

Abstract
Ongoing epidemiological studies estimate that greater than 60% of the adult US population may be categorized as either overweight or obese. There is a growing appreciation that the complications of obesity extend to the central nervous system (CNS) and may result in increased risk for neurological co-morbidities like depressive illness. One potential mechanistic mediator linking obesity and depressive illness is the adipocyte derived hormone leptin. We previously demonstrated that lentivirus-mediated downregulation of hypothalamic insulin receptors increases body weight, adiposity and plasma leptin levels, which is consistent with features of the metabolic syndrome. Using this novel model of obesity, we examined performance in the forced swim test (FST), the sucrose preference test and the elevated plus maze (EPM), approaches that are often used as measures of depressive-like and anxiety-like behaviors, in rats that received third ventricular injections of either an insulin receptor antisense lentivirus (hypo-IRAS) or a control lentivirus (hypo-Con). Hypo-IRAS rats exhibited significant increases in immobility time and corresponding decreases in active behaviors in the FST and exhibited anhedonia as measured by decreased sucrose intake compared to hypo-Con rats. Hypo-IRAS rats also exhibited increases in anxiety-like behaviors in the EPM. Plasma, hippocampal and amygdalar brain-derived neurotrophic factor (BDNF) levels were reduced in hypo-IRAS rats, suggesting that the obesity/hyperleptinemic phenotype may elicit this behavioral phenotype through modulation of neurotrophic factor expression. Collectively, these data support the hypothesis for an increased risk for mood disorders in obesity, which may be related to decreased expression of hippocampal and amygdalar BDNF.
Keywords: obesity, leptin, brain-derived neurotrophic factor, forced swim test, elevated plus maze, anhedonia
1. Introduction
Ongoing epidemiological studies by the Centers for Disease Control estimate that greater than 60% of the adult US population may be categorized as either overweight or obese [1]. In addition to peripheral complications, there is a growing appreciation that the complications of obesity extend to the central nervous system (CNS) [2;3] and may result in increased risk for neurological co-morbidities like depressive illness. In support of this hypothesis, clinical studies indicate that there is an association between obesity and mood disorders [4–15]; this correlation is particularly strong for individuals with a BMI greater than 40 [7]. A number of factors have been suggested as mechanistic links between obesity and mood disorders, including socioeconomic status, genetic predisposition and stress reactivity [8;14]. Another potential mechanistic mediator linking obesity and depressive illness is the adipocyte-derived hormone leptin.
Leptin is synthesized and secreted by adipocytes and is transported across the blood-brain barrier (BBB) via a saturable transport system [16]. In the hypothalamus, leptin is recognized as an important integration factor that regulates food intake, metabolism, body weight and body composition (For review see, [17]). Beyond the hypothalamus, there is a growing literature to support a role for leptin in the facilitation of synaptic plasticity (For reviews see [18–20]). In obesity phenotypes, leptin transport across the blood-brain barrier is impaired [16;21;22]. Moreover, experimental models of obesity are associated with deficits in neuroplasticity [23], which supports the hypothesis that reduced CNS leptin activity may be a mechanistic link between obesity and major depressive illness [24]. However, the wide-range of endocrine and metabolic abnormalities associated with obesity phenotypes makes it challenging to identify the mechanistic links between obesity and depressive illness.
We recently developed a lentivirus vector packaged with an insulin receptor (IR) antisense sequence (IRAS) that when injected into the hypothalamus (hypo-IRAS) selectively decreases IR signaling in hypothalamus without affecting these parameters in the hippocampus [25;26]. Hypo-IRAS rats exhibit increases in body weight, body adiposity, plasma leptin and triglyceride levels, features that are consistent with aspects of the metabolic syndrome. In view of the increased risk for co-morbid depressive illness in obese individuals, the aim of the current studies was to determine whether the obesity phenotype observed in hypo-IRAS rats elicits the development of depressive-like and anxiety-like behaviors.
2. Materials and Methods
2.1. Animal Protocols
Adult male Sprague Dawley rats (CD strain, Charles River) weighing 225–250 g were housed in groups of three with ad libitum access to food and water, in accordance with all guidelines and regulations of The University of South Carolina Animal Care and Use Committee. Animals were maintained in a temperature-controlled room, with a light/dark cycle of 12/12 h (lights on at 0700h). As described previously [25;26], rats were anesthetized, placed in the stereotaxic apparatus and lentivirus was injected into the third ventricle using the following coordinates: AP: −2.6 mm; L: 0.0 mm; DV: −10.0 mm. Rats were injected with lentivirus containing an antisense sequence selective for the IR (LV-IRAS) or control virus (LV-Con). The viral stock (5×106 tu/μl) was injected at a speed of 1 μl/min with a 10 μl Hamilton syringe driven by a motorized stereotaxic injector (Stoelting 53310); the needle was left in place for additional 10 min. Total volume injected was 6 μl. Rats were returned to the housing colony and blood from the tip of the tail was collected 3–4 weeks after surgery to test leptin, triglycerides and BDNF levels. Once the development of the hyperleptinemic phenotype was confirmed, behavioral analyses were performed.
2.2. Elevated Plus Maze Test
The elevated plus maze was used to assess anxiety-like behaviors in hypo-IRAS and hypo-Con rats, as described in our previous studies [27–29]. Behavioral testing took place between 0800h and 1100h in a behavioral suite adjacent to the vivarium room. The plus maze apparatus was made of black Plexiglas had two open arms (56 × 10 × 1 cm) and two closed arms (56 × 10 × 40 cm) and was elevated 50cm above the floor. A white noise generator was used to mask extraneous noises and testing occurred in bright light. Rats were placed in the center of the plus maze facing an open arm and videotaped for 5 minutes. Rats were then removed and returned to their home cage. The maze was cleaned with a 5% ammonium hydroxide solution between subjects. An automated Ethovision tracking system (Noldus Information Technology, Inc, Leesburg, VA) measured time spent in open and closed arms, entries into open and closed arms, center time, and distance traveled. Open arm measures were used as a measure of anxiety and the distance traveled was used as a measure of spontaneous locomotor activity. Percent open arm entries was calculated based number of open arm entries divided by the total number of arm entries (open + closed) times 100.
2.3. Anhedonia
To evaluate anhedonic-like response to a palatable fluid, a sucrose consumption protocol was developed. In order to habituate the animals to the sucrose solution, rats were exposed for 24 hours to two identical bottles containing water and 1% sucrose solution (Sigma Chemical Co., St Louis MO). The next day the rats were water-deprived for 6 hours (1300h–1900h) before testing their preference for sucrose (1%) or water (identical bottles) in a three hour two-bottle choice beginning at 1900h. Consumption of sucrose and water were determined in this period.
2.4. Forced Swim Test
The FST for “behavior despair” was modified from the test originally described by Porsolt [30;31]. Testing for both days was done in the early light hours of the light:dark cycle. Animals were placed in a clear Plexiglas cylinder, 21 cm in diameter and 40.5 cm in height, filled with 30 cm of room temperature water (25°C) in a separate room with no visual or audible stimuli. The first day consisted of a fifteen minute pretest, followed 24 hours later by a five minute test. Animals were viewed and scored later by a scorer blind to the treatment group via video camera for three behaviors: immobility, climbing and swimming. Immobility behaviors were defined as little to no movement of the rat. Climbing was defined as movement in contact with the side of the container. All other movements were defined as swimming. Behaviors were noted by the scorer every three seconds.
2.5. Acute restraint stress test
Hypo-Con and hypo-IRAS rats were subjected to an acute restraint stress session as described in our previous studies [32]. Briefly, rats were subjected to restraint stress in wire mesh restrainers secured at the head and tail ends with clips. Immediately after the rat was secured in the restrainer, a tail bleed was performed to determine baseline corticosterone (CORT) values, following which the rats were returned to their home cage. Thirty minutes after the initiation of stress, the tail was gently massaged to recover blood for peak stress mediated increases in plasma CORT. Upon completion of this tail bleed, rats were released from the restrainers and returned to their home cage. One hour later, the tail was gently messaged to recover blood for post-stress measurement of plasma CORT levels. Plasma CORT levels were determined by enzyme-linked immunosorbent assay.
2.6. Plasma endocrine analysis
Plasma triglycerides were determined using an enzymatic kit (modified Trinder) according to the manufacturer’s instructions (Pointe Scientific, Inc., Canton, MI, USA). Determination of plasma CORT was performed using a commercially-available ELISA kit (Enzo Life Sciences, Inc., Plymouth Meeting, PA). Determination of plasma leptin was performed using a commercially available leptin ELISA kit (Millipore, Corporation, Billerica, MA). Plasma, hippocampal and amygdalar BDNF levels were determined in samples from rats not subjected to behavioral tests, using a commercial available ELISA from Promega (BDNF Emax ImmunoAssay System, Promega Corp., Madison, WI). Hippocampus and amygdala were isolated and prepared for BDNF analysis as described in our previous studies [33]. ELISA plates were analyzed with a BioTek Synergy microplate reader (BioTek Instruments Inc., Winooski, VT), according to the manufacturers’ instructions.
2.7. Statistical analysis
Statistical analysis was performed using an unpaired t-test or a one-way ANOVA, followed by a Student-Newman-Keuls post-hoc test, with P < 0.05 as the criterion for statistical significance.
3. Results
Three weeks following lentivirus administration, hypo-IRAS rats exhibited significant increases in body weight, body adiposity, plasma leptin levels and plasma triglyceride levels, which provide indirect measures of the efficacy of the IRAS construct (Table 1). Successful downregulation of hypothalamic insulin receptors (IRs) was confirmed by western blot analysis; IR expression was similar in the hippocampus and amygdala of hypo-IRAS and hypo-Con rats (Table 1). Since some of the behavioral measures we wished to examine involve stressful stimuli, we wanted to determine whether stress reactivity was altered following downregulation of hypothalamic IRs. To address this question, rats were subjected to an acute (i.e. 30 minute) restraint stress session. In agreement with our previous studies [25], basal levels of plasma corticosterone (CORT) levels did not differ between hypo-IRAS and hypo-Con rats (Figure 1). Additionally, stress-induced increases and post-stress levels of CORT did not differ between the groups, indicating that stress reactivity in response to an acute stress challenge is not affected in hypo-IRAS rats.
Table 1.
Physiological and endocrine parameters in hypo-IRAS and hypo-Con rats.
| Hypo-Con | Hypo-IRAS | |
|---|---|---|
| Plasma leptin (ng/ml) | 8.39 ± 0.1 | 12.69 ± 0.84# |
| Plasma triglycerides (mg/dl) | 125.8 ± 14.0 | 204.0 ± 20.1# |
| Body weight gain (g) | 58.5 ± 4.5 | 77.2 ± 5.2* |
| Hypothalamic IR levels (% of hypo-Con) | 100.0 ± 8.6 | 65.1 ± 9.3* |
| Hippocampal IR levels (% of hypo-Con) | 100.0 ± 14.7 | 110.4 ± 16.7 |
| Amygdalar IR levels (% of hypo-Con) | 100 ± 5.1 | 91.1 ± 4.2 |
P<0.05;
P<0.005
Figure 1.
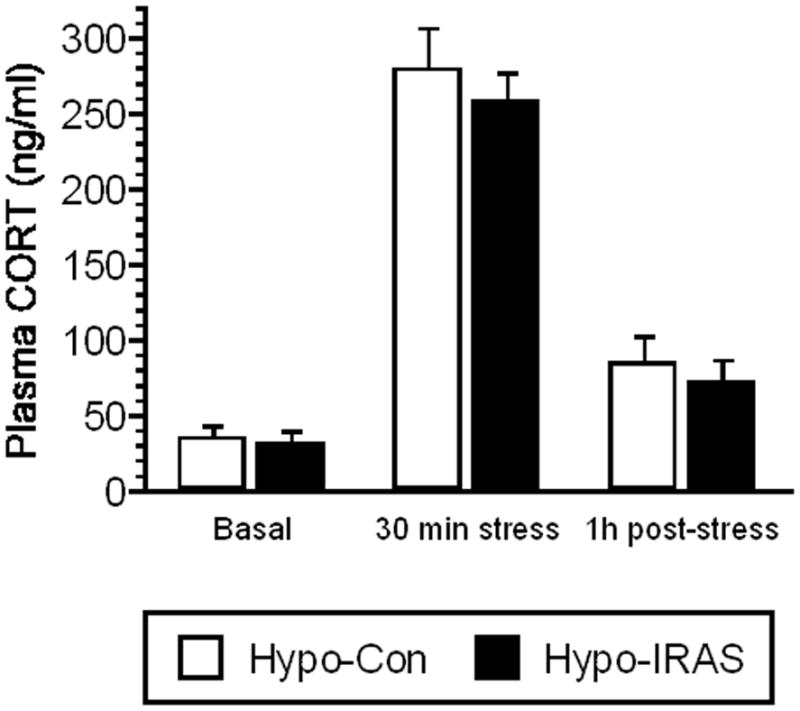
Downregulation of hypothalamic IRs does not affect HPA axis responses to acute stress. Virus-treated rats were subjected to an acute (30 minute) restraint stress challenge to determine if downregulation of hypothalamic IRs affected stress reactivity. Tail bleeds were performed to determine baseline plasma CORT levels (basal), peak restraint stress-induced plasma CORT levels (i.e. 30 min stress) and plasma levels 1 hour following termination of stress (i.e. post-stress). Basal, stress and post-stress plasma CORT levels did not differ in hypo-IRAS rats when compared to hypo-Con rats, suggesting that downregulation of hypothalamic IRs does not affect HPA axis responses to acute stress. Data based upon eight rats per group.
3.1. Evaluation of depressive-like behaviors in hypo-IRAS rats
In an attempt to determine if the obesity/hyperleptinemic phenotype elicits depressive-like symptoms, hypo-IRAS rats and hypo-Con rats were evaluated in the forced swim test (FST) and for sucrose intake. In the FST, immobility behaviors are defined as little to no movement of the rat while active behavior combines climbing and swimming behaviors. An increase in immobility behaviors (and corresponding decrease in active behaviors) is considered a measure of behavioral despair. In the fifteen minute pretest, the levels of immobility and active behavior did not differ between the two groups (Figure 2, Panel A). However, hypo-IRAS rats exhibited significant increases in immobility (t=2.283, p=0.05) and reductions in active behaviors (t=2.263, p=0.05) compared to hypo-Con rats during the 5 min test performed 24 hours later (Figure 2, Panel B). It is important to note that the increases in immobility times observed in hypo-IRAS rats are only apparent during the 5 minute test session performed 24 hours after the pre-test. This indicates that hypo-IRAS rats do not exhibit immobility (i.e. float) in the test phase simply because of their increased adiposity. Rather, the increases in immobility time specific to the test phase of the forced swim test are more likely indicative of behavioral despair.
Figure 2.

Obesity/hyperleptinemic phenotype elicits behavioral despair in the forced swim test (FST). Downregulation of hypothalamic IRs does not affect immobile or active (swimming and climbing) behaviors in the 15 minute pre-test (Panel A). Conversely, when subjected to the 5 minute swim test 24 hours later, hypo-IRAS rats exhibit increases in the time spent immobile and decreases in the time spent in active behaviors when compared to hypo-Con rats. (Panel B). [* = p<0.05 compared to hypo-Con rats; data based upon at least 10 rats/group]
As another measure of depressive-like symptoms, we examined sucrose intake in hypo-Con and hypo-IRAS rats following lentivirus administration. Twenty-four hours following a habituation period, hypo-IRAS rats and hypo-Con rats were provided access to water and a 1% sucrose solution and fluid intake was monitored during the first three hours of the dark cycle. While water consumption did not differ between the groups (data not shown), the ratio of sucrose to water intake was significantly reduced in hypo-IRAS rats compared to hypo-Con rats (Figure 3). Collectively, these data illustrate that hypo-IRAS rats exhibit behavioral despair and anhedonia to a palatable sucrose solution.
Figure 3.
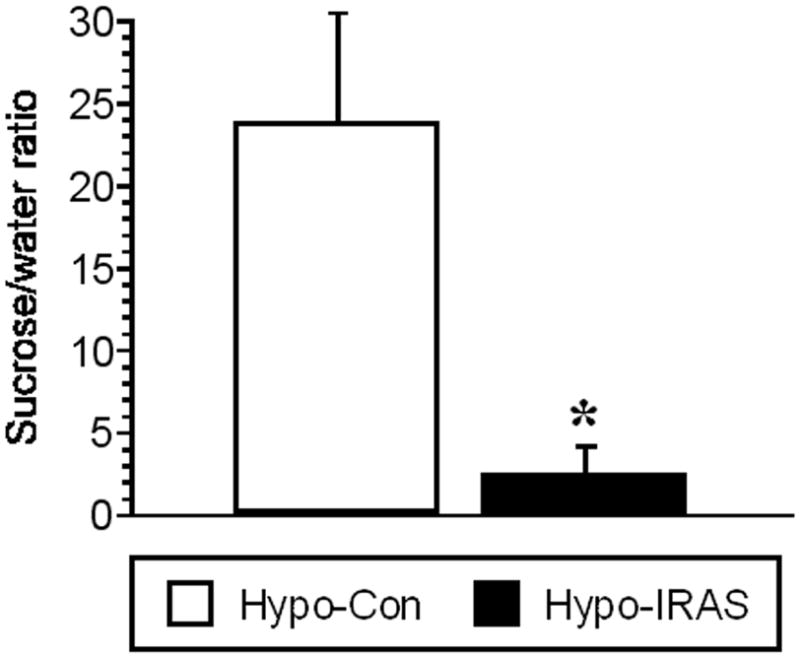
Downregulation of hypothalamic IRs decreases sucrose preference. Following habituation, hypo-Con and hypo-IRAS rats were provided access to sucrose (1% solution) or water for 3 hours. The sucrose to water ratio was significantly decreased in hypo-IRAS rats compared with hypo-Con rats, illustrating that hypo-IRAS rats exhibit anhedonia to a palatable solution. [* = p<0.01 compared to hypo-Con rats; data based upon at least 10 rats/group]
3.2. Anxiety-like behaviors are increased in hypo-IRAS rats
In order to determine the effects of the obesity/hyperleptinemic phenotype upon anxiety-like behaviors, performance in the elevated plus maze was examined in hypo-Con and hypo-IRAS rats. Hypo-IRAS rats exhibited decreases in percent open arm entries (t=2.582, p<0.02) when compared with hypo-Con rats (Figure 4, Panel A). Conversely, activity measures in the plus maze, such as distance traveled, did not differ between groups (t=0.686, p=0.496; Figure 4, Panel B). These data suggest that in addition to eliciting increases in depression-like behaviors, downregulation of hypothalamic IRs also elicits an increase in anxiety-like behaviors without changes in locomotor activity.
Figure 4.

Hypo-IRAS rats exhibit increases in anxiety-like behaviors in the elevated plus maze. Compared to hypo-Con rats, hypo-IRAS rats exhibited significant decreases in percentage of open arm entries (Panel A). These decreases in open arm behaviors were not the result of decreased locomotor activity since the distance traveled in the elevated plus maze did not differ between the groups (Panel B). [* = p<0.02 compared to hypo-Con rats; data based upon at least 10 rats/group]
3.3. Brain-derived neurotrophic factor levels are decreased in hypo-IRAS rats
In view of the proposed role of brain-derived neurotrophic factor (BDNF) in the pathology of depressive illness (for review, see [34]), we examined BNDF levels in hypo-IRAS and hypo-Con rats. Plasma BNDF levels were reduced in hypo-IRAS compared to hypo-Con rats (Figure 5; t=3.17, p<0.01). Similar to observations in mice fed a high-fat diet [35], BNDF protein expression was reduced in the hippocampus (t=2.515, p<0.03), as well as the amygdala (t=4.02, p<0.02) of hypo-IRAS rats compared to hypo-Con rats.
Figure 5.
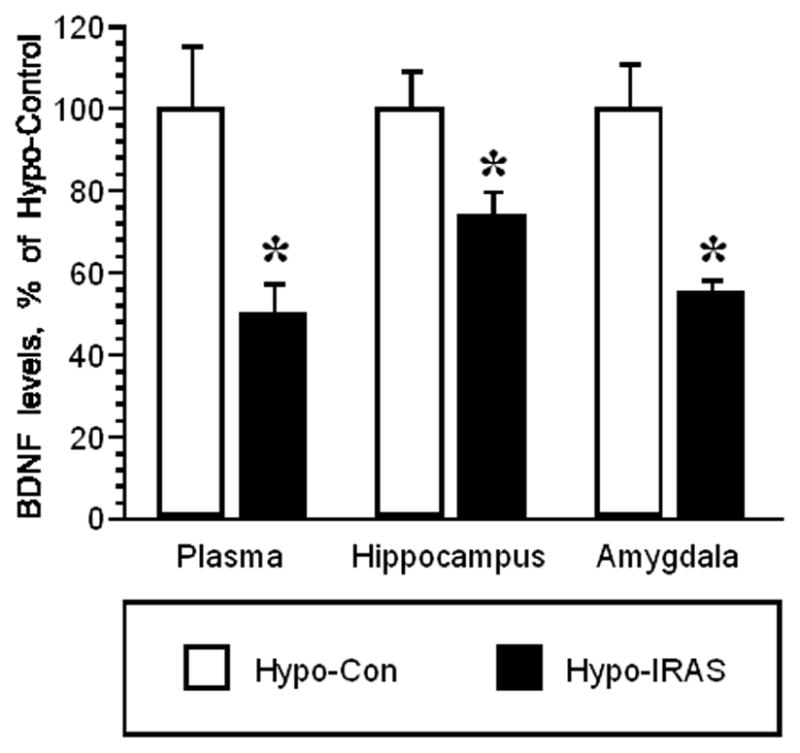
ELISA analysis of BDNF levels in hypo-Con and hypo-IRAS rats. Statistical analysis revealed that plasma, hippocampal and amygdalar BDNF levels are reduced in hypo-IRAS rats compared to hypo-Con rats. Data expressed as a percentage of BDNF levels in hypo-Con rats. Mean hypo-Con levels: plasma = 460.6 ± 69.3 pg/ml; hippocampus = 273.7 ± 24.5 pg/mg protein; amygdala = 111.6 ± 11.9 pg/mg protein [* = p<0.05 compared to hypo-Con rats; data based upon at least 6 rats/group]
4. Discussion
Obesity, defined as a body mass index (BMI) of greater than 30, is associated with a host of co-morbidities, including cardiovascular disease and type 2 diabetes mellitus (T2DM). In addition to peripheral consequences, there is a growing appreciation that the complications associated with obesity extend to the CNS and include increased risk of depressive illness [4–15]. An important question that remains to be addressed is what are the mechanistic mediators that link obesity with depressive illness. The results of the current study illustrate that downregulation of hypothalamic IRs elicits behavioral despair, anhedonia and increased anxiety-like behavior. Mechanistically, these behavioral changes may be mediated by increases in adiposity, as well as increases in plasma leptin and/or triglyceride levels elicited by downregulation of hypothalamic IRs. Indeed, while obese hypo-IRAS rats exhibit significant increases in plasma leptin and triglyceride levels, other metabolic and endocrine measures are unaffected. For example, hypo-IRAS rats exhibit normal stress reactivity in response to an acute stress challenge and exhibit similar responses to an oral glucose tolerance test when compared with hypo-Con rats [25;26]. These observations suggest that the adipocyte-derived hormone leptin may provide a mechanistic link between obesity and depression [24].
In the CNS, the actions of leptin in the hypothalamus are well described and include regulation of feeding, body weight and metabolism ( For review, see [17]). In addition, there is a growing literature to support a role for leptin in the facilitation of hippocampal structure and function [18;36;37]. These structural and functional enhancements of hippocampal plasticity may contribute to leptin’s ability to improve hippocampal-dependent behavioral performance [18;38;39]. Conversely, genetic mutations that result in disrupted leptin signaling such as in the db/db mouse and the Zucker fa/fa rat are associated with impaired performance of hippocampal-dependent tasks [40;41]. Leptin transport across the BBB is impaired under conditions of hyperleptinemia [16;21;22], which has lead to the suggestion that despite chronically increased circulating leptin levels, decreases in CNS leptin signaling and/or BBB leptin transport are associated with decreases in synaptic plasticity [23]. In support of this hypothesis, our recent studies revealed that hypo-IRAS rats exhibit structural and functional deficits in the hippocampus, including decreases in contextual learning, decreases in long-term potentiation and morphological changes that include synaptic reorganization [26;42]. Hypo-IRAS rats also fail to exhibit increased phosphorylation of STAT3 in response to peripherally administered leptin [42]. The lack of activation of this key step in leptin receptor signaling indicate that hypo-IRAS rats are leptin resistant and suggest that the deficits in hippocampal synaptic plasticity observed in hypo-IRAS rats may result from decreases in BBB leptin transport and/or leptin receptor signaling.
In addition to behavioral measures of synaptic plasticity, previous studies have reported that leptin possesses anti-depressant and anxiolytic properties [24]. For example, chronic stress-induced behavioral despair in the forced swim test is reversed by leptin administration in a dose-dependent manner [43]. Leptin administration also reverses chronic stress-induced anhedonia as measured by sucrose intake [43]. In the elevated plus maze, leptin administration increases open arm entries and time spent in the open arm compared to saline-treated controls [44]. Some additional studies support the hypothesis that decreases in brain leptin activity is a mechanistic link between obesity and depressive-like behaviors. In this regard, ob/ob mice, which lack the gene coding for leptin, exhibit increased immobility time in the FST compared to wild-type controls [45]. Additionally, db/db mice, which lack functional leptin receptors, exhibit increased immobility time in the FST and altered anxiety-like behaviors [46]. Streptozotocin-treated mice, an experimental model of type 1 diabetes that have nearly undetectable levels of plasma leptin, exhibit depressive-like [47] and anxiety-like behaviors [48;49]. Interestingly, depressive-like behaviors in STZ mice are inhibited by leptin in a dose-dependent manner [50]. The common feature of these studies is that rodents with impaired leptin signaling (i.e. receptor or ligand deficient) exhibit depressive-like and anxiety-like behaviors. Using these same behavioral measures, namely the FST, sucrose preference and the EPM, the current study revealed that an obesity/hyperleptinemia phenotype of the hypo-IRAS model also elicits behavioral despair and anxiety-like behaviors.
In addition to leptin, there may be an important role for increases in plasma triglycerides in obesity-mediated neuroplasticity deficits. Studies by Banks and colleagues have demonstrated that increases in plasma triglyceride levels inhibit BBB transport of leptin [51]. In addition to decreasing BBB transport of leptin, triglycerides directly impair hippocampal synaptic transmission and behavioral performance [52]. Therefore, obesity-related increases in plasma triglycerides may impair BBB leptin transport, and thereby reduce leptin-mediated synaptic plasticity in the hippocampus. Such results implicate plasma increases of both leptin and triglycerides as mechanistic mediators linking depression and obesity. The results of this study also identify BDNF as a potential mechanistic mediator that links depression and obesity. BDNF is a major regulator of synaptic plasticity in the CNS and contributes to a range of adaptive neuronal responses [53–56]. For example, intrahippocampal administration of BDNF produces antidepressant-like effects [57], supporting the hypothesis that the mechanisms of action of some antidepressant drugs involves upregulation of hippocampal BDNF levels [58]. Depressive illness patients exhibit decreases in plasma BDNF levels [59;60], further supporting the relationship between depressive illness and BDNF expression. Moreover, downregulation of amygdalar BDNF expression using antisense oligonucleotide approaches elicits decreases in open arm entries and open arm time in the elevated plus maze [61]. In the current study, the behavioral changes observed in hypo-IRAS rats are accompanied by decreases in plasma, hippocampal and amygdalar BDNF levels. Such results suggest that there are important interactions between BDNF, leptin and triglycerides that elicit behavioral deficits in obese rodents.
Beyond leptin, triglycerides and BDNF, other putative factors have received greater attention as potential mechanistic mediators linking mood disorders and obesity, in particular stress reactivity and hypothalamic-pituitary-adrenal (HPA) axis function [14]. Indeed, impaired HPA axis function is a common feature of obesity and depressive illness [62]. Stress has been implicated in the etiology and progression of depressive illness [63] and rodents subjected to chronic stress paradigms exhibit a depressive-like phenotype [64]. Moreover, stress paradigms reduce hippocampal BDNF levels [65]. These data support the concept that stress may be an etiological link between obesity and depression. However, the current results suggest that behavioral despair observed in an obesity/hyperleptinemic phenotype can be elicited in the absence of increased basal levels of glucocorticoids or impaired HPA reactivity. Therefore, our data demonstrate that behavioral despair and enhanced anxiety-like behaviors under conditions of increased adiposity may occur in the absence of HPA axis dysfunction.
Conclusions
In summary, the results of the current study indicate that a phenotype that is consistent with aspects of the metabolic syndrome is associated with decreases in hippocampal and amygdalar BDNF levels and elicits behavioral despair, anhedonia and increases in anxiety-like behaviors. While increases in plasma triglycerides and plasma leptin, as well as decreases in BDNF expression, may be factors that link obesity and depression, the relationship between these potential mechanistic mediators remains to be determined. For example, could increases in BDNF expression mediated by antidepressants inhibit the development of a depression-like phenotype even in the absence of changes in plasma leptin and/or triglyceride levels? Could drugs that reduce plasma triglyceride levels and thereby restore BBB transport of leptin [51] reverse and/or inhibit behavioral deficits observed in hypo-IRAS rats? Determination of these relationships between leptin, triglycerides and BDNF would assist in the development of strategies to ameliorate the neurological co-morbidities associated with obesity.
Acknowledgments
Supported by NIH grant numbers NS047728 (LPR), DK017844 (LPR), MH086067 (LPR), MH063344 (MAW), and the University of South Carolina Research Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Ogden CL, Carroll MD, Curtin LR, McDowell MA, Tabak CJ, Flegal KM. Prevalence of overweight and obesity in the United States, 1999–2004. JAMA. 2006;295:1549–1555. doi: 10.1001/jama.295.13.1549. [DOI] [PubMed] [Google Scholar]
- 2.Morley JE, Banks WA. Lipids and cognition. J Alzheimers Dis. 2010;20:737–747. doi: 10.3233/JAD-2010-091576. [DOI] [PubMed] [Google Scholar]
- 3.Benoit SC, Davis JF, Davidson TL. Learned and cognitive controls of food intake. Brain Res. 2010;1350:71–76. doi: 10.1016/j.brainres.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Simon GE, Von Korff M, Saunders K, Miglioretti DL, Crane PK, van Belle G, Kessler RC. Association between obesity and psychiatric disorders in the US adult population. Arch Gen Psychiatry. 2006;63:824–830. doi: 10.1001/archpsyc.63.7.824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Wit L, Luppino F, van Straten A, Penninx B, Zitman F, Cuijpers P. Depression and obesity: A meta-analysis of community-based studies. Psychiatry Res. 2010 doi: 10.1016/j.psychres.2009.04.015. [DOI] [PubMed] [Google Scholar]
- 6.Luppino FS, de Wit LM, Bouvy PF, Stijnen T, Cuijpers P, Penninx BW, Zitman FG. Overweight, obesity, and depression: a systematic review and meta-analysis of longitudinal studies. Arch Gen Psychiatry. 2010;67:220–229. doi: 10.1001/archgenpsychiatry.2010.2. [DOI] [PubMed] [Google Scholar]
- 7.Onyike CU, Crum RM, Lee HB, Lyketsos CG, Eaton WW. Is obesity associated with major depression? Results from the Third National Health and Nutrition Examination Survey. Am J Epidemiol. 2003;158:1139–1147. doi: 10.1093/aje/kwg275. [DOI] [PubMed] [Google Scholar]
- 8.Fabricatore AN, Wadden TA. Obesity. Annu Rev Clin Psychol. 2006;2:357–377. doi: 10.1146/annurev.clinpsy.2.022305.095249. [DOI] [PubMed] [Google Scholar]
- 9.McElroy SL, Kotwal R, Malhotra S, Nelson EB, Keck PE, Nemeroff CB. Are mood disorders and obesity related? A review for the mental health professional. J Clin Psychiatry. 2004;65:634–51. doi: 10.4088/jcp.v65n0507. quiz. [DOI] [PubMed] [Google Scholar]
- 10.Roberts RE, Deleger S, Strawbridge WJ, Kaplan GA. Prospective association between obesity and depression: evidence from the Alameda County Study. Int J Obes Relat Metab Disord. 2003;27:514–521. doi: 10.1038/sj.ijo.0802204. [DOI] [PubMed] [Google Scholar]
- 11.Goldfield GS, Moore C, Henderson K, Buchholz A, Obeid N, Flament MF. Body dissatisfaction, dietary restraint, depression, and weight status in adolescents. J Sch Health. 2010;80:186–192. doi: 10.1111/j.1746-1561.2009.00485.x. [DOI] [PubMed] [Google Scholar]
- 12.Sarwer DB, Wadden TA, Fabricatore AN. Psychosocial and behavioral aspects of bariatric surgery. Obes Res. 2005;13:639–648. doi: 10.1038/oby.2005.71. [DOI] [PubMed] [Google Scholar]
- 13.Andersen JR, Aasprang A, Bergsholm P, Sletteskog N, Vage V, Natvig GK. Anxiety and depression in association with morbid obesity: changes with improved physical health after duodenal switch. Health Qual Life Outcomes. 2010;8:52. doi: 10.1186/1477-7525-8-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stunkard AJ, Faith MS, Allison KC. Depression and obesity. Biol Psychiatry. 2003;54:330–337. doi: 10.1016/s0006-3223(03)00608-5. [DOI] [PubMed] [Google Scholar]
- 15.Malone M, Alger-Mayer SA, Anderson DA. The lifestyle challenge program: a multidisciplinary approach to weight management. Ann Pharmacother. 2005;39:2015–2020. doi: 10.1345/aph.1G287. [DOI] [PubMed] [Google Scholar]
- 16.Banks WA. The many lives of leptin. Peptides. 2004;25:331–338. doi: 10.1016/j.peptides.2004.02.014. [DOI] [PubMed] [Google Scholar]
- 17.Schwartz MW, Woods SC, Porte D, Jr, Seeley RJ, Baskin DG. Central nervous system control of food intake. Nature. 2000;404:661–671. doi: 10.1038/35007534. [DOI] [PubMed] [Google Scholar]
- 18.Harvey J. Leptin regulation of neuronal excitability and cognitive function. Curr Opin Pharmacol. 2007;7:643–647. doi: 10.1016/j.coph.2007.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harvey J, Shanley LJ, O’Malley D, Irving AJ. Leptin: a potential cognitive enhancer? Biochem Soc Trans. 2005;33:1029–1032. doi: 10.1042/BST20051029. [DOI] [PubMed] [Google Scholar]
- 20.Harvey J. Novel actions of leptin in the hippocampus. Ann Med. 2003;35:197–206. doi: 10.1080/07853890310008251. [DOI] [PubMed] [Google Scholar]
- 21.Burguera B, Couce ME, Curran GL, Jensen MD, Lloyd RV, Cleary MP, Poduslo JF. Obesity is associated with a decreased leptin transport across the blood-brain barrier in rats. Diabetes. 2000;49:1219–1223. doi: 10.2337/diabetes.49.7.1219. [DOI] [PubMed] [Google Scholar]
- 22.Banks WA, DiPalma CR, Farrell CL. Impaired transport of leptin across the blood-brain barrier in obesity. Peptides. 1999;20:1341–1345. doi: 10.1016/s0196-9781(99)00139-4. [DOI] [PubMed] [Google Scholar]
- 23.Harvey J, Solovyova N, Irving A. Leptin and its role in hippocampal synaptic plasticity. Progress in Lipid Research. 2006;45:369–378. doi: 10.1016/j.plipres.2006.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu XY. The leptin hypothesis of depression: a potential link between mood disorders and obesity? Curr Opin Pharmacol. 2007;7:648–652. doi: 10.1016/j.coph.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grillo CA, Tamashiro KL, Piroli GG, Melhorn S, Gass JT, Newsom RJ, Reznikov LR, Smith A, Wilson SP, Sakai RR, Reagan LP. Lentivirus-mediated downregulation of hypothalamic insulin receptor expression. Physiol Behav. 2007;92:691–701. doi: 10.1016/j.physbeh.2007.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grillo CA, Piroli GG, Evans AN, Macht VA, Wilson SP, Scott KA, Sakai RR, Mott DD, Reagan LP. Obesity/hyperleptinemic phenotype adversely affects hippocampal plasticity: Effects of dietary restriction. Physiol Behav. 2010 doi: 10.1016/j.physbeh.2010.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wilson MA, Junor L. The role of amygdalar mu-opioid receptors in anxiety-related responses in two rat models. Neuropsychopharmacology. 2008;33:2957–2968. doi: 10.1038/sj.npp.1301675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Primeaux SD, Wilson SP, Cusick MC, York DA, Wilson MA. Effects of altered amygdalar neuropeptide Y expression on anxiety-related behaviors. Neuropsychopharmacology. 2005;30:1589–1597. doi: 10.1038/sj.npp.1300705. [DOI] [PubMed] [Google Scholar]
- 29.Primeaux SD, Wilson SP, McDonald AJ, Mascagni F, Wilson MA. The role of delta opioid receptors in the anxiolytic actions of benzodiazepines. Pharmacol Biochem Behav. 2006;85:545–554. doi: 10.1016/j.pbb.2006.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Porsolt RD, Anton G, Blavet N, Jalfre M. Behavioral despair in rats: a new model sensitive to anti-depressant treatments. Europ J Pharmacol. 1977;47:379. doi: 10.1016/0014-2999(78)90118-8. [DOI] [PubMed] [Google Scholar]
- 31.Porsolt RD, LePichon M, Jalfre M. Depression: a new animal model sensitive to antidepressant treatments. Nature (Lond ) 1977;266:730–732. doi: 10.1038/266730a0. [DOI] [PubMed] [Google Scholar]
- 32.Reagan LP, Rosell DR, Wood GE, Spedding M, Munoz C, Rothstein J, McEwen BS. Chronic restraint stress up-regulates GLT-1 mRNA and protein expression in the rat hippocampus: reversal by tianeptine. Proc Natl Acad Sci U S A. 2004;101:2179–2184. doi: 10.1073/pnas.0307294101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reagan LP, Hendry RM, Reznikov LR, Piroli GG, Wood GE, McEwen BS, Grillo CA. Tianeptine increases brain-derived neurotrophic factor expression in the rat amygdala. Eur J Pharmacol. 2007;565:68–75. doi: 10.1016/j.ejphar.2007.02.023. [DOI] [PubMed] [Google Scholar]
- 34.Duman RS. Synaptic plasticity and mood disorders. Mol Psychiatry. 2002;7 (Suppl 1):S29–S34. doi: 10.1038/sj.mp.4001016. [DOI] [PubMed] [Google Scholar]
- 35.Park HR, Park M, Choi J, Park KY, Chung HY, Lee J. A high-fat diet impairs neurogenesis: involvement of lipid peroxidation and brain-derived neurotrophic factor. Neurosci Lett. 2010;482:235–239. doi: 10.1016/j.neulet.2010.07.046. [DOI] [PubMed] [Google Scholar]
- 36.Shanley LJ, Irving AJ, Harvey J. Leptin enhances NMDA receptor function and modulates hippocampal synaptic plasticity. Journal of Neuroscience. 2001;21:art-RC186. doi: 10.1523/JNEUROSCI.21-24-j0001.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O’Malley D, MacDonald N, Mizielinska S, Connolly CN, Irving AJ, Harvey J. Leptin promotes rapid dynamic changes in hippocampal dendritic morphology. Molecular and Cellular Neuroscience. 2007;35:559–572. doi: 10.1016/j.mcn.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oomura Y, Hori N, Shiraishi T, Fukunaga K, Takeda H, Tsuji M, Matsumiya T, Ishibashi M, Aou S, Li XL, Kohno D, Uramura K, Sougawa H, Yada T, Wayner MJ, Sasaki K. Leptin facilitates learning and memory performance and enhances hippocampal CA1 long-term potentiation and CaMK II phosphorylation in rats. Peptides. 2006;27:2738–2749. doi: 10.1016/j.peptides.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 39.Farr SA, Banks WA, Morley JE. Effects of leptin on memory processing. Peptides. 2006;27:1420–1425. doi: 10.1016/j.peptides.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 40.Winocur G, Greenwood CE, Piroli GG, Grillo CA, Reznikov LR, Reagan LP, McEwen BS. Memory Impairment in Obese Zucker Rats: An Investigation of Cognitive Function in an Animal Model of Insulin Resistance and Obesity. Behav Neurosci. 2005;119:1389–1395. doi: 10.1037/0735-7044.119.5.1389. [DOI] [PubMed] [Google Scholar]
- 41.Li XL, Aou S, Oomura Y, Hori N, Fukunaga K, Hori T. Impairment of long-term potentiation and spatial memory in leptin receptor-deficient rodents. Neuroscience. 2002;113:607–615. doi: 10.1016/s0306-4522(02)00162-8. [DOI] [PubMed] [Google Scholar]
- 42.Grillo CA, Piroli GG, Junor L, Wilson SP, Mott DD, Wilson MA, Reagan LP. Obesity/hyperleptinemic phenotype impairs structural and functional plasticity in the rat hippocampus. Physiol Behav. 2011 doi: 10.1016/j.physbeh.2011.02.028. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lu XY, Kim CS, Frazer A, Zhang W. Leptin: a potential novel antidepressant. Proc Natl Acad Sci U S A. 2006;103:1593–1598. doi: 10.1073/pnas.0508901103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu J, Garza JC, Bronner J, Kim CS, Zhang W, Lu XY. Acute administration of leptin produces anxiolytic-like effects: a comparison with fluoxetine. Psychopharmacology (Berl) 2010;207:535–545. doi: 10.1007/s00213-009-1684-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Collin M, Hakansson-Ovesjo ML, Misane I, Ogren SO, Meister B. Decreased 5-HT transporter mRNA in neurons of the dorsal raphe nucleus and behavioral depression in the obese leptin-deficient ob/ob mouse. Brain Res Mol Brain Res. 2000;81:51–61. doi: 10.1016/s0169-328x(00)00167-4. [DOI] [PubMed] [Google Scholar]
- 46.Sharma AN, Elased KM, Garrett TL, Lucot JB. Neurobehavioral deficits in db/db diabetic mice. Physiol Behav. 2010;101:381–388. doi: 10.1016/j.physbeh.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kamei J, Miyata S, Morita K, Saitoh A, Takeda H. Effects of selective serotonin reuptake inhibitors on immobility time in the tail suspension test in streptozotocin-induced diabetic mice. Pharmacol Biochem Behav. 2003;75:247–254. doi: 10.1016/s0091-3057(03)00080-7. [DOI] [PubMed] [Google Scholar]
- 48.Ramanathan M, Jaiswal AK, Bhattacharya SK. Differential effects of diazepam on anxiety in streptozotocin induced diabetic and non-diabetic rats. Psychopharmacology (Berl) 1998;135:361–367. doi: 10.1007/s002130050523. [DOI] [PubMed] [Google Scholar]
- 49.Jung SW, Han OK, Kim SJ. Increased expression of beta amyloid precursor gene in the hippocampus of streptozotocin-induced diabetic mice with memory deficit and anxiety induction. J Neural Transm. 2010;117:1411–1418. doi: 10.1007/s00702-010-0516-2. [DOI] [PubMed] [Google Scholar]
- 50.Hirano S, Miyata S, Kamei J. Antidepressant-like effect of leptin in streptozotocin-induced diabetic mice. Pharmacol Biochem Behav. 2007;86:27–31. doi: 10.1016/j.pbb.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 51.Banks WA, Coon AB, Robinson SM, Moinuddin A, Shultz JM, Nakaoke R, Morley JE. Triglycerides induce leptin resistance at the blood-brain barrier. Diabetes. 2004;53:1253–1260. doi: 10.2337/diabetes.53.5.1253. [DOI] [PubMed] [Google Scholar]
- 52.Farr SA, Yamada KA, Butterfield DA, Abdul HM, Xu L, Miller NE, Banks WA, Morley JE. Obesity and hypertriglyceridemia produce cognitive impairment. Endocrinology. 2008;149:2628–2636. doi: 10.1210/en.2007-1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ikegaya Y, Ishizaka Y, Matsuki N. BDNF attenuates hippocampal LTD via activation of phospholipase C: implications for a vertical shift in the frequency-response curve of synaptic plasticity. Eur J Neurosci. 2002;16:145–148. doi: 10.1046/j.1460-9568.2002.02051.x. [DOI] [PubMed] [Google Scholar]
- 54.Desai NS, Rutherford LC, Turrigiano GG. BDNF regulates the intrinsic excitability of cortical neurons. Learn Mem. 1999;6:284–291. [PMC free article] [PubMed] [Google Scholar]
- 55.Asztely F, Kokaia M, Olofsdotter K, Ortegren U, Lindvall O. Afferent-specific modulation of short-term synaptic plasticity by neurotrophins in dentate gyrus. Eur J Neurosci. 2000;12:662–669. doi: 10.1046/j.1460-9568.2000.00956.x. [DOI] [PubMed] [Google Scholar]
- 56.Maffei L. Plasticity in the visual system: role of neurotrophins and electrical activity. Arch Ital Biol. 2002;140:341–346. [PubMed] [Google Scholar]
- 57.Shirayama Y, Chen ACH, Nakagawa S, Russell DS, Duman RS. Brain-derived neurotrophic factor produces antidepressant effects in behavioral models of depression. J Neurosci. 2002;22:3251–3261. doi: 10.1523/JNEUROSCI.22-08-03251.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Duman RS, Monteggia LM. A neurotrophic model for stress-related mood disorders. Biol Psychiatry. 2006;59:1116–1127. doi: 10.1016/j.biopsych.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 59.Lee BH, Kim H, Park SH, Kim YK. Decreased plasma BDNF level in depressive patients. J Affect Disord. 2007;101:239–244. doi: 10.1016/j.jad.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 60.Shimizu E, Hashimoto K, Okamura N, Koike K, Komatsu N, Kumakiri C, Nakazato M, Watanabe H, Shinoda N, Okada S, Iyo M. Alterations of serum levels of brain-derived neurotrophic factor (BDNF) in depressed patients with or without antidepressants. Biol Psychiatry. 2003;54:70–75. doi: 10.1016/s0006-3223(03)00181-1. [DOI] [PubMed] [Google Scholar]
- 61.Pandey SC, Zhang H, Roy A, Misra K. Central and medial amygdaloid brain-derived neurotrophic factor signaling plays a critical role in alcohol-drinking and anxiety-like behaviors. J Neurosci. 2006;26:8320–8331. doi: 10.1523/JNEUROSCI.4988-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bornstein SR, Schuppenies A, Wong ML, Licinio J. Approaching the shared biology of obesity and depression: the stress axis as the locus of gene-environment interactions. Mol Psychiatry. 2006;11:892–902. doi: 10.1038/sj.mp.4001873. [DOI] [PubMed] [Google Scholar]
- 63.McEwen BS. Glucocorticoids, depression, and mood disorders: structural remodeling in the brain. Metabolism. 2005;54:20–23. doi: 10.1016/j.metabol.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 64.Dalvi A, Lucki I. Murine models of depression. Psychopharmacology (Berl) 1999;147:14–16. doi: 10.1007/s002130051131. [DOI] [PubMed] [Google Scholar]
- 65.Nair A, Vadodaria KC, Banerjee SB, Benekareddy M, Dias BG, Duman RS, Vaidya VA. Stressor-specific regulation of distinct brain-derived neurotrophic factor transcripts and cyclic AMP response element-binding protein expression in the postnatal and adult rat hippocampus. Neuropsychopharmacology. 2007;32:1504–1519. doi: 10.1038/sj.npp.1301276. [DOI] [PubMed] [Google Scholar]