

Abstract
Through their well described actions in the hypothalamus, appetitive peptides such as insulin, orexin and leptin are recognized as important regulators of food intake, body weight and body composition. Beyond these metabolic activities, these peptides also are critically involved in a wide variety of activities ranging from modulation of immune and neuroendocrine function to addictive behaviors and reproduction. The neurological activities of insulin, orexin and leptin also include facilitation of hippocampal synaptic plasticity and enhancement of cognitive performance. While patients with metabolic disorders such as obesity and diabetes have greater risk of developing cognitive deficits, dementia and Alzheimer’s disease (AD), the underlying mechanisms that are responsible for, or contribute to, age-related cognitive decline are poorly understood. In view of the importance of these peptides in metabolic disorders, it is not surprising that there is a greater focus on their potential role in cognitive deficits associated with aging. The goal of this review is to describe the evidence from clinical and pre-clinical studies implicating insulin, orexin and leptin in the etiology and progression of age-related cognitive decline. Collectively, these studies support the hypothesis that leptin and insulin resistance, concepts normally associated with the hypothalamus, are also applicable to the hippocampus.
Keywords: insulin, leptin, orexin, hippocampus, dementia, Alzheimer’s disease
1. Introduction
Neuroplasticity may be defined as the ability of an organism to adapt to changes in the external or internal milieu and can be assessed using a variety of experimental approaches. In this regard, neuroplasticity can be evaluated through approaches ranging from neuroanatomical analyses to neurochemical and electrophysiological measures to behavior. An important question is what are the underlying molecular, cellular and neurochemical factors that regulate brain plasticity? In addition to neurotransmitters and neurotrophic factors, insulin, leptin and orexin are examples of neuropeptides that promote hippocampal synaptic plasticity (Figure 1). Insulin derived from the pancreas and leptin derived from adipocytes have well described activities in the hypothalamus, which includes regulation of the orexin system. Orexin neurons from the lateral hypothalamus/perfornical area project directly to the hippocampus and to the medial septum, which sends GABAergic, glutamatergic and acetylcholinergic projections to the hippocampus. Indeed, recent studies indicate that activation of these orexigenic pathways facilitates the performance of hippocampal-dependent tasks (Stanley et al., 2012). Beyond these integrative activities, insulin and leptin act directly in the hippocampus to promote measures of hippocampal synaptic plasticity, including enhancement of neuronal morphology, increases in neurogenesis/cell proliferation and increases in synaptic transmission (i.e. long term potentiation; LTP). These neuroanatomical, electrophysiological and neurochemical enhancements will facilitate cognitive/behavioral performance. Conversely, age-related decline in hippocampal neuroplasticity and cognition is associated with impairments in the functional activities of insulin, orexin and leptin. The focus of this review is to highlight how these peptides that are normally associated with the regulation of metabolism and body weight play a critical role in the facilitation of hippocampal neuronal plasticity, as well as how the impairments in the neuroplasticity activities of these appetitive peptides is a contributing factor in age-related cognitive decline.
Figure 1.

The peptides insulin, leptin and orexin work in an integrative fashion to facilitate hippocampal synaptic plasticity. Insulin released from pancreatic β cells and leptin released from adipocytes have well defined metabolic activities in the hypothalamus that include regulation of orexin neuronal populations in the lateral hypothalamus, perifornical area and dorsomedial hypothalamus (blue arrows). Projection sites of these orexin populations include the basal forebrain, where orexin modulates cholinergic, GABAergic and glutamatergic components of septohippocampal neurotransmission (blue arrows). As such, these orexin-driven basal forebrain projections have a significant impact on hippocampal neurochemistry. Beyond these indirect effects, insulin and leptin can directly modulate hippocampal synaptic plasticity (red arrows), including facilitation of synaptic transmission (i.e. LTP), increases in neurogenesis and cell proliferation and promotion of synaptogenesis and dendritic remodeling (morphology). This facilitation of hippocampal synaptic plasticity ultimately leads to the enhancement of cognitive/behavioral function. See text for details.
2. Insulin
2. 1. Role of insulin in promoting plasticity
Insulin is a peptidic hormone secreted by the pancreatic islets of Langerhans, regulated by the gut hormone glucagon-like peptide-1 (GLP-1) in response to meal ingestion. Although insulin receptors were identified in the brain 3 decades ago (Havrankova et al., 1978; Havrankova et al., 1979; Havrankova et al., 1981; Hill et al., 1986; Unger et al., 1989) with functions regulating glucose metabolism, food intake and body weight (Schwartz et al., 1992), only within the last decade or so, the role of insulin in synaptic plasticity and cognitive functions has become a subject of research. Insulin receptors are localized at the synapse where they regulate neurotransmitter release and receptor recruitment (Wan et al., 1997; Jonas et al., 1997; Abbott et al., 1999). Insulin plays a role in regulating synaptic efficacy in the CNS by rapid recruitment of GABAa receptors that potentiate inhibitory synaptic transmission (Wan et al., 1997; Taghibiglou et al., 2009). It has also been shown that insulin enhances NMDA receptor-mediated synaptic transmission (Liu et al., 1995), stimulates the translation of PSD-95 protein (Lee et al., 2005) and promotes internalization of AMPA receptors causing long term depression (LTD) of excitatory synaptic transmission (Huang et al., 2003; Huang et al., 2004). By intracerebroventricular injections, insulin was demonstrated to activate protein kinase B (PKB or Akt) via the phosphatidylinositol 3-kinase (PI3-K) pathway in the rat hippocampus (Grillo et al., 2009). Recently, a GSK3 inhibitor was shown not only to prevent, but also to reverse, Aβ-induced inhibition of LTP in brain slice preparations, suggesting that over-activation of GSK3β, that can result from disrupted insulin signaling but also a number of other pathways including protein phosphatase (Graff et al., 2010; Dineley et al., 2010), is responsible for pathological synaptic plasticity (Jo et al., 2011). More recently, insulin was also shown to increase the frequency of mEPSC (excitatory synapse) and to promote dendritic spine formation via the PI3K/Akt/mTOR pathway, playing a crucial role in structural synaptic plasticity (Lee et al., 2011). Another support of the role of insulin in synaptic plasticity is the recent demonstration that a reduced insulin receptor expression in mouse brain impairs hippocampal late-phase LTP and this was associated with deficits in long term recognition memory (Nistico et al., 2012).
2.2. Role of insulin in age-related cognitive deficits
In this review, we will focus on insulin and age-related cognitive deficits; however insulin appears to play a role independent of age. This is underlined in a recent study demonstrating that cognition is acutely impaired in children with newly diagnosed type 1 diabetes, when insulin regimen is only initiated (Nadebaum et al., 2012). Similarly, metabolic syndrome in adolescents, that included insulin resistance for all subjects, results in reductions in cognitive functions and brain structural integrity, despite a relatively short duration of poor metabolic control and no vascular disease (Yau et al., 2012). These results suggest that insulin resistance without overt hyperglycemia may lead to brain complications. Cognitive deficits generally associated with aging range from mild cognitive impairments (MCI) to dementia, including Alzheimer’s disease (AD). In recent years, the role of insulin in the brain of AD and/or diabetic patients has emerged as a principal contributor to the development of cognitive impairments and has become the focus of detailed studies to understand the mechanisms involved.
Diabetes mellitus is sub-classified into type 1 (insulin-deficient) and type 2 (insulin-resistant) diabetes and cognitive deficits occur in both of these patient populations (Ryan et al., 1985; Rovet et al., 1987; Reaven et al., 1990; Ryan and Williams, 1993; McCarthy et al., 2002; Cukierman et al., 2005; Manschot et al., 2006; Biessels et al., 2008). Studies have found a relative risk of about 1.9 to 2.3 of developing AD for diabetic patients (Ott et al., 1996; Leibson et al., 1997; Ronnemaa et al., 2009), and this risk can increase up to 5.5 in the presence of Apo E4, a risk factor for AD (Peila et al., 2002). More recently, diabetes was shown to increase not only the risk of dementia but also the risk of progression from MCI to AD (Velayudhan et al., 2010). Uncontrolled diabetes (no insulin treatment) was associated with development of AD, while patients with controlled diabetes showed no increased dementia, suggesting a role of impaired insulin signaling in the development of neurodegeneration and AD (Xu et al., 2009). Insulin signaling encompasses assorted signaling pathways, including those mediated by phospholipase Cγ, mitogen-activated protein kinase (MAPK) and PI3-K with the subsequent downstream involvement of GSK3. Recently, we have shown that 9 weeks of peripheral insulin deficiency induced by systemtic injection of streptozotocin is sufficient to cause AD-like features in the brain of type 1 diabetic mice. This pathology was associated with learning and memory deficits and decreased insulin receptor expression and Akt phosphorylation in the brain leading to increased GSK3 activity, increased tau phosphorylation and increased Aβ protein levels (Jolivalt et al., 2008). Several other studies in both type 1 and type 2 diabetes further support impaired brain insulin signaling leading to reduced PI3-K pathway activity and subsequent increased GSK3 activity. Recently, we have shown that type 1 diabetes exacerbates brain aging and increase vascular mediated Aβ depositions in the brain of Senescence prone or resistant mice, a model of accelerated aging (Currais et al., 2012). Interestingly, intranasal insulin delivery in type 1 diabetic mice prevents cognitive decline, cerebral atrophy and the decrease of the PI3-K/Akt signaling pathway (Francis et al., 2008). In a rat model of type 2 diabetes, intranasal insulin administration normalized Akt and GSK3β activities and reduced tau phosphorylation while subcutaneous insulin treatment had only minimal effects (Yang et al., 2012). Metformin, a FDA-approved drug used to treat type 2 diabetic patients, administered to high fat diet fed rats for 21 days improved learning and memory deficits as well as improved insulin resistance (Pintana et al., 2012). However, in db/db mice, which develop insulin resistance, metformin attenuates AD-like neuropathology, such as increased phosphorylated tau but did not improve insulin levels, GSK3 activity or learning behavior (Li et al., 2012a).
AD is characterized by neuronal loss, neurofibrillary tangles enriched with highly phosphorylated tau protein and plaques containing amyloid β derived from cleavage of Amyloid Precursor Protein (APP). It has been suggested that AD is a neuroendocrine disorder of the brain due to deficiency in brain insulin and impaired insulin signaling (de la Monte and Wands, 2005; Steen et al., 2005; Revill et al., 2006). The role of the insulin-signaling pathway and GSK3 in AD brain was suggested by intracerebroventricular-streptozotocin (icv-STZ) rodent models that display AD-type neurodegeneration, including deficits in learning and memory (Plaschke and Hoyer, 1993; Duelli et al., 1994; Lannert and Hoyer, 1998). STZ, when injected directly into the brain, rapidly induced a dramatic reduction of insulin gene expression and subsequent neurodegeneration and cognitive impairments that were accompanied by an increase of activated GSK3, phosphorylated tau, APP mRNA and amyloid β (Steen et al., 2005; Lester-Coll et al., 2006; Plaschke et al., 2010; Santos et al., 2012; Shingo et al., 2012). In addition to the icv-STZ rat model, transgenic mouse models overexpressing active GSK3 also displayed cognitive deficits associated with neuronal loss and tau hyperphosphorylation in contrast to mouse models expressing reduced GSK3 activity [reviewed in (Avila et al., 2010)]. These models strengthen the importance of GSK3 in the neurodegeneration. Also, in vitro studies have shown defective insulin signaling as a result of amyloid β oligomer toxicity, suggesting that restoration of insulin signaling may slow AD pathogenesis (De Felice et al., 2009). The role of insulin signaling in AD brain is further supported by animal studies showing exacerbation of tau deposition into tangles in a mouse model expressing human tau with concomitant insulin-deficient diabetes (Ke et al., 2009) and studies showing that induction of type 1 diabetes with STZ in familial AD transgenic mouse models exaggerated both vascular and AD-like pathology (Burdo et al., 2009; Jolivalt et al., 2010). Further support for the role of impaired insulin signaling as contributor to brain neurodegeneration was demonstrated with insulin-resistance/type 2 diabetes studies showing amyloidosis in a mouse model of AD with concomitant diet-induced insulin-resistance (Ho et al., 2004) and AD transgenic mice APP23 crossbred with type 2 diabetic ob/ob mice that displayed exacerbated cognitive deficits without increased Aβ burden in the brain but cerebral inflammation and amyloid deposit in blood vessels (Takeda et al., 2010).
2.3. Translation of animal models with insulin signaling impairment to the clinical setting
In a cohort of 75 year old men and women, impaired insulin sensitivity, calculated based on fasting insulin and glucose levels, correlates with reduced cognitive performance and decreased brain size (Benedict et al., 2012). Brain insulin resistance in AD patients, assessed ex vivo by insulin stimulation of the hippocampus and Western blots of the insulin signaling pathway proteins, was associated with worse performance on tests of working and episodic memory, suggesting that insulin signaling has a direct association with cognitive status (Talbot et al., 2012). In a recent prospective study following older adults over 9 years, the presence of diabetes mellitus and poor glucose control was associated with worse cognitive function and greater decline than adults without diabetes, suggesting that the severity of diabetes may contribute to accelerated cognitive aging (Yaffe et al., 2012). A recent autopsy study showed decreased activities of the components of the insulin-PI3-K/Akt pathway and increased tau phosphorylation levels in the brain of AD and type 2 diabetic patients, alterations that were more severe in patients with co-morbid AD and type 2 diabetes (Liu et al., 2011). A 4 week diet-induced metabolic syndrome, with insulin resistance, in adults with MCI promoted a decrease of Aβ protein in cerebrospinal fluid that may correspond to the initiation of Aβ deposition in the brain (Bayer-Carter et al., 2011). In contrast, insulin infusion in type 2 diabetic patients reduces APP, presenilin-1and -2 (proteins from the β-secretase complex that cleave APP), and GSK-3β mRNA in peripheral blood mononuclear cells (Dandona et al., 2011). In a recent study to develop a novel diagnostic marker, plasma Aβ levels were increased in AD patients after a glucose load despite similar Aβ levels between AD and non-AD patients (Takeda et al., 2012).
A number of clinical studies with intranasal administration of insulin in patients with diabetes, MCI or AD also underscore the role and mechanism(s) of impaired insulin signaling in the development of cognitive deficits. In the latest review, Shemesh et al. assessed 8 studies performed in the past 8 years that included healthy, obese, MCI or AD patients and concluded that intranasal insulin has a positive effect on cognitive functions despite limited number of patients, different doses and tests used in the reviewed studies (Shemesh et al., 2012). In a recent pilot randomized placebo-controlled trial, 4 month treatment with intranasal insulin resulted in stabilization or improvement of cognition in MCI and AD patients (Craft et al., 2012a).
2.4. Potential therapeutic intervention based on insulin action
Some therapeutic interventions aimed at correcting insulin resistance for AD or MCI patients, such as insulin-sensitizers, insulinotropes, intranasal insulin, antioxidant and anti-inflammatory drugs or drugs targeting GSK3 as treatment for type 2 diabetes and AD are described in recent reviews (de la Monte, 2012; Gao et al., 2012; Craft et al., 2012b). Here, we will summarize only the most recent aspects of intervention based on current results and mechanisms presented above.
Pioglitazone, an insulin-sensitizer, as well as lithium, a GSK3 inhibitor, restored memory impairments in icv-STZ rats, behavioral improvements that were associated with a significant attenuation of GSK3 activity in the hippocampus. Memantine, one of the few approved drug for the treatment of AD (albeit with modest effect on moderate to severe AD) improved memory but not GSK3 activity in the icv-STZ rat model (Ponce-Lopez et al., 2011). Chronic lithium treatment in an aged AD transgenic mouse model decreased the cleavage of APP and senile plaque formation, and this was accompanied by improvement in spatial learning and memory abilities (Zhang et al., 2011). In a recent 12 month-long trial with lithium in MCI patients, cognitive deficits and attention tasks were improved along with decreased levels of phosphorylated tau in the CSF (Forlenza et al., 2011). In our recent work (under revision), the role of GSK3 in learning and memory and synaptic plasticity in type 1 diabetic mice was confirmed by treatment with a GSK3 inhibitor or a neurotrophic factor with GSK3 inhibitory properties. The cognitive improvements were associated with restoration of synaptophysin protein levels in the brain of treated diabetic mice but not with tau phosphorylation or Aβ protein levels, suggesting that, in contrast to synaptic plasticity, tau and Aβ may be late contributors to learning and memory processes in diabetic mice brain. These results also support GSK3 inhibition as a treatment for type 1-associated cognitive deficits and AD.
Glucagon-like peptide-1 (GLP-1) is an incretin hormone regulating energy metabolism and insulin secretion with a short half-life due to degradation. GLP-1, but not its degradation metabolites, has been shown to improve markers of cognitive functions in type 2 diabetes models (Porter et al., 2012). A number of long lasting analogs have been developed, such as 2 drugs on the market to treat type 2 diabetes, exendin-4, commercialized under Byetta™, and liraglutide. Treatment with liraglutide showed neuroprotective effects in mice with high-fat diet-induced obesity and insulin resistance (Porter et al., 2010) and in an AD mouse model APP/PS1 in which it rescued memory deficits and impairments in LTP, reduced Aβ levels and amyloid plaque numbers, protected synapses and reduced inflammation in the brain (McClean et al., 2010; McClean et al., 2011). Similar observations have been made with the long-acting GLP-1 analog (Val8)GLP-1 which protected LTP and reduced the dense-core plaque count in aged APP/PS1 mice (Gengler et al., 2012). In recent studies, GLP-1 or analogs were shown to ameliorate neurodegeneration and cognitive performance in icv-STZ rats with impaired insulin signaling in the brain. Treatment with exedin-4 (Ex-4), a long-acting GLP-1 receptor agonist, or (Val8)GLP-1 improved learning and memory in the Morris water maze and reversed icv-STZ-induced tau phosphorylation (Li et al., 2012b) via down-regulation of GSK3β activity (Chen et al., 2012). Ex-4 was also shown to improve cognitive behavior and neurogenesis in the hippocampus of adult rodents (Isacson et al., 2011) and in a mouse model of obesity and insulin-resistance (Gault et al., 2010). Similarly, in the STZ mouse model of type 1 diabetes, in which we have demonstrated AD-like features and insulin signaling impairment in the brain (Jolivalt et al., 2008), we have shown that Ex-4 significantly prevented memory impairments in the Barnes maze in diabetic mice after 8 weeks of treatment (Figure 2), without affecting plasma glucose or insulin levels (Jolivalt et al., 2011), suggesting that activation of GLP-1 receptor provides neuroprotective effect in the brain of type 1 diabetic mice independent of direct glucose or insulin modulation. These latest data showed that activation of GLP-1 receptor might have beneficial central effects in both types of diabetes as well as in AD.
Figure 2.
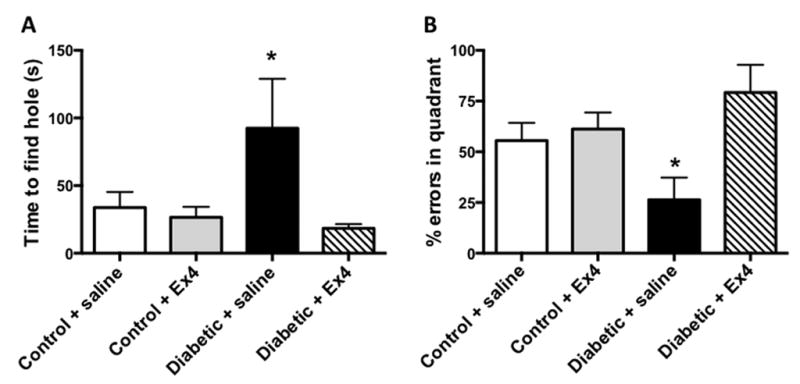
Effect of Exendin-4 (EX4) on memory retention in the Barnes maze for STZ-diabetic mice. Mice were trained to learn the location of the box for 5 consecutive days in the Barnes maze, left untested for 3 day and then placed back on the Barnes maze where the box was removed but the cue was present. Panel A: Data represent the time to find the hole where the box was originally. Panel B: Percent of errors spent in the quadrant where the box was originally located. Data from control mice in white bars, Ex-4-treated control mice in grey bars, from untreated diabetic mice in black bars and from Ex-4-treated diabetic mice in hatched bars are mean+SEM, n=5–6/group. *p<0.05 vs diabetic+Ex-4 by one-way ANOVA followed by Bonferroni’s post-hoc test.
The role of insulin and more particularly of impaired insulin signaling in the brain has been clearly demonstrated in the last decade as a contributor to neurodegeneration and associated cognitive decline. Collectively, these recent data suggest that insulin delivered directly to the brain and modulators of insulin and/or insulin signaling pathway may represent potential therapeutic interventions against impairments of cognitive performance and dementia for patients with diabetes, MCI or AD.
3. Leptin
3.1. Leptin signaling in the regulation of metabolic parameters
Leptin is synthesized and secreted by adipocytes and is transported across the blood-barrier (BBB) via a saturable transport system (Banks, 2004). In the hypothalamus, leptin is recognized as an important integrator for coordinating the peripheral and central signals that regulate food intake, body weight and body composition [For review see, (Schwartz et al., 2000)]. This includes functional connections with brain regions such as the ventral tegmental area [VTA; for review, see (Figlewicz and Benoit, 2009)]. For example, lateral hypothalamic (LH) LepR-containing neurons project to the VTA, where they regulate feeding behavior (Figlewicz et al., 2007; Leinninger et al., 2009). These actions of leptin are mediated through the long form of the LepR (LepRb), which is abundantly expressed in several hypothalamic nuclei including the arcuate nucleus and the ventromedial nucleus (Elmquist et al., 1998; Baskin et al., 1999). In the hypothalamus, activation of the LepRb results in a complex and integrated activation of several signal transduction cascades. Specifically, the LepRb activates the constitutively associated tyrosine kinase Jak2, resulting in autophosphorylation of Jak2, as well as phosphorylation of several tyrosine residues within the intracellular domain of the LepRb. Phosphorylation of the LepRb at Tyr1138 results in the recruitment and phosphorylation of the transcriptional regulator STAT3 (pSTAT3), thereby leading to nuclear translocation and regulation of a variety of genes including suppressor of cytokine signaling 3 (SOCS3). SOCS3 binds to Tyr985 of the LepRb, as well as to Jak2, to mediate feedback inhibition of the LepRb. Interestingly, Try985 also provides a binding site for the tyrosine phosphatase SHP-2, which ultimately leads to the activation of the ERK signaling cascade. Tyr1107 phosphorylation leads to the activation of the transcription factor STAT5. Beyond these Tyr residues on the LepRb, autophosphorylation of Jak2 stimulates the phosphatidylinositol 3-kinase (PI3-K) cascade, leading to the phosphorylation and activation of Akt. [For review of these pathways, see (Munzberg and Myers, Jr., 2005; Myers, Jr. et al., 2010)]. These coordinated signaling cascades are critical for the regulation of food intake and body composition. Conversely, obesity and type 2 diabetes are associated with hypothalamic leptin resistance that results from a combination of decreased LepRb signaling and decreased BBB leptin transport (Banks, 2004). Mechanistically, studies by Banks and Levin demonstrated that decreases in hypothalamic LepRb signaling precedes impaired BBB leptin transport in diet-induced obesity models (Levin et al., 2004). As will be described below, hypothalamic leptin resistance also appears to be a hallmark feature of aging. Beyond the hypothalamus, this section will focus on the role of LepRb in the facilitation of hippocampal synaptic plasticity and will discuss the hypothesis that leptin resistance, a concept normally reserved for the hypothalamus, may be a contributing factor in age-related impairments in hippocampal synaptic plasticity.
3.2. Leptin facilitation of hippocampal neuroplasticity: preclinical studies
Leptin plays a key role in neuronal development (Morrison, 2009) and also facilitates structural and functional activities in the adult central nervous system (CNS). Indeed, previous studies have determined that leptin facilitates hippocampal neuroplasticity in normal control rodents. For example, intrahippocampal administration of leptin elicits dose-dependent effects on hippocampal-dependent learning (Farr et al., 2006). Similarly, intravenous (iv) administration of leptin elicits dose-dependent effects in the passive avoidance learning and spatial learning in the water maze (Oomura et al., 2006). Underlying these enhancements in hippocampal-dependent behaviors, leptin regulates hippocampal plasticity by converting short term potentiation (STP) into LTP (Shanley et al., 2001a), a cellular correlate of learning and memory processes in the mammalian brain. More recent studies illustrate that leptin administration enhances LTP in CA1 pyramidal neurons (Oomura et al., 2006) and dentate gyrus granule neurons (Wayner et al., 2004). Interestingly, NMDA receptor subunit composition appears to dictate the age-dependent effects of leptin on synaptic transmission. Specifically, leptin mediates long term depression (LTD) in neonatal rat hippocampal slices through GluN2B-containing receptors, while eliciting LTP in hippocampal slices from young rats or aged rats through GluN2B containing receptors (Moult and Harvey, 2011). Beyond these electrophysiological measures, leptin modulation of glutamatergic neurotransmission (Shanley et al., 2001b; Durakoglugil et al., 2005; Moult et al., 2009) and glutamate receptor trafficking (Moult and Harvey, 2009; Moult et al., 2010) may also contribute to leptin-mediated enhancement of cognitive function. Confocal immunoflurorescence analyses has determined that leptin regulates the morphological features of primary hippocampal cultures, including the motility of dendritic filopodia and hippocampal synaptic density (O’Malley et al., 2007). Chronic leptin administration increases cell proliferation in the dentate gyrus and protects hippocampal neurons from excitotoxic damage in both the in vivo and in vitro settings (Garza et al., 2008). Collectively, these pre-clinical data support the hypothesis that leptin facilitates morphological, electrophysiology and behavioral measures of hippocampal synaptic plasticity.
Neuroplasticity deficits observed in leptin deficient ob/ob mice and rodents with impaired leptin receptor signaling (db/db mice and Zucker rats) provide further support for the concept that leptin is an important regulator and facilitator of hippocampal synaptic plasticity. This includes deficits in hippocampal long term potentiation (Li et al., 2002; Gerges et al., 2003; Stranahan et al., 2008), morphological changes such as decreases in spine density in dentate gyrus granule neurons (Stranahan et al., 2009), as well as decreases in cell proliferation and neurogenesis in the dentate gyrus (Stranahan et al., 2008). Ultimately, these electrophysiological and morphological deficits contribute to the behavioral deficits observed in db/db mice and Zucker rats (Li et al., 2002; Winocur et al., 2005). In this regard, our prior studies revealed that Zucker rats can perform a learning task in the absence of an inter-trial interval (ITI) or when the ITI is very short. However, when the ITI is increased, which more directly assesses hippocampal-dependent function, these leptin receptor deficient rats perform more poorly when compared to control rats (Winocur et al., 2005). Interestingly, Zucker rats also exhibit increases in AD-like pathology, including increases in hyper-phosphorylated tau immunoreactivity in the dentate gyrus and CA3 region of the hippocampus (Wrighten et al., 2008). Collectively, these data illustrate that hippocampal synaptic plasticity is impaired in leptin-deficient rodents.
One caveat associated with these studies is the identification of the role of leptin receptor deficiency in these models. Indeed, ob/ob mice, db/db mice and Zucker rats exhibit a wide array of metabolic and endocrine changes that include, but are not limited to glucose intolerance, insulin resistance, leptin resistance, hypothalamic-pituitary-adrenal axis dysfunction and alterations in plasma lipid profiles. As a result, the cause and effect relationship of leptin to neuroplasticity deficits described above must be interpreted with caution. However, experimental models of AD and studies in aged rodents provide additional support for a causative role for leptin resistance in age-related cognitive decline. For example, leptin administration to control (six month-old) rats effectively reduces food intake and hypothalamic NPY mRNA expression, but is less effective in aged (30 month-old) senescent rats (Shek and Scarpace, 2000; Scarpace et al., 2000a). These results suggest that aged rats exhibit leptin resistance. Subsequent studies from these same investigators determined that while maximal stimulation of pSTAT3 was similar following peripheral (intravenous) administration, the dose response curve for leptin was shifted to the right in aged rats (Scarpace et al., 2000b). Interestingly, both decreased pSTAT3 activation and the rightward shift of the leptin dose response curve were observed in aged rats following intracerebroventricular leptin administration (Shek and Scarpace, 2000; Scarpace et al., 2001). Moreover, pSTAT3 transcription factor binding activity was significantly reduced in aged rats (Shek and Scarpace, 2000; Scarpace et al., 2001). Decreases in leptin receptor mRNA expression (Fernandez-Galaz et al., 2001) and protein levels (Scarpace et al., 2001; Fernandez-Galaz et al., 2001), increases in SOCS3 mRNA levels (Wang et al., 2001), as well as age-related decreases in BBB leptin transport (Fernandez-Galaz et al., 2001) may also contribute to decreased leptin responsiveness in aged rats. While these studies focused on hypothalamic measures, the take home message is that aged rodents exhibit leptin resistance (although see (Hosoi et al., 2005). An important future direction would be to determine whether age-related leptin resistance is specific for the hypothalamus, or whether leptin resistance extends to other brain regions and contributes to age-related neuroplasticity deficits in the hippocampus.
3.3. Mechanistic basis of leptin-mediated neuroprotection
Studies in experimental models of AD support the notion that leptin provides neuroprotection from the deleterious consequences of aging and have provided insight into the mechanistic basis for this neuroprotection. For example, in the ABPP/PS1 mouse, a transgenenic mouse model of AD, hippocampal administration of a lentiviral vector that overexpresses leptin promotes cell proliferation and neurogenesis in the dentate gyrus and also decreases the density of Fluro-Jade histochemical labeling in the dentate gryus (Perez-Gonzalez et al., 2011). In other transgenic mouse models of AD, leptin infusion has been shown to decrease amyloid-β levels in total brain extracts (Fewlass et al., 2004). This same study determined that leptin treatment dose-dependently decreased Aβ uptake in cloned neuronal cells. Subsequent studies from these same investigators confirmed these in vivo findings and also determined that leptin treatment reduced hippocampal amyloid burden and that chronic leptin administration reversed the impairment in hippocampal-dependent learning in an AD transgenic mouse model (Greco et al., 2010). Mechanistically, these neuroprotective activities of leptin have been proposed to be mediated through LepRb activation of AMP-activated protein kinase (Greco et al., 2009) and STAT5 activation (Marwarha et al., 2011). Recent studies by Doherty and co-workers support the hypothesis that leptin provides protection of hippocampal neurons from AD-mediated neurotoxicity. For example, amyloid-β-induced decreases in LTP and enhancement of LTD in hippocampal slices were inhibited by bath application of leptin. This facilitation of hippocampal synaptic transmission was inhibited by wortmannin and PI828, implicating leptin-induced activation in the PI3 kinase pathway in these activities (Doherty et al., 2013). An obvious question that emerges from these studies in experimental models of aging and AD is whether these observations are translatable to the clinical setting.
3.4. Leptin and age-related cognitive decline: clinical observations
While clinical studies support the concept that cognitive decline and neuroplasticity deficits in patients with metabolic disorders like type 2 diabetes is based in part on leptin resistance (Labad et al., 2012), the picture is less clear regarding a causative role for leptin resistance in age-related cognitive decline. The weaknesses in this causal relationship may be related to a variety of factors, including the potential contribution of age-related changes in endocrine profiles (Rosenbaum et al., 1996; Ostlund, Jr. et al., 1996; Baumgartner et al., 1999; Isidori et al., 2000), as well as age-related changes in body composition and metabolic parameters (Perry, III et al., 1997; Isidori et al., 2000), which includes pre-dementia weight loss (Barrett-Connor et al., 1996; Barrett-Connor et al., 1998). These factors provide a potential explanation for the disparate findings in studies that have measured leptin activity in aging. Generally speaking, these studies suggest that plasma leptin levels decrease with age in women, particularly post-menopausal women and that body mass index (BMI) or fat mass are also important determinants in age-related changes in plasma leptin levels. Although some studies suggest that leptin levels are decreased in AD patients (Power et al., 2001; Bigalke et al., 2011), an important question that these studies cannot address is how these changes in plasma levels impact leptin-mediated neuroplasticity. More simply, can age-related cognitive decline elicited by hippocampal leptin resistance be accurately assessed by measurement of plasma leptin levels?
A number of studies have addressed the relationship of age-related changes in plasma leptin levels to measures of neuroplasticity such as cognitive performance and structural analyses. For example, reductions in executive function, which is proposed to reflect frontal cortical function, was associated with higher plasma leptin levels in otherwise healthy community-dwelling elderly participants (Gunstad et al., 2008). Conversely, higher plasma leptin levels were associated with decreased cognitive decline in a prospective study involving healthy elderly participants (Holden et al., 2009). Similarly, a more recent prospective study reported that higher plasma leptin levels were associated with decreased risk of developing dementia/MCI in elderly women (Zeki et al., 2012). The designs of these studies provide a likely explanation for these dissimilar findings; a single time point measure of cognition versus analysis of cognitive decline over a four-year period. Results from the Framingham Study support this potential explanation and also provided analysis in structural parameters in aged populations. In this regard, elevated plasma leptin levels were associated with decreased incidence of dementia and AD. Moreover, elevated plasma leptin levels were associated with larger total brain volumes, as well as indirect assessments suggestive of larger hippocampal formation volumes (Lieb et al., 2009). Voxel-based morphometric analyses that provided more direct assessment of hippocampal volumes determined that elevated plasma leptin levels were associated with increased grey matter volumes in right hippocampus and left parahippocampus in a small cohort of healthy elderly participants (Narita et al., 2009). Additional support for neurotrophic activities of leptin are provided by studies in leptin-deficient humans receiving leptin replacement (Matochik et al., 2005; Paz-Filho et al., 2008), including morphometric analyses suggesting that leptin replacement in this unique patient population increases grey matter volumes in the right hippocampal formation (London et al., 2011). Taken together, these studies suggest that elevated plasma leptin levels may protect structural and functional measures of neuroplasticity from the deleterious consequences of aging. It is tempting to speculate that the elevated plasma leptin levels in these patients are indicative of increased hippocampal leptin activity, which provides neuroprotection. Conversely, individuals with reductions in plasma leptin levels or those individuals that exhibit hippocampal leptin resistance would not benefit from the neuroprotective activities of leptin and would thereby be more vulnerable to develop MCI and AD. Clarification of these issues would support the concept that enhancement of CNS leptin activity may represent a novel therapeutic option for the treatment of age-related disorders (Harvey, 2010).
4. The orexins/hypocretins: neuromodulatory peptides at the interface of energy balance and cognition
4.1. Discovery
In 1998, two independent research teams reported the discovery of a population of neuropeptide-producing cells whose distribution was limited to the lateral hypothalamus, the perifornical area and contiguous dorsomedial hypothalamus. De Lecea et al. termed these peptides hypocretins (specifically, hypocretin 1 and hypocretin 2) due to their restricted hypothalamic expression and their homology to the incretin family of gut hormone proteins (de Lecea et al., 1998). Meanwhile, Sakurai and colleagues termed these peptides orexin A (OxA) and orexin B (OxB), after the Greek word for appetite (orexis), because they were distributed most densely in the classical ‘feeding center’ of the lateral hypothalamus and initial experiments supported a pro-feeding effect of orexin A upon intracerebroventricular administration (Sakurai et al., 1998). Two G protein-coupled receptors have been described for the hypocretins/orexins (hereafter referred to as orexins for simplicity). The orexin 1 receptor (Ox1R) binds OxA with 10 to 100-fold greater affinity relative to OxB whereas the orexin 2 receptor (Ox2R) binds OxA and OxB with roughly equal affinity (Smart and Jerman, 2002; Ammoun et al., 2003; Scammell and Winrow, 2011).
4.2. Distribution of cell bodies and projections
In rodents and primates, orexins are uniquely localized in a posterior hypothalamic region that includes the lateral hypothalamus, perifornical area and dorsomedial hypothalamus. Despite their restricted localization pattern and fairly limited number, however, orexin neurons give rise to a diffuse pattern of projections within and beyond the diencephalon. Ascending fibers, for example, provide orexin innervation of the cerebral cortex (most prominently, the prefrontal cortex), as well as subcortical limbic structures important for learning, memory, motivation and emotion such as the nucleus accumbens, amygdala, basal forebrain and hippocampus (Peyron et al., 1998). Among thalamic targets, the paraventricular nucleus of the thalamus receives a particularly dense orexin innervation (Kirouac et al., 2005). Descending projections innervate numerous brainstem areas and all levels of the spinal cord (van den Pol, 1999). Of particular interest, orexins provide substantial innervation to nuclei that are the source of diffuse neuromodulatory transmitters, including dopamine neurons of the ventral tegmental area, norepinephrine neurons of the locus coeruleus and cholinergic neurons of the basal forebrain (Fadel and Deutch, 2002; Baldo et al., 2003; Fadel et al., 2005; Espana et al., 2005). Collectively, this pattern of connectivity mediates orexin influences over a wide range of cognitive, behavioral, autonomic and visceral functions, consistent with their description as “physiological integrators” (de Lecea et al., 2002).
Abundant evidence indicates that the orexin neurons are not functionally homogeneous, but differ along a medial-lateral gradient, with medially-located neurons preferentially involved in arousal and stress and the more lateral bank mediating effects on reward-based behaviors, including drug-seeking and feeding (Harris and Aston-Jones, 2006). Functional descriptions of orexin neurons must also take into account that virtually all orexin cells colocalize dynorphin (Chou et al., 2001) and at least some are also glutamatergic (Rosin et al., 2003; Torrealba et al., 2003; Henny et al., 2010).
4.3. Orexins and energy balance
The conditions under which the orexin system promotes food intake are not always straightforward, but the data supporting a pro-feeding role for these peptides remains compelling. Expression of preproorexin and orexin receptors is increased during periods of food restriction (Cai et al., 1999; Lu et al., 2000; Karteris et al., 2005). Intracerebroventricular or intra-hypothalamic administration of orexin increases food intake in rats (Dube et al., 1999; Haynes et al., 1999; Sweet et al., 1999; Baird et al., 2009) and systemic administration of the Ox1R antagonist SB-334867 attenuates food intake (Haynes et al., 2000; Rodgers et al., 2002).
Like other cellular components of hypothalamic feeding circuitry, orexin neurons respond to several peripherally-derived cues whose fluctuations are indicative of metabolic status. Consistent with a pro-feeding role for orexin peptides these neurons are inhibited by leptin, with original reports suggesting a direct effect mediated by leptin receptors expressed on orexin cells (Hakansson et al., 1999; Yamanaka et al., 2003). However, current evidence suggests that leptin inhibition of orexin neurons is mediated transynaptically, as leptin activates a population of LepRb-positive, neurotensin-colocalizing GABAergic LH neurons that give rise to local collaterals to inhibit orexin neurons (Louis et al., 2010; Leinninger et al., 2011). Conversely, orexin neurons are activated by the gut-derived hormone ghrelin and are required for the effects of this hormone on certain reward-related aspects of feeding (Yamanaka et al., 2003; Perello et al., 2010). Perhaps the clearest response of orexin neurons to a more rapidly-fluctuating indicator of energy balance is the activation of these neurons by hypoglycemia and their striking inhibition by elevated—but physiologically-relevant—levels of glucose via activation of tandem-pore potassium channels expressed directly on orexin cells (Moriguchi et al., 1999; Cai et al., 2001; Burdakov et al., 2005; Burdakov et al., 2006). Ethologically, enhanced arousal is necessary during periods of low energy balance in order to promote cognitive and behavioral aspects of food-seeking. Orexin neurons comprise an ideal neural system to integrate these divergent phenomena due to their place at the functional intersection of reward, feeding, stress and arousal. Interestingly, orexins, while serving a “pro-feeding” role, also appear to activate behavioral and metabolic pathways that enhance caloric expenditure, thus maintaining energy balance. For example, orexin is required for brown adipose activation (Sellayah et al., 2011; Tupone et al., 2011) and food-anticipatory behavioral activation (Mieda et al., 2004). These mechanisms may underlie the apparently paradoxical association between decreased food intake in orexin knockout mice and the obese phenotype of these same animals (Hara et al., 2001; Fujiki et al., 2006). Similarly, human narcolepsy patients (a “natural” human model of orexin deficiency) have higher rates of obesity (Lammers et al., 1996; Nishino et al., 2001).
4.4. Narcolepsy and sleep
Pathological correlates of orexin (dys)function are clearly seen in the case of the sleep disorder, narcolepsy. Orexins are deficient (if not completely absent) in most cases of human narcolepsy (Siegel, 1999; Nishino et al., 2000; Thannickal et al., 2000). The narcoleptic phenotype of orexin knockout mice (Chemelli et al., 1999) and the finding of a loss-of-function mutation in Ox2R in a autosomal recessive form of canine narcolepsy (Lin et al., 1999; Reilly, 1999) provide compelling convergent evidence from animal models that orexin deficiency underlies most cases of narcolepsy. Orexin knockout mice also exhibit feeding-induced cataplexy, a sudden loss of muscle tone triggered by strong emotions that is frequently seen in both canine and human forms of narcolepsy (Clark et al., 2009).
4.5. Orexins in cognition and neuroplasticity
Hypothalamic neuropeptides have traditionally been viewed in terms of homeostatic functions mediated largely by modulation of neuroendocrine and autonomic circuitry. However, homeostatic functions such as feeding include cognitive components (interoceptive awareness of hunger or energy balance status; remembering where to obtain food; attention to food-related cues or threats in the environment; etc.). Thus, it is not surprising that many of these peptide systems, including the orexins, have extensive projections to brain areas important for cognition and motivated behavior. One such area is the hippocampal formation, which receives a moderate orexin innervation and expresses both Ox1R and Ox2R. Orexins increase the release of acetylcholine (ACh) in the hippocampus (Stanley and Fadel, 2012). Orexin facilitation of synaptic plasticity has been clearly demonstrated in brain regions associated with drugs of abuse (Borgland et al., 2006). However, orexins also enhance electrophysiological correlates of hippocampal synaptic plasticity (Selbach et al., 2004; Selbach et al., 2010). Indeed, direct administration of OxA into the dentate gyrus enhances long-term potentiation in an Ox1R-dependent manner (Wayner et al., 2004). Conversely, orexin antagonists impair behavioral measures of hippocampal-dependent learning and memory (Akbari et al., 2006; Akbari et al., 2008). Orexin neurons also give rise to a rather dense projection to the medial prefrontal cortex where, in rodents, they activate thalamocortical projections in a manner that corresponds with attentional function and cognitive arousal (Lambe et al., 2005; Huang et al., 2006). Orexins increase release of ACh in the prefrontal cortex (Fadel et al., 2005; Frederick-Duus et al., 2007), and performance in a cholinergic-dependent attentional task is impaired by systemic or basal forebrain administration of an Ox1R antagonist (Boschen et al., 2009). In non-human primates, both systemic and intranasal delivery of OxA ameliorates the negative effects of sleep deprivation on cognitive performance (Deadwyler et al., 2007). Finally, human narcolepsy patients, who presumably lack orexin neurons, show attentional deficits even during period of normal wakefulness (Rieger et al., 2003). Collectively, these observations clearly demonstrate a role for the orexins in cognitive function. Specifically, under conditions in which the orexin system is normally activated, such as enhanced arousal or low energy balance, these peptides are likely to contribute to attention, learning and memory processes in a manner that integrates homeostasis and cognition.
4.6. Orexins and aging
Convergent studies from the human and animal literature demonstrate that the orexin system is negatively impacted in aging. Aged rodents have substantially fewer orexin neurons than their young counterparts (Sawai et al., 2010; Kessler et al., 2011). While melanin concentrating hormone neurons (which are similarly distributed and also play a role in feeding behavior) show some degree of reduction in aged animals, the extent is milder and there is no global age-related neuronal loss in the lateral hypothalamus (Kessler et al., 2011), suggesting that orexin neurons are preferentially, if not exclusively, susceptible to age-related neurodegeneration. Age-related decreases in preproorexin and Ox2R gene expression as well as diminished orexin regulation of feeding and neurotransmitter release have also been reported (Terao et al., 2002; Porkka-Heiskanen et al., 2004; Kotz et al., 2005; Stanley et al., 2011). Aged rats have reduced orexin innervation of medial septal GABAergic neurons (Stanley et al., 2011), a functionally-significant finding given that orexin receptor-expressing medial septal neurons mediate hippocampal theta generation (Gerashchenko et al., 2001). Collectively, these data suggest that not only is the orexin system impacted by aging and peripheral metabolic dysfunction, but orexin-based therapies may have therapeutic efficacy in treating both the central, cognitive manifestations and some of the peripheral correlates of these disorders.
In humans, post-mortem studies have also documented decreases in orexin neurons in some age-related neurodegenerative disorders such as Parkinson’s disease (Thannickal et al., 2007; Fronczek et al., 2008). Perhaps most relevant to age-related cognitive impairment, a recent study documented a significant (~40%) loss of orexin neurons, relative to age-matched controls, in post-mortem brains from AD patients (Fronczek et al., 2012). Because there was not a young control group included in the Alzheimer’s study, it is possible that the disease-specific impact is layered onto an already age-compromised orexin system. When viewed in light of the animal data, the clinical findings support the view that the orexin system is impaired with age and in age-related neurocognitive disorders.
How might the impact of age-related impairments in orexin function be manifest in cognition and homeostasis? Clearly, given the role of orexins in arousal and sleep-wake cycles, loss of these peptides or their receptors could mediate age-related impairments in sleep architecture. However, while overweight during middle age is associated with increased risk for AD, recent clinical data suggest that changes in homeostasis and energy balance in the elderly, including unexplained weight loss, precede and predict the subsequent development of AD (Knittweis, 1998; Johnson et al., 2006). An intriguing possibility is that orexin deficiency late in life mediates changes in sleep and feeding patterns that portend subsequent cognitive decline. Via interactions with neurotransmitters and brain regions classically implicated in senescent dementia, orexin deficiency may also form a mechanistic link between homeostatic dysfunction and cognitive impairment. Because homeostatic alterations in the elderly generally precede the manifestation of frank dementia by a period of months to several years, orexin-based therapies may also represent a novel target for arresting or slowing this progression.
5. Conclusions and future perspectives
As highlighted throughout this review, the clinical and pre-clinical literature clearly indicates that multifactorial mechanisms contribute to age-related deficits in hippocampal plasticity. Disentangling the causative factors responsible for cognitive decline in dementia and AD, as well as determination of the relative contribution of these various causative factors, represents a major obstacle in the development of effective treatment strategies in these patient populations. The presence of co-morbid conditions in patients with AD and dementia, such as obesity and type 2 diabetes, further complicates the identification of the causative factors involved (Wrighten et al., 2008). However, an optimistic viewpoint would contend that these co-morbidities aid in the assessment and identification of potential causative factors in age-related cognitive decline and implicate appetitive peptides like orexin, insulin and leptin in the pathogenesis of age-related deficits in neuroplasticity. An opposing viewpoint would note that while elevated plasma leptin levels provide neuroprotection from MCI and AD, elevated plasma leptin levels are associated with cognitive decline in obesity and type 2 diabetes (Labad et al., 2012). The key distinction in these studies is that plasma measures do not provide insight into the activity of appetitive peptides like leptin and insulin in the CNS. Indeed, it is difficult to predict whether plasma measures of these peripherally-derived peptide hormones effectively assess hippocampal insulin and leptin activity. As described above, the mechanistic basis for leptin and insulin resistance includes decreases in BBB transport, as well as decreases in receptor signaling (Levin et al., 2004). One way to circumvent the deficits in associated with leptin and insulin resistance would be through intranasal administration. The exciting studies related to the therapeutic potential of intranasal insulin in the treatment of AD have been described above. Additionally, studies demonstrating the metabolic effects of intranasal leptin administration may also have therapeutic potential in the treatment cognitive decline observed in obesity, type 2 diabetes and AD (Schulz et al., 2004; Waldrop et al., 2010).
As described above, orexin neurons regulate arousal and cognitive function according to energy balance (Yamanaka et al., 2003)—a phenomenon mediated to a significant extent via modulation of orexin activity by circulating factors such as glucose and leptin. Thus, the impact of leptin resistance on behavioral and cognitive correlates of orexin activity is important to our understanding of the relationship between age-related metabolic alterations and cognitive decline. Another question, of particular relevance to the association between type 2 diabetes and AD, is ‘what is the impact of years of hyperglycemia or phasically dysregulated glycemic control on a neuronal population that is inhibited by elevated glucose concentrations?’ In addition, orexin knockout mice develop insulin resistance in an age-dependent manner (Tsuneki et al., 2008), indicating that central disruptions in orexin function can have peripheral metabolic consequences. Collectively, the role of orexins in regulating feeding, arousal and cognition, and the association between aging and orexin hypofunction suggests that this neuropeptide system serves a key integrative function at the interface between age-related declines in cognitive and homeostatic functions. Orexin dysfunction is likely to contribute to age-related cognitive complications of metabolic disorders such as diabetes and leptin resistance, and direct modulation of the orexin system (e.g. by orexin agonists) may have therapeutic efficacy in ameliorating or reversing age-related cognitive decline in these disorders.
Research Highlights.
Beyond metabolic activities, appetitive peptides facilitate hippocampal plasticity
Insulin and leptin promote plasticity directly and through the orexin system
Orexin projections through the basal forebrain modulate hippocampal neurochemistry
Deficits in these peptide systems contribute to age-related cognitive decline
Appetitive peptides represent novel targets in the treatment of dementia and AD
Acknowledgments
Research in the authors’ labs is supported by: a Career Development Award from the Juvenile Diabetes Research Foundation (JDRF) and the National Institutes of Health (RO1 AG039736; CG Jolivalt); the National Institutes of Health (R01 AG030646) and American Federation for Aging Research (JR Fadel); the National Institutes of Health (RO1 DK017844 and R21 MH086067), the Department of Veterans Affairs (IO1 BX001804-01) and the University of South Carolina Research Foundation (LP Reagan). The authors would like to thank Victoria Macht for assistance in the preparation of the final manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbott MA, Wells DG, Fallon JR. The insulin receptor tyrosine kinase substrate p58/53 and the insulin receptor are components of CNS synapses. The Journal of Neuroscience. 1999;19:7300–7308. doi: 10.1523/JNEUROSCI.19-17-07300.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akbari E, Motamedi F, Naghdi N, Noorbakhshnia M. The effect of antagonization of orexin 1 receptors in CA1 and dentate gyrus regions on memory processing in passive avoidance task. Behavioural Brain Research. 2008;187:172–177. doi: 10.1016/j.bbr.2007.09.019. [DOI] [PubMed] [Google Scholar]
- Akbari E, Naghdi N, Motamedi F. Functional inactivation of orexin 1 receptors in CA1 region impairs acquisition, consolidation and retrieval in Morris water maze task. Behavioural Brain Research. 2006;173:47–52. doi: 10.1016/j.bbr.2006.05.028. [DOI] [PubMed] [Google Scholar]
- Ammoun S, Holmqvist T, Shariatmadari R, Oonk HB, Detheux M, Parmentier M, Akerman KE, Kukkonen JP. Distinct recognition of OX1 and OX2 receptors by orexin peptides. Journal of Pharmacology and Experimental Therapeutics. 2003;305:507–514. doi: 10.1124/jpet.102.048025. [DOI] [PubMed] [Google Scholar]
- Avila J, Wandosell F, Hernandez F. Role of glycogen synthase kinase-3 in Alzheimer’s disease pathogenesis and glycogen synthase kinase-3 inhibitors. Expert Rev Neurother. 2010;10:703–710. doi: 10.1586/ern.10.40. [DOI] [PubMed] [Google Scholar]
- Baird JP, Choe A, Loveland JL, Beck J, Mahoney CE, Lord JS, Grigg LA. Orexin-A hyperphagia: hindbrain participation in consummatory feeding responses. Endocrinology. 2009;150:1202–1216. doi: 10.1210/en.2008-0293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldo BA, Daniel RA, Berridge CW, Kelley AE. Overlapping distributions of orexin/hypocretin- and dopamine-beta-hydroxylase immunoreactive fibers in rat brain regions mediating arousal, motivation, and stress. J Comp Neurol. 2003;464:220–237. doi: 10.1002/cne.10783. [DOI] [PubMed] [Google Scholar]
- Banks WA. The many lives of leptin. Peptides. 2004;25:331–338. doi: 10.1016/j.peptides.2004.02.014. [DOI] [PubMed] [Google Scholar]
- Barrett-Connor E, Edelstein S, Corey-Bloom J, Wiederholt W. Weight loss precedes dementia in community-dwelling older adults. J Nutr Health Aging. 1998;2:113–114. [PubMed] [Google Scholar]
- Barrett-Connor E, Edelstein SL, Corey-Bloom J, Wiederholt WC. Weight loss precedes dementia in community-dwelling older adults. Journal of the American Geriatric Society. 1996;44:1147–1152. doi: 10.1111/j.1532-5415.1996.tb01362.x. [DOI] [PubMed] [Google Scholar]
- Baskin DG, Breininger JF, Schwartz MW. Leptin receptor mRNA identifies a subpopulation of neuropeptide Y neurons activated by fasting in rat hypothalamus. Diabetes. 1999;48:828–833. doi: 10.2337/diabetes.48.4.828. [DOI] [PubMed] [Google Scholar]
- Baumgartner RN, Waters DL, Morley JE, Patrick P, Montoya GD, Garry PJ. Age-related changes in sex hormones affect the sex difference in serum leptin independently of changes in body fat. Metabolism. 1999;48:378–384. doi: 10.1016/s0026-0495(99)90089-6. [DOI] [PubMed] [Google Scholar]
- Bayer-Carter JL, Green PS, Montine TJ, VanFossen B, Baker LD, Watson GS, Bonner LM, Callaghan M, Leverenz JB, Walter BK, Tsai E, Plymate SR, Postupna N, Wilkinson CW, Zhang J, Lampe J, Kahn SE, Craft S. Diet intervention and cerebrospinal fluid biomarkers in amnestic mild cognitive impairment. Archives in Neurology. 2011;68:743–752. doi: 10.1001/archneurol.2011.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedict C, Brooks SJ, Kullberg J, Burgos J, Kempton MJ, Nordenskjold R, Nylander R, Kilander L, Craft S, Larsson EM, Johansson L, Ahlstrom H, Lind L, Schioth HB. Impaired insulin sensitivity as indexed by the HOMA score is associated with deficits in verbal fluency and temporal lobe gray matter volume in the elderly. Diabetes Care. 2012;35:488–494. doi: 10.2337/dc11-2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biessels GJ, Deary IJ, Ryan CM. Cognition and diabetes: a lifespan perspective. Lancet Neurol. 2008;7:184–190. doi: 10.1016/S1474-4422(08)70021-8. [DOI] [PubMed] [Google Scholar]
- Bigalke B, Schreitmuller B, Sopova K, Paul A, Stransky E, Gawaz M, Stellos K, Laske C. Adipocytokines and CD34 progenitor cells in Alzheimer’s disease. PLoS One. 2011;6:e20286. doi: 10.1371/journal.pone.0020286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgland SL, Taha SA, Sarti F, Fields HL, Bonci A. Orexin A in the VTA is critical for the induction of synaptic plasticity and behavioral sensitization to cocaine. Neuron. 2006;49:589–601. doi: 10.1016/j.neuron.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Boschen KE, Fadel JR, Burk JA. Systemic and intrabasalis administration of the orexin-1 receptor antagonist, SB-334867, disrupts attentional performance in rats. Psychopharmacology (Berl) 2009;206:205–213. doi: 10.1007/s00213-009-1596-2. [DOI] [PubMed] [Google Scholar]
- Burdakov D, Gerasimenko O, Verkhratsky A. Physiological changes in glucose differentially modulate the excitability of hypothalamic melanin-concentrating hormone and orexin neurons in situ. The Journal of Neuroscience. 2005;25:2429–2433. doi: 10.1523/JNEUROSCI.4925-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdakov D, Jensen LT, Alexopoulos H, Williams RH, Fearon IM, O’Kelly I, Gerasimenko O, Fugger L, Verkhratsky A. Tandem-pore K+ channels mediate inhibition of orexin neurons by glucose. Neuron. 2006;50:711–722. doi: 10.1016/j.neuron.2006.04.032. [DOI] [PubMed] [Google Scholar]
- Burdo JR, Chen Q, Calcutt NA, Schubert D. The pathological interaction between diabetes and presymptomatic Alzheimer’s disease. Neurobiology of Aging. 2009;30:1910–1917. doi: 10.1016/j.neurobiolaging.2008.02.010. [DOI] [PubMed] [Google Scholar]
- Cai XJ, Evans ML, Lister CA, Leslie RA, Arch JR, Wilson S, Williams G. Hypoglycemia activates orexin neurons and selectively increases hypothalamic orexin-B levels: responses inhibited by feeding and possibly mediated by the nucleus of the solitary tract. Diabetes. 2001;50:105–112. doi: 10.2337/diabetes.50.1.105. [DOI] [PubMed] [Google Scholar]
- Cai XJ, Widdowson PS, Harrold J, Wilson S, Buckingham RE, Arch JR, Tadayyon M, Clapham JC, Wilding J, Williams G. Hypothalamic orexin expression: modulation by blood glucose and feeding. Diabetes. 1999;48:2132–2137. doi: 10.2337/diabetes.48.11.2132. [DOI] [PubMed] [Google Scholar]
- Chemelli RM, Willie JT, Sinton CM, Elmquist JK, Scammell T, Lee C, Richardson JA, Williams SC, Xiong Y, Kisanuki Y, Fitch TE, Nakazato M, Hammer RE, Saper CB, Yanagisawa M. Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell. 1999;98:437–451. doi: 10.1016/s0092-8674(00)81973-x. [DOI] [PubMed] [Google Scholar]
- Chen S, Liu AR, An FM, Yao WB, Gao XD. Amelioration of neurodegenerative changes in cellular and rat models of diabetes-related Alzheimer’s disease by exendin-4. Age (Dordr) 2012;34:1211–1224. doi: 10.1007/s11357-011-9303-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou TC, Lee CE, Lu J, Elmquist JK, Hara J, Willie JT, Beuckmann CT, Chemelli RM, Sakurai T, Yanagisawa M, Saper CB, Scammell TE. Orexin (hypocretin) neurons contain dynorphin. The Journal of Neuroscience. 2001;21:RC168. doi: 10.1523/JNEUROSCI.21-19-j0003.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark EL, Baumann CR, Cano G, Scammell TE, Mochizuki T. Feeding-elicited cataplexy in orexin knockout mice. Neuroscience. 2009;161:970–977. doi: 10.1016/j.neuroscience.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craft S, Baker LD, Montine TJ, Minoshima S, Watson GS, Claxton A, Arbuckle M, Callaghan M, Tsai E, Plymate SR, Green PS, Leverenz J, Cross D, Gerton B. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: a pilot clinical trial. Archives in Neurology. 2012a;69:29–38. doi: 10.1001/archneurol.2011.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craft S, Cholerton B, Baker LD. Insulin and Alzheimer’s Disease: Untangling the Web. J Alzheimers Dis. 2012b doi: 10.3233/JAD-2012-129042. [DOI] [PubMed] [Google Scholar]
- Cukierman T, Gerstein HC, Williamson JD. Cognitive decline and dementia in diabetes--systematic overview of prospective observational studies. Diabetologia. 2005;48:2460–2469. doi: 10.1007/s00125-005-0023-4. [DOI] [PubMed] [Google Scholar]
- Currais A, Prior M, Lo D, Jolivalt C, Schubert D, Maher P. Diabetes exacerbates amyloid and neurovascular pathology in aging-accelerated mice. Aging Cell. 2012 doi: 10.1111/acel.12002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dandona P, Mohamed I, Ghanim H, Sia CL, Dhindsa S, Dandona S, Makdissi A, Chaudhuri A. Insulin suppresses the expression of amyloid precursor protein, presenilins, and glycogen synthase kinase-3beta in peripheral blood mononuclear cells. J Clin Endocrinol Metab. 2011;96:1783–1788. doi: 10.1210/jc.2010-2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Felice FG, Vieira MN, Bomfim TR, Decker H, Velasco PT, Lambert MP, Viola KL, Zhao WQ, Ferreira ST, Klein WL. Protection of synapses against Alzheimer’s-linked toxins: insulin signaling prevents the pathogenic binding of Abeta oligomers. Proc Natl Acad Sci U S A. 2009;106:1971–1976. doi: 10.1073/pnas.0809158106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Monte SM. Therapeutic targets of brain insulin resistance in sporadic Alzheimer’s disease. Front Biosci (Elite Ed) 2012;4:1582–1605. doi: 10.2741/482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Monte SM, Wands JR. Review of insulin and insulin-like growth factor expression, signaling, and malfunction in the central nervous system: relevance to Alzheimer’s disease. J Alzheimers Dis. 2005;7:45–61. doi: 10.3233/jad-2005-7106. [DOI] [PubMed] [Google Scholar]
- de Lecea L, Kilduff TS, Peyron C, Gao X, Foye PE, Danielson PE, Fukuhara C, Battenberg EL, Gautvik VT, Bartlett FS, Frankel WN, van den Pol AN, Bloom FE, Gautvik KM, Sutcliffe JG. The hypocretins: hypothalamus-specific peptides with neuroexcitatory activity. Proc Natl Acad Sci U S A. 1998;95:322–327. doi: 10.1073/pnas.95.1.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lecea L, Sutcliffe JG, Fabre V. Hypocretins/orexins as integrators of physiological information: lessons from mutant animals. Neuropeptides. 2002;36:85–95. doi: 10.1054/npep.2002.0892. [DOI] [PubMed] [Google Scholar]
- Deadwyler SA, Porrino L, Siegel JM, Hampson RE. Systemic and nasal delivery of orexin-A (Hypocretin-1) reduces the effects of sleep deprivation on cognitive performance in nonhuman primates. The Journal of Neuroscience. 2007;27:14239–14247. doi: 10.1523/JNEUROSCI.3878-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dineley KT, Kayed R, Neugebauer V, Fu Y, Zhang W, Reese LC, Taglialatela G. Amyloid-beta oligomers impair fear conditioned memory in a calcineurin-dependent fashion in mice. Journal of Neuroscience Research. 2010;88:2923–2932. doi: 10.1002/jnr.22445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty GH, Beccano-Kelly D, Yan SD, Gunn-Moore FJ, Harvey J. Leptin prevents hippocampal synaptic disruption and neuronal cell death induced by amyloid beta. Neurobiology of Aging. 2013;34:226–237. doi: 10.1016/j.neurobiolaging.2012.08.003. [DOI] [PubMed] [Google Scholar]
- Dube MG, Kalra SP, Kalra PS. Food intake elicited by central administration of orexins/hypocretins: identification of hypothalamic sites of action. Brain Research. 1999;842:473–477. doi: 10.1016/s0006-8993(99)01824-7. [DOI] [PubMed] [Google Scholar]
- Duelli R, Schrock H, Kuschinsky W, Hoyer S. Intracerebroventricular injections of streptozotocin induces discrete local changes in cerebral glucose utilization in rats. International Journal of Developmental Neuroscience. 1994;12:737–743. doi: 10.1016/0736-5748(94)90053-1. [DOI] [PubMed] [Google Scholar]
- Durakoglugil M, Irving AJ, Harvey J. Leptin induces a novel form of NMDA receptor-dependent long-term depression. Journal of Neurochemistry. 2005;95:396–405. doi: 10.1111/j.1471-4159.2005.03375.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmquist JK, Bjorbaek C, Ahima RS, Flier JS, Saper CB. Distributions of leptin receptor mRNA isoforms in the rat brain. J Comp Neurol. 1998;395:535–547. [PubMed] [Google Scholar]
- Espana RA, Reis KM, Valentino RJ, Berridge CW. Organization of hypocretin/orexin efferents to locus coeruleus and basal forebrain arousal-related structures. J Comp Neurol. 2005;481:160–178. doi: 10.1002/cne.20369. [DOI] [PubMed] [Google Scholar]
- Fadel J, Deutch AY. Anatomical substrates of orexin-dopamine interactions: lateral hypothalamic projections to the ventral tegmental area. Neuroscience. 2002;111:379–387. doi: 10.1016/s0306-4522(02)00017-9. [DOI] [PubMed] [Google Scholar]
- Fadel J, Pasumarthi R, Reznikov L. Stimulation of cortical acetylcholine release by orexin A. Neuroscience. 2005;130:541–547. doi: 10.1016/j.neuroscience.2004.09.050. [DOI] [PubMed] [Google Scholar]
- Farr SA, Banks WA, Morley JE. Effects of leptin on memory processing. Peptides. 2006;27:1420–1425. doi: 10.1016/j.peptides.2005.10.006. [DOI] [PubMed] [Google Scholar]
- Fernandez-Galaz C, Fernandez-Agullo T, Campoy F, Arribas C, Gallardo N, Andres A, Ros M, Carrascosa JM. Decreased leptin uptake in hypothalamic nuclei with ageing in Wistar rats. Journal of Endocrinology. 2001;171:23–32. doi: 10.1677/joe.0.1710023. [DOI] [PubMed] [Google Scholar]
- Fewlass DC, Noboa K, Pi-Sunyer FX, Johnston JM, Yan SD, Tezapsidis N. Obesity-related leptin regulates Alzheimer’s A beta. FASEB Journal. 2004;18:1870–1878. doi: 10.1096/fj.04-2572com. [DOI] [PubMed] [Google Scholar]
- Figlewicz DP, Benoit SC. Insulin, leptin, and food reward: update 2008. Am J Physiol Regul Integr Comp Physiol. 2009;296:R9–R19. doi: 10.1152/ajpregu.90725.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figlewicz DP, MacDonald NA, Sipols AJ. Modulation of food reward by adiposity signals. Physiol Behav. 2007;91:473–478. doi: 10.1016/j.physbeh.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forlenza OV, Diniz BS, Radanovic M, Santos FS, Talib LL, Gattaz WF. Disease-modifying properties of long-term lithium treatment for amnestic mild cognitive impairment: randomised controlled trial. British Journal of Psychiatry. 2011;198:351–356. doi: 10.1192/bjp.bp.110.080044. [DOI] [PubMed] [Google Scholar]
- Francis GJ, Martinez JA, Liu WQ, Xu K, Ayer A, Fine J, Tuor UI, Glazner G, Hanson LR, Frey WH, Toth C. Intranasal insulin prevents cognitive decline, cerebral atrophy and white matter changes in murine type I diabetic encephalopathy. Brain. 2008;131:3311–3334. doi: 10.1093/brain/awn288. [DOI] [PubMed] [Google Scholar]
- Frederick-Duus D, Guyton MF, Fadel J. Food-elicited increases in cortical acetylcholine release require orexin transmission. Neuroscience. 2007;149:499–507. doi: 10.1016/j.neuroscience.2007.07.061. [DOI] [PubMed] [Google Scholar]
- Fronczek R, Overeem S, Lee SY, Hegeman IM, van Pelt J, van Duinen SG, Lammers GJ, Swaab DF. Hypocretin (orexin) loss and sleep disturbances in Parkinson’s Disease. Brain. 2008;131:e88. doi: 10.1093/brain/awm222. [DOI] [PubMed] [Google Scholar]
- Fronczek R, van Geest S, Frolich M, Overeem S, Roelandse FW, Lammers GJ, Swaab DF. Hypocretin (orexin) loss in Alzheimer’s disease. Neurobiology of Aging. 2012;33:1642–1650. doi: 10.1016/j.neurobiolaging.2011.03.014. [DOI] [PubMed] [Google Scholar]
- Fujiki N, Yoshida Y, Zhang S, Sakurai T, Yanagisawa M, Nishino S. Sex difference in body weight gain and leptin signaling in hypocretin/orexin deficient mouse models. Peptides. 2006;27:2326–2331. doi: 10.1016/j.peptides.2006.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao C, Holscher C, Liu Y, Li L. GSK3: a key target for the development of novel treatments for type 2 diabetes mellitus and Alzheimer disease. Rev Neurosci. 2012;23:1–11. doi: 10.1515/rns.2011.061. [DOI] [PubMed] [Google Scholar]
- Garza JC, Guo M, Zhang W, Lu XY. Leptin promotes adult hippocampal neurogenesis in vivo and in vitro. Journal of Biological Chemistry. 2008 doi: 10.1074/jbc.M800053200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gault VA, Porter WD, Flatt PR, Holscher C. Actions of exendin-4 therapy on cognitive function and hippocampal synaptic plasticity in mice fed a high-fat diet. Int J Obes (Lond) 2010;34:1341–1344. doi: 10.1038/ijo.2010.59. [DOI] [PubMed] [Google Scholar]
- Gengler S, McClean PL, McCurtin R, Gault VA, Holscher C. Val(8)GLP-1 rescues synaptic plasticity and reduces dense core plaques in APP/PS1 mice. Neurobiology of Aging. 2012;33:265–276. doi: 10.1016/j.neurobiolaging.2010.02.014. [DOI] [PubMed] [Google Scholar]
- Gerashchenko D, Salin-Pascual R, Shiromani PJ. Effects of hypocretin-saporin injections into the medial septum on sleep and hippocampal theta. Brain Research. 2001;913:106–115. doi: 10.1016/s0006-8993(01)02792-5. [DOI] [PubMed] [Google Scholar]
- Gerges NZ, Aleisa AM, Alkadhi KA. Impaired long-term potentiation in obese zucker rats: possible involvement of presynaptic mechanism. Neuroscience. 2003;120:535–539. doi: 10.1016/s0306-4522(03)00297-5. [DOI] [PubMed] [Google Scholar]
- Graff J, Koshibu K, Jouvenceau A, Dutar P, Mansuy IM. Protein phosphatase 1-dependent transcriptional programs for long-term memory and plasticity. Learn Mem. 2010;17:355–363. doi: 10.1101/lm.1766510. [DOI] [PubMed] [Google Scholar]
- Greco SJ, Bryan KJ, Sarkar S, Zhu X, Smith MA, Ashford JW, Johnston JM, Tezapsidis N, Casadesus G. Leptin reduces pathology and improves memory in a transgenic mouse model of Alzheimer’s disease. J Alzheimers Dis. 2010;19:1155–1167. doi: 10.3233/JAD-2010-1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greco SJ, Sarkar S, Johnston JM, Tezapsidis N. Leptin regulates tau phosphorylation and amyloid through AMPK in neuronal cells. Biochemical and Biophysical Research Communications. 2009;380:98–104. doi: 10.1016/j.bbrc.2009.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillo CA, Piroli GG, Hendry RM, Reagan LP. Insulin-stimulated translocation of GLUT4 to the plasma membrane in rat hippocampus is PI3-kinase dependent. Brain Research. 2009;1296:35–45. doi: 10.1016/j.brainres.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunstad J, Spitznagel MB, Keary TA, Glickman E, Alexander T, Karrer J, Stanek K, Reese L, Juvancic-Heltzel J. Serum leptin levels are associated with cognitive function in older adults. Brain Research. 2008;1230:233–236. doi: 10.1016/j.brainres.2008.07.045. [DOI] [PubMed] [Google Scholar]
- Hakansson M, de Lecea L, Sutcliffe JG, Yanagisawa M, Meister B. Leptin receptor- and STAT3-immunoreactivities in hypocretin/orexin neurones of the lateral hypothalamus. Journal of Neuroendocrinology. 1999;11:653–663. doi: 10.1046/j.1365-2826.1999.00378.x. [DOI] [PubMed] [Google Scholar]
- Hara J, Beuckmann CT, Nambu T, Willie JT, Chemelli RM, Sinton CM, Sugiyama F, Yagami K, Goto K, Yanagisawa M, Sakurai T. Genetic ablation of orexin neurons in mice results in narcolepsy, hypophagia, and obesity. Neuron. 2001;30:345–354. doi: 10.1016/s0896-6273(01)00293-8. [DOI] [PubMed] [Google Scholar]
- Harris GC, Aston-Jones G. Arousal and reward: a dichotomy in orexin function. Trends in the Neurological Sciences. 2006;29:571–577. doi: 10.1016/j.tins.2006.08.002. [DOI] [PubMed] [Google Scholar]
- Harvey J. Leptin: the missing link in Alzheimer disease? Clin Chem. 2010;56:696–697. doi: 10.1373/clinchem.2010.144006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havrankova J, Brownstein M, Roth J. Insulin and insulin receptors in rodent brain. Diabetologia. 1981;20(Suppl):268–273. [PubMed] [Google Scholar]
- Havrankova J, Roth J, Brownstein M. Insulin receptors are widely distributed in the central nervous system of the rat. Nature. 1978;272:827–829. doi: 10.1038/272827a0. [DOI] [PubMed] [Google Scholar]
- Havrankova J, Roth J, Brownstein MJ. Concentrations of insulin and insulin receptors in the brain are independent of peripheral insulin levels. Studies of obese and streptozotocin-treated rodents. J Clin Invest. 1979;64:636–642. doi: 10.1172/JCI109504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes AC, Jackson B, Chapman H, Tadayyon M, Johns A, Porter RA, Arch JR. A selective orexin-1 receptor antagonist reduces food consumption in male and female rats. Regul Pept. 2000;96:45–51. doi: 10.1016/s0167-0115(00)00199-3. [DOI] [PubMed] [Google Scholar]
- Haynes AC, Jackson B, Overend P, Buckingham RE, Wilson S, Tadayyon M, Arch JR. Effects of single and chronic intracerebroventricular administration of the orexins on feeding in the rat. Peptides. 1999;20:1099–1105. doi: 10.1016/s0196-9781(99)00105-9. [DOI] [PubMed] [Google Scholar]
- Henny P, Brischoux F, Mainville L, Stroh T, Jones BE. Immunohistochemical evidence for synaptic release of glutamate from orexin terminals in the locus coeruleus. Neuroscience. 2010;169:1150–1157. doi: 10.1016/j.neuroscience.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill JM, Lesniak MA, Pert CB, Roth J. Autoradiographic localization of insulin receptors in rat brain: prominence in olfactory and limbic areas. Neuroscience. 1986;17:1127–1138. doi: 10.1016/0306-4522(86)90082-5. [DOI] [PubMed] [Google Scholar]
- Ho L, Qin W, Pompl PN, Xiang Z, Wang J, Zhao Z, Peng Y, Cambareri G, Rocher A, Mobbs CV, Hof PR, Pasinetti GM. Diet-induced insulin resistance promotes amyloidosis in a transgenic mouse model of Alzheimer’s disease. FASEB Journal. 2004;18:902–904. doi: 10.1096/fj.03-0978fje. [DOI] [PubMed] [Google Scholar]
- Holden KF, Lindquist K, Tylavsky FA, Rosano C, Harris TB, Yaffe K. Serum leptin level and cognition in the elderly: Findings from the Health ABC Study. Neurobiology of Aging. 2009;30:1483–1489. doi: 10.1016/j.neurobiolaging.2007.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoi T, Okuma Y, Ono A, Nomura Y. Alteration of leptin-induced STAT3 activation in the brain of senescence-accelerated mouse (SAM) P8. Biol Pharm Bull. 2005;28:1514–1516. doi: 10.1248/bpb.28.1514. [DOI] [PubMed] [Google Scholar]
- Huang CC, Lee CC, Hsu KS. An investigation into signal transduction mechanisms involved in insulin-induced long-term depression in the CA1 region of the hippocampus. Journal of Neurochemistry. 2004;89:217–231. doi: 10.1111/j.1471-4159.2003.02307.x. [DOI] [PubMed] [Google Scholar]
- Huang CC, You JL, Lee CC, Hsu KS. Insulin induces a novel form of postsynaptic mossy fiber long-term depression in the hippocampus. Mol Cell Neurosci. 2003;24:831–841. doi: 10.1016/s1044-7431(03)00238-0. [DOI] [PubMed] [Google Scholar]
- Huang H, Ghosh P, van den Pol AN. Prefrontal cortex-projecting glutamatergic thalamic paraventricular nucleus-excited by hypocretin: a feedforward circuit that may enhance cognitive arousal. Journal of Neurophysiology. 2006;95:1656–1668. doi: 10.1152/jn.00927.2005. [DOI] [PubMed] [Google Scholar]
- Isacson R, Nielsen E, Dannaeus K, Bertilsson G, Patrone C, Zachrisson O, Wikstrom L. The glucagon-like peptide 1 receptor agonist exendin-4 improves reference memory performance and decreases immobility in the forced swim test. European Journal of Pharmacology. 2011;650:249–255. doi: 10.1016/j.ejphar.2010.10.008. [DOI] [PubMed] [Google Scholar]
- Isidori AM, Strollo F, More M, Caprio M, Aversa A, Moretti C, Frajese G, Riondino G, Fabbri A. Leptin and aging: correlation with endocrine changes in male and female healthy adult populations of different body weights. J Clin Endocrinol Metab. 2000;85:1954–1962. doi: 10.1210/jcem.85.5.6572. [DOI] [PubMed] [Google Scholar]
- Jo J, Whitcomb DJ, Olsen KM, Kerrigan TL, Lo SC, Bru-Mercier G, Dickinson B, Scullion S, Sheng M, Collingridge G, Cho K. Abeta(1–42) inhibition of LTP is mediated by a signaling pathway involving caspase-3, Akt1 and GSK-3beta. Nature Neuroscience. 2011;14:545–547. doi: 10.1038/nn.2785. [DOI] [PubMed] [Google Scholar]
- Johnson DK, Wilkins CH, Morris JC. Accelerated weight loss may precede diagnosis in Alzheimer disease. Archives in Neurology. 2006;63:1312–1317. doi: 10.1001/archneur.63.9.1312. [DOI] [PubMed] [Google Scholar]
- Jolivalt CG, Fineman M, Deacon CF, Carr RD, Calcutt NA. GLP-1 signals via ERK in peripheral nerve and prevents nerve dysfunction in diabetic mice. Diabetes Obes Metab. 2011;13:990–1000. doi: 10.1111/j.1463-1326.2011.01431.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolivalt CG, Hurford R, Lee CA, Dumaop W, Rockenstein E, Masliah E. Type 1 diabetes exaggerates features of Alzheimer’s disease in APP transgenic mice. Experimental Neurology. 2010;223:422–431. doi: 10.1016/j.expneurol.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolivalt CG, Lee CA, Beiswenger KK, Smith JL, Orlov M, Torrance MA, Masliah E. Defective insulin signaling pathway and increased glycogen synthase kinase-3 activity in the brain of diabetic mice: parallels with Alzheimer’s disease and correction by insulin. Journal of Neuroscience Research. 2008;86:3265–3274. doi: 10.1002/jnr.21787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas EA, Knox RJ, Smith TC, Wayne NL, Connor JA, Kaczmarek LK. Regulation by insulin of a unique neuronal Ca2+ pool and of neuropeptide secretion. Nature. 1997;385:343–346. doi: 10.1038/385343a0. [DOI] [PubMed] [Google Scholar]
- Karteris E, Machado RJ, Chen J, Zervou S, Hillhouse EW, Randeva HS. Food deprivation differentially modulates orexin receptor expression and signaling in rat hypothalamus and adrenal cortex. Am J Physiol Endocrinol Metab. 2005;288:E1089–E1100. doi: 10.1152/ajpendo.00351.2004. [DOI] [PubMed] [Google Scholar]
- Ke YD, Delerue F, Gladbach A, Gotz J, Ittner LM. Experimental diabetes mellitus exacerbates tau pathology in a transgenic mouse model of Alzheimer’s disease. PLoS One. 2009;4:e7917. doi: 10.1371/journal.pone.0007917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler BA, Stanley EM, Frederick-Duus D, Fadel J. Age-related loss of orexin/hypocretin neurons. Neuroscience. 2011;178:82–88. doi: 10.1016/j.neuroscience.2011.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirouac GJ, Parsons MP, Li S. Orexin (hypocretin) innervation of the paraventricular nucleus of the thalamus. Brain Research. 2005;1059:179–188. doi: 10.1016/j.brainres.2005.08.035. [DOI] [PubMed] [Google Scholar]
- Knittweis J. Weight loss precedes Alzheimer’s disease symptoms: a case study. Journal of the American Geriatric Society. 1998;46:540–541. doi: 10.1111/j.1532-5415.1998.tb02487.x. [DOI] [PubMed] [Google Scholar]
- Kotz CM, Mullett MA, Wang C. Diminished feeding responsiveness to orexin A (hypocretin 1) in aged rats is accompanied by decreased neuronal activation. Am J Physiol Regul Integr Comp Physiol. 2005;289:R359–R366. doi: 10.1152/ajpregu.00717.2004. [DOI] [PubMed] [Google Scholar]
- Labad J, Price JF, Strachan MW, Deary IJ, Seckl JR, Sattar N, Reynolds RM. Serum leptin and cognitive function in people with Type 2 diabetes. Neurobiology of Aging. 2012;33:2938–2941. doi: 10.1016/j.neurobiolaging.2012.02.026. [DOI] [PubMed] [Google Scholar]
- Lambe EK, Olausson P, Horst NK, Taylor JR, Aghajanian GK. Hypocretin and nicotine excite the same thalamocortical synapses in prefrontal cortex: correlation with improved attention in rat. The Journal of Neuroscience. 2005;25:5225–5229. doi: 10.1523/JNEUROSCI.0719-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammers GJ, Pijl H, Iestra J, Langius JA, Buunk G, Meinders AE. Spontaneous food choice in narcolepsy. Sleep. 1996;19:75–76. doi: 10.1093/sleep/19.1.75. [DOI] [PubMed] [Google Scholar]
- Lannert H, Hoyer S. Intracerebroventricular administration of streptozotocin causes long-term diminutions in learning and memory abilities and in cerebral energy metabolism in adults rats. Behavioral Neuroscience. 1998;112:1199–1208. doi: 10.1037//0735-7044.112.5.1199. [DOI] [PubMed] [Google Scholar]
- Lee CC, Huang CC, Hsu KS. Insulin promotes dendritic spine and synapse formation by the PI3K/Akt/mTOR and Rac1 signaling pathways. Neuropharmacology. 2011;61:867–879. doi: 10.1016/j.neuropharm.2011.06.003. [DOI] [PubMed] [Google Scholar]
- Lee CC, Huang CC, Wu MY, Hsu KS. Insulin stimulates postsynaptic density-95 protein translation via the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway. Journal of Biological Chemistry. 2005;280:18543–18550. doi: 10.1074/jbc.M414112200. [DOI] [PubMed] [Google Scholar]
- Leibson CL, Rocca WA, Hanson VA, Cha R, Kokmen E, O’Brien PC, Palumbo PJ. Risk of dementia among persons with diabetes mellitus: a population-based cohort study. Am J Epidemiol. 1997;145:301–308. doi: 10.1093/oxfordjournals.aje.a009106. [DOI] [PubMed] [Google Scholar]
- Leinninger GM, Jo YH, Leshan RL, Louis GW, Yang H, Barrera JG, Wilson H, Opland DM, Faouzi MA, Gong Y, Jones JC, Rhodes CJ, Chua S, Jr, Diano S, Horvath TL, Seeley RJ, Becker JB, Munzberg H, Myers MG., Jr Leptin acts via leptin receptor-expressing lateral hypothalamic neurons to modulate the mesolimbic dopamine system and suppress feeding. Cell Metab. 2009;10:89–98. doi: 10.1016/j.cmet.2009.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinninger GM, Opland DM, Jo YH, Faouzi M, Christensen L, Cappellucci LA, Rhodes CJ, Gnegy ME, Becker JB, Pothos EN, Seasholtz AF, Thompson RC, Myers MG., Jr Leptin action via neurotensin neurons controls orexin, the mesolimbic dopamine system and energy balance. Cell Metab. 2011;14:313–323. doi: 10.1016/j.cmet.2011.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lester-Coll N, Rivera EJ, Soscia SJ, Doiron K, Wands JR, de la Monte SM. Intracerebral streptozotocin model of type 3 diabetes: relevance to sporadic Alzheimer’s disease. J Alzheimers Dis. 2006;9:13–33. doi: 10.3233/jad-2006-9102. [DOI] [PubMed] [Google Scholar]
- Levin BE, Dunn-Meynell AA, Banks WA. Obesity-prone rats have normal blood-brain barrier transport but defective central leptin signaling before obesity onset. Am J Physiol Regul Integr Comp Physiol. 2004;286:R143–R150. doi: 10.1152/ajpregu.00393.2003. [DOI] [PubMed] [Google Scholar]
- Li J, Deng J, Sheng W, Zuo Z. Metformin attenuates Alzheimer’s disease-like neuropathology in obese, leptin-resistant mice. Pharmacology Biochemistry and Behavior. 2012a;101:564–574. doi: 10.1016/j.pbb.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Zhang ZF, Holscher C, Gao C, Jiang YH, Liu YZ. (Val(8)) glucagon-like peptide-1 prevents tau hyperphosphorylation, impairment of spatial learning and ultra-structural cellular damage induced by streptozotocin in rat brains. European Journal of Pharmacology. 2012b;674:280–286. doi: 10.1016/j.ejphar.2011.11.005. [DOI] [PubMed] [Google Scholar]
- Li XL, Aou S, Oomura Y, Hori N, Fukunaga K, Hori T. Impairment of long-term potentiation and spatial memory in leptin receptor-deficient rodents. Neuroscience. 2002;113:607–615. doi: 10.1016/s0306-4522(02)00162-8. [DOI] [PubMed] [Google Scholar]
- Lieb W, Beiser AS, Vasan RS, Tan ZS, Au R, Harris TB, Roubenoff R, Auerbach S, Decarli C, Wolf PA, Seshadri S. Association of plasma leptin levels with incident Alzheimer disease and MRI measures of brain aging. Journal of the American Medical Society. 2009;302:2565–2572. doi: 10.1001/jama.2009.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Faraco J, Li R, Kadotani H, Rogers W, Lin X, Qiu X, de Jong PJ, Nishino S, Mignot E. The sleep disorder canine narcolepsy is caused by a mutation in the hypocretin (orexin) receptor 2 gene. Cell. 1999;98:365–376. doi: 10.1016/s0092-8674(00)81965-0. [DOI] [PubMed] [Google Scholar]
- Liu L, Brown JC, III, Webster WW, Morrisett RA, Monaghan DT. Insulin potentiates N-methyl-D-aspartate receptor activity in Xenopus oocytes and rat hippocampus. Neuroscience Letters. 1995;192:5–8. doi: 10.1016/0304-3940(95)11593-l. [DOI] [PubMed] [Google Scholar]
- Liu Y, Liu F, Grundke-Iqbal I, Iqbal K, Gong CX. Deficient brain insulin signalling pathway in Alzheimer’s disease and diabetes. J Pathol. 2011;225:54–62. doi: 10.1002/path.2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- London ED, Berman SM, Chakrapani S, Delibasi T, Monterosso J, Erol HK, Paz-Filho G, Wong ML, Licinio J. Short-term plasticity of gray matter associated with leptin deficiency and replacement. J Clin Endocrinol Metab. 2011;96:E1212–E1220. doi: 10.1210/jc.2011-0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis GW, Leinninger GM, Rhodes CJ, Myers MG., Jr Direct innervation and modulation of orexin neurons by lateral hypothalamic LepRb neurons. The Journal of Neuroscience. 2010;30:11278–11287. doi: 10.1523/JNEUROSCI.1340-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu XY, Bagnol D, Burke S, Akil H, Watson SJ. Differential distribution and regulation of OX1 and OX2 orexin/hypocretin receptor messenger RNA in the brain upon fasting. Horm Behav. 2000;37:335–344. doi: 10.1006/hbeh.2000.1584. [DOI] [PubMed] [Google Scholar]
- Manschot SM, Brands AM, van der GJ, Kessels RP, Algra A, Kappelle LJ, Biessels GJ. Brain magnetic resonance imaging correlates of impaired cognition in patients with type 2 diabetes. Diabetes. 2006;55:1106–1113. doi: 10.2337/diabetes.55.04.06.db05-1323. [DOI] [PubMed] [Google Scholar]
- Marwarha G, Prasanthi JR, Schommer J, Dasari B, Ghribi O. Molecular interplay between leptin, insulin-like growth factor-1, and beta-amyloid in organotypic slices from rabbit hippocampus. Mol Neurodegener. 2011;6:41. doi: 10.1186/1750-1326-6-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matochik JA, London ED, Yildiz BO, Ozata M, Caglayan S, Depaoli AM, Wong ML, Licinio J. Effect of leptin replacement on brain structure in genetically leptin-deficient adults. J Clin Endocrinol Metab. 2005;90:2851–2854. doi: 10.1210/jc.2004-1979. [DOI] [PubMed] [Google Scholar]
- McCarthy AM, Lindgren S, Mengeling MA, Tsalikian E, Engvall JC. Effects of diabetes on learning in children. Pediatrics. 2002;109:E9. doi: 10.1542/peds.109.1.e9. [DOI] [PubMed] [Google Scholar]
- McClean PL, Gault VA, Harriott P, Holscher C. Glucagon-like peptide-1 analogues enhance synaptic plasticity in the brain: a link between diabetes and Alzheimer’s disease. European Journal of Pharmacology. 2010;630:158–162. doi: 10.1016/j.ejphar.2009.12.023. [DOI] [PubMed] [Google Scholar]
- McClean PL, Parthsarathy V, Faivre E, Holscher C. The diabetes drug liraglutide prevents degenerative processes in a mouse model of Alzheimer’s disease. The Journal of Neuroscience. 2011;31:6587–6594. doi: 10.1523/JNEUROSCI.0529-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mieda M, Williams SC, Sinton CM, Richardson JA, Sakurai T, Yanagisawa M. Orexin neurons function in an efferent pathway of a food-entrainable circadian oscillator in eliciting food-anticipatory activity and wakefulness. The Journal of Neuroscience. 2004;24:10493–10501. doi: 10.1523/JNEUROSCI.3171-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriguchi T, Sakurai T, Nambu T, Yanagisawa M, Goto K. Neurons containing orexin in the lateral hypothalamic area of the adult rat brain are activated by insulin-induced acute hypoglycemia. Neuroscience Letters. 1999;264:101–104. doi: 10.1016/s0304-3940(99)00177-9. [DOI] [PubMed] [Google Scholar]
- Morrison CD. Leptin signaling in brain: A link between nutrition and cognition? Biochim Biophys Acta. 2009;1792:401–408. doi: 10.1016/j.bbadis.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moult PR, Cross A, Santos SD, Carvalho AL, Lindsay Y, Connolly CN, Irving AJ, Leslie NR, Harvey J. Leptin regulates AMPA receptor trafficking via PTEN inhibition. The Journal of Neuroscience. 2010;30:4088–4101. doi: 10.1523/JNEUROSCI.3614-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moult PR, Harvey J. Regulation of glutamate receptor trafficking by leptin. Biochem Soc Trans. 2009;37:1364–1368. doi: 10.1042/BST0371364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moult PR, Harvey J. NMDA receptor subunit composition determines the polarity of leptin-induced synaptic plasticity. Neuropharmacology. 2011;61:924–936. doi: 10.1016/j.neuropharm.2011.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moult PR, Milojkovic B, Harvey J. Leptin reverses long-term potentiation at hippocampal CA1 synapses. Journal of Neurochemistry. 2009;108:685–696. doi: 10.1111/j.1471-4159.2008.05810.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munzberg H, Myers MG., Jr Molecular and anatomical determinants of central leptin resistance. Nature Neuroscience. 2005;8:566–570. doi: 10.1038/nn1454. [DOI] [PubMed] [Google Scholar]
- Myers MG, Jr, Leibel RL, Seeley RJ, Schwartz MW. Obesity and leptin resistance: distinguishing cause from effect. Trends Endocrinol Metab. 2010;21:643–651. doi: 10.1016/j.tem.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadebaum C, Scratch SE, Northam EA, Cameron FJ. Clinical utility of mental state screening as a predictor of intellectual outcomes 6 months after diagnosis of type 1 diabetes. Pediatr Diabetes. 2012 doi: 10.1111/j.1399-5448.2012.00870.x. [DOI] [PubMed] [Google Scholar]
- Narita K, Kosaka H, Okazawa H, Murata T, Wada Y. Relationship between plasma leptin level and brain structure in elderly: a voxel-based morphometric study. Biological Psychiatry. 2009;65:992–994. doi: 10.1016/j.biopsych.2008.10.006. [DOI] [PubMed] [Google Scholar]
- Nishino S, Ripley B, Overeem S, Lammers GJ, Mignot E. Hypocretin (orexin) deficiency in human narcolepsy. The Lancet. 2000;355:39–40. doi: 10.1016/S0140-6736(99)05582-8. [DOI] [PubMed] [Google Scholar]
- Nishino S, Ripley B, Overeem S, Nevsimalova S, Lammers GJ, Vankova J, Okun M, Rogers W, Brooks S, Mignot E. Low cerebrospinal fluid hypocretin (Orexin) and altered energy homeostasis in human narcolepsy. Annals of Neurology. 2001;50:381–388. doi: 10.1002/ana.1130. [DOI] [PubMed] [Google Scholar]
- Nistico R, Cavallucci V, Piccinin S, Macri S, Pignatelli M, Mehdawy B, Blandini F, Laviola G, Lauro D, Mercuri NB, D’Amelio M. Insulin Receptor beta-Subunit Haploinsufficiency Impairs Hippocampal Late-Phase LTP and Recognition Memory. Neuromolecular Med. 2012 doi: 10.1007/s12017-012-8184-z. [DOI] [PubMed] [Google Scholar]
- O’Malley D, MacDonald N, Mizielinska S, Connolly CN, Irving AJ, Harvey J. Leptin promotes rapid dynamic changes in hippocampal dendritic morphology. Mol Cell Neurosci. 2007;35:559–572. doi: 10.1016/j.mcn.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oomura Y, Hori N, Shiraishi T, Fukunaga K, Takeda H, Tsuji M, Matsumiya T, Ishibashi M, Aou S, Li XL, Kohno D, Uramura K, Sougawa H, Yada T, Wayner MJ, Sasaki K. Leptin facilitates learning and memory performance and enhances hippocampal CA1 long-term potentiation and CaMK II phosphorylation in rats. Peptides. 2006;27:2738–2749. doi: 10.1016/j.peptides.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Ostlund RE, Jr, Yang JW, Klein S, Gingerich R. Relation between plasma leptin concentration and body fat, gender, diet, age, and metabolic covariates. J Clin Endocrinol Metab. 1996;81:3909–3913. doi: 10.1210/jcem.81.11.8923837. [DOI] [PubMed] [Google Scholar]
- Ott A, Stolk RP, Hofman A, van Harskamp F, Grobbee DE, Breteler MM. Association of diabetes mellitus and dementia: the Rotterdam Study. Diabetologia. 1996;39:1392–1397. doi: 10.1007/s001250050588. [DOI] [PubMed] [Google Scholar]
- Paz-Filho GJ, Babikian T, Asarnow R, Delibasi T, Esposito K, Erol HK, Wong ML, Licinio J. Leptin replacement improves cognitive development. PLoS One. 2008;3:e3098. doi: 10.1371/journal.pone.0003098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peila R, Rodriguez BL, Launer LJ. Type 2 diabetes, APOE gene, and the risk for dementia and related pathologies: The Honolulu-Asia Aging Study. Diabetes. 2002;51:1256–1262. doi: 10.2337/diabetes.51.4.1256. [DOI] [PubMed] [Google Scholar]
- Perello M, Sakata I, Birnbaum S, Chuang JC, Osborne-Lawrence S, Rovinsky SA, Woloszyn J, Yanagisawa M, Lutter M, Zigman JM. Ghrelin increases the rewarding value of high-fat diet in an orexin-dependent manner. Biological Psychiatry. 2010;67:880–886. doi: 10.1016/j.biopsych.2009.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Gonzalez R, Antequera D, Vargas T, Spuch C, Bolos M, Carro E. Leptin induces proliferation of neuronal progenitors and neuroprotection in a mouse model of Alzheimer’s disease. J Alzheimers Dis. 2011;24(Suppl 2):17–25. doi: 10.3233/JAD-2011-102070. [DOI] [PubMed] [Google Scholar]
- Perry HM, III, Morley JE, Horowitz M, Kaiser FE, Miller DK, Wittert G. Body composition and age in African-American and Caucasian women: relationship to plasma leptin levels. Metabolism. 1997;46:1399–1405. doi: 10.1016/s0026-0495(97)90138-4. [DOI] [PubMed] [Google Scholar]
- Peyron C, Tighe DK, van den Pol AN, de Lecea L, Heller HC, Sutcliffe JG, Kilduff TS. Neurons containing hypocretin (orexin) project to multiple neuronal systems. The Journal of Neuroscience. 1998;18:9996–10015. doi: 10.1523/JNEUROSCI.18-23-09996.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pintana H, Apaijai N, Pratchayasakul W, Chattipakorn N, Chattipakorn SC. Effects of metformin on learning and memory behaviors and brain mitochondrial functions in high fat diet induced insulin resistant rats. Life Sci. 2012;91:409–414. doi: 10.1016/j.lfs.2012.08.017. [DOI] [PubMed] [Google Scholar]
- Plaschke K, Hoyer S. Action of the diabetogenic drug streptozotocin on glycolytic and glycogenolytic metabolism in adult rat brain cortex and hippocampus. International Journal of Developmental Neuroscience. 1993;11:477–483. doi: 10.1016/0736-5748(93)90021-5. [DOI] [PubMed] [Google Scholar]
- Plaschke K, Kopitz J, Siegelin M, Schliebs R, Salkovic-Petrisic M, Riederer P, Hoyer S. Insulin-resistant brain state after intracerebroventricular streptozotocin injection exacerbates Alzheimer-like changes in Tg2576 AbetaPP-overexpressing mice. J Alzheimers Dis. 2010;19:691–704. doi: 10.3233/JAD-2010-1270. [DOI] [PubMed] [Google Scholar]
- Ponce-Lopez T, Liy-Salmeron G, Hong E, Meneses A. Lithium, phenserine, memantine and pioglitazone reverse memory deficit and restore phospho-GSK3beta decreased in hippocampus in intracerebroventricular streptozotocin induced memory deficit model. Brain Research. 2011;1426:73–85. doi: 10.1016/j.brainres.2011.09.056. [DOI] [PubMed] [Google Scholar]
- Porkka-Heiskanen T, Alanko L, Kalinchuk A, Heiskanen S, Stenberg D. The effect of age on prepro-orexin gene expression and contents of orexin A and B in the rat brain. Neurobiology of Aging. 2004;25:231–238. doi: 10.1016/s0197-4580(03)00043-5. [DOI] [PubMed] [Google Scholar]
- Porter D, Faivre E, Flatt PR, Holscher C, Gault VA. Actions of incretin metabolites on locomotor activity, cognitive function and in vivo hippocampal synaptic plasticity in high fat fed mice. Peptides. 2012;35:1–8. doi: 10.1016/j.peptides.2012.03.014. [DOI] [PubMed] [Google Scholar]
- Porter DW, Kerr BD, Flatt PR, Holscher C, Gault VA. Four weeks administration of Liraglutide improves memory and learning as well as glycaemic control in mice with high fat dietary-induced obesity and insulin resistance. Diabetes Obes Metab. 2010;12:891–899. doi: 10.1111/j.1463-1326.2010.01259.x. [DOI] [PubMed] [Google Scholar]
- Power DA, Noel J, Collins R, O’Neill D. Circulating leptin levels and weight loss in Alzheimer’s disease patients. Dement Geriatr Cogn Disord. 2001;12:167–170. doi: 10.1159/000051252. [DOI] [PubMed] [Google Scholar]
- Reaven GM, Thompson LW, Nahum D, Haskins E. Relationship between hyperglycemia and cognitive function in older NIDDM patients. Diabetes Care. 1990;13:16–21. doi: 10.2337/diacare.13.1.16. [DOI] [PubMed] [Google Scholar]
- Reilly CE. I. Mutation in the hypocretin (orexin) receptor 2 gene causes canine narcolepsy. J Neurol. 1999;246:985–986. doi: 10.1007/s004150050501. [DOI] [PubMed] [Google Scholar]
- Revill P, Moral MA, Prous JR. Impaired insulin signaling and the pathogenesis of Alzheimer’s disease. Drugs Today (Barc) 2006;42:785–790. doi: 10.1358/dot.2006.42.12.1032059. [DOI] [PubMed] [Google Scholar]
- Rieger M, Mayer G, Gauggel S. Attention deficits in patients with narcolepsy. Sleep. 2003;26:36–43. [PubMed] [Google Scholar]
- Rodgers RJ, Ishii Y, Halford JC, Blundell JE. Orexins and appetite regulation. Neuropeptides. 2002;36:303–325. doi: 10.1016/s0143-4179(02)00085-9. [DOI] [PubMed] [Google Scholar]
- Ronnemaa E, Zethelius B, Sundelof J, Sundstrom J, Degerman-Gunnarsson M, Lannfelt L, Berne C, Kilander L. Glucose metabolism and the risk of Alzheimer’s disease and dementia: a population-based 12 year follow-up study in 71-year-old men. Diabetologia. 2009;52:1504–1510. doi: 10.1007/s00125-009-1393-9. [DOI] [PubMed] [Google Scholar]
- Rosenbaum M, Nicolson M, Hirsch J, Heymsfield SB, Gallagher D, Chu F, Leibel RL. Effects of gender, body composition, and menopause on plasma concentrations of leptin. J Clin Endocrinol Metab. 1996;81:3424–3427. doi: 10.1210/jcem.81.9.8784109. [DOI] [PubMed] [Google Scholar]
- Rosin DL, Weston MC, Sevigny CP, Stornetta RL, Guyenet PG. Hypothalamic orexin (hypocretin) neurons express vesicular glutamate transporters VGLUT1 or VGLUT2. J Comp Neurol. 2003;465:593–603. doi: 10.1002/cne.10860. [DOI] [PubMed] [Google Scholar]
- Rovet JF, Ehrlich RM, Hoppe M. Intellectual deficits associated with early onset of insulin-dependent diabetes mellitus in children. Diabetes Care. 1987;10:510–515. doi: 10.2337/diacare.10.4.510. [DOI] [PubMed] [Google Scholar]
- Ryan C, Vega A, Drash A. Cognitive deficits in aldolescents who developed diabetes early in life. Pediatrics. 1985;75:921–927. [PubMed] [Google Scholar]
- Ryan CM, Williams TM. Effects of insulin-dependent diabetes on learning and memory efficiency in adults. Journal of Clinical and Experimental Neuropsychology. 1993;15:685–700. doi: 10.1080/01688639308402589. [DOI] [PubMed] [Google Scholar]
- Sakurai T, Amemiya A, Ishii M, Matsuzaki I, Chemelli RM, Tanaka H, Williams SC, Richardson JA, Kozlowski GP, Wilson S, Arch JR, Buckingham RE, Haynes AC, Carr SA, Annan RS, McNulty DE, Liu WS, Terrett JA, Elshourbagy NA, Bergsma DJ, Yanagisawa M. Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell. 1998;92:573–585. doi: 10.1016/s0092-8674(00)80949-6. [DOI] [PubMed] [Google Scholar]
- Santos TD, Mazucanti CH, Xavier GF, Torrao AD. Early and late neurodegeneration and memory disruption after intracerebroventricular streptozotocin. Physiol Behav. 2012;107:401–413. doi: 10.1016/j.physbeh.2012.06.019. [DOI] [PubMed] [Google Scholar]
- Sawai N, Ueta Y, Nakazato M, Ozawa H. Developmental and aging change of orexin-A and -B immunoreactive neurons in the male rat hypothalamus. Neuroscience Letters. 2010;468:51–55. doi: 10.1016/j.neulet.2009.10.061. [DOI] [PubMed] [Google Scholar]
- Scammell TE, Winrow CJ. Orexin receptors: pharmacology and therapeutic opportunities. Annu Rev Pharmacol Toxicol. 2011;51:243–266. doi: 10.1146/annurev-pharmtox-010510-100528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarpace PJ, Matheny M, Moore RL, Tumer N. Impaired leptin responsiveness in aged rats. Diabetes. 2000a;49:431–435. doi: 10.2337/diabetes.49.3.431. [DOI] [PubMed] [Google Scholar]
- Scarpace PJ, Matheny M, Shek EW. Impaired leptin signal transduction with age-related obesity. Neuropharmacology. 2000b;39:1872–1879. doi: 10.1016/s0028-3908(00)00014-9. [DOI] [PubMed] [Google Scholar]
- Scarpace PJ, Matheny M, Tumer N. Hypothalamic leptin resistance is associated with impaired leptin signal transduction in aged obese rats. Neuroscience. 2001;104:1111–1117. doi: 10.1016/s0306-4522(01)00142-7. [DOI] [PubMed] [Google Scholar]
- Schulz C, Paulus K, Lehnert H. Central nervous and metabolic effects of intranasally applied leptin. Endocrinology. 2004;145:2696–2701. doi: 10.1210/en.2003-1431. [DOI] [PubMed] [Google Scholar]
- Schwartz MW, Figlewicz DP, Baskin DG, Woods SC, Porte D., Jr Insulin in the brain: A hormonal regulator of energy balance. Endocrinology Reviews. 1992;13:387–414. doi: 10.1210/edrv-13-3-387. [DOI] [PubMed] [Google Scholar]
- Schwartz MW, Woods SC, Porte D, Jr, Seeley RJ, Baskin DG. Central nervous system control of food intake. Nature. 2000;404:661–671. doi: 10.1038/35007534. [DOI] [PubMed] [Google Scholar]
- Selbach O, Bohla C, Barbara A, Doreulee N, Eriksson KS, Sergeeva OA, Haas HL. Orexins/hypocretins control bistability of hippocampal long-term synaptic plasticity through co-activation of multiple kinases. Acta Physiol (Oxf) 2010;198:277–285. doi: 10.1111/j.1748-1716.2009.02021.x. [DOI] [PubMed] [Google Scholar]
- Selbach O, Doreulee N, Bohla C, Eriksson KS, Sergeeva OA, Poelchen W, Brown RE, Haas HL. Orexins/hypocretins cause sharp wave- and theta-related synaptic plasticity in the hippocampus via glutamatergic, gabaergic, noradrenergic, and cholinergic signaling. Neuroscience. 2004;127:519–528. doi: 10.1016/j.neuroscience.2004.05.012. [DOI] [PubMed] [Google Scholar]
- Sellayah D, Bharaj P, Sikder D. Orexin is required for brown adipose tissue development, differentiation, and function. Cell Metab. 2011;14:478–490. doi: 10.1016/j.cmet.2011.08.010. [DOI] [PubMed] [Google Scholar]
- Shanley LJ, Irving AJ, Harvey J. Leptin enhances NMDA receptor function and modulates hippocampal synaptic plasticity. J Neurosci. 2001a;21:RC186. doi: 10.1523/JNEUROSCI.21-24-j0001.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanley LJ, Irving AJ, Harvey J. Leptin enhances NMDA receptor function and modulates hippocampal synaptic plasticity. Journal of Neuroscience. 2001b;21:art-RC186. doi: 10.1523/JNEUROSCI.21-24-j0001.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shek EW, Scarpace PJ. Resistance to the anorexic and thermogenic effects of centrally administrated leptin in obese aged rats. Regul Pept. 2000;92:65–71. doi: 10.1016/s0167-0115(00)00151-8. [DOI] [PubMed] [Google Scholar]
- Shemesh E, Rudich A, Harman-Boehm I, Cukierman-Yaffe T. Effect of intranasal insulin on cognitive function: a systematic review. J Clin Endocrinol Metab. 2012;97:366–376. doi: 10.1210/jc.2011-1802. [DOI] [PubMed] [Google Scholar]
- Shingo AS, Kanabayashi T, Murase T, Kito S. Cognitive decline in STZ-3V rats is largely due to dysfunctional insulin signalling through the dentate gyrus. Behavioural Brain Research. 2012;229:378–383. doi: 10.1016/j.bbr.2012.01.034. [DOI] [PubMed] [Google Scholar]
- Siegel JM. Narcolepsy: a key role for hypocretins (orexins) Cell. 1999;98:409–412. doi: 10.1016/s0092-8674(00)81969-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart D, Jerman J. The physiology and pharmacology of the orexins. Pharmacol Ther. 2002;94:51–61. doi: 10.1016/s0163-7258(02)00171-7. [DOI] [PubMed] [Google Scholar]
- Stanley EM, Fadel J. Aging-related deficits in orexin/hypocretin modulation of the septohippocampal cholinergic system. Synapse. 2012;66:445–452. doi: 10.1002/syn.21533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley EM, Fadel JR, Mott DD. Interneuron loss reduces dendritic inhibition and GABA release in hippocampus of aged rats. Neurobiology of Aging. 2011 doi: 10.1016/j.neurobiolaging.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley EM, Wilson MA, Fadel JR. Hippocampal neurotransmitter efflux during one-trial novel object recognition in rats. Neuroscience Letters. 2012;511:38–42. doi: 10.1016/j.neulet.2012.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steen E, Terry BM, Rivera EJ, Cannon JL, Neely TR, Tavares R, Xu XJ, Wands JR, de la Monte SM. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease--is this type 3 diabetes? J Alzheimers Dis. 2005;7:63–80. doi: 10.3233/jad-2005-7107. [DOI] [PubMed] [Google Scholar]
- Stranahan AM, Arumugam TV, Cutler RG, Lee K, Egan JM, Mattson MP. Diabetes impairs hippocampal function through glucocorticoid-mediated effects on new and mature neurons. Nature Neuroscience. 2008;11:309–317. doi: 10.1038/nn2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranahan AM, Lee K, Martin B, Maudsley S, Golden E, Cutler RG, Mattson MP. Voluntary exercise and caloric restriction enhance hippocampal dendritic spine density and BDNF levels in diabetic mice. Hippocampus. 2009;19:951–961. doi: 10.1002/hipo.20577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweet DC, Levine AS, Billington CJ, Kotz CM. Feeding response to central orexins. Brain Research. 1999;821:535–538. doi: 10.1016/s0006-8993(99)01136-1. [DOI] [PubMed] [Google Scholar]
- Taghibiglou C, Bradley CA, Gaertner T, Li Y, Wang Y, Wang YT. Mechanisms involved in cholesterol-induced neuronal insulin resistance. Neuropharmacology. 2009;57:268–276. doi: 10.1016/j.neuropharm.2009.05.013. [DOI] [PubMed] [Google Scholar]
- Takeda S, Sato N, Uchio-Yamada K, Sawada K, Kunieda T, Takeuchi D, Kurinami H, Shinohara M, Rakugi H, Morishita R. Diabetes-accelerated memory dysfunction via cerebrovascular inflammation and Abeta deposition in an Alzheimer mouse model with diabetes. Proc Natl Acad Sci U S A. 2010;107:7036–7041. doi: 10.1073/pnas.1000645107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda S, Sato N, Uchio-Yamada K, Yu H, Moriguchi A, Rakugi H, Morishita R. Oral Glucose Loading Modulates Plasma beta-Amyloid Level in Alzheimer’s Disease Patients: Potential Diagnostic Method for Alzheimer’s Disease. Dement Geriatr Cogn Disord. 2012;34:25–30. doi: 10.1159/000338704. [DOI] [PubMed] [Google Scholar]
- Talbot K, Wang HY, Kazi H, Han LY, Bakshi KP, Stucky A, Fuino RL, Kawaguchi KR, Samoyedny AJ, Wilson RS, Arvanitakis Z, Schneider JA, Wolf BA, Bennett DA, Trojanowski JQ, Arnold SE. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest. 2012;122:1316–1338. doi: 10.1172/JCI59903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terao A, Apte-Deshpande A, Morairty S, Freund YR, Kilduff TS. Age-related decline in hypocretin (orexin) receptor 2 messenger RNA levels in the mouse brain. Neuroscience Letters. 2002;332:190–194. doi: 10.1016/s0304-3940(02)00953-9. [DOI] [PubMed] [Google Scholar]
- Thannickal TC, Lai YY, Siegel JM. Hypocretin (orexin) cell loss in Parkinson’s disease. Brain. 2007;130:1586–1595. doi: 10.1093/brain/awm097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thannickal TC, Moore RY, Nienhuis R, Ramanathan L, Gulyani S, Aldrich M, Cornford M, Siegel JM. Reduced number of hypocretin neurons in human narcolepsy. Neuron. 2000;27:469–474. doi: 10.1016/s0896-6273(00)00058-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrealba F, Yanagisawa M, Saper CB. Colocalization of orexin a and glutamate immunoreactivity in axon terminals in the tuberomammillary nucleus in rats. Neuroscience. 2003;119:1033–1044. doi: 10.1016/s0306-4522(03)00238-0. [DOI] [PubMed] [Google Scholar]
- Tsuneki H, Murata S, Anzawa Y, Soeda Y, Tokai E, Wada T, Kimura I, Yanagisawa M, Sakurai T, Sasaoka T. Age-related insulin resistance in hypothalamus and peripheral tissues of orexin knockout mice. Diabetologia. 2008;51:657–667. doi: 10.1007/s00125-008-0929-8. [DOI] [PubMed] [Google Scholar]
- Tupone D, Madden CJ, Cano G, Morrison SF. An orexinergic projection from perifornical hypothalamus to raphe pallidus increases rat brown adipose tissue thermogenesis. The Journal of Neuroscience. 2011;31:15944–15955. doi: 10.1523/JNEUROSCI.3909-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unger J, McNeill TH, Moxley RT, III, White M, Moss A, Livingston JN. Distribution of insulin receptor-like immunoreactivity in the rat forebrain. Neuroscience. 1989;31:143–157. doi: 10.1016/0306-4522(89)90036-5. [DOI] [PubMed] [Google Scholar]
- van den Pol AN. Hypothalamic hypocretin (orexin): robust innervation of the spinal cord. The Journal of Neuroscience. 1999;19:3171–3182. doi: 10.1523/JNEUROSCI.19-08-03171.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velayudhan L, Poppe M, Archer N, Proitsi P, Brown RG, Lovestone S. Risk of developing dementia in people with diabetes and mild cognitive impairment. British Journal of Psychiatry. 2010;196:36–40. doi: 10.1192/bjp.bp.109.067942. [DOI] [PubMed] [Google Scholar]
- Waldrop MA, Leinung MC, Lee DW, Grasso P. Intranasal delivery of mouse [D-Leu-4]-OB3, a synthetic peptide amide with leptin-like activity, improves energy balance, glycaemic control, insulin sensitivity and bone formation in leptin-resistant C57BLK/6-m db/db mice. Diabetes Obes Metab. 2010;12:871–875. doi: 10.1111/j.1463-1326.2010.01243.x. [DOI] [PubMed] [Google Scholar]
- Wan Q, Xiong ZG, Man HY, Ackerley CA, Braunton J, Lu WY, Becker LE, MacDonald JF, Wang YT. Recruitment of functional GABA(A) receptors to postsynaptic domains by insulin. Nature. 1997;388:686–690. doi: 10.1038/41792. [DOI] [PubMed] [Google Scholar]
- Wang ZW, Pan WT, Lee Y, Kakuma T, Zhou YT, Unger RH. The role of leptin resistance in the lipid abnormalities of aging. FASEB Journal. 2001;15:108–114. doi: 10.1096/fj.00-0310com. [DOI] [PubMed] [Google Scholar]
- Wayner MJ, Armstrong DL, Phelix CF, Oomura Y. Orexin-A (Hypocretin-1) and leptin enhance LTP in the dentate gyrus of rats in vivo. Peptides. 2004;25:991–996. doi: 10.1016/j.peptides.2004.03.018. [DOI] [PubMed] [Google Scholar]
- Winocur G, Greenwood CE, Piroli GG, Grillo CA, Reznikov LR, Reagan LP, McEwen BS. Memory Impairment in Obese Zucker Rats: An Investigation of Cognitive Function in an Animal Model of Insulin Resistance and Obesity. Behavioral Neuroscience. 2005;119:1389–1395. doi: 10.1037/0735-7044.119.5.1389. [DOI] [PubMed] [Google Scholar]
- Wrighten SA, Piroli GG, Grillo CA, Reagan LP. A look inside the diabetic brain: Contributors to diabetes-induced brain aging. Biochim Biophys Acta. 2008 doi: 10.1016/j.bbadis.2008.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu WL, von Strauss E, Qiu CX, Winblad B, Fratiglioni L. Uncontrolled diabetes increases the risk of Alzheimer’s disease: a population-based cohort study. Diabetologia. 2009;52:1031–1039. doi: 10.1007/s00125-009-1323-x. [DOI] [PubMed] [Google Scholar]
- Yaffe K, Falvey C, Hamilton N, Schwartz AV, Simonsick EM, Satterfield S, Cauley JA, Rosano C, Launer LJ, Strotmeyer ES, Harris TB. Diabetes, glucose control, and 9-year cognitive decline among older adults without dementia. Archives in Neurology. 2012;69:1170–1175. doi: 10.1001/archneurol.2012.1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka A, Beuckmann CT, Willie JT, Hara J, Tsujino N, Mieda M, Tominaga M, Yagami K, Sugiyama F, Goto K, Yanagisawa M, Sakurai T. Hypothalamic orexin neurons regulate arousal according to energy balance in mice. Neuron. 2003;38:701–713. doi: 10.1016/s0896-6273(03)00331-3. [DOI] [PubMed] [Google Scholar]
- Yang Y, Ma D, Wang Y, Jiang T, Hu S, Zhang M, Yu X, Gong CX. Intranasal Insulin Ameliorates Tau Hyperphosphorylation in a Rat Model of Type 2 Diabetes. J Alzheimers Dis. 2012 doi: 10.3233/JAD-2012-121294. [DOI] [PubMed] [Google Scholar]
- Yau PL, Castro MG, Tagani A, Tsui WH, Convit A. Obesity and metabolic syndrome and functional and structural brain impairments in adolescence. Pediatrics. 2012;130:e856–e864. doi: 10.1542/peds.2012-0324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeki AH, Stone KL, Haan MN, Yaffe K. Leptin, Mild Cognitive Impairment, and Dementia Among Elderly Women. J Gerontol A Biol Sci Med Sci. 2012 doi: 10.1093/gerona/gls155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Heng X, Li T, Li L, Yang D, Zhang X, Du Y, Doody RS, Le W. Long-term treatment with lithium alleviates memory deficits and reduces amyloid-beta production in an aged Alzheimer’s disease transgenic mouse model. J Alzheimers Dis. 2011;24:739–749. doi: 10.3233/JAD-2011-101875. [DOI] [PubMed] [Google Scholar]