppendorf, Inc.). Primers used were as follows: TNF-α (5′-CAGCCTCTTCTCATTCCTGC-3′, 5′-GGTCTGGGCCATAGAACTGA-3′), IL-1β (5′-GCAACTGTTCCTGAACTCAACT-3′, 5′-ATCTTTTGGGGTCCGTCAACT-3′), IL-6 (5′-CCGGAGAGGAGACTTCACAGA-3′, 5′-CAGAATTGCCATTGCACAAC-3′), ICAM-1 (5′-TCCGCTGTGCTTTGAGAACT-3′, 5′-AGGGTGAGGTCCTTGCCTAC-3′), NOS1 (5′-GCGGAGAACAGGATGAACTG-3′, 5′-AGGTCTCTGTCCACCTGGATT-3′), NOS2 (5′-GTTCTCAGCCCAACAATACAAGA-3′, 5′-GTGGACGGGTCGATGTCAC-3′) and 18S rRNA (5′-CGGCTACCACATCCAAGGAA-3′, 5′-GCTGGAATTACCGCGGCT-3′). Changes in the relative gene expression normalized to levels of 18S rRNA were determined using the relative CT method. The mean value of samples from control WT mice was set as 1.
Measurements of tissue leukocyte infiltration
Quantification of leukocyte recruitment to the heart and liver 22h after CASP was performed on paraffin sections stained with anti-CD45 monoclonal antibodies (purified rat anti-mouse CD45, BD Biosciences, San Jose, CA). The number of CD45+ cells was manually counted by an investigator blinded to the genotype or treatment in three serial sections at mid-papillary level of the heart and left lobe of the liver per mouse (n=3 for sham-operated and n=5 for mice subjected to CASP for each genotype). The average number of leukocytes per square millimeter of tissue area was reported.
Measurements of cardiac function
For measurement of cardiac function 22h after CASP or sham surgery, mice were anesthetized by intraperitoneal injection with ketamine (100 mg/kg), fentanyl (50 μg/kg), and pancuronium (2 mg/kg), intubated, and mechanically ventilated (fraction of inspired O2 = 1, 10 μl/g, 120 breaths/min). The chest was opened, and a pressure-volume conductance catheter (model SPR-839, Millar Instruments, Houston, TX) was introduced through the apex into the left ventricle (LV), as described previously (9).
Measurement of contractility and intracellular Ca2+ concentration in isolated cardiomyocytes
LV cardiomyocytes were enzymatically isolated from WT and NOS3KO mice 22 h after CASP, as described previously (9). Cardiomyocytes isolated from healthy mice without surgical intervention were used as controls. Cardiomyocytes were loaded with 1 μM fura-2/AM (Invitrogen) and superfused with a buffer containing 137 mM NaCl, 5.4 mM KCl, 1.2 mM CaCl2, 0.5 mM MgCl2, 10 mM HEPES, 5.5 mM glucose and 0.5 mM probenecid, pH 7.4 at 32°C. Cardiomyocytes were paced at 2 Hz, and sarcomere length (SL) and fura-2 fluorescence were recorded simultaneously using the IonWizard video-edge detection and fluorescence system (IonOptix, Milton MA). A subset of cardiomyocytes from each genotype was utilized to calibrate the fura-2 fluorescence ratio to intracellular Ca2+ concentration values in situ.
Measurement of protein expression levels and phosphorylation of phospholamban
Protein extracts from LV tissue homogenates were microcentrifuged for 20 min at 20,000g. Supernatant proteins (30 μg) were fractionated on 10 % SDS-PAGE gels and transferred to nitrocellulose membranes. Membranes were blocked for 1h in 5 % non-fat milk and incubated overnight with primary antibodies against sarcoplasmic reticulum (SR) Ca2+-ATPase2a (Cell Signaling Technology Inc.), total phospholamban (PLN) (Affinity Bioreagents, Inc.), PLN phosphorylated at Ser16 (Upstate Group LLC) and PLN phosphorylated at Thr17 (Badrilla Ltd.). Bound antibodies were detected with a horseradish peroxidase-linked antibody against mouse IgG (Promega Corp.) or rabbit IgG (Cell Signaling Technology Inc.) and were visualized using chemiluminescence with ECL Plus (Amersham Biosciences Corp.).
Isolation of heart mitochondria
Mouse heart mitochondria were isolated 22 h after CASP or sham operation by differential centrifugation using the MITO-ISO1 kit (Sigma-Aldrich, Saint Louis, MO) following the manufacturer’s instruction. A series of centrifugations were performed at 600 g and 6000 g. The mitochondrial pellet was washed once and resuspended in a buffer containing 200 mM mannitol, 70 mM sucrose and 10 mM HEPES (pH 7.4). Protein concentrations were determined by the BCA assay (Pierce, Rockford, IL) using BSA as the standard. Mitochondrial suspensions were kept on ice until use or frozen at − 80 °C for later measurements of respiratory chain enzyme activity.
Measurements of mitochondrial permeability transition (MPT)
MPT pore opening was measured by monitoring the decrease in absorbance at 540 nm associated with mitochondrial swelling (11). Mitochondrial suspensions were diluted to a final concentration of 0.5 mg/ml in a buffer containing 200 mM mannitol, 70 mM sucrose, 10 mM HEPES and 5 mM succinate (pH 7.4). After 5 minutes of initial stabilization, 400 μM CaCl2 was added to induce MPT. The decrease in absorbance was monitored for 1000 seconds at room temperature with a spectrophotometer (BioRad, SmartSpec). The time in seconds was calculated as t1/2 at which one-half of the absorbance change was observed and used as an index of MPT.
Measurement of ATP production by mitochondria
The ability of mitochondria to produce ATP was determined with a luminescence-based assay using the ATP Determination Kit (Invitrogen) according to the manufacturer’s instruction. Aliquots of mitochondria were diluted to a final concentration of 0.002 mg/ml in the reaction solution at room temperature. After a 3-minute stabilization period, 25 μM ADP was added to initiate oxidative phosphorylation. Samples (100 μl each) were transferred to a 96-well microplate and luminescence was monitored for 2 minutes at room temperature using a Synergy 2 microplate reader (BioTek Instruments, Inc., Winooski, VT). The standard curve was generated using known concentrations of ATP and was used to calculate the rate of ATP production.
Complex I activity assay
Mitochondrial respiratory chain complex I activity was determined by spectrophotometrically monitoring the oxidation of NADH in the presence of coenzyme Q1. Aliquots of thawed mitochondria were diluted to a final protein concentration of 0.1 mg/ml in the assay buffer containing 50 mM KCl, 10 mM Tris-HCl (pH 7.4), 1 mM EDTA, 2 mM KCN, 10 μM antimycin A and 100 μM coenzyme Q1 in the presence or absence of 2 μM rotenone. Samples (200 μl each) were transferred to a 96-well microplate and reactions were initiated by addition of 100 μM NADH. The decrease in absorbance at 340 nm was monitored at room temperature for 3 minutes using a Synergy 2 microplate reader. Complex I activity was calculated from the difference between the NADH oxidation rate in the presence and in the absence of rotenone, using an extinction coefficient of 6.22 mM−1·cm−1 at 340 nm (12).
Complex IV activity assay
Mitochondrial respiratory chain complex IV activity was determined by spectrophotometrically monitoring the oxidation of reduced cytochrome c. Aliquots of thawed mitochondria were diluted to a final protein concentration of 0.5 μg/ml in the assay buffer containing 50 mM KH2PO4 (pH 7.2), 0.5 mM dodecyl maltoside. Reactions were initiated by adding 50 μM of reduced cytochrome c (reduced with 1mM DTT). The decrease in absorbance at 550 nm was monitored at room temperature using a spectrophotometer (BioRad, SmartSpec). Complex IV activity was calculated from the initial rate of cytochrome c oxidation, using an extinction coefficient of 18.5 mM−1·cm−1 at 550 nm (13).
Statistics
All data are presented as means ± SE. Data were analyzed by ANOVA using SigmaStat statistical software package (Systat Software, Inc.). Analysis for gene expression levels was performed after log transformation when appropriate. Student-Newman-Keuls post hoc test was performed as required. Kaplan-Meier survival analysis was performed using the Gehan-Breslow method.
RESULTS
NOS3 deficiency shortened survival time after CASP
All mice recovered from anesthesia uneventfully after CASP or sham operation. All mice subjected to CASP developed clinical signs of sepsis including decreased mobility and depressed level of alertness within 12 h after the operation, whereas mice subjected to sham operation did not. While all mice died by 48h after CASP, NOS3KO mice exhibited a shorter survival time than did WT mice (22±1 vs 31±3 h, P=0.023, Kaplan-Meier survival analysis using the Gehan-Breslow method).
NOS3 deficiency worsens systemic inflammation after CASP
To elucidate the impact of NOS3 on systemic inflammation after CASP, we examined expression levels of genes encoding inflammatory cytokines in the lung and liver at 10h and in the heart at 10 and 22h after CASP. Levels of TNF-α, IL-1β, IL-6, ICAM-1, and NOS2 mRNA in the lung and liver were increased at 10h after CASP in both genotypes (Figure 1). NOS3 deficiency markedly augmented the induction of IL-1β, IL-6, ICAM-1, and NOS2 both in the lung and liver at 10h after CASP. Similarly, levels of IL-1β, IL-6, and ICAM-1 mRNA were increased in the heart at 10h after CASP in both genotypes (Figure 2). NOS3 deficiency markedly enhanced the induction of ICAM-1 at 10h and IL-1β and IL-6 in the heart at 22h after CASP. Of note, in the heart, TNF-α was induced only after 22h after CASP, and the expression of NOS1 and NOS2 did not significantly change after CASP. These results suggest that NOS3 deficiency worsens systemic inflammation after CASP.
Figure 1.

Relative gene expression levels in the liver (A) and lung (B) tissue from WT and NOS3KO mice at baseline and 10 h after CASP operation. Gene expression was normalized to 18S rRNA expression level and the mean values for baseline WT mice were set to 1. TNFα, tumor necrosis factor α; IL-1β, interleukin 1β; IL-6, interleukin 6; ICAM-1, intercellular adhesion molecule 1; NOS2, nitric oxide synthase 2. N = 3 – 6 for each group. *P<0.05 vs baseline mice of the same genotype, **P<0.01 vs baseline mice of the same genotype, #P<0.05 vs WT mice after CASP.
Figure 2.

Relative gene expression levels in cardiac tissue from WT and NOS3KO mice at baseline and 10 and 22 h after CASP. Gene expression was normalized to 18S rRNA expression level and the mean values for baseline WT mice were set to 1. A: TNFα, tumor necrosis factor α, B: IL-1β, interleukin 1β, C: IL-6, interleukin 6, D: ICAM-1, intercellular adhesion molecule 1, E: NOS1, nitric oxide synthase 1, F: NOS2, nitric oxide synthase 2. N = 4 – 8 for each group. *P<0.05 vs baseline mice of the same genotype, **P<0.01 vs baseline mice of the same genotype, #P<0.05 vs WT mice after CASP.
NOS3-deficiency increased leukocyte infiltration in heart and liver 22h after CASP
The number of CD45+ cells in the heart and liver did not differ between WT and NOS3KO mice at baseline. In both heart and liver tissues, CASP markedly increased the number of CD45+ cells in NOS3KO mice but not in WT mice (Figure 3). Of note, CD45+ cells were found predominantly in the intravascular space in the liver but more CD45+ cells appeared to be located in the extravascular compartment in the heart of NOS3KO mice after CASP.
Figure 3.
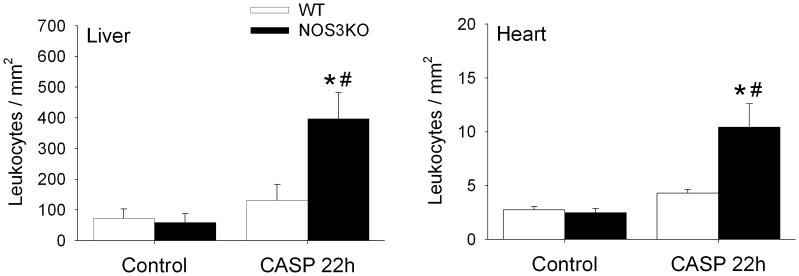
Number of CD45+ cells accumulating in the liver or heart at baseline and 22h after CASP in WT (open bar) and NOS3KO (black bar) mice. *P<0.05 vs same genotype at baseline. #P<0.05 vs WT mice after CASP.
NOS3 deficiency worsens cardiac dysfunction after CASP
Cardiac function parameters after sham operation were similar between both genotypes, except that LV end-systolic pressure (LVESP) was higher in NOS3KO (Table 2). CASP markedly depressed cardiac output (CO) only in NOS3KO but not in WT. CASP decreased the maximum rate of developed LV pressure (dP/dtmax) and ejection fraction (EF) in both genotypes, but the magnitude of reduction was greater in NOS3KO than in WT. Parameters of load-independent LV contractility including dP/dtmax divided by instantaneous pressure (dP/dtmax/IP) and end-systolic elastance (Ees) were markedly depressed in NOS3KO but not in WT after CASP. Myocardial mechanical efficiency (estimated by the ratio of arterial elastance to end-systolic elastance [Ea/Ees]) was impaired (increased ratio) in NOS3KO but not in WT after CASP. As for diastolic function, CASP induced a greater increase of LV end-diastolic pressure (LVEDP) in NOS3KO than in WT, and the time constant of isovolumic relaxation (τ) was markedly prolonged after CASP in NOS3KO but not in WT (Table 2). These results indicate that NOS3 deficiency impairs both systolic and diastolic myocardial functions after CASP.
Table 2.
Hemodynamic measurements 22h after CASP or sham operation
| WT | NOS3KO | |||
|---|---|---|---|---|
| n | sham 5 |
CASP 8 |
sham 6 |
CASP 7 |
| HR, bpm | 567±11 | 567±11 | 538±13 | 547±21 |
| LVESP, mmHg | 111±4 | 77±6 | 131±6# | 84±10* |
| LVEDP, mmHg | 2±0 | 5±1 | 3±0 | 8±2*# |
| dP/dtmax, mmHg/s | 19768±1084 | 11667±1726* | 17637±369 | 6527±795*# |
| dP/dtmin, mmHg/s | −9974±875 | −9809±1071 | −11363±1505 | −5013±728*# |
| EF, % | 61±4 | 45±3* | 69±4 | 34±4*# |
| CO, ml/min | 14.8±1.6 | 17.5±1.8 | 13.5±2.1 | 8.1±1.3*# |
| dP/dtmax/IP, s−1 | 222±8 | 219±8 | 201±15 | 135±21*# |
| Ea, mmHg/μL | 3±2 | 3±1 | 5±1 | 6±1# |
| Ees, mmHg/μL | 27±6 | 16±3 | 30±6 | 10±1* |
| Ea/Ees | 0.1±0.1 | 0.2±0.1 | 0.2±0.1 | 0.6±0.1*# |
| τ, msec | 5.5±0.2 | 4.6±0.2 | 5.5±0.1 | 8.5±1.2*# |
Values are means ± SE. HR, heart rate; LVESP, left ventricular end-systolic pressure; LVEDP, left ventricular end-diastolic pressure; dP/dtmax, maximum rate of developed left ventricular pressure; dP/dtmin, minimum rate of developed left ventricular pressure; EF, ejection fraction; CO, cardiac output; dP/dtmax/IP, dP/dtmax divided by instantaneous pressure; Ea, arterial elastance; Ees, left ventricular end-systolic elastance; Ea/Ees, ratio of Ea to Ees; τ, time constant of isovolumic relaxation.
P<0.05 vs sham-operated mice of the same genotype.
P<0.05 vs WT mice with the same operation.
NOS3 deficiency worsens the contractile function and Ca2+ handling of cardiomyocytes isolated from mice subjected to CASP
To further evaluate myocardial dysfunction after CASP in the absence of extracardiac factors, we studied the contractile function and Ca2+ handling of isolated cardiomyocytes. The yield of calcium-tolerant cardiomyocyte (40–50%) did not differ between WT and NOS3KO with or without CASP. While CASP decreased sarcomere length (SL) shortening and peak amplitude of intracellular Ca2+ transients (Δ [Ca2+]i) of isolated cardiomyocytes in both genotypes, the magnitude of decrease was greater in NOS3KO than in WT (Figure 4 A–C). Although CASP prolonged the time constants for SL relengthening and [Ca2+]i decay in both genotypes, the degree of prolongation was more marked in NOS3KO than in WT (Figure 4D, 4E). CASP decreased SL shortening and peak [Ca2+]i in a parallel manner between both genotypes (Figure 4F). These observations suggest that more marked depression of cardiomyocyte function in NOS3KO mice is due to a greater impairment of Ca2+ handling but not to altered myofilament sensitivity to calcium (Figure 4F).
Figure 4.

Contractility and Ca2+ handling of isolated cardiomyocytes 22h after CASP. A: Representative tracings of changes in sarcomere length (SL) and intracellular Ca2+ transients ([Ca2+]i) of isolated cardiomyocytes paced at 2 Hz. B–E: Effects of CASP on the contractile function and Ca2+ handling of isolated cardiomyocytes paced at 2 Hz. Data were summarized for peak percent SL shortening (B), peak amplitude of [Ca2+]i (Δ[Ca2+]i) (C), the time constant for SL relengthening (D) and the time constant for [Ca2+]i decay (E). 20–25 cells from 3 – 5 mice were analyzed for each group. *P<0.05 vs cardiomyocytes from control mice of the same genotype, #P<0.05 vs cardiomyocytes from WT mice after CASP. F: Effects of CASP on relationship of peak systolic [Ca2+] and %SL shortening.
NOS3 deficiency does not affect SR Ca2+-ATPase protein levels and phospholamban phosphorylation
Sarcoplasmic reticulum (SR) Ca2+-ATPase activity is a major determinant of cardiomyocyte calcium handling and is inhibited by phospholamban. Phosphorylation of phospholamban releases the inhibition of SR Ca2+-ATPase activity. Protein expression levels of SR Ca2+-ATPase did not differ between WT and NOS3KO with or without CASP (Figure 5A). Because we found a greater impairment of Ca2+ handling after CASP in NOS3KO heart, impact of CASP on phosphorylation of phospholamban at Ser16 and Thr17 in cardiac tissue extracts was examined. There was no difference in the fraction of PLN phosphorylated at Ser16 or Thr17 between genotypes after CASP or sham operation (Figure 5B, 5C). These results suggest that deleterious effects of NOS3 deficiency on SR Ca2+ handling after CASP are not mediated by altered PLN phosphorylation.
Figure 5.

Immunoblot analysis. Protein expression of SR Ca2+-ATPase2a (A), fractions of phospholamban phosphorylated at Ser16 (PLN Ser16 (B)) or Thr17 (PLN Thr17 (C)) in cardiac tissue extracts from WT and NOS3KO mice 22 h after CASP or sham operation are shown with loading control (β-tubulin). Data were normalized to WT sham groups. Representative blots from 2 – 4 mice for each group are shown. N = 6 – 8 for each group.
CASP impairs cardiac mitochondrial integrity in NOS3KO mice
Ca2+ sequestration by SR Ca2+-ATPase is an energy consuming process that depends on local energy levels primarily maintained by mitochondrial ATP production. To examine whether or not mitochondrial dysfunction contributes to the sepsis-induced Ca2+ mishandling of cardiomyocytes in our model, we investigated cardiac mitochondrial integrity and function 22h after CASP. CASP decreased t1/2 of the Ca2+-induced relative absorbance change in NOS3KO but not in WT mitochondria. There was a trend towards a faster occurrence of MPT in the mitochondria of NOS3KO than in WT after CASP (P = 0.094, WT CASP vs NOS3KO CASP; Figure 6A, 6B).
Figure 6.

Measurements of Mitochondrial Permeability Transition (MPT) in isolated mitochondria 22h after CASP or sham operation. A: Representative tracings of absorbance change. Absorbance values at the beginning and the end of measurement were set to 1 and 0, respectively. B: Effects of CASP on the time to one-half of the absorbance change (t1/2) in WT and NOS3KO after CASP or sham operation. N = 9 – 10 for each group. *P<0.05 vs NOS3KO after sham operation.
CASP impairs the ability of mitochondria to produce ATP
To further investigate the impact of sepsis on mitochondrial function, we studied mitochondrial ATP production in cardiac mitochondria isolated from mice subjected to CASP or sham operation. The yield of mitochondrial protein per tissue weight did not differ between WT and NOS3KO with or without CASP. There was no difference in the ability to produce ATP between mitochondria isolated from WT and NOS3KO after sham operation (Figure 7A). In contrast, NOS3 deficiency markedly impaired the ability of isolated mitochondria to produce ATP after CASP.
Figure 7.

Inhibition of oxidative phosphorylation in cardiac mitochondria after CASP. A: ATP production ability in cardiac mitochondria 22 h after CASP or sham operation. N = 7 – 9 for CASP groups and N = 3 for sham groups. B and C: Respiratory chain enzyme activities (complex I (B) and complex IV (C)) in cardiac mitochondria 22 h after CASP or sham operation. N= 5 – 7 for each group. *P<0.05 vs WT after sham operation, **P<0.01 vs NOS3KO after sham operation, #P<0.05 vs WT after CASP.
To elucidate the mechanism responsible for the CASP-induced inhibition of mitochondrial ATP production, we measured the activities of mitochondrial respiratory chain complex I and complex IV in a subgroup of mice 22 h after CASP or sham operation. Although mitochondria isolated 22 h after sham operation showed comparable complex I activity in both genotypes, it was lower in NOS3KO than in WT 22 h after CASP (Figure 7B). On the other hand, complex IV activity did not change after CASP in both genotypes (Figure 7C). These results indicate that NOS3 deficiency worsens the ability of mitochondria to produce ATP after CASP at least in part via a greater inhibition of complex I, but not of complex IV activity.
DISCUSSION
The current study revealed that NOS3 deficiency worsens sepsis-induced systemic inflammation in a murine model of peritonitis-induced polymicrobial sepsis. The pronounced systemic inflammation in NOS3KO mice was associated with worsened myocardial function after CASP compared to WT mice. The detrimental effects of NOS3-deficiency on myocardial function appear to be caused by impaired Ca2+ handling of isolated cardiomyocytes obtained from mice subjected to CASP. Depressed Ca2+ handling of cardiomyocytes of NOS3KO mice were associated with impairment of mitochondrial integrity and marked depression of the ability of mitochondria to produced ATP, a determinant of the function of the SR Ca2+-ATPase pump. Taken together, these observations suggest that endogenous level of NOS3 confer protection against polymicrobial sepsis-induced inflammation and myocardial dysfunction.
Generalized endothelial dysfunction, with exaggerated coagulation and inflammation, is a hallmark of severe sepsis and septic shock. Since NO has been shown to inhibit activation of platelets and neutrophils (14, 15), role of NOS3 in sepsis has been intensively studied. Although the use of genetically-modified mouse models greatly advanced our understanding of the impact of NOS3 in sepsis, majority of the studies have been done in murine endotoxin challenge models. Endotoxin, the lipopolysaccharide (LPS) associated with outer membrane of Gram-negative microorganisms, is a homogeneous bacterial product that triggers inflammatory reaction in a dose-dependent and reproducible manner (16). However, the overall response to polymicrobial infection, such as that caused by ruptured bowel and trauma, is far more complex than the effects of LPS alone, raising the question of the validity of murine LPS challenge as a model of human sepsis (17). In fact, majority of the results obtained from mouse LPS challenge models were not reproduced in human clinical trials (18).
In the current study, we therefore opted to use the colon ascendens stent peritonitis (CASP) model. Polymicrobial (Gram-positive and Gram-negative) fecal peritonitis induced by CASP closely mimics the clinical course of diffuse peritonitis with early and steadily increasing systemic infection and inflammation (10). We chose the CASP model over another peritonitis model, cecum ligation and perforation (CLP), because the former model has been shown to cause more severe diffuse peritonitis in a more reproducible manner than CLP (19). We found CASP induced inflammatory cytokine expression in the heart, liver, and lung in mice. In the heart, inflammatory cytokine expression increased in a time-dependent manner over 22hr after CASP. NOS3 deficiency markedly enhanced the pro-inflammatory cytokine induction after CASP. The pro-inflammatory effects of NOS3 deficiency were confirmed by the increased leukocyte infiltration in the liver and heart of NOS3KO, but not in WT mice, 22h after CASP. These results show that endogenous levels of NOS3 confer anti-inflammatory effects in a clinically-relevant polymicrobial model of sepsis.
Interestingly, NOS2 gene expression was markedly increased in the liver and lung 10h after CASP and it was further enhanced by NOS3 deficiency. These results contradict with the reports of Connelly and colleagues who demonstrated that LPS-induced NOS2 protein expression in the liver, lung, heart, and aorta was attenuated by NOS3 deficiency (6). Reasons for this discrepancy are unknown but likely to be multifactorial. For example, time courses of cytokine induction after bolus administration of LPS and peritonitis-induced sepsis are different; the former induces cytokines much earlier than the latter (19). It is possible that NO derived from NOS3 may have pro-inflammatory effects early after the onset of endotoxemia while exert anti-inflammatory impact in late phases of sepsis. It is also conceivable that NO derived from NOS3 plays different roles in infectious process caused by Gram-negative vs Gram-positive bacteria. A recent human study suggested that a loss-of-function NOS3 polymorphism (G→T, Glu298Asp) has differing hemodynamic effects during Gram-negative and Gram-positive bacteremia (20).
Pro-inflammatory cytokines including IL-1β have been implicated as cardio-depressants responsible for septic cardiomyopathy, and their plasma levels correlate with patient survival during sepsis (21). The enhanced pro-inflammatory cytokine expression in the heart of NOS3KO mice in response to polymicrobial infection is likely to be one of the mechanisms that contribute to the depressed myocardial function and shortened survival time in NOS3KO mice after CASP. Although precise mechanisms by which cytokines depress myocardial function during sepsis remain incompletely understood, a number of hypothesis have been proposed including impaired Ca2+ cycling (i.e., depressed Ca2+ release and uptake) (22), decreased myofilament sensitivity to Ca2+ (23), and decreased ATP production by mitochondria (24). In the current study, we found parallel reductions of Ca2+ transient amplitude and sarcomere length change in isolated cardiomyocytes of both genotypes after CASP, but the magnitudes of reduction in both parameters were greater in NOS3KO than in WT cardiomyocytes (see Figure 4F). These observations suggest that deleterious effects of NOS3 deficiency on cardiomyocyte contraction during sepsis are primarily caused by more marked impairment of Ca2+ cycling, but not by a greater depression in myofilament sensitivity to Ca2+.
The SR Ca2+-ATPase is the central regulator of Ca2+ cycling in cardiomyocytes. Although we found that NOS3 deficiency does not affect the phosphorylation of PLN and SR Ca2+-ATPase protein levels in cardiac tissue extracts after CASP, SR Ca2+-ATPase activity can be modulated by multiple factors including local energy state (i.e., ATP supply/demand balance)(25). In fact, energy deficiency caused by reduced mitochondrial content has been shown to play a significant role in the pathogenesis of cardiac dysfunction (26). To elucidate the mechanism responsible for the depressed cardiomyocyte function in NOS3KO after CASP, we therefore examined the impact of NOS3 on cardiac mitochondorial integrity and function.
Mitochondrial permeability transition has been implicated as a cause of organ dysfunction and cell death in sepsis (27). The current study demonstrates that marked cardiac dysfunction in NOS3KO mice after CASP is associated with an accelerated MPT. These observations are in line with the findings of Larche and colleagues who showed that sepsis-induced cardiac dysfunction was associated with accelerated MPT, and inhibition of MPT with cyclosporine A improved cardiac function after sepsis (28). The loss of mitochondrial integrity demonstrated as accelerated MPT in septic NOS3KO mice is also consistent with the markedly reduced ability of NOS3-deficient mitochondria to produced ATP after CASP. It is conceivable that the impairment of mitochondrial integrity and function depressed local ATP supply leading to impair the Ca2+ cycling function of the SR Ca2+-ATPase in NOS3-deficient cardiomyocytes.
The decreased ability to produce ATP in NOS3KO mice was at least in part attributable to depressed complex I but not complex IV activity in the current study. Mitochondrial respiratory chain complexes can be inhibited by reactive oxygen species (ROS) and/or reactive nitrogen species (RNS) (29). Although NO can directly or indirectly inhibit complex I, NO derived from NOS1 or NOS2 is unlikely to be the primary cause of the greater inhibition of complex I activity in NOS3KO than in WT mice after CASP; CASP did not affect cardiac NOS1 and NOS2 expression (see Figure 2) and nitrate/nitrite levels in the heart were similar in both genotypes after CASP (data not shown). Alternatively, it is possible that CASP induced higher levels of ROS in the hearts of NOS3KO than in WT, leading to a greater inhibition of complex I activity (30). Damaged mitochondria in NOS3KO hearts, as evidenced by an accelerated MPT, is a possible source of increased ROS production after CASP. In addition, the increased number of leukocytes in the NOS3KO hearts after CASP is likely to be another source of increased ROS production.
How does NOS3 protect heart during sepsis? NOS3 primarily resides in the plasmalemmal caveolae in the vicinity of SR-T-tubule junction and modulates functions of plasmalemmal ion channels (e.g., L-type Ca2+ channel) and SR calcium channels (e.g., ryanodine receptor) by protein S-nitrosylation (31). Inflammation and ROS are thought to depress myocardial contractility by causing oxidative modifications of ion channels of cardiomyocytes (32). It is tempting to speculate that S-nitrosylation induced by NOS3 protects ion channels from the harmful oxidative modifications. Alternatively, while it is controversial, NOS3 has been suggested as a candidate for mitochondrial NOS (33). It is possible that NOS3 plays important role in regulating MPT and ROS production in mitochondria. The molecular mechanisms responsible for the protective effects of NOS3 on cardiomyocyte function during sepsis remains to be determined.
One limitation in the current study is that we measured mitochondrial functions in isolated mitochondria. The differential centrifugation technique only isolates mitochondria of known density and size (34). Mitochondria damaged and swollen during the inflammatory process in sepsis might be largely excluded, leaving only relatively normal mitochondria. Therefore, our measurements are likely to underestimate the CASP-induced mitochondrial dysfunction. In addition, several studies have demonstrated that NOS3 deficiency reduces mitochondrial mass in the heart tissue (35). This implicates that a similar degree of mitochondrial dysfunction could have a greater impact on myocardial dysfunction in NOS3KO than in WT mice. These factors may partly explain that the observed difference between genotypes in CASP-induced mitochondrial dysfunction was relatively small compared with that in isolated cell functions or hemodynamic study in vivo.
In summary, the current study demonstrates NOS3 deficiency aggravates sepsis-induced systemic inflammation and myocardial dysfunction in murine polymicrobial sepsis. NOS3 deficiency worsens Ca2+ handling of cardiomyocytes after sepsis and that is associated with depressed mitochondrial ATP production in the heart. These results demonstrate a protective role for NOS3 in clinically-relevant polymicrobial sepsis, and implicate that the selective modulation of NOS3 function could be a therapeutic target for prevention of sepsis-induced systemic inflammation, myocardial dysfunction, and death.
Acknowledgments
Funding information: This work was supported by grants from National Institute of Health HL71987 and GM79360 to Dr. Ichinose.
Authors thank Dr. Kenneth D. Bloch for valuable comments.
References
- 1.Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29:1303–1310. doi: 10.1097/00003246-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Moncada S, Higgs A. The L-arginine-nitric oxide pathway. N Engl J Med. 1993;329:2002–2012. doi: 10.1056/NEJM199312303292706. [DOI] [PubMed] [Google Scholar]
- 3.Ullrich R, Scherrer-Crosbie M, Bloch KD, Ichinose F, Nakajima H, Picard MH, Zapol WM, Quezado ZM. Congenital deficiency of nitric oxide synthase 2 protects against endotoxin-induced myocardial dysfunction in mice. Circulation. 2000;102:1440–1446. doi: 10.1161/01.cir.102.12.1440. [DOI] [PubMed] [Google Scholar]
- 4.Lopez A, Lorente JA, Steingrub J, Bakker J, McLuckie A, Willatts S, Brockway M, Anzueto A, Holzapfel L, Breen D, Silverman MS, Takala J, Donaldson J, Arneson C, Grove G, Grossman S, Grover R. Multiple-center, randomized, placebo-controlled, double-blind study of the nitric oxide synthase inhibitor 546C88: effect on survival in patients with septic shock. Crit Care Med. 2004;32:21–30. doi: 10.1097/01.CCM.0000105581.01815.C6. [DOI] [PubMed] [Google Scholar]
- 5.Shesely EG, Maeda N, Kim HS, Desai KM, Krege JH, Laubach VE, Sherman PA, Sessa WC, Smithies O. Elevated blood pressures in mice lacking endothelial nitric oxide synthase. PNAS. 1996;93:13176–13181. doi: 10.1073/pnas.93.23.13176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Connelly L, Madhani M, Hobbs AJ. Resistance to Endotoxic Shock in Endothelial Nitric-oxide Synthase (eNOS) Knock-out Mice: A PRO-INFLAMMATORY ROLE FOR eNOS-DERIVED NO IN VIVO. Journal of Biological Chemistry. 2005;280:10040–10046. doi: 10.1074/jbc.M411991200. [DOI] [PubMed] [Google Scholar]
- 7.Yamashita T, Kawashima S, Ohashi Y, Ozaki M, Ueyama T, Ishida T, Inoue N, Hirata Ki, Akita H, Yokoyama M. Resistance to Endotoxin Shock in Transgenic Mice Overexpressing Endothelial Nitric Oxide Synthase. Circulation. 2000;101:931–937. doi: 10.1161/01.cir.101.8.931. [DOI] [PubMed] [Google Scholar]
- 8.Fink MP, Heard SO. Laboratory models of sepsis and septic shock. J Surg Res. 1990;49:186–196. doi: 10.1016/0022-4804(90)90260-9. [DOI] [PubMed] [Google Scholar]
- 9.Ichinose F, Buys ES, Neilan TG, Furutani EM, Morgan JG, Jassal DS, Graveline AR, Searles RJ, Lim CC, Kaneki M, Picard MH, Scherrer-Crosbie M, Janssens S, Liao R, Bloch KD. Cardiomyocyte-Specific Overexpression of Nitric Oxide Synthase 3 Prevents Myocardial Dysfunction in Murine Models of Septic Shock. Circ Res. 2007;100:130–139. doi: 10.1161/01.RES.0000253888.09574.7a. [DOI] [PubMed] [Google Scholar]
- 10.Zantl N, Uebe A, Neumann B, Wagner H, Siewert JR, Holzmann B, Heidecke CD, Pfeffer K. Essential role of gamma interferon in survival of colon ascendens stent peritonitis, a novel murine model of abdominal sepsis. Infect Immun. 1998;66:2300–2309. doi: 10.1128/iai.66.5.2300-2309.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Van Remmen H, Williams MD, Guo Z, Estlack L, Yang H, Carlson EJ, Epstein CJ, Huang TT, Richardson A. Knockout mice heterozygous for Sod2 show alterations in cardiac mitochondrial function and apoptosis. AJP - Heart and Circulatory Physiology. 2001;281:H1422–H1432. doi: 10.1152/ajpheart.2001.281.3.H1422. [DOI] [PubMed] [Google Scholar]
- 12.Merker MP, Audi SH, Lindemer BJ, Krenz GS, Bongard RD. Role of mitochondrial electron transport complex I in coenzyme Q1 reduction by intact pulmonary arterial endothelial cells and the effect of hyperoxia. Am J Physiol Lung Cell Mol Physiol. 2007;293:L809–L819. doi: 10.1152/ajplung.00448.2006. [DOI] [PubMed] [Google Scholar]
- 13.Zhao X, Chen YR, He G, Zhang A, Druhan LJ, Strauch AR, Zweier JL. Endothelial nitric oxide synthase (NOS3) knockout decreases NOS2 induction, limiting hyperoxygenation and conferring protection in the postischemic heart. AJP - Heart and Circulatory Physiology. 2007;292:H1541–H1550. doi: 10.1152/ajpheart.00264.2006. [DOI] [PubMed] [Google Scholar]
- 14.Mellion BT, Ignarro LJ, Myers CB, Ohlstein EH, Ballot BA, Hyman AL, Kadowitz PJ. Inhibition of human platelet aggregation by S-nitrosothiols. Heme- dependent activation of soluble guanylate cyclase and stimulation of cyclic GMP accumulation. Mol Pharmacol. 1983;23:653–664. [PubMed] [Google Scholar]
- 15.Clancy RM, Leszczynska-Piziak J, Abramson SB. Nitric oxide, an endothelial cell relaxation factor, inhibits neutrophil superoxide anion production via a direct action on the NADPH oxidase. J Clin Invest. 1992;90:1116–1121. doi: 10.1172/JCI115929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ulevitch RJ. Molecular mechanisms of innate immunity. Immunol Res. 2000;21:49–54. doi: 10.1385/IR:21:2-3:49. [DOI] [PubMed] [Google Scholar]
- 17.Redl H, Bahrami S, Schlag G, Traber DL. Clinical detection of LPS and animal models of endotoxemia. Immunobiology. 1993;187:330–345. doi: 10.1016/S0171-2985(11)80348-7. [DOI] [PubMed] [Google Scholar]
- 18.Rittirsch D, Hoesel LM, Ward PA. The disconnect between animal models of sepsis and human sepsis. J Leukoc Biol. 2007;81:137–143. doi: 10.1189/jlb.0806542. [DOI] [PubMed] [Google Scholar]
- 19.Maier S, Traeger T, Entleutner M, Westerholt A, Kleist B, Huser N, Holzmann B, Stier A, Pfeffer K, Heidecke CD. Cecal ligation and puncture versus colon ascendens stent peritonitis: two distinct animal models for polymicrobial sepsis. Shock. 2004;21:505–511. doi: 10.1097/01.shk.0000126906.52367.dd. [DOI] [PubMed] [Google Scholar]
- 20.Huttunen R, Hurme M, Laine J, Eklund C, Vuento R, Aittoniemi J, Huhtala H, Syrjanen J. Endothelial nitric oxide synthase G894T (GLU298ASP) polymorphism is associated with hypotension in patients with E. coli bacteremia but not in bacteremia caused by a gram-positive organism. Shock. 2009;31:448–453. doi: 10.1097/SHK.0b013e318188e58e. [DOI] [PubMed] [Google Scholar]
- 21.Parrillo JE. Pathogenetic mechanisms of septic shock. N Engl J Med. 1993;328:1471–1477. doi: 10.1056/NEJM199305203282008. [DOI] [PubMed] [Google Scholar]
- 22.Dong LW, Wu LL, Ji Y, Liu MS. Impairment of the ryanodine-sensitive calcium release channels in the cardiac sarcoplasmic reticulum and its underlying mechanism during the hypodynamic phase of sepsis. Shock. 2001;16:33–39. doi: 10.1097/00024382-200116010-00007. [DOI] [PubMed] [Google Scholar]
- 23.Kumar A, Krieger A, Symeoneides S, Kumar A, Parrillo JE. Myocardial dysfunction in septic shock: Part II. Role of cytokines and nitric oxide. J Cardiothorac Vasc Anesth. 2001;15:485–511. doi: 10.1053/jcan.2001.25003. [DOI] [PubMed] [Google Scholar]
- 24.Tavener SA, Long EM, Robbins SM, McRae KM, Van Remmen H, Kubes P. Immune Cell Toll-Like Receptor 4 Is Required for Cardiac Myocyte Impairment During Endotoxemia. Circ Res. 2004;95:700–707. doi: 10.1161/01.RES.0000144175.70140.8c. [DOI] [PubMed] [Google Scholar]
- 25.Levy RJ. Mitochondrial dysfunction, bioenergetic impairment, and metabolic down-regulation in sepsis. Shock. 2007;28:24–28. doi: 10.1097/01.shk.0000235089.30550.2d. [DOI] [PubMed] [Google Scholar]
- 26.Joubert F, Wilding JR, Fortin D, Domergue-Dupont Vr, Novotova M, Ventura-Clapier Re, Veksler V. Local energetic regulation of sarcoplasmic and myosin ATPase is differently impaired in rats with heart failure. The Journal of Physiology. 2008;586:5181–5192. doi: 10.1113/jphysiol.2008.157677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 28.Larche J, Lancel S, Hassoun SM, Favory R, Decoster B, Marchetti P, Chopin C, Neviere R. Inhibition of Mitochondrial Permeability Transition Prevents Sepsis-Induced Myocardial Dysfunction and Mortality. Journal of the American College of Cardiology. 2006;48:377–385. doi: 10.1016/j.jacc.2006.02.069. [DOI] [PubMed] [Google Scholar]
- 29.Brown GC, Borutaite V. Nitric oxide and mitochondrial respiration in the heart. Cardiovasc Res. 2007;75:283–290. doi: 10.1016/j.cardiores.2007.03.022. [DOI] [PubMed] [Google Scholar]
- 30.Frost MT, Wang Q, Moncada S, Singer M. Hypoxia accelerates nitric oxide-dependent inhibition of mitochondrial complex I in activated macrophages. Am J Physiol Regul Integr Comp Physiol. 2005;288:R394–R400. doi: 10.1152/ajpregu.00504.2004. [DOI] [PubMed] [Google Scholar]
- 31.Petroff MGV, Kim SH, Pepe S, Dessy C, Marban E, Balligand JL, Sollott SJ. Endogenous nitric oxide mechanisms mediate the stretch dependence of Ca2+ release in cardiomyocytes. Nat Cell Biol. 2001;3:867–873. doi: 10.1038/ncb1001-867. [DOI] [PubMed] [Google Scholar]
- 32.Terentyev D, Gyorke I, Belevych AE, Terentyeva R, Sridhar A, Nishijima Y, Carcache de Blanco E, Khanna S, Sen CK, Cardounel AJ, Carnes CA, Gyorke S. Redox Modification of Ryanodine Receptors Contributes to Sarcoplasmic Reticulum Ca2+ Leak in Chronic Heart Failure. Circ Res. 2008;103:1466–1472. doi: 10.1161/CIRCRESAHA.108.184457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zanella B, Giordano E, Muscari C, Zini M, Guarnieri C. Nitric oxide synthase activity in rat cardiac mitochondria. Basic Res Cardiol. 2004;99:159–164. doi: 10.1007/s00395-003-0454-3. [DOI] [PubMed] [Google Scholar]
- 34.Singer M. Mitochondrial function in sepsis: acute phase versus multiple organ failure. Crit Care Med. 2007;35:S441–S448. doi: 10.1097/01.CCM.0000278049.48333.78. [DOI] [PubMed] [Google Scholar]
- 35.Nisoli E, Clementi E, Paolucci C, Cozzi V, Tonello C, Sciorati C, Bracale R, Valerio A, Francolini M, Moncada S, Carruba MO. Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science. 2003;299:896–899. doi: 10.1126/science.1079368. [DOI] [PubMed] [Google Scholar]