

Abstract
A broad variety of organisms display circadian rhythms (i.e., oscillations with 24-hr periodicities) in many aspects of their behavior, physiology and metabolism. These rhythms are under genetic control and are generated endogenously at the cellular level. In mammals, the core molecular mechanism of the oscillator consists of two transcriptional activators, CLOCK and BMAL1, and their transcriptional targets, CRYPTOCHROMES (CRYS) and PERIODS (PERS). The CRY and PER proteins function as negative regulators of CLOCK/BMAL1 activity, thus forming the major circadian autoregulatory feedback loop. It is believed that the circadian clock system regulates daily variations in output physiology and metabolism through periodic activation/repression of the set of clock-controlled genes that are involved in various metabolic pathways. Importantly, circadian-controlled pathways include those that determine in vivo responses to genotoxic stress. By using circadian mutant mice deficient in different components of the molecular clock system, we have established genetic models that correlate with the two opposite extremes of circadian cycle as reflected by the activity of the CLOCK/BMAL1 transactivation complex. Comparison of the in vivo responses of these mutants to the chemotherapeutic drug, cyclophosphamide (CY), has established a direct correlation between drug toxicity and the functional status of the CLOCK/BMAL1 transcriptional complex. We have also demonstrated that CLOCK/BMAL1 modulates sensitivity to drug-induced toxicity by controlling B cell responses to active CY metabolites. These results suggest that the sensitivity of cells to genotoxic stress induced by anticancer therapy may be modulated by CLOCK/BMAL1 transcriptional activity. Further elucidation of the molecular mechanisms of circadian control as well as identification of specific pharmacological modulators of CLOCK/BMAL1 activity are likely to lead to the development of new anti-cancer treatment schedules with increased therapeutic index and reduced morbidity.
Keywords: Circadian, CLOCK, BMAL1, transcription, anticancer therapy
ANTICANCER THERAPY—A DOUBLE-EDGED SWORD
Two major types of anticancer therapy—chemotherapy and radiation—were introduced into the clinic several decades ago and have demonstrated significant success in treating a variety of tumors. However, both types of anticancer therapy are compromised by accompanying nonspecific damage to normal tissues that often results in debilitating side effects. Well-known examples of such complications include the hematopoietic syndrome (induced by radiation and some DNA-damaging chemotherapeutic drugs, such as cyclophosphamide), direct neurotoxicity (induced by vinca alkaloids), damage to the oral mucosa, kidney and bladder, radiation-induced damage to intestinal epithelia, and many others. All of these side effects not only enhance the morbidity associated with treatment, but also increase the risk of bacterial, viral and fungal infections in immuno-compromised patients. The severity of side effects depends on a variety of factors including the particular type of therapy, the dosage and schedule used and individual responses to treatment. To estimate the relative effect of the positive and negative aspects of chemotherapy and radiation, each type of therapy is characterized by a therapeutic index, which reflects the level of tumor response to treatment and the severity of side effects (the amount of damage caused to normal tissues). Improving the ratio of desired to undesired effects of therapy, the therapeutic index, is a major goal of cancer therapy.
Various approaches have been used to increase the therapeutic index for treating different types of cancer. Some of them are directed towards development of more specific means of drug delivery, while others exploit the differences in cellular responses and metabolic requirements between tumor and normal cells. For example, the use of chemotherapy in combination with monoclonal antibody against HER2 surface antigen significantly increased the therapeutic index of treating metastatic breast cancers that overexpress HER2.1 Various hormone-based therapies exploit their selective action on particular types of cells and are widely used to inhibit the growth of breast and prostate cancer cells.2,3 Because many tumor cells develop resistance to drugs in the course of treatment, the efficacy of the treatment could be improved by using the anticancer agents in combination with inhibitors of multi-drug transporters.4
There are also a variety of gene therapy strategies aimed at enhancing the effects of radiation and drug therapy through direct targeting of individual components of specific pathways,5 such as PI3-Akt,6 angiogenesis7 or p53.8 The latter is based on a well-known fact that a variety of tumor cells are resistant to apoptosis due to the loss of critical components of cell death inducing pathways, such as p53. Therefore, temporal suppression of p53 by chemical inhibitors helps to reduce radiation damage to normal tissues without affecting the sensitivity of tumor cells.9,10
Among known metabolic regulators of cellular responses to genotoxic drugs that could be used for modulation of stress response are inhibitors of glycolysis (2-deoxy-D-glucose), which could specifically modify the responses of cancer cells, particularly to DNA damaging therapeutic agents.11 In addition, dietary supplements like glutamine may decrease the incidence and/or severity of chemotherapy-associated mucositis, irinotecan-associated diarrhea and paclitaxel-induced neuropathy and may enhance the therapeutic index by protecting normal tissues from radiation-related injury.12
All of the strategies described above assume that neoplastic and normal tissues respond to various treatments in a certain constant manner and the understanding of the underlying mechanisms of response can be used for improvement of therapy. However, observations made over 30 years ago in clinical practice and in experimental model systems conclusively demonstrated that in vivo responses to various therapies might display significant variations depending upon time of treatment. Based upon these observations, it has been proposed that together with many other physiological and metabolic parameters, an organism’s response to different treatments could be controlled by the circadian clock system and that introduction of chronomodulated schedules could further enhance the efficacy of anticancer therapy.
CHRONOTHERAPY—BENEFICIAL BUT INCONVENIENT FOR PRACTICAL APPLICATIONS
Historically, clinical studies testing whether cancer chemotherapy timing affects therapeutic index started with the observation that the circadian pattern of Ara-C administration determines both its toxicity and antitumor activity.13 This initial observation was later extended to testing the effect of timed administration of over 30 anticancer agents in mice and rats.14 For most of them, the differences in toxicity ranged from 2-to 8-fold depending upon the time of their administration. Although less experimental data has been generated for chronomodulated radiation therapy, there have been some reports on circadian variations in sensitivity to whole body radiation in mice (reviewed in ref. 15).
Taken together, these findings led to the concept of chronotherapy, wherein medication or other treatment is delivered at the optimal time of day to maximize efficacy and minimize morbidity. These studies formed the basis for several clinical trials in which the efficiency of a given treatment was compared using either conventional or chronomodulated delivery schedules. The first of these studies, performed for doxorubicin/cisplatin treatment of advanced ovarian cancer, demonstrated a significant advantage of timed drug administration. Patients that received both drugs at specific times associated with reduced toxicity in experimental systems demonstrated better performance and showed half as many complications with the same therapeutic effect16. Other widely known and well-documented examples of the successful use of a timed drug delivery schedule are the chronotherapy of metastatic colorectal cancer with 5-FU in combination with leucovorin and oxaliplatin (reviewed in ref. 17) and 5-FU treatment of bladder cancer.18
Despite the existence of compelling experimental data from animal studies and the strong support of several successful clinical trials, the chronomodulated approach has not become accepted in clinical practice. Empirically designed optimal treatment schedules are typically inconvenient both for patients and medical personnel, as they often dictate treatments at times that are normally considered rest times. In addition, the lack of a clear mechanistic rationale behind the experimental observations did not allow standardization of these procedures in clinical practice. However, recent progress in the identification of circadian clock genes and the elucidation of the molecular mechanisms underlying circadian control of various physiological processes have generated novel tools and approaches allowing a reassessment of the basic concepts of chronotherapy at a mechanistic level.
CIRCADIAN CLOCKS—A UNIVERSAL TIMING MECHANISM CONTROLLING RESPONSES TO VARIOUS TREATMENTS
Living organisms have developed many systems that ensure proper adaptation to constantly changing environmental conditions. One of the most powerful conditions that affect a broad range of organisms, from bacteria and fungi to plants and animals, is the daily light-dark cycle caused by the Earth’s rotation. To ensure proper adaptation to this constantly changing environment, many organisms have developed a molecular system called the circadian system (from Latin “circa”—about and “diem”—day), capable of generating 24-hour periodicities in various physiological and behavioral processes. In mammals, such fundamental parameters as the sleep-wake cycle, blood pressure, body temperature, hormone secretion, and immune system activity all demonstrate daily fluctuations that can persist with a period close to 24-hr even in the absence of environmental cues. It has been recognized that these rhythms are generated endogenously and are capable of responding to external stimuli to coordinate the phases of the endogenous clocks with the environment.
During the past decade, enormous progress has been made in understanding the molecular mechanisms underlying various circadian processes. Thus, it has been established that in mammals molecular clocks are present not only in the specialized neurons of the suprachiasmatic nucleus (SCN) of the anterior hypothalamus, but also in multiple other tissues of the body (reviewed in ref. 19). In all tissues studied, clock function at the molecular level is determined by interlocking transcription/translation-based feedback loops that drive rhythmic rNA and protein expression of key clock components (reviewed in Ref. 20). The major loop is regulated by two positive activators, CLOCK and BMAL1, which belong to the family of bHLH-PAS domain transcription factors. Upon forming a heterodimer, they regulate gene expression by interacting with E-box enhancer elements (CACGTG) in the promoters of target genes. CLOCK/BMAL1 target genes include those encoding several repressors, such as PERIOD proteins, PER1, PER2 and the CRYPTOCHROME molecules, CRY1 and CRY2, which function to inhibit the CLOCK/BMAL1 complex (Fig. 1). The CLOCK/BMAL1 heterodimer also controls transcription of the Rev-erba gene, whose protein product feeds back to periodically repress expression of Bmal121. Positive and negative elements of these regulatory loops function to coordinate transcriptional events at appropriate times to provide 24-hour periodicities (Fig. 1). Posttranslational mechanisms such as protein phosphorylation of negative (PER and CRY22–24) and positive (CLOCK and BMAL125, 26) components also play important roles in core clock activity.
Figure 1.
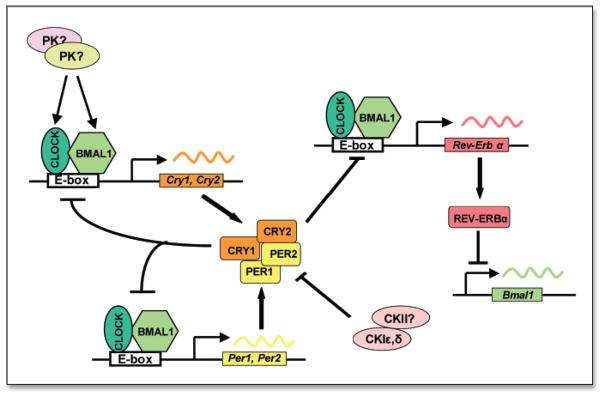
Schematic representation of mammalian circadian autoregulatory feedback loop. Positive elements of the loop (CLOCK and BMAL1) dimerize to activate rhythmic transcription of Per and Cry genes through specific enhancer elements. The nuclear-localized CRY and PER proteins interact with CLOCK and BMAL1 to negatively regulate CLOCK:BMAL1-mediated transcription. Rev-erbα transcription is regulated by the same components that control Per and Cry transcription, and the resulting circadian accumulation of REV-ERBα leads to periodic repression of Bmal1 transcription.
How do oscillations in the expression of clock genes translate into physiological periodicities? Insight into this question was made possible by the development of microarray technology and its application to the circadian system. When applied to various circadian models, this approach identified a large number of genes showing 24-hr periodicities in their expression profiles27–32 indicating that they may be under direct or indirect control of the CLOCK/BMAL1 transactivation complex. Altogether, the microarray studies demonstrated several important points. First, they showed that a significant portion of the genome (in mammals, approximately 3–10% of detectably expressed transcripts) is under circadian control30. Second, they demonstrated remarkable tissue-specificity of circadian regulation with less then 5–10% overlap in clock-controlled genes between different tissues. In many cases, tissue-specific clock-controlled genes were recognized to be involved in rate-limiting steps that were distinct for the organ function30–31. Finally, these studies presented the possibility that profiling of additional tissues might reveal more circadian regulated genes, and that the list of clock-controlled genes might include some that modulate sensitivity to genotoxic stress induced by cancer treatment (chemotherapy and radiation). Such clock-controlled fluctuations in gene expression levels could potentially provide the mechanistic background for the daily variations in therapeutic effects observed in various animals systems and in clinical practice.
CIRCADIAN MUTANT MICE AS A MODEL SYSTEM FOR TWO EXTREME STATES OF CIRCADIAN CLOCK FUNCTION
Most traditional in vivo chronobiological studies have been performed on animals that were synchronized to a light:dark cycle 12:12 (12 hrs of light, 12 hrs of darkness) and subjected to certain treatments or sample collection at different times of the 24-hr cycle. This experimental approach, however, is compromised by multiple variable physiological and behavioral factors for which it is not possible to control. The most obvious of these complications is that animal husbandry typically occurs during daytime because it is convenient for laboratory personnel, yet this is normally the rest time for nocturnal rodents. More definitive experimental models have recently been established due to identification of clock genes and an emerged understanding of molecular clock function. Based upon our current view on the molecular mechanism of clock system functions, we can mimic the two opposite extremes of the activity of the CLOCK/BMAL1 transactivation complex using three mouse genetic models—Clock/Clock mutant, Bmal1−/− knockout and Cry1−/−Cry2−/− knockout animals. Phenotypically, all three mutant mouse models are characterized by disrupted circadian rhythmicity at the behavioral level33–35. However, at the molecular level, these experimental models correlate with different temporal states of circadian clock function and may approximate two opposite extremes in relation to the activity of the CLOCK/BMAL1 transactivation complex. Thus, mutant mice with targeted disruption of the Bmal1 gene or deficiency in CLOCK transactivation function due to mutation represent the minimal transcriptional activity of the complex due to the deficiency of transcriptional activators35–37. In contrast, mice with targeted disruption of both Cryptochrome genes contain CLOCK/BMAL1 in its maximally active state since they lack the CRY1/CRY2 circadian inhibitors38, 39. At the same time, in wild-type mice, circadian variations in CLOCK/BMAL1 activity occur with a normal 24-hour cycle (Fig. 2). To explore possible molecular links between response to chemotherapeutic agents in vivo and the status of the major circadian transactivation complex, we compared the sensitivity of these three types of mutant mice to a widely used chemotherapeutic drug—cyclophosphamide (CY).
Figure 2.

Schematic representation of the molecular consequences of mutations in circadian genes. (A) Normal circadian pattern of expression. Both core clock genes (Periods and Cryptochromes) and multiple clock-controlled target genes are rhythmically expressed due to periodic activation and inhibition of the CLOCK/BMAL1 transcriptional complex. (B) Either mutation within the Clock gene or disruption of the Bmal1 gene blocks activation of both core clock genes and clock-controlled target genes, resulting in reduced transcript abundance. (C) Deficiency in both CRYPTOCHROMEs results in constant activation of CLOCK/BMAL1 target genes. PERIODs cannot effectively inhibit CLOCK/BMAL1 in the absence of CRY1 and CRY2 for two reasons: (1) PERs are not as potent inhibitors as CRYs;74 and (2) CRYs are likely to promote PER nuclear translocation.75 The lack of CRY inhibition is expected to lead to constant medium-to-high levels of target gene transcripts, as demonstrated for Per transcripts.38,39
SENSITIVITY OF NORMAL CELLS TO GENOTOXIC STRESS DEPENDS ON THE FUNCTIONAL STATUS OF THE CLOCK/BMAL1 TRANSACTIVATION COMPLEX
As expected based upon previous studies, normal mice show a robust rhythm in sensitivity to CY. Both drug-induced mortality and morbidity were the lowest when CY was administered during late afternoon/early night hours, which corresponds to the time of day when transcriptional activity of CLOCK/BMAL1 is at its highest daily level in most peripheral tissues tested40. Consistent with this, Clock/Clock mutant mice and Bmal1−/− mice, which are characterized by constant low levels of CLOCK/BMAL1 transcriptional activity, both demonstrated increased drug sensitivity at all times tested. In contrast, Cry1−/−Cry2−/− mice, which are deficient in the CRY transcriptional repressors, were more resistant to the toxicity of CY treatment than wild-type mice. Thus, comparison of wild-type mice to three different models of circadian mutant mice showed that both survival and morbidity after administration of sublethal doses of CY were directly correlated with CLOCK/BMAL1 functional activity. These results confirm the involvement of the circadian system in regulating sensitivity to drug-induced toxicity in vivo. Moreover, they suggest that drug sensitivity does not depend upon persistence of rhythmicity per se but rather is guided by functional activity of the major circadian transcriptional complex, CLOCK/BMAL1. This implies that CLOCK/BMAL1 may directly control the molecular determinants of drug sensitivity at the transcriptional level.
Traditionally, daily variations in sensitivity to chemotherapy have been attributed to rhythms in the activity of multiple drug metabolizing enzymes14,41. This point of view is supported by the results of global gene expression analysis of circadian transcriptional output, which demonstrated that many genes encoding drug metabolizing enzymes display 24-hr periodicities in their expression pattern in the liver30,31. That is why the results of our detailed pharmacokinetic analysis of CY metabolites in the plasma of mice of different circadian genotypes after CY administration were quite surprising. Performed in various experimental setups, they did not reveal any genotype- or time-of-administration-dependent changes that could account for the dramatic differences in drug sensitivity observed in our in vivo experiments40. These data are also supported by the measurements of CY activation rate by hepatocytes isolated from wild-type, Clock/Clock and Bmal1−/− mice. Taken together, these results suggest that in contrast to traditional view, the circadian clock system modulates response to chemotherapeutic drug through controlling the sensitivity of target cells to drug-induced toxicity rather than by modulating the pharmacokinetics of CY.
There are several cellular targets of CY-induced toxicity that have been described in animal models and clinical studies. Among others, side effects of CY therapy include liver and lung damage due to endothelial cell injury42,43, bladder toxicity, hemorrhagic intestinal injury and cardiomyopathy. However the major target for CY toxicity, which is considered to be the primary dose-limiting factor of CY use in the clinic, is the hematopoietic system44. In mice, the peak of CY-induced myelosupression occurs at ~ day 3–5 (depending on the strain) after CY treatment and is followed by a period of rebound neutrophilia45. This time course corresponds closely to the body weight loss profile observed in our experiments—wild-type mice reached their drug-induced body weight minimum at days 3–5, after which they go through a dose-dependent recovery process. In contrast, CY-sensitive circadian mutants lost significantly more weight by this time and were unable to recover40.
The measurement of basic hematological parameters performed at different times after CY administration in wild-type and Clock/Clock mutant mice demonstrated a severe reduction in the number of circulating lymphocytes, which was more pronounced in the CY-sensitive Clock/Clock mutants than in wild type mice both at the peak time of bone marrow suppression and during the recovery period. Flow cytometric analysis of bone marrow and spleen cells with fluorescently-labeled anti-B220 antibody showed that, in both tissues, the reduction in circulating lymphocytes was due to loss of B cells. Interestingly, all of the circadian mutant mice tested showed no differences in basal hematological parameters under normal conditions. However, following exposure to CY, wild-type and Cry1−/−Cry2−/− mice retained significantly higher numbers of lymphocytes when compared to Bmal1−/− and Clock/Clock mutant mice and their overall WBC parameters were less distorted40. These data conclusively demonstrate that circadian control of drug response in vivo is mediated through CLOCK/BMAL1-dependent modulation of B cell survival/recovery.
Although the exact mechanism of circadian control of drug-induced toxicity still remains to be clarified, the fact that it is not determined simply by metabolic differences but rather involves regulation of a component of the immune system underscores the important role of the circadian system in global physiology. It is quite likely that such control is not limited to the effects of cyclophosphamide, but may be involved in responses to other classes of therapeutic agents.
MOLECULAR CLOCKS IN TUMORS
The direct correlation between CLOCK/BMAL1 function and in vivo drug sensitivity demonstrated in Gorbacheva et al40 opens up an important prospective application of the intrinsic clock mechanism, in which existing therapeutic schedules might be rationally modulated to increase the therapeutic index of a given cancer treatment. If cellular responses to genotoxic stress can be modulated by the functional status of a molecular clock, it might be possible to use this mechanism to specifically increase the sensitivity of tumor cells to chemotherapeutic drugs and radiation. Such an approach would have an important advantage if used in combination with conventional protocols, allowing a similar therapeutic effect to be achieved while using lower doses of drugs and thus reducing the morbidity associated with the treatment. There are, however, a number of important questions that, for the most part, still remain unanswered:
Are circadian genes expressed in tumors and if so, are they functional?
Does the activity of CLOCK/BMAL1 correlate with tumor sensitivity to anticancer treatment?
Are tumor clocks synchronized with those in other tissues of the organism?
Tumors and tumor-derived immortalized cell lines display multiple genetic abnormalities. The most prominent among them are the loss of tumor suppressor gene expression and enhanced expression of oncogenes, which, in turn, may affect various pathways. To date, there have been only a few reports on the expression levels of clock genes in tumors and tumor cell lines. The results of the most comprehensive study, in which global gene expression levels were measured in different human tumor cell lines, are summarized on a publicly available database.46 Analysis of these data demonstrates that a variety of tumor cell lines of different origin have detectable expression levels of all clock genes. Similar results were obtained through analysis of a variety of mouse tumor cell lines using real-time PCR with probes specific to major clock genes (Antoch M, unpublished data).
If clock genes are not lost in tumors, are they still capable of sustaining circadian fluctuations in target gene expression? Circadian rhythms in cell culture were first detected in rat-1 fibroblasts and the H35 hepatoma cell line.47,48 These studies demonstrated that molecular clocks are present in various cells and tissues and most likely are capable of sustaining oscillations at a single cell level;49,50 however, to display coordinated oscillation, they have to be synchronized. Such synchronization occurs in vivo through a signal coming from the SCN and in vitro can be achieved by short treatment with high serum concentrations or a wide variety of chemicals that affect different signaling pathways, including activators of protein kinase A, protein kinase C, MAP kinase, dexamethazone, glucose, endothelin and retinoic acid receptor.47,48,51–54 Several lines of evidence suggest that at least some cultured tumor cells retain functional clock activity and thus can display rhythmic expression under certain conditions. For example, neuroblastoma cells SH-SY5Y, when induced by high serum concentration, demonstrated circadian oscillation of hPer1 expression.55 However, in other cell lines derived from glioblastomas and osteosarcomas, hPer1 expression was induced by high serum but failed to display circadian oscillations in expression. Rhythmic expression of core clock genes has been also detected in sarcoma 180 and melanoma B16 tumors implanted in nude mice.56 Interestingly, the phases of circadian gene expression oscillation in these tumors were similar to those in the normal tissues (liver) of the animal. Finally, rhythmic expression of the clock-controlled gene encoding methionine aminopeptidase 2 has recently been reported in three types of transplanted tumors—sarcoma 180, Lewis lung carcinoma, and B16 melanoma—and in all cases, the rhythmic expression of the target gene reflected the CLOCK/BMAL1 phase in peripheral tissues.57 Nevertheless, it still remains to be determined if, similar to what has been described for normal cells, sensitivity to genotoxic stress in tumor cell correlates with CLOCK/BMAL1 functional activity.
MOLECULAR DETERMINANTS OF SENSITIVITY TO ANTICANCER DRUGS
In order to exploit circadian-controlled drug sensitivity to improve treatment of patients, one must first identify the molecular targets of this control both in normal and tumor cells. In general, the primary targets of the toxic effects of anticancer therapy are cells with high levels of proliferation such as hematopoietic cells, vascular endothelial cells and intestinal epithelial cells. Several groups have reported daily or/and circadian rhythms in proliferation activity in these tissues.58–60 Since cell sensitivity to anticancer agents is known to depend upon cell cycle stage, these observations provide a possible explanation for the time-of-day variations in drug-induced toxicity. Indeed, daily variations in radiation-induced apoptosis in mouse small intestine have been reported as a major determinant of circadian control of radiation response.61 In addition, several examples of cellular rhythms in enzymatic activities and cell cycle regulation were described as major determinants of anticancer drug pharmacology.62–64 Our own study identified B-cells as a major target of CLOCK/BMAL1-controlled CY toxicity.40
Although rhythms in cellular proliferation have been observed, the molecular basis underlying these rhythms remains unknown. It is possible that the CLOCK/BMAL1 complex may be directly involved in regulating expression of genes involved in cell cycle regulation and apoptosis. Indeed, some of these genes demonstrate circadian variation in their transcript levels and their expression patterns are disrupted in the tissues of circadian mutant mice. Low-amplitude circadian variations in the expression profiles of some pro-and anti-apoptotic genes have been described in mouse bone marrow and tumors;65 however, it is still not clear how these fluctuations may be mechanistically translated into differences in cell survival rate. Recently, two examples of possible molecular connections between the circadian system and cell proliferation/apoptosis were reported. One study used a liver regeneration model to determine the effect of clock genes on the timing of the G2/M transition. The results show that timing of the G2/M transition is set by the circadian clock through CLOCK/BMAL1-controlled expression of the Wee1 kinase that is involved in phosphorylation of the crucial regulator of the cell cycle, CDC2. Furthermore, the rate of liver regeneration depended on the activity of CRYPTOCHROMEs, since in Cry double-knockout mice regeneration occurred more slowly than in wild-type mice.66 The second report demonstrates that mice with mutations in the circadian Period2 gene are more resistant to low doses of ionizing radiation, which could be detected at the level of individual thymocytes.67 These findings suggest that circadian control may be based on the CLOCK/BMAL1-dependent regulation of the balance of pro-and anti-apoptotic factors at the single-cell level.
In contrast to cultured cells, at the organismal level, cell survival following genotoxic stress may be determined not only by cell-intrinsic mechanisms, but also by extracellular signaling pathways that may be modulated by CLOCK/BMAL1. It is well known that treatment with different growth factors and cytokines can prevent cell death induced by a variety of cytotoxic agents. Some of these agents, such as fibroblast growth factor, epidermal growth factor, and transforming growth factor are known to display daily variations in their plasma concentrations.68–70 It is likely that circadian modulation of responses to genotoxic stress in vivo will reflect the complex superposition of multiple factors and levels of regulation. Clearly, more experiments need to be done to decipher each of these levels of regulation.
CONCLUDING REMARKS
The recent progress in our understanding of the molecular link between the circadian system and stress response pathways has focused on the role of circadian clock genes in modulating sensitivity to anticancer drugs. However, there are other aspects of host-tumor interactions that may be modulated by CLOCK/BMAL1 and may be potentially important in therapeutic applications. For example, there is growing evidence that disruption of circadian rhythmicity increases the rate of tumor progression both in experimental mouse models71 and in clinical studies.72,73 The only mechanistic link between the circadian system and tumorigenesis that has been proposed to date is based upon the observation that mice with mutations in the circadian Period2 gene were more cancer-prone than wild-type mice.67 It is not clear, however, if the observed predisposition is due to direct regulation of genes affecting cell death and proliferation within individual cells or if it reflects the long-term effect of disruption of circadian rhythmicity at the level of the organism. In the latter case, Period2 would qualify as a gene involved in tumor prevention rather than as a classical tumor suppressor gene.
An improved understanding of the molecular link between the circadian system and genotoxic stress response pathways and the identification of more direct targets of CLOCK/BMAL1 that could modulate cell survival in response to stress both at the cellular and organismal levels promise to improve treatment of human cancer patients. Circadian-modulated therapeutic regimens could be based on two approaches: they may either be rationally adjusted to specific times that prove to be less toxic to normal tissues or may be pharmacologically modulated by specific drugs acting through or on circadian system regulators. In this respect, direct correlation of drug response in vivo with CLOCK/BMAL1 transcriptional activity recognizes this complex as a potential direct target for drug discovery. The identification of such drugs is an extremely important goal as, ideally, it will allow clinicians to specifically target tumor cells while sparing normal cells, independent of the time of treatment.
References
- 1.Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, Fleming T, Eiermann W, Wolter J, Pegram M, Baselga J, Norton L. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783–92. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 2.McLeod DG. Hormonal therapy: Historical perspective to future directions. Urology. 2003;61:3–7. doi: 10.1016/s0090-4295(02)02393-2. [DOI] [PubMed] [Google Scholar]
- 3.Jones KL, Buzdar AU. A review of adjuvant hormonal therapy in breast cancer. Endocr Relat Cancer. 2004;11:391–406. doi: 10.1677/erc.1.00594. [DOI] [PubMed] [Google Scholar]
- 4.Thomas H, Coley HM. Overcoming multidrug resistance in cancer: An update on the clinical strategy of inhibiting p-glycoprotein. Cancer Control. 2003;10:159–65. doi: 10.1177/107327480301000207. [DOI] [PubMed] [Google Scholar]
- 5.Collis SJ, DeWeese TL. Enhanced radiation response through directed molecular targeting approaches. Cancer Metastasis Rev. 2004;23:277–92. doi: 10.1023/B:CANC.0000031767.30730.d1. [DOI] [PubMed] [Google Scholar]
- 6.Lu Y, Wang H, Mills GB. Targeting PI3K-AKT pathway for cancer therapy. Rev Clin Exp Hematol. 2003;7:205–28. [PubMed] [Google Scholar]
- 7.Wachsberger P, Burd R, Dicker AP. Improving tumor response to radiotherapy by targeting angiogenesis signaling pathways. Hematol Oncol Clin North Am. 2004;18:1039–57. viii. doi: 10.1016/j.hoc.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 8.Gudkov AV, Komarova EA. The role of p53 in determining sensitivity to radiotherapy. Nat Rev Cancer. 2003;3:117–29. doi: 10.1038/nrc992. [DOI] [PubMed] [Google Scholar]
- 9.Komarov PG, Komarova EA, Kondratov RV, Christov-Tselkov K, Coon JS, Chernov MV, Gudkov AV. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science. 1999;285:1733–7. doi: 10.1126/science.285.5434.1733. [DOI] [PubMed] [Google Scholar]
- 10.Komarova EA, Gudkov AV. Chemoprotection from p53-dependent apoptosis: Potential clinical applications of the p53 inhibitors. Biochem Pharmacol. 2001;62:657–67. doi: 10.1016/s0006-2952(01)00733-x. [DOI] [PubMed] [Google Scholar]
- 11.Maschek G, Savaraj N, Priebe W, Braunschweiger P, Hamilton K, Tidmarsh GF, De Young LR, Lampidis TJ. 2-deoxy-D-glucose increases the efficacy of adriamycin and paclitaxel in human osteosarcoma and nonsmall cell lung cancers in vivo. Cancer Res. 2004;64:31–4. doi: 10.1158/0008-5472.can-03-3294. [DOI] [PubMed] [Google Scholar]
- 12.Savarese DM, Savy G, Vahdat L, Wischmeyer PE, Corey B. Prevention of chemotherapy and radiation toxicity with glutamine. Cancer Treat Rev. 2003;29:501–13. doi: 10.1016/s0305-7372(03)00133-6. [DOI] [PubMed] [Google Scholar]
- 13.Haus E, Halberg F, Pauly JE, Cardoso S, Kuhl JF, Sothern RB, Shiotsuka RN, Hwang DS. Increased tolerance of leukemic mice to arabinosyl cytosine with schedule adjusted to circadian system. Science. 1972;177:80–2. doi: 10.1126/science.177.4043.80. [DOI] [PubMed] [Google Scholar]
- 14.Levi F. Handbook of Experimental Pharmacology. Berlin: Springer-Verlag; 1997. Chronopharmacology of anticancer agents; pp. 299–331. [Google Scholar]
- 15.Haus E. Chronobiology of the mammalian response to ionizing radiation potential applications in oncology. Chronobiol Int. 2002;19:77–100. doi: 10.1081/cbi-120002592. [DOI] [PubMed] [Google Scholar]
- 16.Hrushesky WJ. Circadian timing of cancer chemotherapy. Science. 1985;228:73–5. doi: 10.1126/science.3883493. [DOI] [PubMed] [Google Scholar]
- 17.Giacchetti S. Chronotherapy of colorectal cancer. Chronobiol Int. 2002;19:207–19. doi: 10.1081/cbi-120002602. [DOI] [PubMed] [Google Scholar]
- 18.Kobayashi M, Wood PA, Hrushesky WJ. Circadian chemotherapy for gynecological and genitourinary cancers. Chronobiol Int. 2002;19:237–51. doi: 10.1081/cbi-120002600. [DOI] [PubMed] [Google Scholar]
- 19.Reppert SM, Weaver DR. Coordination of circadian timing in mammals. Nature. 2002;418:935–41. doi: 10.1038/nature00965. [DOI] [PubMed] [Google Scholar]
- 20.Lowrey PL, Takahashi JS. Mammalian circadian biology: Elucidating genome-wide levels of temporal organization. Annu Rev Genomics Hum Genet. 2004;5:407–41. doi: 10.1146/annurev.genom.5.061903.175925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Preitner N, Damiola F, Lopez-Molina L, Zakany J, Duboule D, Albrecht U, Schibler U. The orphan nuclear receptor REV-ERBalpha controls circadian transcription within the positive limb of the mammalian circadian oscillator. Cell. 2002;110:251–60. doi: 10.1016/s0092-8674(02)00825-5. [DOI] [PubMed] [Google Scholar]
- 22.Lowrey PL, Shimomura K, Antoch MP, Yamazaki S, Zemenides PD, Ralph MR, Menaker M, Takahashi JS. Positional syntenic cloning and functional characterization of the mammalian circadian mutation tau. Science. 2000;288:483–92. doi: 10.1126/science.288.5465.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eide EJ, Virshup DM. Casein kinase I: Another cog in the circadian clockworks. Chronobiol Int. 2001;18:389–98. doi: 10.1081/cbi-100103963. [DOI] [PubMed] [Google Scholar]
- 24.Lee C, Weaver DR, Reppert SM. Direct association between mouse PERIOD and CKIepsilon is critical for a functioning circadian clock. Mol Cell Biol. 2004;24:584–94. doi: 10.1128/MCB.24.2.584-594.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee C, Etchegaray JP, Cagampang FR, Loudon AS, Reppert SM. Posttranslational mechanisms regulate the mammalian circadian clock. Cell. 2001;107:855–67. doi: 10.1016/s0092-8674(01)00610-9. [DOI] [PubMed] [Google Scholar]
- 26.Kondratov RV, Chernov MV, Kondratova AA, Gorbacheva VY, Gudkov AV, Antoch MP. BMAL1-dependent circadian oscillation of nuclear CLOCK: Posttranslational events induced by dimerization of transcriptional activators of the mammalian clock system. Genes Dev. 2003;17:1921–32. doi: 10.1101/gad.1099503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grundschober C, Delaunay F, Puhlhofer A, Triqueneaux G, Laudet V, Bartfai T, Nef P. Circadian regulation of diverse gene products revealed by mRNA expression profiling of synchronized fibroblasts. J Biol Chem. 2001;276:46751–8. doi: 10.1074/jbc.M107499200. [DOI] [PubMed] [Google Scholar]
- 28.Harmer SL, Hogenesch JB, Straume M, Chang HS, Han B, Zhu T, Wang X, Kreps JA, Kay SA. Orchestrated transcription of key pathways in Arabidopsis by the circadian clock. Science. 2000;290:2110–3. doi: 10.1126/science.290.5499.2110. [DOI] [PubMed] [Google Scholar]
- 29.McDonald MJ, Rosbash M. Microarray analysis and organization of circadian gene expression in Drosophila. Cell. 2001;107:567–78. doi: 10.1016/s0092-8674(01)00545-1. [DOI] [PubMed] [Google Scholar]
- 30.Panda S, Antoch MP, Miller BH, Su AI, Schook AB, Straume M, Schultz PG, Kay SA, Takahashi JS, Hogenesch JB. Coordinated transcription of key pathways in the mouse by circadian clock. Cell. 2002;109:307–320. doi: 10.1016/s0092-8674(02)00722-5. [DOI] [PubMed] [Google Scholar]
- 31.Storch KF, Lipan O, Leykin I, Viswanathan N, Davis FC, Wong WH, Weitz CJ. Extensive and divergent circadian gene expression in liver and heart. Nature. 2002;417:78–83. doi: 10.1038/nature744. [DOI] [PubMed] [Google Scholar]
- 32.Ueda HR, Matsumoto A, Kawamura M, Iino M, Tanimura T, Hashimoto S. Genome-wide transcriptional orchestration of circadian rhythms in Drosophila. J Biol Chem. 2002;277:14048–52. doi: 10.1074/jbc.C100765200. (Epub 2002 Feb 19) [DOI] [PubMed] [Google Scholar]
- 33.Vitaterna MH, King DP, Chang AM, Kornhauser JM, Lowrey PL, McDonald JD, Dove WF, Pinto LH, Turek FW, Takahashi JS. Mutagenesis and mapping of a mouse gene, Clock, essential for circadian behavior. Science. 1994;264:719–25. doi: 10.1126/science.8171325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van der Horst GT, Muijtjens M, Kobayashi K, Takano R, Kanno S, Takao M, de Wit J, Verkerk A, Eker AP, van Leenen D, Buijs R, Bootsma D, Hoeijmakers JH, Yasui A. Mammalian Cry1 and Cry2 are essential for maintenance of circadian rhythms [see comments] Nature. 1999;398:627–30. doi: 10.1038/19323. [DOI] [PubMed] [Google Scholar]
- 35.Bunger MK, Wilsbacher LD, Moran SM, Clendenin C, Radcliffe LA, Hogenesch JB, Simon MC, Takahashi JS, Bradfield CA. Mop3 is an essential component of the master circadian pacemaker in mammals. Cell. 2000;103:1009–17. doi: 10.1016/s0092-8674(00)00205-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.King DP, Zhao Y, Sangoram AM, Wilsbacher LD, Tanaka M, Antoch MP, Steeves TD, Vitaterna MH, Kornhauser JM, Lowrey PL, Turek FW, Takahashi JS. Positional cloning of the mouse circadian clock gene. Cell. 1997;89:641–53. doi: 10.1016/s0092-8674(00)80245-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gekakis N, Staknis D, Nguyen HB, Davis FC, Wilsbacher LD, King DP, Takahashi JS, Weitz CJ. Role of the CLOCK protein in the mammalian circadian mechanism. Science. 1998;280:1564–9. doi: 10.1126/science.280.5369.1564. [DOI] [PubMed] [Google Scholar]
- 38.Okamura H, Miyake S, Sumi Y, Yamaguchi S, Yasui A, Muijtjens M, Hoeijmakers JH, van der Horst GT. Photic induction of mPer1 and mPer2 in cry-deficient mice lacking a biological clock. Science. 1999;286:2531–4. doi: 10.1126/science.286.5449.2531. [DOI] [PubMed] [Google Scholar]
- 39.Vitaterna MH, Selby CP, Todo T, Niwa H, Thompson C, Fruechte EM, Hitomi K, Thresher RJ, Ishikawa T, Miyazaki J, Takahashi JS, Sancar A. Differential regulation of mammalian period genes and circadian rhythmicity by cryptochromes 1 and 2. Proc Natl Acad Sci USA. 1999;96:12114–9. doi: 10.1073/pnas.96.21.12114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gorbacheva VY, Kondratov RV, Zhang R, Cherukuri S, Gudkov AV, Takahashi JS, Antoch MP. From the cover: Circadian sensitivity to the chemotherapeutic agent cyclophosphamide depends on the functional status of the CLOCK/BMAL1 transactivation complex. Proc Natl Acad Sci USA. 2005;102:3407–12. doi: 10.1073/pnas.0409897102. (Epub 2005 Feb 2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lemmer B. Chronopharmacokinetics: Implications for drug treatment. J Pharm Pharmacol. 1999;51:887–90. doi: 10.1211/0022357991773294. [DOI] [PubMed] [Google Scholar]
- 42.DeLeve LD, Wang X, Kuhlenkamp JF, Kaplowitz N. Toxicity of azathioprine and monocrotaline in murine sinusoidal endothelial cells and hepatocytes: The role of glutathione and relevance to hepatic venoocclusive disease. Hepatology. 1996;23:589–99. doi: 10.1002/hep.510230326. [DOI] [PubMed] [Google Scholar]
- 43.Kachel DL, Martin IInd WJ. Cyclophosphamide-induced lung toxicity: Mechanism of endothelial cell injury. J Pharmacol Exp Ther. 1994;268:42–6. [PubMed] [Google Scholar]
- 44.Colvin OM. An overview of cyclophosphamide development and clinical applications. Curr Pharm Des. 1999;5:555–60. [PubMed] [Google Scholar]
- 45.Watters JW, Kloss EF, Link DC, Graubert TA, McLeod HL. A mouse-based strategy for cyclophosphamide pharmacogenomic discovery. J Appl Physiol. 2003;95:1352–60. doi: 10.1152/japplphysiol.00214.2003. [DOI] [PubMed] [Google Scholar]
- 46.Su AI, Wiltshire T, Batalov S, Lapp H, Ching KA, Block D, Zhang J, Soden R, Hayakawa M, Kreiman G, Cooke MP, Walker JR, Hogenesch JB. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci USA. 2004;101:6062–7. doi: 10.1073/pnas.0400782101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Balsalobre A, Brown SA, Marcacci L, Tronche F, Kellendonk C, Reichardt HM, Schutz G, Schibler U. Resetting of circadian time in peripheral tissues by glucocorticoid signaling. Science. 2000;289:2344–7. doi: 10.1126/science.289.5488.2344. [DOI] [PubMed] [Google Scholar]
- 48.Balsalobre A, Damiola F, Schibler U. A serum shock induces circadian gene expression in mammalian tissue culture cells. Cell. 1998;93:929–37. doi: 10.1016/s0092-8674(00)81199-x. [DOI] [PubMed] [Google Scholar]
- 49.Nagoshi E, Saini C, Bauer C, Laroche T, Naef F, Schibler U. Circadian gene expression in individual fibroblasts: Cell-autonomous and self-sustained oscillators pass time to daughter cells. Cell. 2004;119:693–705. doi: 10.1016/j.cell.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 50.Welsh DK, Yoo SH, Liu AC, Takahashi JS, Kay SA. Bioluminescence imaging of individual fibroblasts reveals persistent, independently phased circadian rhythms of clock gene expression. Curr Biol. 2004;14:2289–95. doi: 10.1016/j.cub.2004.11.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Akashi M, Nishida E. Involvement of the MAP kinase cascade in resetting of the mammalian circadian clock. Genes Dev. 2000;14:645–9. [PMC free article] [PubMed] [Google Scholar]
- 52.Hirota T, Okano T, Kokame K, Shirotani-Ikejima H, Miyata T, Fukada Y. Glucose down-regulates Per1 and Per2 mRNA levels and induces circadian gene expression in cultured Rat-1 fibroblasts. J Biol Chem. 2002;277:44244–51. doi: 10.1074/jbc.M206233200. (Epub 2002 Sep 3) [DOI] [PubMed] [Google Scholar]
- 53.Yagita K, Okamura H. Forskolin induces circadian gene expression of rPer1, rPer2 and dbp in mammalian rat-1 fibroblasts. FEBS Lett. 2000;465:79–82. doi: 10.1016/s0014-5793(99)01724-x. [DOI] [PubMed] [Google Scholar]
- 54.Yagita K, Tamanini F, van Der Horst GT, Okamura H. Molecular mechanisms of the biological clock in cultured fibroblasts. Science. 2001;292:278–81. doi: 10.1126/science.1059542. [DOI] [PubMed] [Google Scholar]
- 55.Maronde E, Motzkus D. Oscillation of human period 1 (hPER1) reporter gene activity in human neuroblastoma cells in vivo. Chronobiol Int. 2003;20:671–81. doi: 10.1081/cbi-120022413. [DOI] [PubMed] [Google Scholar]
- 56.Koyanagi S, Kuramoto Y, Nakagawa H, Aramaki H, Ohdo S, Soeda S, Shimeno H. A molecular mechanism regulating circadian expression of vascular endothelial growth factor in tumor cells. Cancer Res. 2003;63:7277–83. [PubMed] [Google Scholar]
- 57.Nakagawa H, Koyanagi S, Takiguchi T, Kuramoto Y, Soeda S, Shimeno H, Higuchi S, Ohdo S. 24-hour oscillation of mouse methionine aminopeptidase2, a regulator of tumor progression, is regulated by clock gene proteins. Cancer Res. 2004;64:8328–33. doi: 10.1158/0008-5472.CAN-04-2122. [DOI] [PubMed] [Google Scholar]
- 58.Smaaland R, Sothern RB, Laerum OD, Abrahamsen JF. Rhythms in human bone marrow and blood cells. Chronobiol Int. 2002;19:101–27. doi: 10.1081/cbi-120002594. [DOI] [PubMed] [Google Scholar]
- 59.Bjarnason GA, Jordan RC, Wood PA, Li Q, Lincoln DW, Sothern RB, Hrushesky WJ, Ben-David Y. Circadian expression of clock genes in human oral mucosa and skin: Association with specific cell-cycle phases. Am J Pathol. 2001;158:1793–801. doi: 10.1016/S0002-9440(10)64135-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bjarnason GA, Jordan R. Rhythms in human gastrointestinal mucosa and skin. Chronobiol Int. 2002;19:129–40. doi: 10.1081/cbi-120002595. [DOI] [PubMed] [Google Scholar]
- 61.Ruifrok AC, Weil MM, Thames HD, Mason KA. Diurnal variations in the expression of radiation-induced apoptosis. Radiat Res. 1998;149:360–5. [PubMed] [Google Scholar]
- 62.Li XM, Metzger G, Filipski E, Boughattas N, Lemaigre G, Hecquet B, Filipski J, Levi F. Pharmacologic modulation of reduced glutathione circadian rhythms with buthionine sul foximine: Relationship with cisplatin toxicity in mice. Toxicol Appl Pharmacol. 1997;143:281–90. doi: 10.1006/taap.1996.8088. [DOI] [PubMed] [Google Scholar]
- 63.Zhang R, Lu Z, Liu T, Soong SJ, Diasio RB. Relationship between circadian-dependent toxicity of 5-fluorodeoxyuridine and circadian rhythms of pyrimidine enzymes: Possible relevance to fluoropyrimidine chemotherapy. Cancer Res. 1993;53:2816–22. [PubMed] [Google Scholar]
- 64.Ohdo S, Makinosumi T, Ishizaki T, Yukawa E, Higuchi S, Nakano S, Ogawa N. Cell cycle-dependent chronotoxicity of irinotecan hydrochloride in mice. J Pharmacol Exp Ther. 1997;283:1383–8. [PubMed] [Google Scholar]
- 65.Granda TG, Liu XH, Smaaland R, Cermakian N, Filipski E, Sassone-Corsi P, Levi F. Circadian regulation of cell cycle and apoptosis proteins in mouse bone marrow and tumor. Faseb J. 2005;19:304–6. doi: 10.1096/fj.04-2665fje. (Epub 2004 Nov 15) [DOI] [PubMed] [Google Scholar]
- 66.Matsuo T, Yamaguchi S, Mitsui S, Emi A, Shimoda F, Okamura H. Control mechanism of the circadian clock for timing of cell division in vivo. Science. 2003;302:255–9. doi: 10.1126/science.1086271. (Epub 2003 Aug 21) [DOI] [PubMed] [Google Scholar]
- 67.Fu L, Pelicano H, Liu J, Huang P, Lee C. The circadian gene Period2 plays an important role in tumor suppression and DNA damage response in vivo. Cell. 2002;111:41–50. doi: 10.1016/s0092-8674(02)00961-3. [DOI] [PubMed] [Google Scholar]
- 68.Haus E, Dumitriu L, Nicolau GY, Bologa S, Sackett-Lundeen L. Circadian rhythms of basic fibroblast growth factor (bFGF), epidermal growth factor (EGF), insulin-like growth factor-1 (IGF-1), insulin-like growth factor binding protein-3 (IGFBP-3), cortisol, and melatonin in women with breast cancer. Chronobiol Int. 2001;18:709–27. doi: 10.1081/cbi-100106083. [DOI] [PubMed] [Google Scholar]
- 69.Holzheimer RG, Curley P, Saporoschetz IB, Doherty JM, Mannick JA, Rodrick ML. Circadian rhythm of cytokine secretion following thermal injury in mice: Implications for burn and trauma research. Shock. 2002;17:527–9. doi: 10.1097/00024382-200206000-00015. [DOI] [PubMed] [Google Scholar]
- 70.Liu JH. Circadian variations of transforming growth factor-beta2 and basic fibroblast growth factor in the rabbit aqueous humor. Curr Eye Res. 2002;24:75–80. doi: 10.1076/ceyr.24.1.75.5435. [DOI] [PubMed] [Google Scholar]
- 71.Filipski E, King VM, Li X, Granda TG, Mormont MC, Liu X, Claustrat B, Hastings MH, Levi F. Host circadian clock as a control point in tumor progression. J Natl Cancer Inst. 2002;94:690–7. doi: 10.1093/jnci/94.9.690. [DOI] [PubMed] [Google Scholar]
- 72.Schernhammer ES, Laden F, Speizer FE, Willett WC, Hunter DJ, Kawachi I, Fuchs CS, Colditz GA. Night-shift work and risk of colorectal cancer in the nurses’ health study. J Natl Cancer Inst. 2003;95:825–8. doi: 10.1093/jnci/95.11.825. [DOI] [PubMed] [Google Scholar]
- 73.Schernhammer ES, Laden F, Speizer FE, Willett WC, Hunter DJ, Kawachi I, Colditz GA. Rotating night shifts and risk of breast cancer in women participating in the nurses’ health study. J Natl Cancer Inst. 2001;93:1563–8. doi: 10.1093/jnci/93.20.1563. [DOI] [PubMed] [Google Scholar]
- 74.Kume K, Zylka MJ, Sriram S, Shearman LP, Weaver DR, Jin X, Maywood ES, Hastings MH, Reppert SM. mCRY1 and mCRY2 are essential components of the negative limb of the circadian clock feedback loop. Cell. 1999;98:193–205. doi: 10.1016/s0092-8674(00)81014-4. [DOI] [PubMed] [Google Scholar]
- 75.Yagita K, Tamanini F, Yasuda M, Hoeijmakers JH, van der Horst GT, Okamura H. Nucleocytoplasmic shuttling and mCRY-dependent inhibition of ubiquitylation of the mPER2 clock protein. EMBO J. 2002;21:1301–14. doi: 10.1093/emboj/21.6.1301. [DOI] [PMC free article] [PubMed] [Google Scholar]