

Abstract
Altered expression of structural and functional molecules expressed by astrocytes may play a role in the pathophysiology of schizophrenia. We investigated the hypothesis that the astrocytic enzyme glutamine synthetase, involved in maintaining the glutamate-glutamine cycle, and the cytoskeletal molecule glial fibrillary acidic protein (GFAP) are abnormally expressed in schizophrenia. We used Western blot analysis to measure levels of glutamine synthetase and GFAP in several brain regions of subjects with schizophrenia and a comparison group. We found that glutamine synthetase protein expression was significantly decreased in the superior temporal gyrus, and both glutamine synthetase and GFAP were significantly reduced in the anterior cingulate cortex in schizophrenia. Neither molecule demonstrated altered expression in the dorsolateral prefrontal cortex, primary visual cortex, or hippocampus. Chronic treatment with haloperidol did not alter the expression of these molecules in the rat brain, suggesting that our findings are not due to a medication effect. These data support an astrocytic component to the pathophysiology of schizophrenia and suggest that astrocytic molecules involved in enzymatic activity and cytoskeletal integrity may have a role in disease-related abnormalities in this illness.
Keywords: anterior cingulate cortex, astrocyte, glutamate, haloperidol, postmortem, superior temporal gyrus
1. Introduction
The search for potential astrocytic abnormalities in schizophrenia represents a relatively new line of investigation, as the majority of work to date has focused on neuronal dysfunction in this illness. One of the preliminary human postmortem findings that suggested a role for astrocytes in schizophrenia was altered cell density in several brain regions thought to be involved in the pathophysiology of the illness, including the prefrontal, anterior cingulate, and motor cortices (Benes et al., 1986, Cotter et al., 2001, Cotter et al., 2002, Stark et al., 2004). Thus, it was hypothesized that a loss of astrocytes or compromised astrocytic functioning in those particular brain regions could contribute to brain dysfunction in schizophrenia. Although, subsequent research has failed to show prominent changes in astrocyte density in select brain regions in schizophrenia, an alternate hypothesis suggests that key structural and functional astrocytic molecules are abnormally expressed and may contribute to prominent dysfunction in the brains of patients with schizophrenia.
A glutamate hypothesis of schizophrenia arose when phencyclidine (PCP), which had been used as an anesthetic, was shown to induce symptoms resembling the positive, negative and cognitive symptoms of schizophrenia in non-psychiatrically ill patients, and magnify those symptoms in patients with schizophrenia (Javitt, 1987). PCP was shown to act as a noncompetitive antagonist of the N-methyl-D-aspartate (NMDA) receptor. This finding, together with the fact that other NMDA receptor antagonists, including ketamine, also elicit a “schizophrenia-like” syndrome, helped establish a model of NMDA receptor hypofunction in schizophrenia (Lahti et al., 1995; Coyle, 1996). Many human postmortem studies have since reported altered expression of the NMDA receptor, including receptor subunits and binding sites, as well as associated postsynaptic density proteins which facilitate intracellular signaling (Clinton et al., 2003; Laruelle et al., 2003; Meador-Woodruff et al., 2003; and for review see Kristiansen et al., 2007). Furthermore, NMDA receptor hypofunction is supported by data demonstrating that co-agonists of the receptor, which facilitate receptor function, can attenuate some symptoms of schizophrenia (Heresco-Levy et al., 2005). Recent research suggests that glutamatergic dysfunction and NMDA receptor-related abnormalities that are prominent in schizophrenia may have an underlying astrocytic component as well.
One of the ways in which astrocytes participate in glutamatergic transmission is by maintaining a glutamate-glutamine cycle. Within the glutamatergic synapse, presynaptic neurons package glutamate into vesicles for release into the synapse where it may bind to and activate receptors found on pre- and postsynaptic neurons and astrocytes (Bellocchio et al., 2000, Hollmann and Heinemann, 1994, Takamori et al., 2000). The subsequent removal of glutamate from the synapse, an event critical to regulating synaptic activity and preventing excitotoxicity, is accomplished primarily by astrocytes which express high affinity excitatory amino acid transporters (EAATs), mainly types 1 and 2 (Kim et al., 2003, Sonnewald et al., 2002). Recovered glutamate may be converted to glutamine by the astrocytic enzyme glutamine synthetase and subsequently transported back to the presynaptic neuron for use as a metabolic substrate or transmitter molecule (Rothman et al., 1999). The role of glutamine synthetase in regulating the glutamate-glutamine cycle and makes it a high yield target for investigating astrocyte-related glutamate dysfunction in schizophrenia.
The ability of astrocytes to monitor and respond to changing conditions in the glutamatergic synapse is made possible, in part, by extensive process branching. Astrocyte processes contain the cytoskeletal molecule glial fibrillary acidic protein (GFAP), the assembly of which into the cytoskeleton facilitates changes to astrocyte morphology (Rodnight et al., 1997). In vivo and in vitro experiments of altered GFAP expression demonstrate that loss of this cytoskeletal protein leads to decreased branching, abnormal synaptic functioning, and to aberrant behavior in rodents (Chen and Liem, 1994, McCall et al., 1996, Shibuki et al., 1996, Weinstein et al., 1991). Abnormal assembly of the cytoskeleton caused by mutations in GFAP, as seen in Alexander disease, often leads to neurodegeneration and premature death (Mignot et al., 2004, Perng et al., 2006). Since GFAP is necessary for cytoskeletal integrity and process branching, abnormalities in the expression of this molecule could lead to marked dysfunction.
Astrocytes provide a multitude of crucial functions in the brain and there is growing evidence to suggest that astrocytes have a contributing role in the pathophysiology of schizophrenia. A recent human postmortem gene array profile established that the genes most frequently altered in schizophrenia were related to glial cell function (Sugai et al., 2005). Examining the molecular components of astrocytes that facilitate brain function will help determine whether there is a relationship between the expression and function of astrocytes and the dysfunction seen in schizophrenia.
Early on, the prefrontal cortex emerged as a major site of dysfunction in schizophrenia (Weinberger et al., 1986). Normal functioning of this area depends on reciprocal connections with other structures including, but not limited to, the superior temporal gyrus, cingulate cortex, hippocampus, and primary visual cortex. In vivo imaging studies in patients with schizophrenia have implicated all of these corticolimbic structures in the pathophysiology of schizophrenia by volumetric changes, abnormal activation, and aberrant functional connectivity (Assaf et al., 2006; Garrity et al., 2007). Within these brain regions, astrocytes express molecules that are critical to normal brain function, including enzymes involved in the glutamate-glutamine cycle and cytoskeletal proteins that enable branching of astrocytic processes. We hypothesize that glutamine synthetase and GFAP protein are abnormally expressed in this illness, although it is unclear whether these abnormalities are confined to specific brain regions or whether a global astrocytic lesion exists. Thus, we measured these molecules in an elderly cohort with schizophrenia and a non-psychiatrically ill comparison group using Western blot analysis in several brain areas associated with complex cognitive functions that are often impaired in schizophrenia, including the dorsolateral prefrontal cortex (DLPFC), anterior cingulate cortex (ACC), superior temporal gyrus (STG), primary visual cortex (PVC), and hippocampus (HC).
The majority of our subjects with schizophrenia were treated with typical antipsychotics, which could potentially affect the expression of astrocytic molecules. To address this possibility in an animal model, the expression of glutamine synthetase and GFAP was also investigated in rats chronically administered haloperidol, to determine whether results from our postmortem studies might be attributable to a medication effect.
2. Materials and Methods
2.1 Acquisition and processing of human brain tissue
Fifty subjects from the Mount Sinai Medical Center and Bronx Veterans Administration Medical Center were utilized for this study, consisting of 27 non-psychiatrically ill individuals and 23 patients with schizophrenia (Table 1). Consent for autopsy and use of brain tissue for research was obtained from the legal next of kin of each donor. Subjects were diagnosed with schizophrenia if the following criteria were fulfilled: 1) the presence of schizophrenic symptoms could be documented before age 40; 2) the medical records contained evidence of psychotic symptoms and at least 10 years of psychiatric hospitalization with a diagnosis of schizophrenia; 3) a DSM-III-R diagnosis of schizophrenia was agreed upon by two experienced clinicians; and 4) neuropathologic examination did not reveal Alzheimer’s disease or other discernable neuropathologic abnormalities.
Brains obtained at autopsy were divided mid-sagittally at the time of extraction. The right half was fixed in 4% formaldehyde and used for neuropathological characterization. The left half was sectioned in 6–8 mm coronal slabs, snap-frozen, and stored at −80°C. The areas of interest were identified by gross anatomical landmarks and dissected from the frozen slabs from the following areas: dorsolateral prefrontal cortex, primary visual cortex, superior temporal gyrus, anterior cingulate cortex, and hippocampus (Dracheva et al., 2006, Dracheva et al., 2004, Katsel et al., 2005). Dissected, never-thawed tissues were pulverized at −190°C into a fine powder, aliquoted into individual Eppendorf tubes, and stored at −80°C.
2.2 Acquisition and processing of rodent brain tissue
Twenty-two adult, male Sprague-Dawley rats (250g) were housed 2–3 to a cage, with food and water ad libitum. Animals were treated with daily intramuscular injections of haloperidol dissolved in Dimethyl sulfoxide (DMSO) (Fisher Scientific, Fair Lawn, NJ, USA) (1mg/kg/day, n=11), or vehicle (DMSO) (n=11) for 28 days (Halim et al., 2004, Schmitt et al., 2004, Spurney et al., 1999). Twenty-four hours after the last injection, the animals were sacrificed and brains were immediately removed, dissected, and frozen in isopentane. The frontal, cingulate, occipital, and temporal cortices, and hippocampus were dissected using landmarks from The Rat Brain in Stereotaxic Coordinates (Paxinos and Watson, 1986). The brains were stored at −80°C until assayed. All animal experiments were approved by the University Committee on the Use and Care of Animals at the University of Michigan and were performed according to the guidelines for The Care and Use of Laboratory Animals of the National Institutes of Health.
2.3 Tissue preparation
Tissue specimens (human 50mg, rodent 20mg) were homogenized in 5mM Tris–HCl (pH 7.4), containing Complete, mini, EDTA-free protease inhibitor cocktail tablets (Roche Applied Science, Indianapolis, IN, USA) (1 tablet/10mls) for 30 seconds with a PowerGen 125 homogenizer (Fisher Scientific International, Inc., Hampton NH, USA). Total protein concentration was determined with a BCA Protein Assay Kit (Pierce Biotechnology, Inc., Rockford IL, USA), and homogenates were stored at −80° C.
2.4 Western blot analysis
Samples were prepared by combining tissue homogenate with sample buffer (62.5mM Tris-HCl, 20% glycerol, 2% sodium dodecyl sulfate, 5% β-mercaptoethanol, pH 6.8), and then heated at 95° C for four minutes. Samples (20μg protein/well) and a Kaleidoscope pre-stained standard (Bio-Rad Laboratories, Richmond, CA, USA) were loaded on pre-cast 10% polyacrylamide Tris-HCl gels (Bio-Rad Laboratories, Richmond, CA, USA) in duplicate. To correct for variability in transblotting and loading that can occur within blots, we normalized each sample to a β-Tubulin value. Samples were run in SDS/Tris/glycine buffer (25mM Tris-HCl, 192mM glycine, 0.1% SDS, pH 8.3) at 130 mV for about one hour. Separated proteins were then transferred to nitrocellulose membranes overnight in in Tris-glycine buffer (25mM Tris, 192mM glycine, pH 8.3), in the same apparatus to avoid between blot variability. Following transblotting, all gels were stained to verify effective transfer of proteins. The concentration of protein needed for visualization of immunopositive bands of glutamine synthetase, GFAP, and β-Tubulin (20μg protein/well) was experimentally pre-determined to give a signal that was linear over a large range of detected protein. For each antibody, optimized protocols for washing buffer, dry milk and antibody concentrations were identified before actual experiments.
Blots were blocked with powdered milk in 5% TBS with 0.01% Tween-20 (TBST) (Fisher Scientific, Fair Lawn, NJ, USA) (pH 7.4) for one hour at room temperature and then agitated with mouse anti-glutamine synthetase monoclonal antibody (BD Biosciences, San Diego, CA) (1:1000), or mouse anti-glial fibrillary acidic protein monoclonal antibody (1:500,000) (Chemicon International, Temecula, CA, USA), both of which recognize the human epitope, in 5% powdered milk in TBST for two hours. Blots were washed four times in TBST and incubated with a horseradish peroxidase-coupled goat anti-mouse secondary antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) (1:5000) for two hours on a shaker at room temperature. Following four washes in TBST and two washes in distilled water, enhanced chemiluminescence (ECL) was used for detection. Blots were saturated with ECL reagent (Amersham, Piscataway, NJ, USA), covered in plastic wrap and exposed to ECL film (Amersham, Piscataway, NJ, USA). To ensure there was no between blot variability, all blots from a particular experiment were treated with ECL at the same time, wrapped together, and exposed to the same piece of film. Film was developed and digitally captured with a CCD based imaging system using Scion Imaging software 4.0.3. Gray scale values were obtained for protein bands at the expected molecular weights, membrane background was subtracted, and the adjusted gray scale values from duplicate samples in adjacent lanes were averaged and converted to optical density. Membranes were stripped and re-blotted with secondary antibody to ensure the complete removal of primary antibody, and then re-blotted for β-Tubulin (Upstate, lake Placid, NY, USA). Each sample was later normalized to a β-Tubulin value to correct for within and between blot variability due to possible differences in loading and transblotting. As there were no differences detected for diagnosis and β-Tubulin levels, the mean ratio of optical density of glutamine synthetase or GFAP/β-Tubulin was used for data analysis. No protein loss was detected due to stripping procedures.
This study was designed as five separate experiments, one for each brain region studied. We loaded gels and transblotted the samples to membranes as separate groups from each brain region. Additionally, the proteins of interest varied across brain regions, which necessitated different experimental conditions, such as ECL exposure time to film. Thus, we also analyzed the brain regions separately. The planned comparisons for the dependent variables under study (glutamine synthetase, GFAP) included examining differences in diagnostic groups (schizophrenia vs. controls), but not comparisons of different brain regions. Thus, we did not include region as a grouping factor as it would not have been valid given our experimental design and methodological limitations noted above.
2.5 Statistical analysis
Statistical analysis was carried out using Statistica software (StatSoft, Tulsa, OK). Analysis of variance (ANOVA) was performed with diagnosis as the independent variable and mean optical density ratio as the dependent variable for both human and rodent data. Each brain region was run on a separate membrane and had varying film exposure times depending on the region and molecule examined, thus each was analyzed separately rather than making multiple comparisons. Analysis of human data included a secondary analysis for sex differences and regression analysis for age, postmortem interval (PMI), and pH. In instances where age, PMI, or pH were found to significantly correlate with a diagnosis analysis of covariance (ANCOVA) was used. For all tests α = 0.05.
3. Results
3.1 Protein expression of glutamine synthetase and GFAP in schizophrenia
We used Western blot analysis to measure the expression of glutamine synthetase and GFAP in the DLPFC, ACC, STG, PVC, and HC. Using antibodies specific for these molecules, we detected prominent bands at the expected molecular weights of 45 kDa for glutamine synthetase, and 51 kDa for GFAP (Figure 1).
Figure 1.
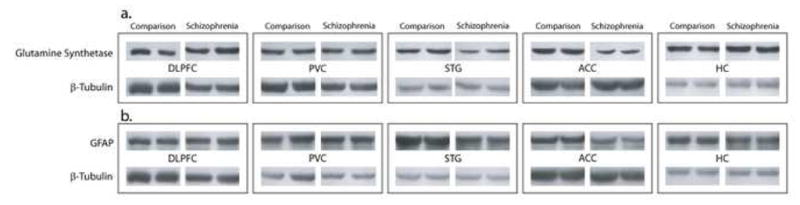
Western blots of representative subjects from a nonpsychiatrically ill comparison group and those with schizophrenia, demonstrating expression of the astrocytic molecules (a) glutamine synthetase, and (b) GFAP in the dorsolateral prefrontal cortex, primary visual cortex, superior temporal gyrus, anterior cingulate cortex, and hippocampus. β-Tubulin was measured as a loading control and subsequent analyses report a ratio of this molecule with the astrocytic proteins measured.
Regression analysis showed no associations between glutamine synthesis expression and age, postmortem interval or pH in any of the areas examined. We did not detect differences by sex in any of the areas studied. Using ANOVA, we found a main effect for diagnosis for glutamine synthetase expression in the STG (F(1, 30) = 6.44; p = 0.02) and ACC (F(1, 30) = 6.99; p = 0.01,) but not in the DLPFC, PVN, or HC (Figure 2). Glutamine synthetase expression in the STG was reduced by 32% in schizophrenia versus the comparison group. In the ACC, there was a 19% decrease in the expression of glutamine synthetase in schizophrenia.
Figure 2.
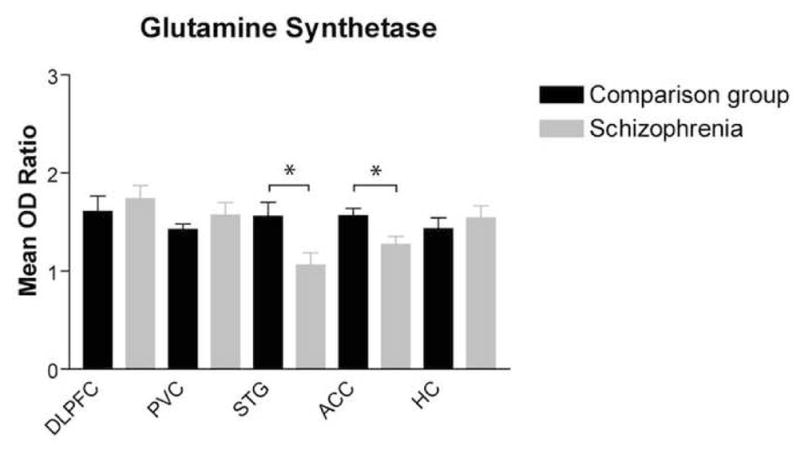
Glutamine synthetase protein expression (mean OD ratio ± SEM) in the dorsolateral prefrontal cortex, primary visual cortex, superior temporal gyrus, anterior cingulate cortex, and hippocampus was analyzed by diagnosis (comparison group and schizophrenia). Glutamine synthetase was significantly decreased in the superior temporal gyrus in schizophrenia (F(1,30) = 6.44; *p < 0.05 and ACC (F(1,30) = 6.99; p < 0.05).
Regression analysis showed no associations between GFAP expression and age, postmortem interval or pH in the DLPFC, ACC, or PVC. In the STG and HC, there was an association between GFAP expression, age, and PMI: STG (age: r = 0.443, p = 0.003; PMI: r = 0.465, p = .0002), and HC (age: r = 0.329, p = 0.041; PMI: r = 0.409, p = 0.009). Using ANCOVA with age and PMI as co-variates, we did not detect an effect for diagnosis on GFAF expression in either the STG or HC. We did not detect differences by sex in any of the areas studied. Using ANOVA, we found a main effect for diagnosis for GFAP expression in the ACC (F(1, 43) = 5.68; p = 0.02), but not in the DLPFC, or PVC (Figure 3). The expression of GFAP in the ACC was reduced in schizophrenia by 13% versus the comparison group.
Figure 3.
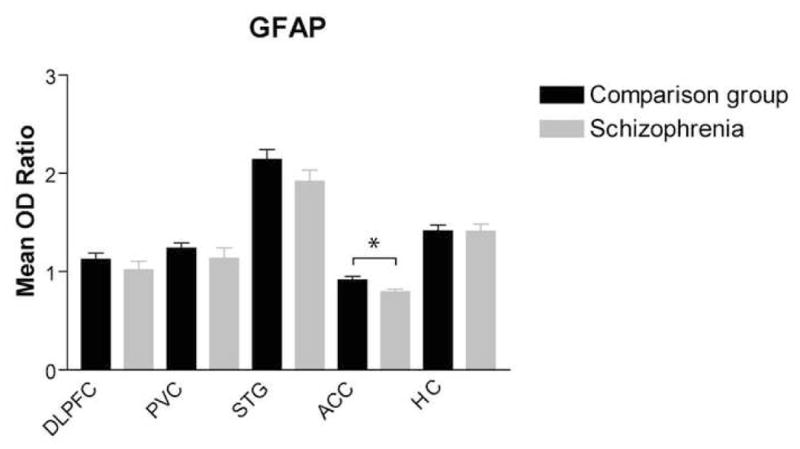
GFAP protein expression (mean OD ratio) in the dorsolateral prefrontal cortex, primary visual cortex, superior temporal gyrus, anterior cingulate cortex, and hippocampus was analyzed by diagnosis (comparison group and schizophrenia). GFAP was significantly decreased in the anterior cingulate cortex in schizophrenia [F(1,43) = 5.68; p < 0.05).
We performed regression analysis to examine the effect of age on protein expression of glutamine synthetase and GFAP in these five brain regions. We did not detect a significant effect for age on glutamine synthetase expression in any of the regions studied, whether data were analyzed by diagnosis or pooled (data not shown). GFAP expression was not associated with age in any of the brain areas when subjects from each diagnostic group where analyzed separately. When data were pooled, there was a significant association between GFAP expression and age in the STG (p = 0.003), and HC (p = 0.04) (Data not shown). Using pooled data, there was no association between age and GFAP expression in the DLPFC, PVC, or ACC.
3.2 Effects of haloperidol treatment on protein expression of glutamine synthetase and GFAP in the rat brain
The majority of subjects with schizophrenia in this study were treated with typical antipsychotics. We used Western blot analysis to measure the expression of glutamine synthetase and GFAP in rats chronically treated (1mg/kg/day) for 28 days with haloperidol. We did not detect differences in protein expression for either molecule in any of the brain regions studied (Figure 4).
Figure 4.

Protein expression of (a) glutamine synthetase and (b) GFAP in the frontal cortex (FC), primary visual cortex, superior temporal gyrus, anterior cingulate cortex, and hippocampus of rats treated chronically treated with haloperidol or vehicle. There were no treatment effects detected.
4. Discussion
We tested the hypothesis that critical components of astrocytes, including molecules involved with the glutamate-glutamine cycle and cytoskeletal structure, are abnormally expressed in schizophrenia. Astrocytes are involved in a cycle of synthesis and degradation of glutamate. The enzyme glutamine synthetase contributes to this cycle by converting recovered glutamate to glutamine, which is then shuttled back to the presynaptic terminal. Thus, altered expression and/or function of this enzyme might have profound effects on the glutamate-glutamine cycle and potentially on glutamatergic transmission. We found decreased expression of this enzyme in the ACC and STG in schizophrenia, suggesting a decreased capacity to cycle glutamate in these regions.
The cytoskeletal integrity of astrocytes is vital for normal brain functioning. Changes in GFAP expression leading to abnormal structure and function of astrocytes, including decreased process branching, have been linked to impairments in synaptic functioning and abnormal behavior. We found decreased GFAP protein in the ACC in schizophrenia, suggesting that astrocyte-mediated brain functions requiring astrocytic process extension may be impaired in this brain region in schizophrenia.
4.1 Expression of glutamine synthetase in schizophrenia
Glutamine synthetase is beginning to emerge as an astrocytic molecule that is altered in schizophrenia. Hashimoto et al. has reported an altered glutamine to glutamate ratio in the CSF of patients with schizophrenia (Hashimoto et al., 2005). Furthermore, glutamine synthetase expression changes have also been found in post mortem brain tissue. Our present results of decreased glutamine synthetase are consistent with those of Burbaeva et al., who reported deceased protein expression in area 10 of the PFC (Burbaeva et al., 2003). A proteomic study by Prabakaran et al. also described reduced glutamine synthetase in schizophrenia in area 9 of the frontal cortex (Prabakaran et al., 2004). Consistent with reports of multiple isoforms of glutamine synthetase (Chakrabarti et al., 1995; Boksha et al., 2000), the study by Burbaeva et al. also showed that protein expression of an enzyme similar in activity and immunoreactivity to glutamine synthetase, termed glutamine synthetase like protein (GSLP), was increased in this brain region. In addition to protein levels, this study measured the transferase activity of glutamine synthetase and GSLP; however, no changes were detected. Taken together, unchanged enzyme activity and expression changes of glutamine synthetase and GSLP in opposite directions suggest that these enzymes may be differentially regulated in disease states by astrocytes. Furthermore, these data suggest that the study of glutamine synthetase may be confounded by the presence of multiple distinct isoforms and that past reports of unchanged glutamine synthetase activity may have been due to a summation of activity from multiple isoforms (Gluck et al., 2002).
Abnormal glutamine synthetase expression in the ACC and STG may be the result of glutamatergic dysfunction in other brain regions, such as the thalamus, a relay station of sensory inputs that processes information from multiple cortical regions. Some of the sensory and cognitive disturbances seen in schizophrenia are consistent with glutamatergic dysfunction of thalamic circuitry. Transcript expression studies of glutamatergic molecules in the thalamus by our laboratory have revealed many changes in this illness, including increased vesicular glutamate transporters, decreased NMDA receptor subunits and proteins involved in receptor trafficking, increased astrocytic EAATs 1 and 2, and increased EAAT interacting proteins (Clinton et al., 2006, Clinton and Meador-Woodruff, 2004, Huerta et al., 2006, Ibrahim et al., 2000, Meador-Woodruff et al., 2003, Smith et al., 2001a, Smith et al., 2001b). We have also demonstrated that glutamine synthetase transcript levels in the thalamus are increased in several nuclei, including those that project to the ACC and STG (Bruneau et al., 2005). Increased transcript expression of thalamic glutamine synthetase may be a compensatory response to decreased glutamine synthetase protein or to abnormal protein assembly that has lead to decreased enzyme activity. Reduced glutamate recycling in the thalamus could cause abnormal glutamatergic transmission and potentially affect astrocyte gene expression in areas that receive thalamic efferent projections, such as the ACC and STG.
The enzymatic activity of glutamine synthetase may be another factor influencing protein expression of this molecule. NMDA receptor activation in neighboring neurons, in part, modulates glutamine synthetase activity (Watts et al., 2005). Following presynaptic glutamate release and subsequent postsynaptic receptor activation, neurons release nitric oxide (NO), a key biological messenger and neurotransmitter, which can diffuse to bordering astrocytes and decrease glutamine synthetase activity (Kosenko et al., 2003). Conversely, decreased NMDA receptor activation reduces inhibition of glutamine synthetase. Abnormalities of NMDA receptor expression, assembly, cellular localization, and binding have been shown in the ACC and STG in schizophrenia (Corre et al., 2000, Grimwood et al., 1999, Humphries et al., 1996, Kristiansen et al., 2006), so it is possible that illness-related changes in NMDA receptor activation could keep glutamine synthetase in a chronically active state. In turn, this could cause astrocytes to decrease production of glutamine synthetase or to increase protein disassembly and degradation to correct for abnormally high activity (Kosenko et al., 2003). So far, studies have not shown changes to glutamine synthetase activity in the brain in schizophrenia (Burbaeva et al., 2003, Gluck et al., 2002).
Changes to the expression of this enzyme may reflect a decrease in astrocyte density, as has been reported in several of the cortical areas compromised in schizophrenia, including the PFC, ACC, and motor cortex (Benes et al., 1986, Cotter et al., 2001, Cotter et al., 2002, Stark et al., 2004). Alternatively, more subtle expression and activity changes may actually be present, but masked by loss of astrocyte density. This account might explain some results from past studies, which did not find changes in glutamine synthetase mRNA or protein expression (Beasley et al., 2006, Toro et al., 2006).
The functional significance of regional changes in glutamine synthetase expression is not obvious. Decreased glutamine synthetase expression suggests a decreased capacity to cycle glutamate that may be secondary to abnormal expression of other astrocytic molecules, such as GFAP. Such changes may reflect region-specific abnormalities in both the structure and function of astrocytes in schizophrenia.
4.2 Expression of GFAP in schizophrenia
Our data show decreased expression of GFAP in the ACC, which is consistent with previous reports of altered expression of this cytoskeletal protein. GFAP expression in the PFC, as measured by immunohistochemical labeling, has been shown to be significantly reduced in schizophrenia (Knable et al., 2001). Additionally, studies demonstrating lamina-specific reductions in the area occupied by GFAP labeled cells have been interpreted as evidence of decreased processes branching in this illness (Rajkowska et al., 2002). Consistent with loss of process branching, phosphorylated isoforms of GFAP protein are also decreased, suggesting that mechanisms regulating GFAP assembly and disassembly may also be functionally abnormal (Johnston-Wilson et al., 2000). Taken together, these studies demonstrate that loss of process branching due to decreased GFAP expression may be contributing to astrocytic dysfunction, effectively causing widespread dysfunction in schizophrenia. Alternatively, loss of astrocyte density in the ACC, as has been previously shown, is another possible explanation for the current results (Cotter et al., 2001, Stark et al., 2004).
Several studies have demonstrated that GFAP mRNA and protein increase with age without an accompanying change in astrocyte density (David et al., 1997, Hansen et al., 1987, Kohama et al., 1995, Nichols et al., 1993). Consistent with these findings, our analysis of pooled data, which included both diagnostic groups (schizophrenia and comparison), showed that GFAP expression increased with age in all of the brain regions studied. There was a positive correlation between GFAP expression and age that reached statistical significance in the STG and HC and marginal significance in the ACC. However, when subjects were analyzed separately by diagnosis none of the measures reached statistical significance.
One study found increased GFAP protein expression in elderly patients with schizophrenia also exhibiting dementia (Arnold et al., 1996). This, taken with our present data from elderly subjects, suggests that GFAP expression varies not only by region and diagnosis, but may also delineate subgroups of persons with schizophrenia, particularly associated with advanced age and end stage schizophrenia. Supporting this hypothesis, several studies that have used younger subjects found GFAP expression unchanged in schizophrenia; however most of these studies did not seek to examine the effects of age on GFAP expression (Dean et al., 2006, Fatemi et al., 2004, Karson et al., 1999, Perrone-Bizzozero et al., 1996). Recently, a study found increased GFAP immunoreactivity in the prefrontal cortex (Toro 2006), however, because these results were drawn from the combined analysis of tissue from multiple Brodmann areas it is difficult to reconcile with our data that shows region-specific differences in GFAP expression.
As a whole, these data suggest that decreased GFAP in the ACC in schizophrenia may be evidence of: 1) a loss of process branching, 2) region-specific compensatory gene regulation and, 3) illness-related expression changes inconsistent with predicted changes caused by aging.
4.3 Effects of haloperidol on expression of glutamine synthetase and GFAP in the rat brain
The majority of our subjects with schizophrenia were treated with typical antipsychotics, which could potentially affect the expression of glutamine synthetase and/or GFAP. We investigated the protein expression of these molecules in rats chronically administered haloperidol. We did not find any changes in glutamine synthetase expression in any of the brain regions of the treated rats, suggesting that our findings of decreased glutamine synthetase are not likely due to a medication effect.
There are few reports describing the effects of antipsychotic treatment on glutamine synthetase expression. Studies measuring glutamine synthetase protein levels in the brain do not find changes that can be related to antipsychotic treatment (Burbaeva et al., 2003, Toro et al., 2006). However, recently it was reported that peripheral protein levels of GSLP, an isoform of glutamine synthetase, may be altered by treatment with antipsychotics (Burbaeva et al., 2006). Burbaeva and colleagues demonstrated that increased platelet expression of GSLP in schizophrenia is reduced following treatment with the atypical antipsychotic, olanzapine. In addition to expression, the activity of this enzyme may also be influenced by antipsychotic medication. A study found that glutamine synthetase activity was increased in various regions of the rat brain after a single dose of the typical antipsychotic chlorpromazine, and was increased in the cerebral cortex following long-term administration (Chandrakala et al., 1987).
We did not find changes in GFAP expression in rats treated with haloperidol. This is consistent with a recent study that failed to detect changes in GFAP protein expression in rat frontal cortex following chronically administered haloperidol (Dean et al., 2006). The literature surrounding acute and chronic treatment with haloperidol does not resolve whether changes in astrocyte density or the expression of astrocytic molecules such as GFAP can be attributed to antipsychotics. Reportedly, rats given a single dose of haloperidol did not demonstrate astrocyte activation as measured by double immunofluorescence labeling of c-fos and GFAP (Ma et al., 2003). Another study reported that MK801-induced increases in astrocyte density and GFAP immunoreactivity in the rat cingulate and retrosplenial cortices could be prevented by the atypical antipsychotic clozapine, but not by haloperidol (Arif et al., 2007).
5. Conclusions
In this study, we report decreased glutamine synthetase in ACC and STG and decreased GFAP protein in the ACC in schizophrenia. Further study is needed to clarify whether multi-regional glutamatergic dysfunction results in a decreased capacity to cycle glutamate or whether loss of process branching that limits the reuptake of synaptic glutamate, consistent with our findings of GFAP expression changes, underlie altered glutamine synthetase expression. Our data support the hypothesis that astrocytes contribute to the pathophysiology of schizophrenia and that astrocytic molecules involved in glutamatergic function and cytoskeletal integrity are compromised in this illness.
5.1 Limitations and Future Studies
Western blot analysis, while a useful means of analyzing protein expression changes within a brain region is not designed to determine whether discrete populations of astrocytes are exhibiting expression changes. Astrocytes are a heterogeneous cell type displaying different phenotypes and physiologies, thus, it would be advantageous to identify the specific population(s) of astrocytes involved in these molecular changes and in the pathophysiology of schizophrenia.
Table I.
Subject Characteristics
| Diagnosis | Sex | Age | PMI (hours) | pH | Neuroleptic | Cause of Death |
|---|---|---|---|---|---|---|
| Comparison Group | F | 62 | 7.0 | 6.6 | acute myocardial infarction | |
| F | 73 | 3.4 | 6.3 | acute myocardial infarction | ||
| F | 74 | 3.0 | 6.0 | cardiopulmonary arrest | ||
| F | 75 | 6.5 | 6.0 | cardiopulmonary arrest | ||
| F | 80 | 4.8 | 6.2 | sepsis | ||
| F | 82 | 5.7 | 6.1 | cardiopulmonary arrest | ||
| F | 83 | 6.2 | 6.8 | cardiopulmonary arrest | ||
| F | 84 | 18.5 | 6.2 | myocardial infarction | ||
| F | 86 | 4.7 | 6.5 | acute myocardial infarction | ||
| F | 88 | 5.1 | 6.4 | chronic obstructive pulmonary disease | ||
| F | 89 | 2.3 | 6.7 | bronchopneumonia, chronic obstructive pulmonary disease | ||
| F | 90 | 4.2 | 6.0 | pneumonia | ||
| F | 98 | 1.4 | 6.6 | myocardial infarction, aortic aneurysm | ||
| M | 59 | 20.4 | 6.7 | coronary arterial disease | ||
| M | 60 | 28.8 | 6.6 | acute myocardial infarction | ||
| M | 64 | 4.2 | 6.4 | acute myocardial infarction | ||
| M | 65 | 3.8 | 6.8 | renal failure | ||
| M | 66 | 7.6 | 6.6 | acute myocardial infarction | ||
| M | 69 | 4.3 | 6.3 | cancer of lung | ||
| M | 69 | 7.4 | 6.7 | septic shock | ||
| M | 74 | 16.6 | 6.7 | cardiopulmonary arrest | ||
| M | 76 | 2.9 | 6.3 | bronchopneumonia | ||
| M | 85 | 5.3 | 6.5 | transitional cell cancer of bladder | ||
| M | 92 | 20.0 | 6.4 | arrhythmia | ||
| M | 93 | 4.2 | 6.3 | acute myocardial infarction | ||
| M | 95 | 4.1 | 6.5 | chronic renal failure | ||
| M | 101 | 4.7 | 6.8 | congestive heart failure | ||
| 14M, 13F | 79 ± 12 | 7.7 ± 6.8 | 6.5 ± 0.2 | |||
|
| ||||||
| Schizophrenia | F | 69 | 13.7 | 6.2 | Mellaril | cardiopulmonary arrest |
| F | 74 | 7.0 | 6.3 | Haldol | Cardiopulmonary Arrest | |
| F | 77 | 9.7 | 6.0 | Risperidol | cardiopulmonary arrest | |
| F | 79 | 9.9 | 6.8 | Haldol | cardiac arrest | |
| F | 81 | 12.5 | 5.9 | Risperidol | acute myocardial infarction | |
| F | 81 | 15.1 | 6.7 | UKN | cardiopulmonary arrest | |
| F | 83 | 20.4 | 7.1 | Mellaril | cardiopulmonary arrest, cancer of pancreas | |
| M | 52 | 29.5 | 5.9 | Mellaril | cardiopulmonary arrest | |
| M | 56 | 13.5 | 6.5 | Haldol | cardiopulmonary arrest | |
| M | 57 | 30.3 | 6.1 | Haldol | expired during open heart surgery | |
| M | 57 | 21.4 | 6.4 | Haldol | small cell cancer of lung | |
| M | 58 | 6.7 | 6.2 | Haldol | cardiopulmonary arrest | |
| M | 58 | 13.3 | 6.9 | Haldol | cardiopulmonary arrest | |
| M | 63 | 6.2 | 6.3 | Thorazine | cardiopulmonary arrest | |
| M | 68 | 17.3 | 6.6 | Haldol | congestive heart failure, coronary artery disease | |
| M | 69 | 40.2 | 6.7 | UKN | acute renal failure | |
| M | 73 | 7.9 | 6.5 | Haldol | cardiopulmonary arrest | |
| M | 76 | 16.6 | 6.7 | Risperidone | coronary artery disease, congestive heart failure | |
| M | 84 | 6.2 | 6.5 | Haledol | cardiopulmonary arrest | |
| M | 85 | 5.3 | 6.3 | Thorazine | chronic obstructive pulmonary disease, pneumonia | |
| M | 86 | 7.0 | 6.3 | Mellaril | cardiopulmonary arrest | |
| M | 86 | 14.1 | 6.7 | Mellaril | artheroschlerotic heart disease | |
| M | 87 | 11.2 | 6.5 | Thorazine | cardiopulmonary arrest | |
| 16M, 7F | 72 ± 12 | 14.6 ± 8.9 | 6.5 ± 0.3 | 18Typ, 3 Atyp | ||
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allen N, Barres B. Signaling between glia and neurons: Focus on synaptic plasticity. Current Opinion in Neurobiology. 2005;15 (5):542–548. doi: 10.1016/j.conb.2005.08.006. [DOI] [PubMed] [Google Scholar]
- Arif M, Chikuma T, Ahmed M, Yoshida S, Kato T. Suppressive effect of clozapine but not haloperidol on the increases of neuropeptide-degrading enzymes and glial cells in MK-801-treated rat brain regions. Neuroscience Research. 2007;57 (2):248–258. doi: 10.1016/j.neures.2006.10.021. [DOI] [PubMed] [Google Scholar]
- Arnold S, Franz B, Trojanowski J, Moberg P, Gur R. Glial fibrillary acidic protein-immunoreactive astrocytosis in elderly patients with schizophrenia and dementia. Acta Neuropathologica. 1996;91:269–277. doi: 10.1007/s004010050425. [DOI] [PubMed] [Google Scholar]
- Assaf M, Rivkin P, Kuzu C, Calhoun V, Kraut M, Groth K, Yassa M, Jr, JH, Pearlson G. Abnormal object recall and anterior cingulate overactivation correlate with formal thought disorder in schizophrenia. Biological Psychiatry. 2006;59 (5):452–459. doi: 10.1016/j.biopsych.2005.07.039. [DOI] [PubMed] [Google Scholar]
- Beasley C, Pennington K, Behan A, Wait R, Dunn M, Cotter D. Proteomic analysis of the anterior cingulate cortex in the major psychiatric disorders: Evidence for disease-associated changes. Proteomics. 2006;6 (11):3414–3425. doi: 10.1002/pmic.200500069. [DOI] [PubMed] [Google Scholar]
- Bellocchio E, Reimer R, Fremeau R, Edwards R. Uptake of glutamate into synaptic vesicles by an inorganic phosphate transporter. Science. 2000;289 (5481):957–960. doi: 10.1126/science.289.5481.957. [DOI] [PubMed] [Google Scholar]
- Benes F, Davidson J, Bird E. Quantitative cytoarchitectural studies of the cerebral cortex of schizophrenics. Archives of General Psychiatry. 1986;43:31–35. doi: 10.1001/archpsyc.1986.01800010033004. [DOI] [PubMed] [Google Scholar]
- Boksha I, Tereshkina E, Burbaeva GS. Glutamine synthetase and glutamine synthetase-like protein from human brain. Purification and comparative characterization. Journal of Neurochemistry. 2000;75:2574–2582. doi: 10.1046/j.1471-4159.2000.0752574.x. [DOI] [PubMed] [Google Scholar]
- Bruneau E, McCullumsmith R, Haroutunian V, Davis K, Meador-Woodruff J. Increased expression of glutaminase and glutamine synthetase mRNA in the thalamus in schizophrenia. Schizophrenia Research. 2005;75 (1):27–34. doi: 10.1016/j.schres.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Burbaeva G, Boksha I, Turishcheva M, Vorobyeva E, Savushkina O, Tereshkina E. Glutamine synthetase and glutamate dehydrogenase in the prefrontal cortex of patients with schizophrenia. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2003;27 (4):675–680. doi: 10.1016/s0278-5846(03)00078-2. [DOI] [PubMed] [Google Scholar]
- Burbaeva GS, Boksha I, Tereshkina E, Savushkina O, Turishcheva M, Starodubtseva L, Brusov O, Morozova M. Effect of olanzapine treatment on platelet glutamine synthetase-like protein and glutamate dehydrogenase immunoreactivity in schizophrenia. World Journal of Biological Psychiatry. 2006;7 (2):75–81. doi: 10.1080/15622970510029957. [DOI] [PubMed] [Google Scholar]
- Chakrabarti R, McCracken J, Chakrabarti D, Souba W. Detection of a functional promoter/enhancer in an intronless human gene encoding a GS-like enzyme. Gene. 1995;153:163–169. doi: 10.1016/0378-1119(94)00751-d. [DOI] [PubMed] [Google Scholar]
- Chandrakala M, Marcus S, Nadiger H, Sadasivudu B. Acute and long-term effects of chlorpromazine on glutamine synthetase and glutaminase in rat brain. Journal of Neurochemistry. 1987;49 (1):32–34. doi: 10.1111/j.1471-4159.1987.tb03389.x. [DOI] [PubMed] [Google Scholar]
- Chen W, Liem R. Reexpression of glial fibrillary acidic protein rescues the ability of astrocytoma cells to form processes in response to neurons. Journal of Cell Biology. 1994;127 (3):813–823. doi: 10.1083/jcb.127.3.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clinton S, Haroutunian V, Meador-Woodruff J. Up-regulation of NMDA receptor subunit and post-synaptic density protein expression in the thalamus of elderly patients with schizophrenia. Journal of Neurochemistry. 2006;98 (4):1114–1125. doi: 10.1111/j.1471-4159.2006.03954.x. [DOI] [PubMed] [Google Scholar]
- Clinton S, Meador-Woodruff J. Thalamic dysfunction in schizophrenia: neurochemical, neuropathological, and in vivo imaging abnormalities. Schizophrenia Research. 2004;69 (2–3):237–253. doi: 10.1016/j.schres.2003.09.017. [DOI] [PubMed] [Google Scholar]
- Clinton S, Haroutunian V, Davis K, Meador-Woodruff J. Altered transcript expression of NMDA receptor-associated postsynaptic proteins in the thalamus of subjects with schizophrenia. The American Journal of Psychiatry. 2003;160 (6):1100–1109. doi: 10.1176/appi.ajp.160.6.1100. [DOI] [PubMed] [Google Scholar]
- Cornell-Bell A, Finkbeiner S, Cooper M, Smith S. Glutamate induces calcium waves in cultured astrocytes: long-range glial signaling. Science. 1990;247:470–473. doi: 10.1126/science.1967852. [DOI] [PubMed] [Google Scholar]
- Corre SL, Harper C, Lopez P, Ward P, Catts S. Increased levels of expression of an NMDARI splice variant in the superior temporal gyrus in schizophrenia. Neuroreport. 2000;11 (5):983–986. doi: 10.1097/00001756-200004070-00017. [DOI] [PubMed] [Google Scholar]
- Cotter D, Mackay D, Chana G, Beasley C, Landau S, Everall I. Reduced neuronal size and glial cell density in area 9 of the dorsolateral prefrontal cortex in subjects with major depressive disorder. Cerebral Cortex. 2002;12:386–394. doi: 10.1093/cercor/12.4.386. [DOI] [PubMed] [Google Scholar]
- Cotter D, Mackay D, Landau S, Kerwin R, Everall I. Reduced glial cell density and neuronal size in the anterior cingulate cortex in major depressive disorder. Archives of General Psychiatry. 2001;58:545–553. doi: 10.1001/archpsyc.58.6.545. [DOI] [PubMed] [Google Scholar]
- Coyle J. The glutamatergic dysfunction hypothesis for schizophrenia. Harvard Review of Psychiatry. 1996;3:21–36. doi: 10.3109/10673229609017192. [DOI] [PubMed] [Google Scholar]
- David J, Ghozali F, Fallet-Bianco C, Wattez A, Delaine S, Boniface B, Menza CD, Delacourte A. Glial reaction in the hippocampal formation is highly correlated with aging in human brain. Neuroscience Letters. 1997;235 (1–2):53–56. doi: 10.1016/s0304-3940(97)00708-8. [DOI] [PubMed] [Google Scholar]
- Dean B, Gray L, Scarr E. Regionally specific changes in levels of cortical S100beta in bipolar 1 disorder but not schizophrenia. Australian and New Zealand Journal of Psychiatry. 2006;40 (3):217–224. doi: 10.1080/j.1440-1614.2006.01777.x. [DOI] [PubMed] [Google Scholar]
- Dracheva S, Davis K, Chin B, Woo D, Schmeidler J, Haroutunian V. Myelin-associated mRNA and protein expression deficits in the anterior cingulate cortex and hippocampus in elderly schizophrenia patients. Neurobiology of Disease. 2006;21 (3):531–540. doi: 10.1016/j.nbd.2005.08.012. [DOI] [PubMed] [Google Scholar]
- Dracheva S, Elhakem S, McGurk S, Davis K, Haroutunian V. GAD67 and GAD65 mRNA and protein expression in cerebrocortical regions of elderly patients with schizophrenia. Journal of Neuroscience Research. 2004;76 (4):581–592. doi: 10.1002/jnr.20122. [DOI] [PubMed] [Google Scholar]
- Fatemi S, Laurence J, Araghi-Niknam M, Stary J, Schulz S, Lee S, Gottesman I. Glial fibrillary acidic protein is reduced in cerebellum of subjects with major depression, but not schizophrenia. Schizophrenia Research. 2004;69:317–323. doi: 10.1016/j.schres.2003.08.014. [DOI] [PubMed] [Google Scholar]
- Garrity A, Pearlson G, McKiernan K, Lloyd D, Kiehl K, Calhoun V. Aberrant “default mode” functional connectivity in schizophrenia. American Journal of Psychiatry. 2007;164 (3):450–457. doi: 10.1176/ajp.2007.164.3.450. [DOI] [PubMed] [Google Scholar]
- Gluck M, Thomas R, Davis K, Haroutunian V. Implications for altered glutamate and GABA metabolism in the dorsolateral prefrontal cortex of aged schizophrenic patients. American Journal of Psychiatry. 2002;159 (7):1165–1173. doi: 10.1176/appi.ajp.159.7.1165. [DOI] [PubMed] [Google Scholar]
- Goff D, Coyle J. The emerging role of glutamate in the pathophysiology and treatment of schizophrenia. The American Journal of Psychiatry. 2001;158 (9):1367–1377. doi: 10.1176/appi.ajp.158.9.1367. [DOI] [PubMed] [Google Scholar]
- Grimwood S, Slater P, Deakin J, Hutson P. NR2B-containing NMDA receptors are up-regulated in temporal cortex in schizophrenia. Neuroreport. 1999;10 (3):461–465. doi: 10.1097/00001756-199902250-00004. [DOI] [PubMed] [Google Scholar]
- Halim N, Weickert C, McClintock B, Weinberger D, Lipska B. Effects of chronic haloperidol and clozapine treatment on neurogenesis in the adult rat hippocampus. Neuropsychopharmacology. 2004;29 (6):1063–1069. doi: 10.1038/sj.npp.1300422. [DOI] [PubMed] [Google Scholar]
- Hansen L, Armstrong D, Terry R. An immunohistochemical quantification of fibrous astrocytes in the aging human cerebral cortex. Neurobiology of Aging. 1987;8 (1):1–6. doi: 10.1016/0197-4580(87)90051-0. [DOI] [PubMed] [Google Scholar]
- Hashimoto A, Yoshikawa M, Niwa A, Konno R. Mice lacking D-amino acid oxidase activity display marked attenuation of stereotypy and ataxia induced by MK-801. Brain Research. 2005;1033 (2):210–215. doi: 10.1016/j.brainres.2004.11.031. [DOI] [PubMed] [Google Scholar]
- Heresco-Levy U, Javitt D, Ebstein R, Vass A, Lichtenberg P, Bar G, Catinari S, Ermilov M. D-serine efficacy as add-on pharmacotherapy to risperidone and olanzapine for treatment-refractory schizophrenia. Biological Psychiatry. 2005;57 (6):577–585. doi: 10.1016/j.biopsych.2004.12.037. [DOI] [PubMed] [Google Scholar]
- Hollmann M, Heinemann S. Cloned glutamate receptors. Annual Review of Neuroscience. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- Huerta I, McCullumsmith R, Haroutunian V, Gimenez-Amaya J, Meador-Woodruff J. Expression of excitatory amino acid transporter interacting protein transcripts in the thalamus in schizophrenia. Synapse. 2006;59 (7):394–402. doi: 10.1002/syn.20250. [DOI] [PubMed] [Google Scholar]
- Humphries C, Mortimer A, Hirsch S, Belleroche Jd. NMDA receptor mRNA correlation with antemortem cognitive impairment in schizophrenia. Neuroreport. 1996;7 (12):2051–2055. doi: 10.1097/00001756-199608120-00040. [DOI] [PubMed] [Google Scholar]
- Ibrahim H, Jr, AH, Healy D, Haroutunian V, Davis K, Meador-Woodruff J. Ionotropic glutamate receptor binding and subunit mRNA expression in thalamic nuclei in schizophrenia. American Journal of Psychiatry. 2000;157 (11):1811–1823. doi: 10.1176/appi.ajp.157.11.1811. [DOI] [PubMed] [Google Scholar]
- Javitt D. Negative schizophrenic symptomatology and the PCP (phencyclidine) model of schizophrenia. Hillside Journal of Clinical Psychiatry. 1987;9 (1):12–35. [PubMed] [Google Scholar]
- Johnston-Wilson NL, Sims CD, Hofmann JP, Anderson L, Shore AD, Torrey EF, Yolken RH. Disease-specific alterations in frontal cortex brain proteins in schizophrenia, bipolar disorder, and major depressive disorder. Molecular Psychiatry. 2000;5 (2):142–149. doi: 10.1038/sj.mp.4000696. [DOI] [PubMed] [Google Scholar]
- Karson C, Mrak R, Schluterman K, Sturner W, Sheng J, Griffin W. Alterations in synaptic proteins and their encoding mRNAs in prefrontal cortex in schizophrenia: a possible neurochemical basis for ‘hypofrontality’. Molecular Psychiatry. 1999;4:39–45. doi: 10.1038/sj.mp.4000459. [DOI] [PubMed] [Google Scholar]
- Katsel P, Davis K, Gorman J, Haroutunian V. Variations in differential gene expression patterns across multiple brain regions in schizophrenia. Schizophrenia Research. 2005;77 (2–3):241–252. doi: 10.1016/j.schres.2005.03.020. [DOI] [PubMed] [Google Scholar]
- Kim S, Choi S, Chao W, Volsky D. Transcriptional regulation of human excitatory amino acid transporter 1 (EAAT1): cloning of the EAAT1 promoter and characterization of its basal and inducible activity in human astrocytes. Journal of Neurochemistry. 2003;87 (6):1485–1498. doi: 10.1046/j.1471-4159.2003.02128.x. [DOI] [PubMed] [Google Scholar]
- Knable M, Torrey E, Webster M, Bartko J. Multivariate analysis of prefrontal cortical data from the Stanley Foundation Neuropathology Consortium. Brain Research Bulletin. 2001;55 (5):651–659. doi: 10.1016/s0361-9230(01)00521-4. [DOI] [PubMed] [Google Scholar]
- Kohama S, Goss J, Finch C, McNeill T. Increases of glial fibrillary acidic protein in the aging female mouse brain. Neurobiology of Aging. 1995;16 (1):59–67. doi: 10.1016/0197-4580(95)80008-f. [DOI] [PubMed] [Google Scholar]
- Kosenko E, Llansola M, Montoliu C, Monfort P, Rodrigo R, Hernandez-Viadel M, Erceg S, Sanchez-Perez A, Felipo V. Glutamine synthetase activity and glutamine content in brain: Modulation by NMDA receptors and nitric oxide. Neurochemistry International. 2003;43 (4–5):493. doi: 10.1016/s0197-0186(03)00039-1. [DOI] [PubMed] [Google Scholar]
- Kristiansen L, Huerta I, Beneyto M, Meador-Woodruff J. NMDA receptors and schizophrenia. Current Opinion in Pharmacology. 2007;7(1):48–55. doi: 10.1016/j.coph.2006.08.013. [DOI] [PubMed] [Google Scholar]
- Kristiansen L, Beneyto M, Haroutunian V, Meador-Woodruff J. Changes in NMDA receptor subunits and interacting PSD proteins in dorsolateral prefrontal and anterior cingulate cortex indicate abnormal regional expression in schizophrenia. Molecular Psychiatry. 2006;11 (8):737–747. doi: 10.1038/sj.mp.4001844. [DOI] [PubMed] [Google Scholar]
- Lahti A, Koffel B, LaPorte D, Tamminga C. Subanesthetic doses of ketamine stimulate psychosis in schizophrenia. Neuropsychopharmacology. 1995;13:9–19. doi: 10.1016/0893-133X(94)00131-I. [DOI] [PubMed] [Google Scholar]
- Laruelle M, Kegeles L, Abi-Dargham A. Glutamate, Dopamine, and Schizophrenia: From Pathophysiology to Treatment. Annals of the New York Academy of Sciences. 2003;1003 (1):138–158. doi: 10.1196/annals.1300.063. [DOI] [PubMed] [Google Scholar]
- Ma J, Ye N, Lange N, Cohen B. Dynorphinergic GABA neurons are a target of both typical and atypical antipsychotic drugs in the nucleus accumbens shell, central amygdaloid nucleus and thalamic central medial nucleus. Neuroscience. 2003;121 (4):991–998. doi: 10.1016/s0306-4522(03)00397-x. [DOI] [PubMed] [Google Scholar]
- Matute C, Melone M, Vallejo-Illarramendi A, Conti F. Increased expression of the astrocytic glutamate transporter GLT-1 in the prefrontal cortex of schizophrenics. Glia. 2005;49:451–455. doi: 10.1002/glia.20119. [DOI] [PubMed] [Google Scholar]
- McCall M, Gregg R, Behringer R, Brenner M, Delaney C, Galbreath E, Zhang C, Pearce R, Chiu S, Messing A. Targeted deletion in astrocyte intermediate filament (GFAP) alters neuronal physiology. Proceedings of the National Academy of Sciences. 1996;93 (13):6361–6366. doi: 10.1073/pnas.93.13.6361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meador-Woodruff J, Clinton S, Beneyto M, McCullumsmith R. Molecular abnormalities of the glutamate synapse in the thalamus in schizophrenia. Annals of the New York Academy of Sciences. 2003;1003 (1):75–93. doi: 10.1196/annals.1300.005. [DOI] [PubMed] [Google Scholar]
- Mignot C, Boespflug-Tanguy O, Gelot A, Dautigny A, Pham-Dinh D, Rodriguez D. Alexander disease: putative mechanisms of an astrocytic encephalopathy. Cellular and Molecular Life Sciences. 2004;61 (3):369–385. doi: 10.1007/s00018-003-3143-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols N, Day J, Laping N, Johnson S, Finch C. GFAP mRNA increases with age in rat and human brain. Neurobiology of Aging. 1993;14 (5):421–429. doi: 10.1016/0197-4580(93)90100-p. [DOI] [PubMed] [Google Scholar]
- Parpura V, Basarsky TA, Liu F, Jeftinija K, Jeftinija S, Haydon PG. Glutamate-mediated astrocyte-neuron signalling. Nature. 1994;369 (6483):744–747. doi: 10.1038/369744a0. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. Academic Press; New York: 1986. [Google Scholar]
- Perng M, Su M, Wen S, Li R, Gibbon T, Prescott A, Brenner M, Quinlan R. The Alexander disease–causing glial fibrillary acidic protein mutant, R416W, accumulates into rosenthal fibers by a pathway that involves filament aggregation and the association of αB-crystallin and HSP27. The American Journal of Human Genetics. 2006;79 (2):197–213. doi: 10.1086/504411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrone-Bizzozero N, Sower A, Bird E, Benowitz L, Ivins K, Neve R. Levels of the growth-associated protein GAP-43 are selectively increased in association cortices in schizophrenia. Proceedings of the National Academy of Sciences. 1996;93:14182–14187. doi: 10.1073/pnas.93.24.14182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabakaran S, Swatton J, Ryan M, Huffaker S, Huang J, Griffin J, Wayland M, Freeman T, Dudbridge F, Lilley K, Karp N, Hester S, Tkachev D, Mimmack M, Yolken R, Webster M, Torrey E, Bahn S. Mitochondrial dysfunction in schizophrenia: evidence for compromised brain metabolism and oxidative stress. Molecular Psychiatry. 2004;9:684–697. doi: 10.1038/sj.mp.4001511. [DOI] [PubMed] [Google Scholar]
- Rajkowska G, Miguel-Hidalgo J, Makkos Z, Meltzer H, Overholser J, Stockmeier C. Layer-specific reductions in GFAP-reactive astroglia in the dorsolateral prefrontal cortex in schizophrenia. Schizophrenia Research. 2002;57 (2–3):127–138. doi: 10.1016/s0920-9964(02)00339-0. [DOI] [PubMed] [Google Scholar]
- Rodnight R, Gonçalves C, Wofchuk S, Leal R. Control of the phosphorylation of the astrocyte marker glial fibrillary acidic protein (GFAP) in the immature rat hippocampus by glutamate and calcium ions: possible key factor in astrocytic plasticity. Brazilian Journal of Medical and Biological Research. 1997;30:3325–338. doi: 10.1590/s0100-879x1997000300005. [DOI] [PubMed] [Google Scholar]
- Rothman D, Sibson N, Hyder F, Shen J, Behar K, Shulman R. In vivo nuclear magnetic resonance spectroscopy studies of the relationship between the glutamate-glutamine neurotransmitter cycle and functional neuroenergetics. Philosophical Transactions of the Royal Society B: Biological Sciences. 1999;354 (1387):1165–1177. doi: 10.1098/rstb.1999.0472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schell M, Molliver M, Snyder S. D-serine, an endogenous synaptic modulator: localization to astrocytes and glutamate-stimulated release. Proceedings of the National Academy of Sciences. 1995;92 (9):3948–3952. doi: 10.1073/pnas.92.9.3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt A, Weber S, Jatzko A, Braus D, Henn F. Hippocampal volume and cell proliferation after acute and chronic clozapine or haloperidol treatment. Journal of Neural Transmission. 2004;111 (1):91–100. doi: 10.1007/s00702-003-0070-2. [DOI] [PubMed] [Google Scholar]
- Shibuki K, Gomi H, Chen L, Bao S, Kim J, Wakatsuki H, Fujisaki T, Fujimoto K, Katoh A, Ikeda T. Deficient cerebellar long-term depression, impaired eyeblink conditioning, and normal motor coordination in GFAP mutant mice. Neuron. 1996;16 (3):587–599. doi: 10.1016/s0896-6273(00)80078-1. [DOI] [PubMed] [Google Scholar]
- Smith R, Haroutunian V, Davis K, Meador-Woodruff J. Expression of excitatory amino acid transporter transcripts in the thalamus of subjects with schizophrenia. American Journal of Psychiatry. 2001;158 (9):1393–1399. doi: 10.1176/appi.ajp.158.9.1393. [DOI] [PubMed] [Google Scholar]
- Smith R, Haroutunian V, Davis K, Meador-Woodruff J. Vesicular glutamate transporter transcript expression in the thalamus in schizophrenia. Neuroreport. 2001;12 (13):2885–2887. doi: 10.1097/00001756-200109170-00026. [DOI] [PubMed] [Google Scholar]
- Sonnewald U, Qu H, Aschner M. Pharmacology and toxicology of astrocyte-neuron glutamate transport and cycling. J Pharmacol Exp Ther. 2002;301 (1):1–6. doi: 10.1124/jpet.301.1.1. [DOI] [PubMed] [Google Scholar]
- Spurney C, Baca S, Murray A, Jaskiw G, Kleinman J, Hyde T. Differential effects of haloperidol and clozapine on ionotropic glutamate receptors in rats. Synapse. 1999;34 (4):266–276. doi: 10.1002/(SICI)1098-2396(19991215)34:4<266::AID-SYN3>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Stark A, Uylings H, Sanz-Arigita E, Pakkenberg B. Glial cell loss in the anterior cingulate cortex, a subregion of the prefrontal cortex, in subjects with schizophrenia. American Journal of Psychiatry. 2004;161:882–888. doi: 10.1176/appi.ajp.161.5.882. [DOI] [PubMed] [Google Scholar]
- Takamori S, Rhee J, Rosenmund C, Jahn R. Identification of a vesicular glutamate transporter that defines a glutamatergic phenotype in neurons. Nature. 2000;407 (6801):189–194. doi: 10.1038/35025070. [DOI] [PubMed] [Google Scholar]
- Toro C, Hallak J, Dunham J, Deakin J. Glial fibrillary acidic protein and glutamine synthetase in subregions of prefrontal cortex in schizophrenia and mood disorder. Neuroscience Letters. 2006;404 (3):276–281. doi: 10.1016/j.neulet.2006.05.067. [DOI] [PubMed] [Google Scholar]
- Watts J, Fowler L, Whitton P, Pearce B. Release of arginine, glutamate and glutamine in the hippocampus of freely moving rats: Involvement of nitric oxide. Brain Research Bulletin. 2005;65 (6):521. doi: 10.1016/j.brainresbull.2005.03.011. [DOI] [PubMed] [Google Scholar]
- Weinberger D, Berman K, Zec R. Physiologic dysfunction of dorsolateral prefrontal cortex in schizophrenia. I. Regional cerebral blood flow evidence. Archives of General Psychiatry. 1986;43 (2):114–124. doi: 10.1001/archpsyc.1986.01800020020004. [DOI] [PubMed] [Google Scholar]
- Weinstein D, Shelanski M, Liem R. Suppression by antisense mRNA demonstrates a requirement for the glial fibrillary acidic protein in the formation of stable astrocytic processes in response to neurons. Journal of Cell Biology. 1991;112 (6):1205–1213. doi: 10.1083/jcb.112.6.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]