

Abstract
Background
Autosomal dominant hypercholesterolemia (ADH), characterized by elevated plasma levels of low density lipoprotein-cholesterol (LDL-C), is caused by variants in at least three different genes:LDL receptor (LDLR), apolipoprotein B-100 (APOB), and proprotein convertase subtilisin-like kexin type 9 (PCSK9). There is paucity of data about the molecular basis of ADH among ethnic groups other than those of European or Japanese descent. Here, we examined the molecular basis of ADH in a multi-ethnic patient cohort from lipid clinics in a large urban U.S. city.
Methods and Results
A total of 38 males and 53 females, age 22 to 76 years, met modified Simon-Broome criteria for ADH and were screened for mutations in the exons and consensus splice sites of LDLR, and in selected exons of APOB and PCSK9. Deletions and duplications of LDLR exons were detected with multiplex ligation-dependent probe amplification. Heterozygous variants in LDLR were identified in 30 patients and in APOB in one patient. The remaining 60 patients (65%) had “unexplained ADH.” A higher proportion of African Americans (77%) than either non-Hispanic whites (57%) or Hispanics (53%) had “unexplained ADH.” As compared to patients with LDLR variants, those with “unexplained ADH” had lower levels of LDL-C (292 ± 47vs 239 ± 42 mg/dL, respectively; p < 0.0001) and higher levels of HDL-cholesterol (45 ± 12vs 54 ± 13 mg/dL, respectively, p = 0.003).
Conclusions
Our findings suggest that additional loci may contribute to ADH, especially in understudied populations such as African Americans.
Keywords: familial hypercholesterolemia, genetics, lipids, lipoproteins
Introduction
Elevated plasma levels of low-density lipoprotein cholesterol (LDL-C) are an important contributor to the pathogenesis of coronary heart disease (CHD), one of the major causes of morbidity and mortality in the U. S. 1. Approximately 50% of the inter-individualvariation in plasma levels of LDL-C isattributable to genetic variation2, 3. The major portionof this genetic variation is polygenic, reflecting thecumulative effects of multiple sequence variants in anygiven individual2, 4.
However, a subset of patients have monogenic forms of hypercholesterolemia with an autosomal dominant inheritance pattern 5. Patients with autosomal dominant hypercholesterolemia (ADH) present withvery high LDL-C levels accompanied by the accumulation of cholesterol in tendons (tendon xanthomas) and premature CHD. ADH is genetically heterogeneous and is caused by variants in at least three different genes. Those with mutations in LDL receptor (LDLR) are termedfamilial hypercholesterolemia (FH, OMIM # 143890), those with apolipoprotein B-100 mutations (APOB, OMIM # 107730)are termed familial defective apolipoprotein B-100 (FDB), and those with gain-of-function mutations in proprotein convertase subtilisin-like kexin type 9 (PCSK9), which encodes a secreted protease that mediates degradation of the LDLR by interacting with the extracellular domain6, are termedfamilial hypercholesterolemia 3 (FH3, OMIM # 603776).
Systematic screenings to identify mutations in the known genes causing ADH have been undertaken in several countries through nationwide screening programs and lipid clinic networks. These studies reported between 43–90% of the ADH patients had mutationsin LDLR, 3.75–12% in APOB, and 0.7–1.7%in PCSK97–15. Approximately 5%–53% of patients with a clinical phenotype of ADH do not have mutations in any of the three known loci responsible for ADH. The frequency of finding the responsible mutation was much greater in children from the Netherlands, where a gene defect was identified in 95% of the subjects14. In addition, populations such as the Afrikaners 16, Christian Lebanese 17, French-Canadians 18, Ashkenazi Jews 19, and Old-Order Amish 20 have a high prevalence of LDLR or APOB variants due to founder effects. Based on studies with the most comprehensive protocols, Varet et al21estimated that 15%–25% of ADH patients in the general population do not have mutations in the known loci.
Most of thesescreening studies have been performed in Europe or in Japan andare from relatively homogenous populations with regards to ethnicity and race. As a result, there is a paucity of data in subjects of African origin and other minorities. Therefore, we examined the molecular basis of ADH in a multi-ethnic cohortfrom a large urban U.S. city.
Methods
Patients
All patients and their family members gave written informed consent, and the Institutional Review Board of UT Southwestern Medical Center approved the protocol. ADH patients were ascertained from specialty lipid clinics in the Dallas, Texas area according to modified Simon-Broome criteria 22. Specifically, pretreatment LDL-Cwas greater than the 95th percentile for age and sex, with one of the two following criteria:1) tendon xanthoma (proband or 1st degree relative), or 2) either 1st degree relative with premature CHD (less than 55 years in males or 65 years in females) or pretreatment LDL-Cgreater than the 95th percentile for age and sex. When 1st degree relatives were not available to participate in the study, an autosomal dominant inheritance pattern was inferred based on family history of hypercholesterolemia in more than one generation.
Ethnicity/race was self-reported according to the guidelines of the National Institutes of Health. In addition, patients were allowed to report their maternal and paternal ancestral origins.
Detailed family historyregarding hypercholesterolemia, CHD, cerebrovascular accidents, and xanthomas was obtained. Patients were examined for the presence of arcussenilisand tendon xanthomas. A grading scheme was used for arcussenilisas previously described23. Xanthomas were considered to be present if tendons appeared diffusely enlarged or had focal nodularity. The Achilles tendon width was calculated from the mean of measurements using Lange calipers (Beta Technology Incorporated, Cambridge, Maryland, USA) in the superior, middle, and inferior aspects bilaterally.
Exclusion criteria for the study included:pretreatment serum triglyceride concentrationsof >500 mg/dL, or any secondary cause of the dyslipidemia (i.e.uncontrolled diabetes mellitus, obstructive liver disease, hypothyroidism or nephrotic syndrome).
DNA Isolation
Genomic DNA was isolated fromwhole blood, using the Easy-DNA kit (Invitrogen, Carlsbad, California, USA).
Candidate Gene Analysis
All 18 exons and the flanking intronic regions of LDLR, exon 26 of APOB, and exons 2, 4, and 7 of PCSK9 were amplified and sequenced using Applied Biosystems 3730xl DNA Analyzer (Applied Biosystems, Carlsbad, California, USA) Deletions and duplications of one or more exons of LDLRwere detected with the SALSA Multiplex Ligation-dependent Probe Amplification (MLPA) kit (MRC-Holland, the Netherlands) as previously described24, and data were analyzed with Gene Marker software from Soft Genetics Version 1.95 (State College, PA, USA). The genes were sequenced sequentially: LDLR, APOB, and then the search for a deletion/duplication of LDLR. Finally, PCSK9was sequenced.
Apolipoprotein E Genotyping
Specific 5′-nucleotidase assays for the ApoE sequence variations were developed using the TaqMan system (Applied Biosystems). The assays were performed on an HT7900 Real-Time PCR system with probes and reagents purchased from Applied Biosystems. For the ApoE_112 variant, two primers (forward 5′-CGGAACTGGAGGAACAACTGA-3′ and reverse 5′-GGTGCTCTGGCCGAGCAT-3′) and two fluorogenic MGB probes (fam-TGGAGGACGTGCGC and vic-ATGGAGGACGTGTGC) were used to amplify either the wild type or mutant allele. Similarly, for the ApoE_158 variant, two primers (forward 5′-TCCCACCTGCGCAAGCT-3′ and reverse 5′-CCCCGGCCTGGTACACT-3′) and two fluorogenic MGB probes (fam-CTGCAGAAGTGCC and vic-CTGCAGAAGCGCCT) were used. For both variants, amplification consisted of 2 min at 50 degrees, 10 min at 95 degrees, and 40 cycles of 95 degrees for 15 sec and 60 degrees for 1 min.
Lipids and Lipoproteins
Medical charts were reviewed to obtain untreated lipid levels. All centers measured fasting total cholesterol, triglycerides and high density lipoprotein (HDL)-cholesterol using enzymatic assays in commercial laboratories. LDL-C was estimated with the Friedewald equationwhen serum triglycerides were less than 500 mg/dL.
Historical untreated serum lipid values were not available in six patients; however, their clinical picture was consistent with ADH, and therefore, they were included in candidate gene analysis but excluded from statistical analyses of untreated lipid levels.
Plasma PCSK9 measurement
Plasma concentrations of PCSK9 were determined as previously described 25.
In vitro PCSK9(D129N) Experiments
Construction of PCSK9(D129N) Expression Vector
All DNA manipulations were performed using standard molecular biology techniques. A D129N change was introduced into the pCMV-PCSK9-FLAG vector by site-directed mutagenesis using the primer 5’-GGCTTCCTGGTGAAGATGAGTGGCAACCTGCTGGAGCTGGCCTTGAAGTTG-3’ as described26; the resulting plasmid was designated pCMV-PCSK9(D129N)-FLAG.
Tissue Culture Medium
Medium A contained Dulbecco’s Modified Eagle Medium (Cellgro) supplemented with 100 units/ml penicillin, 100 μg/ml streptomycin sulfate and 1 g/L glucose. Medium B contained Medium A supplemented with 10% (v/v) fetal calf serum . Medium C contained Medium A supplemented with 2.5% (v/v) newborn calf lipoprotein-deficient serum, 10 μM sodium compactin and 50 μM sodium mevalonate.
Purification of Epitope-tagged PCSK9 proteins
For stable lines expressing PCSK9(D129N), HEK 293S cells were cultured in Medium B in 60-mm dishes and transfected with 0.2 μg of pCMV-PCSK9 using FuGENE 6 transfection reagent (Roche Applied Science). Colonies surviving neomycin selection were assessed for PCSK9 secretion by immunoblot analysis of the medium using an anti-FLAG M2 antibody. FLAG-tagged human PCSK9(WT), PCSK9(D129N), PCSK9(D374Y) proteins were purified from the stably transfected HEK 293S cells as described26.
PCSK9 protein activity in HepG2 Cells
HepG2 cells (ATCC, HB-8065) were plated at 4 x 105 cells/60-mm dish in Medium B on day 0. On day 3, Medium B was replaced with Medium C. After 18 h, cells were treated with purified FLAG-tagged PCSK9 proteins for 4 h. Following incubation with exogenous PCSK9, cell surface proteins were biotinylated and isolated from whole cell extracts as described 26. Mouse anti-human LDLR IgG HL-1 and mouse anti-human transferrin receptor (TFR) IgG (Invitrogen, Carlsbad, California) were used for immunoblot analysis. An IRDye800-conjugated donkey anti-mouse antibody (LI-COR Biosciences) was used as the secondary antibody. Proteins were visualized and quantified using the LI-COR Odyssey infrared imaging system (LI-COR Biosciences) as described27. Transferrin receptor was used as the loading control for the calculation of relative LDLR protein levels.
Statistical analysis
Comparisons of quantitative variables were made with two-way analysis of variance models to simultaneously test for group and gender effects and for interaction between group and gender. Categorical variable comparisons between groups were made with Cochran-Mantel-Haenszel stratifying by gender; homogeneity of strata was assessed with the Breslow-Day test. Cox proportional hazard models were used for comparing risk of premature CHD and censoring age at 55 years for males and 65 years for females (ages corresponding to the definition of premature CHD). Serum triglycerides values were log transformed prior to analysis. Continuous variables are summarized as mean (SD) and median unless otherwise specified. A two-sided p-value < 0.05 was considered statistically significant. Statistical analyses were performed using SAS version 9.2 (SAS Institute, Cary, NC).
Results
Ninety-one unrelated patients with ADH (38 males and 53 females) met entry criteria and agreed to participate. Ages ranged from 22 to 76 years, and body mass index from 16 to 58 kg/m2. Total cholesterol ranged from 254 to 557 mg/dL; LDL-C from 175 to 397 mg/dL;triglycerides from 52 to 498 mg/dL, and HDL-C from26 to 95 mg/dL.
Candidate Gene Analysis
Heterozygous LDLR mutations were found in 27 patients via Sanger sequencing. MLPA detected 3 patients with small heterozygous LDLRdeletions(Figure 1). Overall, 28 different heterozygous mutations were represented among these 30 patients (Table 1). Of the six patients with missinguntreated serum lipids values, fourhadLDLR mutations. Four new LDLR variants were detected; these variants were not present in University College London LDLRFH database (www.ucl.ac.uk/ldlr/LOVDv.1.1.0/)28. A 44-year-old non-Hispanic African-American man with an untreated LDL-C of 234 mg/dL, premature arcussenilis, tendon xanthomas, and history of ischemic stroke harbored a heterozygous p.F11S (c.95T>C) variant. Another heterozygous variant p.Q324P (c.1034A>C) was found in a 54-year-old non-Hispanic white woman with premature arcussenilis, tendon xanthomas, and a family history of premature CHD. Although her untreated serum lipid values were not available, her LDL-C was 171 mg/dL while taking maximum doses of colesevelam and atorvastatin. A heterozygous variant, p.L435P (c.1367T>C) was found in a 63-year-old African-American man with an untreated LDL-C of 279 mg/dL and arcussenilis. Finally, heterozygous p.D339H (c.1078G>C) mutation was found in a 72-year-old Hispanic woman with an untreated LDL-C of 250 mg/dL and history of ischemic stroke. None of these LDLR variants were found in SNP databases.
Figure 1.
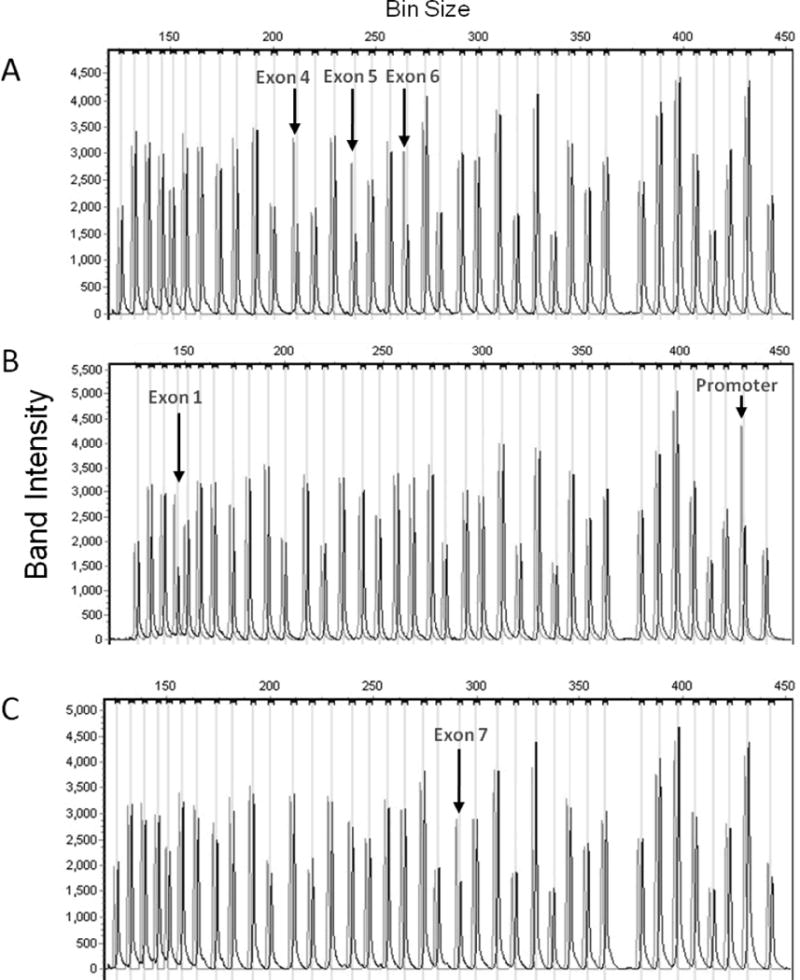
Capillary electrophoresis of Multiplex Ligation-dependent Probe Amplification LDLR probe products in the three patients found to have heterozygous deletions. Graylines show control data, while black linesshow data from the patients. Heterozygous deletions are indicated by peaks that are half as high as control peaks.A. A 25-year-old Hispanic woman with untreated LDL-C 339 mg/dL and premature arcussenilis was found to have heterozygous deletion of exons 4–6. B. A 47-year-old white man with xanthomas (untreated lipids not available) was found to have heterozygous deletion of promoter and exon 1. C. A 49-year-old African American man with untreated LDL-C 271 mg/dL, tendon xanthomas, and premature coronary heart disease was found to have heterozygous deletion of exon 7.
Table 1.
List of heterozygous LDLRvariants by ethnicity and race:
| Ethnicity | n | Variants | Exon | |
|---|---|---|---|---|
|
| ||||
| Protein Alteration** | Nucleotide*** | |||
| African American | 1 | F11S* | 95C>T | 2 |
| 1 | D283N | 910G>A | 6 | |
| 1 | NA | Exon 7 deletion | 7 | |
| 1 | NA | IVS6-4G>A | – | |
| 1 | L435P* | 1367T>C | 10 | |
| 2 | C646R | 1999T>C | 14 | |
| 1 | P678L | 2096C>T | 14 | |
| Non-Hispanic White | 1 | NA | Promoter, exon 1 deletion | Promoter, 1 |
| 1 | NA | IVS4+1G>T | 4 | |
| 1 | D206fsX12 | 680_681delAC | 4 | |
| 1 | D245E | 798T>A | 5 | |
| 1 | Q324P* | 1034A>C | 7 | |
| 1 | V408M | 1285G>A | 9 | |
| 1 | G571E | 1775G>A | 12 | |
| 1 | NA | IVS16-2A>G | – | |
| Non-Hispanic White and Native American | ||||
| 2 | NA | IVS3+1G>A | – | |
| 1 | G457R | 1432G>A | 10 | |
| Hispanic | 1 | A29S | 148G>T | 2 |
| 1 | Exon 4,5,6 deletion | 4,5,6 | ||
| 1 | C176Y | 590G>A | 4 | |
| 1 | H190Y | 631C>T | 4 | |
| 1 | C331Y | 1055G>A | 7 | |
| 1 | D339H* | 1078G>C | 8 | |
| 1 | C343R | 1090T>C | 8 | |
| 1 | C347Y | 1103G>A | 8 | |
| 1 | Q427X | 1342C>T | 9 | |
| South East Asian (Bangladeshi) | 1 | V776M | 2389G>A | 16 |
| Middle Eastern (Lebanese) | 1 | C660X | 2043C>A | 14 |
NA denotes not available
Not present in University College London low density lipoprotein receptor familial hypercholesterolemia database (www.ucl.ac.uk/ldlr/LOVDv.1.1.0/)28
Note: Numbering system for exonic variations does not include 21 amino acids in the signal sequence (original amino acid number)
Mutation at DNA level. Nucleotide numbering is according to the genomic reference sequence (http://www.ucl.ac.uk/ldlr/LOVDv.1.1.0/refseq/LDLR_codingDNA.html) and includes the promoter region
Sequencing of APOB revealed one patient, who was of German descent, with heterozygousc.10580G>A; p.R3527Q (also known as p.R3500Q according to the previous numbering system) mutation. Another patient, of African American descent, was heterozygous for another missense mutation, p.W3594R (c.10780T>C), which has not been reported previously. However, her brother, who was also hypercholesterolemic, did not inherit the mutation (Figure 2A).
Figure 2.
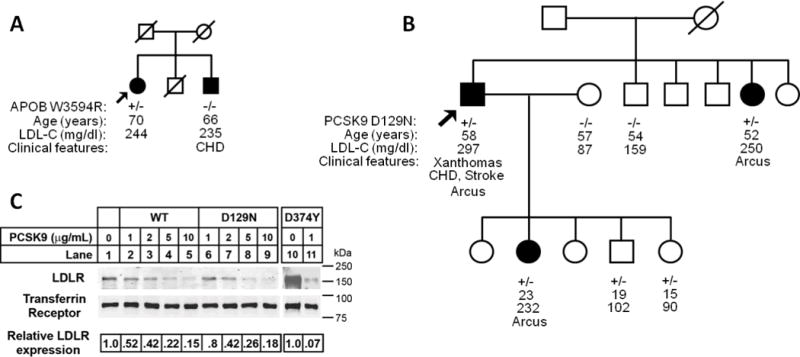
Lack of co-segregation with the hypercholesterolemia phenotype in families found to have APOB and PCSK9 variants. Arrows indicate probands. Filled circle and square designate female and male (respectively) with hypercholesterolemia.A. African American pedigree with APOB p.W3594R heterozygous variant. No disease-causing variants were found in LDLR or PCSK9.B. Hispanic pedigree with PCSK9 p.D129N heterozygous variant. No disease-causing variants were found in LDLR or APOB. C. Degradation of LDLR by exogenous PCSK9 proteins. HepG2 cells were cultured in sterol-supplemented medium (see “Experimental Procedures”) and incubated with the indicated amount of wild-type PCSK9 (WT), PCSK9(D129N) and PCSK9(D374Y) protein for 4 h. Following biotinylation, isolated cell surface protein lysates were subjected to SDS-PAGE and immunoblot analysis. LDLR was detected with an anti-LDLR mAb (HL-1). Transferrin Receptor (TFR) was detected as a control for equal protein loading. Secondary detection used an infrared dye (IRDye800)-labeled antibody. Blots were visualized and quantified using the LI-COR Odyssey infrared imaging system. LDLR levels were normalized to TFR expression. For these data, results shown are from representative experiments repeated at least three times with similar results. CHD denotes coronary heart disease
Sequencing of PCSK9 revealed one proband with themissense mutation p.D129N(c.385 G>A). This PCSK9 variant had been previously identified in a single individual with hypercholesterolemia. Functional studies in cells transfected with PCSK9(D129N) suggested that it degraded LDLRs more efficiently than wild-type PCSK9, although the effect was modest29. Here, we found that this variant did not cosegregate with the ADH phenotype in the family (Figure 2B). To further study the activity this PCSK9 variant, recombinant PCSK9(D129N) protein was purified and the activity of this protein was compared to wild-type PCSK9 and PCSK9(D374Y), a confirmed gain-of-function mutation30. As shown in Figure 2C, the ability of PCSK9(D129N) to degrade LDLRs in HepG2 cells was equivalent to that of wild-type PCSK9, and both were significantly less potent that PCSK9(D374Y).
A total of 66% patients lacked any mutation in the 3 candidate genes screened which included27 men and 33 women. Ethnic and racial breakdown of the patients is shown in Figure 3. Out of 35 African American patients, only eight (23%) had mutations in the known ADH genes. “Unexplained ADH” was found in 57% of non-Hispanic whites, 50% American Indians, and 53% of Hispanic patients in our study. Only two out of nine patients of Asian origin were found to have disease-causing mutations, and one Ethiopian male did not have any disease-causing mutation.
Figure 3.
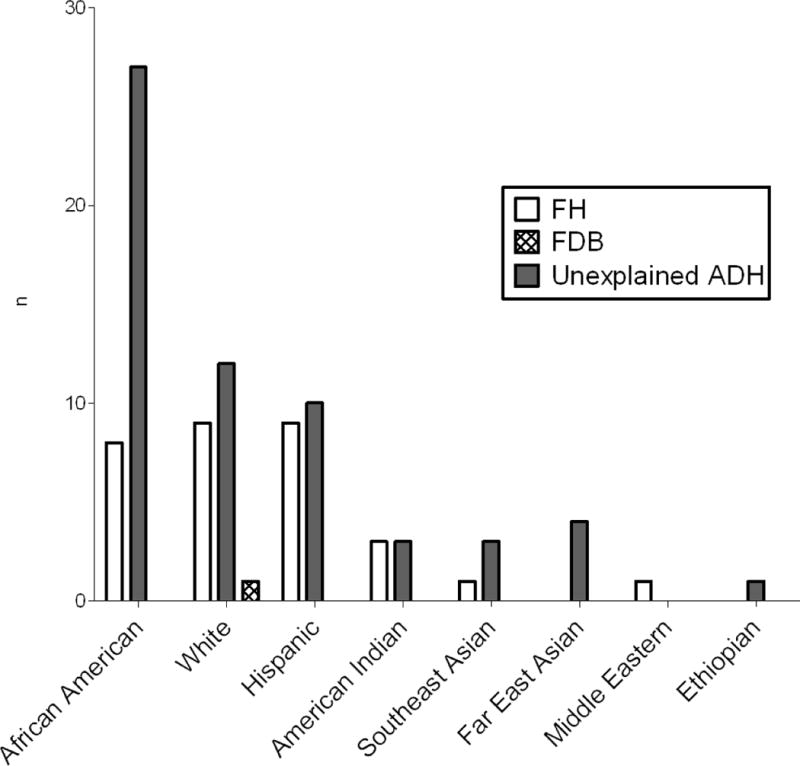
Self-reported ethnicity/race based of 91 patients with autosomal dominant hypercholesterolemia (ADH). Maternal and paternal ancestor’s origins were used if provided by the patient. FH (familial hypercholesterolemia) denotes patients found to have LDLR mutations. FDB (familial defective apolipoprotein B) denotes patients found to have APOB mutations. Unexplained ADH denotes patients with ADH but no mutations in the known loci. ‘n’ indicates number of subjects.
Apolipoprotein E Genotyping
No patients had the E2/E2 genotype. Using reference populations31, we tested whether E4 allele is more prevalent in the African Americans and whites, respectively, and found no significant difference from the reference populations.
Comparison of FH and “Unexplained ADH”
We compared the clinical characteristics and laboratory data between those with LDLR mutations (FH) and those in whom no mutations were found (“unexplained ADH”) (Table 2). As compared to patients with LDLR variants, those with “unexplained ADH” had lower levels of LDL-C (292 ± 47 vs 239 ± 42 mg/dL, respectively; p < 0.0001) and higher levels of HDL-cholesterol (45 ± 12 vs 54 ± 13 mg/dL, respectively, p = 0.003) (Table 2 and Figure 4A and B). Serum triglycerides were higher in men with FH compared with the “unexplained ADH” group, but the opposite was observed in women (p for interaction 0.005). Frequency of tendon xanthomas was nearly double in those with FH. There were no significant differences in the Achilles tendon width in the two groups, but Achilles tendon width had good predictive ability for the presence of xanthomas (area under the curve ~0.85). Also, the two groups were not different with regards to the frequency ofarcussenilis, grade of arcussenilis, premature CHD in first-degree relatives, andplasma PCSK9 levels. There was no difference in the prevalence of premature CHD in male probands; however, women with “unexplained ADH” had a lower prevalence of premature CHD than women with LDLR mutations and men with “unexplained ADH” (Figure 4C and D). Both groups showed a similar response to LDL lowering medications, and the use of lipid lowering medications was similar in both the groups (Supplementary Tables S1 and S2).
Table 2.
Comparison of clinical features and lipid and lipoprotein values in patients with Familial Hypercholesterolemia versus “unexplained ADH.”
| FH (LDLR) Patients | Unexplained ADH Patients | Group x Gender Interaction p-value | |||
|---|---|---|---|---|---|
|
| |||||
| Male | Female | Male | Female | ||
| N* | 11 | 19 | 27 | 33 | |
| Age (years) | 53 (14) | 55 (13) a | 54 (11) e | 61 (6) | 0.27 |
| Body Mass Index (kg/m2) | 30.6 (5.4) | 31.1 (7.3) | 31.0 (4.3) | 31.9 (7.4) | 0.88 |
| Ethnicity (%) | |||||
| African-American | 18.2 | 31.6 | 44.4 | 45.5 | |
| Caucasian, non-Hispanic | 63.6 | 26.3 | 18.5 | 30.3 | |
| Hispanic | 18.2 | 36.8 | 18.5 | 15.2 | |
| other | 0 | 5.3 | 18.5 | 9.1 | |
| Total Cholesterol (mg/dL) | 374 (79) b | 375 (40) b | 326 (49) | 329 (35) | 0.89 |
| LDL-C (mg/dL) | 285 (55) a | 295 (42) c | 244 (47) | 237 (38) | 0.45 |
| TG (mg/dL) | 231 (97) ad | 152 (57) | 167 (71) | 193 (86) | 0.005 |
| HDL-C (mg/dL) | 36.7 (7.0) be | 49.0 (11.7) a | 50.6 (10.9) | 56.5 (14.1) | 0.28 |
| PCSK9 (ng/mL) | 633 (269) | 780 (225) | 554 (326) d | 672 (306) | 0.92 |
| Achilles Tendon Width (mm) | 17.8 (3.6) | 16.2 (4.1) | 15.8 (2.1) | 15.4 (3.1) | 0.44 |
| Arcussenilis (%) | 72.7 | 84.2 | 76.9 | 72.7 | 0.40 |
| Tendon xanthomas (%) | 81.8 a | 52.6 | 36.0 | 28.1 | 0.33 |
| Lipid treatment (%) g | 100 | 100 | 92.6 | 87.9 | 1.0 |
| Premature CHD (%)h | 36.4 | 36.8 a | 44.4 f | 18.2 | 0.17 |
| Age of onset of CHD (years) | 44 (1) n=4 | 48 (10) n=7 | 45 (7) n=12 | 56 (5) n=6 | |
| Premature CHD in relatives (%) | 100 e | 43.8 | 59.1 | 44.8 | 0.05 |
Continuous variables are summarized as mean (SD). FH denotes familial hypercholesterolemia. ADH denotes autosomal dominant hypercholesterolemia. LDL-C denotes low density lipoprotein-cholesterol; TG denotes triglycerides; HDL-C denotes high density lipoprotein-cholesterol; PCSK9 denotes proprotein convertase subtilisin/kexin 9; CHD denotes coronary heart disease
P-values are from two-way ANOVA for continuous variables, Breslow-Day test for categorical variables, and Cox proportional hazard models for comparing premature CHD onset.
Untreated lipid and lipoprotein values were available in 9 FH (LDLR) males, 17 FH (LDLR) females, 25 “unexplained ADH” males, and 33 “unexplained ADH” females.
One patient with APOB heterozygous mutation not included
p<0.05,
p<0.01,
p<0.001, FH(LDLR) versus unexplained ADH for the same gender.
p<0.05,
p<0.01,
p<0.001, Male versus Female for the same group
Includes statins, ezetimibe, bile acid sequestrants, niacin, fish oil, and/or sitostanol
Premature CHD is defined as the diagnosis of CHD at ages less than 55 years in males or 65 years in females.
Figure 4.
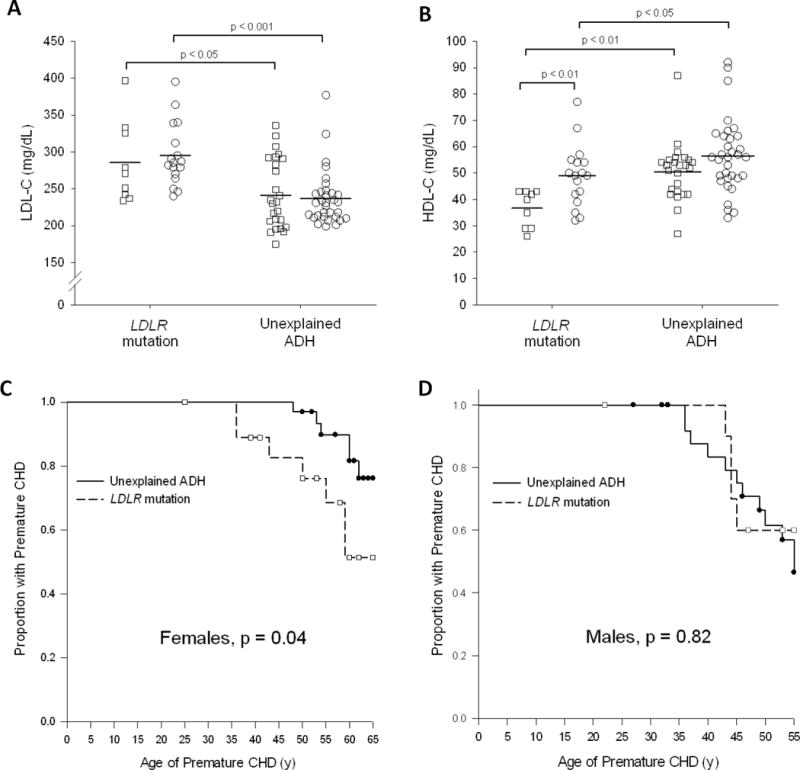
A,B: LDL-C (A) and HDL-C (B) values in Familial Hypercholesterolemia (LDLR mutation)vs Unexplained ADH. Squares (□) represent male subjects, circles (○) represent female subjects. Horizontal bar indicates median values. C,D: Kaplan-Meier curves depicting the age of onset of premature coronary heart disease (CHD) in the females (C) and males (D), respectively. Each step down represents the occurrence of the patient’s coronary heart disease diagnosis. Symbols represent censored observations. Premature CHD is defined as the diagnosis of CHD at ages less than 55 years in males or 65 years in females.
Discussion
The most striking finding of our study was our inability to identify a disease-causing mutation in LDLR, APOBor PCSK9 in 66% of ourpatients with ADH. In examining the molecular basis of ADH in a multi-ethnic patient cohort from lipid clinics in a large U.S. city, we find that a higher proportion of African Americans (77%) than either non-Hispanic whites (57%) or Hispanics (53%) had “unexplained ADH.”Our findings suggest that new ADH loci exist, especially among understudied populations such as African Americans. Nearly all patients with a gene defect had FH, and differences in clinical features were observed between FH and “Unexplained ADH.”
African American adults are more likely than non-Hispanic whites to be diagnosed with coronary heart disease, and they are more likely to die from heart disease (2009 National Healthcare Quality & Disparities Reports, www.ahrq.gov/qual/qrdr09.htm). This disparity has been linked to hypertension, but only limited data are available about the frequency of LDLR disease-causing variants in ADH patients of African origin. Previously, Thiartet al32reported data on 16 unrelated Africans from South Africa and found 10 with mutations in LDLR, but four shared the same 6 bp deletion in exon 2. These four Africans were not from the same tribe, andthe authors excluded the likelihood of a recurrent mutational event, suggesting that there is a common ancestry. Four individuals of African/Afro-Caribbean descent were included in a study from the UK 7, and only one harbored an LDLR mutation. Thus, our finding of a low frequency of LDLR mutations (23%) in African Americans suggests that other mechanisms may be a common cause of the ADH phenotype in patients of African origin.
Our mutation detection rate among Hispanics corresponds well to prior small studies that showed 40–61% of Mexican ADH patients have mutations in LDLRor APOB33. In addition, our study included eight patients of Asian origin, only one of whom was found to have a mutation. Previous small studies of Asian cohorts have also found low mutation detection rates. In Malaysian ADH cohorts, only 26–42% of patients have been found to have LDLR mutations 34, 35. Studies of patients from other Asian origins (Taiwanese, Philippines, Asian/Indian origin living in UK), have found mutation detection rates between 20% and 60% 7, 36, 37. Non-Hispanic whites in our study had a mutation detection rate lower than expected, perhaps due to admixture in US citizens who self-report to be white.
Mutations in LDLR account for the majority of ADH cases and more than 1000 unique disease-causing variants have been reported (www.ucl.ac.uk/ldlr/LOVDv.1.1.0/)28. Worldwide, the prevalence of heterozygous FH due to LDLR variants is estimated to be 1 in 5003. Consistent with previous observations, LDLR mutations accounted for nearly all patients with ADH for whom a molecular basis was ascertained in our study. Even though more that 1000 disease causing mutations in LDLR have been reported, we found four new missense variants: p.F11S, p.Q324P, p.D339H, and p.L435P. Of note, p.F11C, p.Q324X, and p. L435Hhave been previously reported to cause FH 38. Also, in our cohort, there were only two recurrences of mutations.
The most common FDB mutation in APOB occurs in exon 26 (p.R3527Q), with the majority of affected patients being of Central European descent and an estimated frequency of 1/1000 3. While some other variants in APOB have been reported in patients with high LDL-C levels, there has not been a robust documentation of segregation with the ADH phenotype within pedigrees and proof of decreased LDLR binding affinity in functional assays39. Similarly, we found a p.W3594R APOBvariant that did not cosegregate with hypercholesterolemia in the family. The molecular mechanism of ADH in this pedigree (Figure 1A) remains unknown.
Since exons 2, 4, and 7 harbor the well-defined FH3 causing variants (p.S127R, p.F216L, p.D374Y), we chose to sequence only these. We identified a family with heterozygous PCSK9p.D129N variant. This mutation was previously reported in a single individual with extremely elevated cholesterol 40. However, the D129N variant did not cosegregatewith hypercholesterolemia in our family, and our functional studies using recombinant PCSK9(D129N) and wild-type PCSK9 protein showed that there was no significant difference in their ability to degraded LDLRs. Therefore, the molecular mechanism of ADH in this pedigree (Figure 1B) also remains unknown.
Our study suggests that the “unexplained ADH” group may be distinct compared to the FH group. Clinically, those with “unexplained ADH” had lower LDL-C levels, higher HDL-C levels, and reduced frequency of tendon xanthomas (Table 2). Also, men with FH had the highest triglycerides despite BMIs that were similar to the other groups.
Although differences in LDL-C and xanthoma prevalence are consistent with multiple prior reports, HDL-C differences have not been widely reported in prior systematic screening studies 7, 12–14, 21, 34. In a French ADH study 12, in which only 19% of ADH patients were unexplained, similar differences in HDL-C were noted. HDL particle size and the ratio of HDL cholesterol to apolipoprotein A1 is significantly lower in FH patients than that in normal subjects 41, 42. Prior studies have shown an increased fractional catabolic rate of apolipoprotein A143 and higher cholesterol ester transfer protein (CETP) levels42. Alternatively, low HDL-C may be secondary to the decreased uptake of TG-rich lipoproteins by deficient the LDLR.
Remarkably, men in the “unexplained ADH” group had similar onset of premature CHD as the FH group, indicating that men with severe hypercholesterolemia should be treated aggressively with regards to preventing CHD regardless of any knowledge of LDLR defects. The CHD rate in the “Unexplained ADH” females, however, was about 2 fold lower than that seen in the “Unexplained ADH” males and FH females. It is not clear why this group has lower CHD. Similar to the general population, CHD in women with ADH tends to lag behind men with ADH by 10 years44.
Limitations of this study are that we did not sequence the LDLR promoter or introns for cryptic mutations, and we limited the sequencing of APOB and PCSK9 to exons with known disease-causing variants. While the prevalence of LDLR cryptic mutations is not clear, a recent study of LDLR promoter variants in a large cohort of European (Spanish) patients with ADH found their prevalence to be less than 1% 45. Future studies with next generation sequencing techniques, such as whole-genome sequencing, may help elucidate the contribution of cryptic LDLRmutations in ADH patients. Unlike LDLR variants, only a few variants inAPOB and PCSK9cause ADH, accounting for less than 14% of patients,7–15, 46. The probability of novel mutations in non-sequenced exons of either of these genes is extremely unlikelyand may not significantly affectthe frequency of “unexplained ADH.”
Our results have the lowest mutation detection rate when compared to previous studies, likely due to the diverse genetic background of our patients, particularly inclusion of many African American patients. Other possibilities could be inclusion of phenocopies such as familial combined hyperlipidemia (FCHL), type 3 hyperlipoproteinemia, secondary hypercholesterolemia, and non-genetic factors. However, all patients were carefully screened for secondary hypercholesterolemias, and none of our patients had the apolipoprotein E2/E2 genotype that can result in type 3 hyperlipoproteinemia. The likelihood of FCHL is minimal because serum triglyceride levels were not significantly different between those with and without LDLRmutations. Non-genetic factors that may be causing elevations in LDL-C include age, post menopausal status, diet and obesity. For example, diets high in saturated fats, trans fats, and cholesterol may cluster in families and incorrectly suggest inheritence. “Unexplained ADH” may also be related to increased function of PCSK9 due to non-genetic influences47 but has not been clinically reported.
Another possibility for the low mutation detection rateis that a polygenic inheritance is responsible for the ADH phenotype. This may be particularly true among those individuals in our cohort who had LDL-C greater than the 95th percentile but less than 230 mg/dL, since the mutation detection rate was 0 out of 26 in this subgroup (Figure 3). Since there is at least a 5% chance that any first degree relative will have LDL-C above the 95th percentile, collecting data on large number of relatives to show nearly 50% prevalence of high LDL-C may provide hard evidence for autosomal dominant inheritance pattern in the “unexplained ADH” group. However, we are unable to ascertain the precise prevalence of high LDL-C levels among the relatives of the “Unexplained ADH” group for lack of data. Regardless, the majority of the “unexplained ADH” group had severe hypercholesterolemia with LDL-Cgreater than230 mg/dLand included many patients with tendon xanthomas and evidence of autosomal dominant inheritance.
In conclusion, we found a low mutation detection rate of the known genes that cause ADH among a multi-ethnic cohort of patients who met widely accepted criteria for a clinical diagnosis of ADH. Our findings suggest the existence of new ADH loci, especially in understudied populations such as African Americans. The discovery of new ADH genes would lead to further insight into lipid metabolism and new therapeutic targets.
Supplementary Material
Acknowledgments
We thank Scott Grundy, MD, PhD, for ongoing support and guidance; Jean Wilson, MD, for critical review of the manuscript; Jay D. Horton, MD, Jonathan Cohen, PhD; and Helen Hobbs, MD; for their support and advice;Claudia Quittner and Barbara Gilbert forcollecting and processing samples;Drs. Frederick Dunn, Amit Khera, and David Balis for referring patients; and Tommy Hyatt, Sarah Masood, Rashmin Asher, Tuyet Dang, and Norma Anderson for technical assistance.
Funding Sources: The work was partly supported by grants from Department of Internal Medicine, UT Southwestern and from the National Institutes of Health HL020948.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Disclosures: A.G. is a consultant for Pfizer, on the speaker bureau for Merck, and has grant support from Amylin, Inc.
References
- 1.Lloyd-Jones D, Adams RJ, Brown TM, Carnethon M, Dai S, De Simone G, et al. Executive summary: heart disease and stroke statistics--2010 update: a report from the American Heart Association. Circulation. 2010;121:948–954. doi: 10.1161/CIRCULATIONAHA.109.192666. [DOI] [PubMed] [Google Scholar]
- 2.Heller DA, de Faire U, Pedersen NL, Dahlen G, McClearn GE. Genetic and environmental influences on serum lipid levels in twins. N Engl J Med. 1993;328:1150–1156. doi: 10.1056/NEJM199304223281603. [DOI] [PubMed] [Google Scholar]
- 3.Rader DJ, Cohen J, Hobbs HH. Monogenic hypercholesterolemia: new insights in pathogenesis and treatment. J Clin Invest. 2003;111:1795–1803. doi: 10.1172/JCI18925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466:707–713. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garg A, Simha V. Update on dyslipidemia. J Clin Endocrinol Metab. 2007;92:1581–1589. doi: 10.1210/jc.2007-0275. [DOI] [PubMed] [Google Scholar]
- 6.Horton JD, Cohen JC, Hobbs HH. Molecular biology of PCSK9: its role in LDL metabolism. Trends Biochem Sci. 2007;32:71–77. doi: 10.1016/j.tibs.2006.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Taylor A, Wang D, Patel K, Whittall R, Wood G, Farrer M, et al. Mutation detection rate and spectrum in familial hypercholesterolaemia patients in the UK pilot cascade project. Clin Genet. 2010;77:572–580. doi: 10.1111/j.1399-0004.2009.01356.x. [DOI] [PubMed] [Google Scholar]
- 8.Medeiros AM, Alves AC, Francisco V, Bourbon M. Update of the Portuguese Familial Hypercholesterolaemia Study. Atherosclerosis. 2010;212:553–558. doi: 10.1016/j.atherosclerosis.2010.07.012. [DOI] [PubMed] [Google Scholar]
- 9.Chmara M, Wasag B, Zuk M, Kubalska J, Wegrzyn A, Bednarska-Makaruk M, et al. Molecular characterization of Polish patients with familial hypercholesterolemia: novel and recurrent LDLR mutations. J Appl Genet. 2010;51:95–106. doi: 10.1007/BF03195716. [DOI] [PubMed] [Google Scholar]
- 10.Lombardi MP, Redeker EJ, van Gent DH, Smeele KL, Weerdesteijn R, Mannens MM. Molecular genetic testing for familial hypercholesterolemia in the Netherlands: a stepwise screening strategy enhances the mutation detection rate. Genet Test. 2006;10:77–84. doi: 10.1089/gte.2006.10.77. [DOI] [PubMed] [Google Scholar]
- 11.Dedoussis GV, Skoumas J, Pitsavos C, Choumerianou DM, Genschel J, Schmidt H, et al. FH clinical phenotype in Greek patients with LDL-R defective vs. negative mutations. Eur J Clin Invest. 2004;34:402–409. doi: 10.1111/j.1365-2362.2004.01351.x. [DOI] [PubMed] [Google Scholar]
- 12.Marduel M, Carrie A, Sassolas A, Devillers M, Carreau V, Di Filippo M, et al. Molecular spectrum of autosomal dominant hypercholesterolemia in France. Hum Mutat. 2010;31:E1811–1824. doi: 10.1002/humu.21348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Civeira F, Ros E, Jarauta E, Plana N, Zambon D, Puzo J, et al. Comparison of genetic versus clinical diagnosis in familial hypercholesterolemia. Am J Cardiol. 2008;102:1187–1193. 1193–e1181. doi: 10.1016/j.amjcard.2008.06.056. [DOI] [PubMed] [Google Scholar]
- 14.van der Graaf A, Avis HJ, Kusters DM, Vissers MN, Hutten BA, Defesche JC, et al. Molecular basis of autosomal dominant hypercholesterolemia: assessment in a large cohort of hypercholesterolemic children. Circulation. 2011;123:1167–1173. doi: 10.1161/CIRCULATIONAHA.110.979450. [DOI] [PubMed] [Google Scholar]
- 15.Miyake Y, Yamamura T, Sakai N, Miyata T, Kokubo Y, Yamamoto A. Update of Japanese common LDLR gene mutations and their phenotypes: Mild type mutation L547V might predominate in the Japanese population. Atherosclerosis. 2009;203:153–160. doi: 10.1016/j.atherosclerosis.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 16.Jenkins T, Nicholls E, Gordon E, Mendelsohn D, Seftel HC, Andrew MJ. Familial hypercholesterolaemia--a common genetic disorder in the Afrikaans population. S Afr Med J. 1980;57:943–947. [PubMed] [Google Scholar]
- 17.Khachadurian AK, Uthman SM. Experiences with the homozygous cases of familial hypercholesterolemia. A report of 52 patients. Nutr Metab. 1973;15:132–140. doi: 10.1159/000175431. [DOI] [PubMed] [Google Scholar]
- 18.Moorjani S, Roy M, Gagne C, Davignon J, Brun D, Toussaint M, et al. Homozygous familial hypercholesterolemia among French Canadians in Quebec Province. Arteriosclerosis. 1989;9:211–216. doi: 10.1161/01.atv.9.2.211. [DOI] [PubMed] [Google Scholar]
- 19.Seftel HC, Baker SG, Jenkins T, Mendelsohn D. Prevalence of familial hypercholesterolemia in Johannesburg Jews. Am J Med Genet. 1989;34:545–547. doi: 10.1002/ajmg.1320340418. [DOI] [PubMed] [Google Scholar]
- 20.Abifadel M, Rabes JP, Boileau C, Varret M. [After the LDL receptor and apolipoprotein B, autosomal dominant hypercholesterolemia reveals its third protagonist: PCSK9] Ann Endocrinol (Paris) 2007;68:138–146. doi: 10.1016/j.ando.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 21.Varret M, Abifadel M, Rabes JP, Boileau C. Genetic heterogeneity of autosomal dominant hypercholesterolemia. Clinical genetics. 2008;73:1–13. doi: 10.1111/j.1399-0004.2007.00915.x. [DOI] [PubMed] [Google Scholar]
- 22.Risk of fatal coronary heart disease in familial hypercholesterolaemia. Scientific Steering Committee on behalf of the Simon Broome Register Group. BMJ. 1991;303:893–896. doi: 10.1136/bmj.303.6807.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Winder AF, Jolleys JC, Day LB, Butowski PF. Corneal arcus, case finding and definition of individual clinical risk in heterozygous familial hypercholesterolaemia. Clin Genet. 1998;54:497–502. doi: 10.1111/j.1399-0004.1998.tb03770.x. [DOI] [PubMed] [Google Scholar]
- 24.Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002;30:e57. doi: 10.1093/nar/gnf056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lakoski SG, Lagace TA, Cohen JC, Horton JD, Hobbs HH. Genetic and metabolic determinants of plasma PCSK9 levels. J Clin Endocrinol Metab. 2009;94:2537–2543. doi: 10.1210/jc.2009-0141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lagace TA, Curtis DE, Garuti R, McNutt MC, Park SW, Prather HB, et al. Secreted PCSK9 decreases the number of LDL receptors in hepatocytes and in livers of parabiotic mice. J Clin Invest. 2006;116:2995–3005. doi: 10.1172/JCI29383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McNutt MC, Kwon HJ, Chen C, Chen JR, Horton JD, Lagace TA. Antagonism of secreted PCSK9 increases low density lipoprotein receptor expression in HepG2 cells. J Biol Chem. 2009;284:10561–10570. doi: 10.1074/jbc.M808802200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leigh SE, Foster AH, Whittall RA, Hubbart CS, Humphries SE. Update and analysis of the University College London low density lipoprotein receptor familial hypercholesterolemia database. Ann Hum Genet. 2008;72:485–498. doi: 10.1111/j.1469-1809.2008.00436.x. [DOI] [PubMed] [Google Scholar]
- 29.Fasano T, Sun XM, Patel DD, Soutar AK. Degradation of LDLR protein mediated by ‘gain of function’ PCSK9 mutants in normal and ARH cells. Atherosclerosis. 2009;203:166–171. doi: 10.1016/j.atherosclerosis.2008.10.027. [DOI] [PubMed] [Google Scholar]
- 30.Abifadel M, Varret M, Rabes JP, Allard D, Ouguerram K, Devillers M, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003;34:154–156. doi: 10.1038/ng1161. [DOI] [PubMed] [Google Scholar]
- 31.Hallman DM, Boerwinkle E, Saha N, Sandholzer C, Menzel HJ, Csazar A, et al. The apolipoprotein E polymorphism: a comparison of allele frequencies and effects in nine populations. Am J Hum Genet. 1991;49:338–349. [PMC free article] [PubMed] [Google Scholar]
- 32.Thiart R, Scholtz CL, Vergotine J, Hoogendijk CF, de Villiers JN, Nissen H, et al. Predominance of a 6 bp deletion in exon 2 of the LDL receptor gene in Africans with familial hypercholesterolaemia. J Med Genet. 2000;37:514–519. doi: 10.1136/jmg.37.7.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Robles-Osorio L, Huerta-Zepeda A, Ordonez ML, Canizales-Quinteros S, Diaz-Villasenor A, Gutierrez-Aguilar R, et al. Genetic heterogeneity of autosomal dominant hypercholesterolemia in Mexico. Archives of medical research. 2006;37:102–108. doi: 10.1016/j.arcmed.2005.04.018. [DOI] [PubMed] [Google Scholar]
- 34.Khoo KL, van Acker P, Defesche JC, Tan H, van de Kerkhof L, Heijnen-van Eijk SJ, et al. Low-density lipoprotein receptor gene mutations in a Southeast Asian population with familial hypercholesterolemia. Clin Genet. 2000;58:98–105. doi: 10.1034/j.1399-0004.2000.580202.x. [DOI] [PubMed] [Google Scholar]
- 35.Al-Khateeb A, Zahri MK, Mohamed MS, Sasongko TH, Ibrahim S, Yusof Z, et al. Analysis of sequence variations in low-density lipoprotein receptor gene among Malaysian patients with familial hypercholesterolemia. BMC Med Genet. 2011;12:40. doi: 10.1186/1471-2350-12-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang KC, Su YN, Shew JY, Yang KY, Tseng WK, Wu CC, et al. LDLR and ApoB are major genetic causes of autosomal dominant hypercholesterolemia in a Taiwanese population. J Formos Med Assoc. 2007;106:799–807. doi: 10.1016/S0929-6646(08)60044-3. [DOI] [PubMed] [Google Scholar]
- 37.Punzalan FE, Sy RG, Santos RS, Cutiongco EM, Gosiengfiao S, Fadriguilan E, et al. Low density lipoprotein--receptor (LDL-R) gene mutations among Filipinos with familial hypercholesterolemia. J Atheroscler Thromb. 2005;12:276–283. doi: 10.5551/jat.12.276. [DOI] [PubMed] [Google Scholar]
- 38.Day IN, Whittall RA, O’Dell SD, Haddad L, Bolla MK, Gudnason V, et al. Spectrum of LDL receptor gene mutations in heterozygous familial hypercholesterolemia. Hum Mutat. 1997;10:116–127. doi: 10.1002/(SICI)1098-1004(1997)10:2<116::AID-HUMU4>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 39.Rabes JP, Varret M, Devillers M, Aegerter P, Villeger L, Krempf M, et al. R3531C mutation in the apolipoprotein B gene is not sufficient to cause hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2000;20:E76–82. doi: 10.1161/01.atv.20.10.e76. [DOI] [PubMed] [Google Scholar]
- 40.Marduel M, Carrie A, Sassolas A, Devillers M, Carreau V, Di Filippo M, et al. Molecular spectrum of autosomal dominant hypercholesterolemia in France. Human mutation. 2010;31:E1811–1824. doi: 10.1002/humu.21348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kajinami K, Mabuchi H, Koizumi J, Takeda R. Serum apolipoproteins in heterozygous familial hypercholesterolemia. Clin Chim Acta. 1992;211:93–99. doi: 10.1016/0009-8981(92)90108-3. [DOI] [PubMed] [Google Scholar]
- 42.Hogue JC, Lamarche B, Gaudet D, Tremblay AJ, Despres JP, Bergeron J, et al. Association of heterozygous familial hypercholesterolemia with smaller HDL particle size. Atherosclerosis. 2007;190:429–435. doi: 10.1016/j.atherosclerosis.2006.02.023. [DOI] [PubMed] [Google Scholar]
- 43.Frenais R, Ouguerram K, Maugeais C, Marchini JS, Benlian P, Bard JM, et al. Apolipoprotein A-I kinetics in heterozygous familial hypercholesterolemia: a stable isotope study. J Lipid Res. 1999;40:1506–1511. [PubMed] [Google Scholar]
- 44.Stone NJ, Levy RI, Fredrickson DS, Verter J. Coronary artery disease in 116 kindred with familial type II hyperlipoproteinemia. Circulation. 1974;49:476–488. doi: 10.1161/01.cir.49.3.476. [DOI] [PubMed] [Google Scholar]
- 45.De Castro-Oros I, Pampin S, Bolado-Carrancio A, De Cubas A, Palacios L, Plana N, et al. Functional analysis of LDLR promoter and 5’UTR mutations in subjects with clinical diagnosis of familial hypercholesterolemia. Hum Mutat. 2011;32:868–872. doi: 10.1002/humu.21520. [DOI] [PubMed] [Google Scholar]
- 46.Wang J, Huff E, Janecka L, Hegele RA. Low density lipoprotein receptor (LDLR) gene mutations in Canadian subjects with familial hypercholesterolemia, but not of French descent. Hum Mutat. 2001;18:359. doi: 10.1002/humu.1205. [DOI] [PubMed] [Google Scholar]
- 47.Baass A, Dubuc G, Tremblay M, Delvin EE, O’Loughlin J, Levy E, et al. Plasma PCSK9 is associated with age, sex, and multiple metabolic markers in a population-based sample of children and adolescents. Clin Chem. 2009;55:1637–1645. doi: 10.1373/clinchem.2009.126987. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.