

Abstract
In response to injury, the highly specialized and terminally differentiated glomerular visceral epithelial cell, or podocyte, may undergo several cell fates, including dedifferentiation and proliferation, persistent cell cycle arrest, hypertrophy, apoptosis, or necrosis. Common to these potential outcomes of injury is their ultimate regulation at the level of the cell cycle. There is now a large body of literature confirming the importance of cell cycle regulatory proteins in the cellular response to injury. Although CDK inhibitor p21 levels increase in podocytes following injury, the role of p21 is unclear in focal segmental glomerulosclerosis (FSGS), in part because its function depends heavily on the cytotoxic stimulus and the cellular context. Adriamycin (ADR) is a podocyte toxin used to induce experimental FSGS. The purpose of this study was to define the role of p21 in ADR-induced podocyte injury. BALB/c mice, a strain carrying the recessive ADR susceptibility gene, were backcrossed against c57B6 p21−/− mice to yield a 12th generation BALB/c p21−/− strain. Experimental FSGS was induced by injection of ADR 12 mg/kg × 2 doses (n = 8/group), with mice killed at 1, 2, 8, and 11 wk. Diseased p21−/− mice demonstrated worse albuminuria, more widespread glomerulosclerosis, and higher blood urea nitrogen compared with diseased p21+/+ mice. In diseased p21−/− mice vs. p21+/+ mice, apoptosis [measured by TdT-mediated dUTP nick end labeling (TUNEL) assay] was increased, and podocyte number (measured by WT-1 immunostaining) was decreased. To validate these findings in vitro, we utilized differentiated mouse podocytes, p21−/− and p21+/+, exposed to 0.125 μg/ml ADR. Apoptosis, measured by Hoechst 33342 staining and TUNEL assay, was greater in cultured p21−/− podocytes compared with p21+/+ podocytes. Reconstitution of p21 via retroviral transfection rescued the p21−/− podocytes from apoptosis. We conclude that p21 is prosurvival in the podocyte's response to ADR-induced injury. Ongoing studies are defining the mechanisms of this protective effect as it relates to DNA damage and apoptosis.
Keywords: apoptosis, adriamycin nephropathy, focal glomerulosclerosis
chronic kidney disease (CKD) constitutes a major threat to public health. In the United States, there are ∼400,000 end-stage renal disease (ESRD) patients and 20 million CKD patients. By the year 2015, it is predicted that there will be 136,000 patients with incident ESRD and 712,000 patients with prevalent disease, increasing the public and private expenditure for dialysis to $32 billion per year (10, 22). Studies of kidney biopsy archives suggest that focal segmental glomerulosclerosis (FSGS) is now the most common primary glomerular disease leading to ESRD, and the incidence has increased dramatically over the last 20 years (31). FSGS is a clinicopathologic syndrome exemplified by proteinuria, elevated creatinine, and segmental glomerular scarring, a process by which extracellular matrix replaces normal architecture, eventually leading to CKD (7). FSGS may be primary or secondary to conditions including hypertension, obesity, viruses (including HIV), drugs, and reduced nephron mass (62). Regardless of the cause, emerging data support a strong correlation between injury to the glomerular visceral epithelial cell, or podocyte, and development of FSGS (53).
The highly specialized and terminally differentiated podocyte is a critical component of the glomerular filtration barrier and functions to prevent urinary protein leakage and to provide structural support to the capillary wall, thereby counteracting the hydrostatic pressure within the glomerulus (46). Common to many human kidney diseases and experimental animal models is a strong association between podocyte injury and the development of progressive kidney disease. In fact, there is compelling evidence that a decline in podocyte number strongly correlates with, and likely underlies, development of FSGS in humans and animals (30, 41, 45, 66). Defining the causal mechanisms of reduced podocyte number is fundamental to developing therapies for proteinuria and FSGS.
In response to injury, podocytes may undergo several cell fates, including proliferation, dedifferentiation, hypertrophy, apoptosis, and necrosis. Common to these disparate outcomes of renal injury is their ultimate regulation at the level of the cell cycle. There is now an expanding body of literature confirming the importance of cell cycle regulatory proteins in the cellular response to injury (39). Although cyclin-dependent kinase (CDK) inhibitor p21 levels increase in podocytes following injury (27, 32, 54), the function of p21 can vary substantially, based on its concentration and subcellular localization, the cell type and cellular context, and the cytotoxic stimulus (1, 9, 15, 29). In nonimmune-mediated FSGS, the role of p21 is unknown and is the focus of our current study.
Adriamycin (ADR), an anthracycline antibiotic, is a podocyte toxin used to induce experimental FSGS (6, 47, 65) in susceptible murine strains (67, 68). Despite the common clinical use of ADR for its antitumor effects, its cytotoxic mechanisms are not well-understood. Proposed mechanisms include DNA intercalation, reactive oxygen species generation (12), and/or topoisomerase II inhibition (21). The purpose of this study is to define the role of CDK inhibitor p21 in ADR-induced podocyte injury, both in vitro and in vivo. We hypothesize that the presence of the CDK inhibitor p21 is protective in the setting of ADR-induced podocyte injury. One mechanism whereby p21 may be protective in the setting of ADR-induced podocyte injury is in its capacity as a cell cycle checkpoint to maintain growth arrest in the setting of genotoxic stress. We hypothesize that without p21, cell cycle arrest cannot be maintained and the accumulation of ADR-induced damaged DNA leads to apoptosis.
MATERIALS AND METHODS
ADR nephropathy model of experimental FSGS.
In mice, the ADR nephropathy model has proven to be a robust experimental analog of human focal glomerulosclerosis. Therefore, the ADR nephropathy model was chosen to examine more closely the role of CDK inhibitor p21 in the initiation and propagation of FSGS. BALB/c mice, a strain carrying the recessive ADR susceptibility gene (67, 68), were backcrossed against c57B6 p21−/− mice (4) to yield a 12th generation BALB/c p21−/− strain. After a preliminary dose-finding study, ADR nephropathy was induced in male p21+/+ and p21−/− mice, aged 12 wk, by tail vein injection of ADR 12 mg/kg body wt × 2 (n = 8/group), following a protocol that was a modification of methods previously described (6). Similar to findings by others (63), in our preliminary studies, a one-time dose induced only transient proteinuria, peaking at day 14. For predictable and reproducible induction of sustained proteinuria and progressive glomerulosclerosis, two doses, separated by 4 wk, were utilized in our studies. During a pilot and feasibility study, we discovered that the mice experienced a significant weight loss by ∼11 wk in the disease course. Therefore, we determined that the mice would be killed at the 11-wk time point. Control animals were injected with equal volume of vehicle only (0.9% NaCl).
Weekly and before death, a weight was recorded and urine was collected from each mouse in metabolic cages to determine 24-h albumin excretion. Due to the small urine volumes of BALB/c mice, urine albumin was measured by radial immunodiffusion assay following a protocol that was a modification of methods previously described (37). Briefly, rabbit anti-mouse albumin antibody at 1:75 dilution (Accurate Chemical, Westbury, NY) and 4% rabbit serum (Pel-Freez, Rogers, AR) were incorporated into a thin layer of 1.5% type I, low EEO agarose gel (Sigma) in 0.5 M veronal buffer, poured into Integrid 100 × 15-mm square petri dish on a leveling platform (VWR, West Chester, PA). A small volume of urine was placed in a well cut into the agar layer. As the antigens diffuse from the well, only the specific antigen (albumin) reacted with its antibody in the agar. The reaction formed a halo of precipitation around the well. A measurement of the halo after it had reached maximal size was related directly to antigen concentration, with reference made to a calibration curve prepared from known concentrations of purified fraction V mouse albumin standards (MP Biomedicals, Irvine, CA) tested under identical conditions. Creatinine was measured in the urine via a colorimetric assay (Cayman Chemical, Ann Arbor, MI) and an albumin-to-creatinine ratio was calculated.
Death was induced by cervical dislocation. Blood samples obtained at time of death were centrifuged (12,000 g for 5 min) and plasma was collected for measurement of blood urea nitrogen (BUN) via QuantiChrom Urea Assay kit (Bioassay Systems, Hayward, CA). Tissues were obtained for renal biopsies. Histopathology was examined after periodic acid-Schiff staining. The animal care committee of the University of Washington School of Medicine reviewed and approved the experimental protocol for the adriamycin nephropathy mouse model of experimental FSGS. All animal procedures were conducted in accord with the Institutional Animal Care and Use Committee.
Immunohistochemistry.
Indirect immunoperoxidase immunostaining for WT-1, Bcl-2, and phospho-ERK was performed on formalin-fixed paraffin-embedded kidney specimens. Briefly, 4-μm tissue sections were deparaffinized in Histo-Clear (National Diagnostics, Atlanta, GA) and rehydrated in graded ethanol. Endogenous peroxidases were blocked with 3% H2O2, followed by overnight incubation with primary antibody for WT-1, diluted 1:1,000 (Santa Cruz Biotechnology, Santa Cruz, CA), Bcl-2, diluted 1:250 (Cell Signaling, Beverly, MA), and phospho-ERK, diluted 1:250 (Cell Signaling), in 1% BSA in PBS. After being washed in PBS, sections were incubated with biotinylated secondary antibody, diluted in 1% BSA in PBS, for 1 h at room temperature. ABC-Elite reagent (Vector Laboratories, Burlingame, CA) was used for signal amplification and 3,3′-diaminobenzidine (DAB; Sigma) catalyzed by NiCl2 was used as chromagen. Slides were counterstained with methyl green or hematoxylin, dehydrated, and coverslipped.
Apoptosis detection.
TdT-mediated dUTP nick end labeling (TUNEL) staining was utilized to determine whether apoptosis occurred following exposure to ADR. The protocol was a modification of methods previously described (20). TUNEL staining was performed on 4-μm formalin-fixed paraffin-embedded tissue sections. Following deparaffinization and rehydration, endogenous peroxidases were inactivated with 3% H2O2. Tissue sections were treated with citric acid to enhance antigen retrieval. Nuclei were permeabilized by incubation with proteinase K for 20 min at room temperature. Following incubation in One-Phor-All buffer (GE Healthcare, Piscataway, NJ), fragmented DNA was labeled by exposure of sections to diluted TdT (GE Healthcare) and biotin-14-dATP (Invitrogen, Grand Island, NY) for 60 min. The reaction was terminated with PBS. ABC-Elite reagent (Vector Laboratories) was used for signal amplification. DAB (Sigma) with NiCl2 was used as chromagen. Slides were counterstained with methyl green, dehydrated, and coverslipped.
Mouse podocytes in culture.
A conditionally immortalized mouse podocyte cell line isolated from H-2Kb-tsA58 transgenic mice kidneys (Jackson Laboratory, Bar Harbor, ME) was used in vitro, as we previously reported (28, 42, 55). In brief, in this cell line, γ-interferon-inducible H-2Kb promoter controls a temperature-sensitive SV40 large T cell antigen. To induce proliferation, cells were grown on Primaria plates (VWR International) coated with collagen I (BD Biosciences) at 33°C in RPMI 1640 media (Life Technologies, Gaithersburg, MD) supplemented with 9% FCS (Summit Biotechnology, Fort Collins, CO), with added recombinant mouse γ-interferon 50 U/ml (Roche, Indianapolis, IN) (23). To induce the quiescent, differentiated phenotype, cells were grown at 37°C without γ-interferon (growth-restrictive condition). Experiments were performed at least three times, on growth-restricted day 12–14, utilizing more than one cell clone to verify reproducibility of results. The p21+/+ and p21−/− cell lines were derived in our lab, identified, and characterized by immunostaining and RT-PCR, as previously described (64).
Immunocytochemistry.
Given the importance of actin cytoskeletal disruption and reorganization in the development of foot process effacement characteristic of FSGS, F-actin staining was performed on podocytes in culture. Podocytes from p21−/− and p21+/+ cell lines were plated in 24-well plates. After 14 days in culture under growth-restrictive conditions, the cells were exposed to ADR (or the appropriate control) for 48 h. Following ADR exposure, the podocytes were fixed with 2% paraformaldehyde (Sigma) containing 4% sucrose for 15 min. To visualize F-actin, fixed cells were incubated with FITC-conjugated phalloidin diluted 1:100 (Sigma) for 30 min, and then mounted with Vectashield containing 4′,6-diamidino-2-phenylindole (Vector Laboratories). Other investigators reported that normal podocytes are characterized by a central group of thick and well-defined actin stress fibers. Following injury, podocytes lose these stress fibers and form lamellipodia. An actin score was obtained, as a modified calculation of what has been previously described in the literature, using the following scale for the appearance of actin stress fibers based on intensity of staining: 0 = absence of stress fibers, 1 = <25% of normal, 2 = 25–50% of normal, 3 = 50–75% of normal, and 4 = 75–100% of normal (33).
Apoptosis detection in vitro following ADR exposure.
Following preliminary dose-defining studies, growth-restricted heat-sensitive mouse podocytes were exposed to 0.125 μg/ml ADR for 6, 18, 24, and 48 h, with appropriate negative control. First, after growing podocytes to 80% confluence on plastic plates, apoptosis was measured by staining with Hoechst 33342 (Sigma, St. Louis, MO), as previously described (26). Briefly, at the above time points following exposure to ADR, DNA-binding dye Hoechst 33342, which stains the condensed chromatin of apoptotic cells more brightly than the looser chromatin of living cells, was added to the media at a 1:100 dilution. After incubation for 5 min, Hoechst-positive cells (i.e., those with nuclear condensation) were quantitated. The number of Hoechst-positive cells was determined by counting at least 300 cells per well. All experiments were performed a minimum of three times.
To confirm the presence of apoptosis, TUNEL staining was next performed. The protocol was a modification of methods previously described (20). Based on the results from the Hoechst 33342 staining, only the 48-h time point was examined. Briefly, following exposure to ADR for 48 h, cultured podocytes were fixed with 10% buffered formalin overnight at 4°C. Following rehydration with PBS, the above protocol was followed. Cells were counterstained with Camco Quik stain (Cambridge Diagnostic, Fort Lauderdale, FL).
DNA damage detection in vitro following ADR exposure.
To determine whether the presence or absence of p21 affected the degree of ADR-induced DNA damage, the single-cell gel electrophoresis assay (CometAssay; Trevigen, Gaithersburg, MD) was utilized, as previously described (38). Briefly, following exposure to ADR for 48 h, podocytes were gently harvested and embedded in an agarose layer on a microscope slide. Cells were lysed to remove cellular proteins and DNA was uncoiled under alkaline conditions. After unwinding, DNA was electrophoresed for 20 min in 1× TBE buffer and stained with fluorescent dye. In contrast to undamaged and supercoiled DNA that remains within the nuclear membrane, the podocytes with damaged DNA, which migrates away from the nucleus, demonstrated the characteristic long “comet tail.” The extent of DNA liberated from the head of the “comet” was directly proportional to DNA damage. The number of positive cells with long comet tails was determined by counting at least 200 cells per well. All experiments were performed a minimum of three times.
Reconstitution of p21 in p21−/− podocytes by retroviral transduction.
Reconstitution of p21 protein expression in p21−/− mouse podocytes in culture was achieved using a retroviral transduction system. The pBabe vectors encoding full-length human p21 or GFP were transfected into Phoenix packaging cells to generate retrovirus (8, 56). The retrovirus-containing media were harvested and filtered onto 50% confluent, proliferating, undifferentiated p21−/− podocytes. Following 48-h selection with puromycin (2.5 μg/ml), podocytes were passaged and transferred to growth-restricted conditions to initiate growth arrest and differentiation, as previously described (24). Western blotting for p21 to show successful transfection with reexpression of p21 in the p21−/− podocytes was performed, as previously described (11). p21−/− Podocytes with reconstituted p21 were exposed to ADR and apoptosis and DNA damage were measured using Hoechst 33342, TUNEL staining, and CometAssay, as detailed above.
Statistical analysis.
For comparison of mean values between two groups, the unpaired t-test was used. All values are means ± SD or SE, except where otherwise indicated. Statistical significance was evaluated using GraphPad Prism version 5.02 (GraphPad Software, La Jolla, CA). The experimental findings were considered statistically significant if P < 0.05. All photomicrographs were made at similar intensity and background. During data analysis, the observer was blinded to the treatment categories.
RESULTS
Weight loss was worse in p21−/− mice immediately following ADR injection.
Before ADR exposure, the mean body weights for p21+/+ and p21−/− mice were similar. Following the first injection of ADR, the p21−/− mice lost more weight than the p21+/+ mice and this difference reached significance (P = 0.005; Fig. 1). The weights reached a nadir 7–14 days after the first injection, after which both groups of mice began to regain weight and the differences between the means were no longer statistically significant. Following the second injection of ADR, the p21−/− mice once again lost more weight than the p21+/+ mice and this difference reached significance by week 6 (P = 0.020). However, at all subsequent measurements, the mean weights between the two groups were not statistically significantly different. The more significant weight loss following the ADR injections in the p21−/− mice supports the hypothesis that p21 is protective in the setting of ADR-induced disease.
Fig. 1.
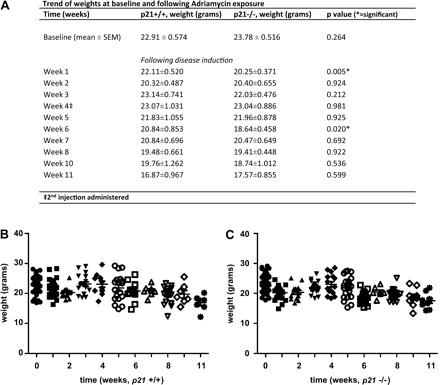
Trend of weights in p21+/+ and p21−/− mice following treatment with Adriamycin (ADR). A: weight loss trend. At baseline, before disease induction, there is no statistically significant difference between the mean body weights of p21+/+ and p21−/− mice. Following the injections, by weeks 1 and 6, p21−/− mice lost more weight to a significant degree. Other time points do not demonstrate significant differences. B and C: weights of individual mice at each time point during the 11-wk study. B: p21+/+ animals. C: p21−/− animals.
Worsened renal function and albuminuria in diseased p21−/− mice in experimental FSGS.
Following administration of ADR to induce experimental FSGS, biochemical parameters were assessed to determine severity of renal dysfunction in p21+/+ vs. p21−/− mice, and the results are shown in Fig. 2A. At baseline and in animals that received vehicle only, there was no difference in plasma BUN between p21+/+ and p21−/− mice. By 8 wk following disease induction in the ADR nephropathy model of experimental FSGS, there was a statistically significant increase in plasma BUN in the p21−/− mice compared with the p21+/+ mice (52.38 vs. 34.56 mg/dl, P = 0.027). The difference between diseased p21−/− and p21+/+ mice increased further by 11 wk following disease induction (96.21 vs. 52.70 mg/dl, respectively, P = 0.002; Fig. 2A).
Fig. 2.
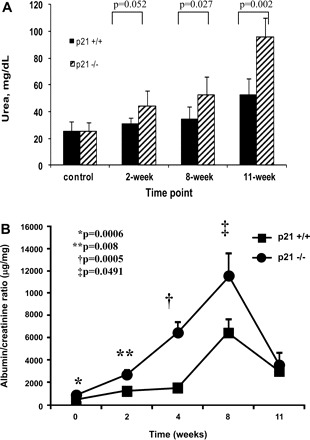
Plasma blood urea nitrogen and urinary albumin-to-creatinine ratio in p21+/+ and p21−/− mice following treatment with ADR. A: plasma blood urinary nitrogen. By 8 wk, diseased p21−/− mice had a statistically significantly greater plasma blood urinary nitrogen level than diseased p21+/+ mice. The difference remained statistically significant at 11 wk following disease induction (P < 0.05). B: urinary albumin-to-creatinine ratio. At baseline, 2 wk, 4 wk, and 8 wk, diseased p21−/− mice demonstrated statistically significantly greater levels of albumin-to-creatinine ratio than diseased p21+/+ mice (P < 0.05). By 11 wk, the difference was no longer statistically significant.
The albumin-to-creatinine ratio results are shown in Fig. 2B. At time points 0, 2 wk, 4 wk, and 8 wk, there was a statistically significant difference in urinary albumin-to-creatinine ratio, with p21−/− mice exhibiting higher ratios compared with p21+/+ mice (time point 0: 797.1 vs. 399.4 μg/mg, P = 0.0006; time point 2 wk: 2,644 vs. 1,161 μg/mg, P = 0.008; time point 4 wk: 6,385 vs. 1,476 μg/mg, P = 0.0005; time point 8 wk: 4,250 vs. 2,965 μg/mg, P = 0.0491). By 11 wk, there was no longer a statistically significant difference between the albumin-to-creatinine ratio of p21−/− and p21+/+ mice.
Together, these results demonstrate that treatment with ADR caused renal dysfunction, characterized by elevated plasma BUN and urinary albumin-to-creatinine ratio, and this renal dysfunction was worse in the p21−/− mice compared with p21+/+ mice.
Increased glomerulosclerosis in diseased p21−/− mice with ADR nephropathy.
The histopathology from mice that received ADR was typified by early glomerular vacuolization, tuft collapse, mesangial expansion, and tubulointerstitial collagen deposition, with later development of focal glomerulosclerosis and interstitial fibrosis, as previously described (65) (Fig. 3, A–D). To assess semiquantitatively the severity of glomerular injury, a glomerular sclerosis index (GSI) was calculated by examining histopathologic sections stained with periodic acid-Schiff, as described previously, (38, 44) using the following scale: 0 = normal glomerulus, 1 = sclerotic area <25% of glomerular area, 2 = sclerotic area 25–50% of glomerular area, 3 = sclerotic area 50–75% of glomerular area, and 4 = sclerotic area >75% of glomerular area or global sclerosis. The GSI was calculated by multiplying the number of glomeruli with a sclerosis score of 1 by one, 2 by two, and so on. These values were summed to obtain the final GSI. At baseline and in animals that received vehicle only, there was no statistically significant difference in GSI between the p21+/+ and p21−/− mice. As expected, the GSI increased in both p21+/+ and p21−/− mice at 8 and 11 wk following disease induction with ADR administration. However, there was a statistically significant difference in GSI by 11 wk in the p21−/− mice compared with the p21+/+ mice (GSI score 196.78 vs. 141.29, respectively, P = 0.045; Fig. 3E), which was consistent with the urinary albumin-to-creatinine ratio and plasma BUN data. These results provide supporting data that there was worse disease in the p21−/− mice in the ADR nephropathy model of focal glomerulosclerosis.
Fig. 3.
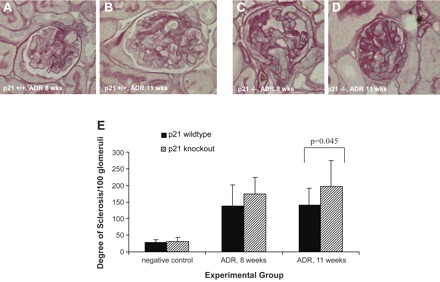
Histopathologic changes by periodic acid-Schiff (PAS) staining in p21+/+ and p21−/− mice following treatment with ADR. A and B: histopathology (original magnification ×40). Representative micrographs of PAS staining of tissues from p21+/+ mice are shown, time points 8 and 11 wk following disease induction. C and D: histopathology (original magnification ×40). Representative micrographs of PAS staining of tissues from p21−/− mice are shown, time points 8 and 11 wk following disease induction. Tissue from diseased p21−/− mice exhibited greater disease severity than tissue from diseased p21+/+ mice. E: sclerosis index. Tissue from diseased p21−/− mice exhibited statistically significantly greater disease severity compared with tissue from diseased p21+/+ mice, as semiquantitatively assessed by sclerosis index, at 11 wk following disease induction (P < 0.05).
Decreased podocyte number in diseased p21−/− mice with ADR nephropathy.
To determine whether podocyte number was reduced following disease induction by ADR, immunostaining for Wilms′ tumor protein (WT-1), a surrogate marker for podocyte number, was performed. By WT-1 staining, there was no significant difference in the baseline complement of podocytes between p21+/+ and p21−/− mice before disease induction. In mice receiving vehicle and at all early time points (1, 2, and 8 wk) following disease induction by ADR, there was no statistically significant difference in the number of WT-1-positive cells in glomeruli of p21+/+ and p21−/− mice. By 11 wk following disease induction by ADR, there was a statistically significant decrease in WT-1-positive cells in the glomeruli of p21−/− compared with the p21+/+ mice (5.16 vs. 6.77 per glomerulus, respectively, P = 0.003; Fig. 4). These results show that, at the later time point, in glomeruli that lacked CDK inhibitor p21, there was a loss of WT-1-positive cells following ADR-induced injury.
Fig. 4.
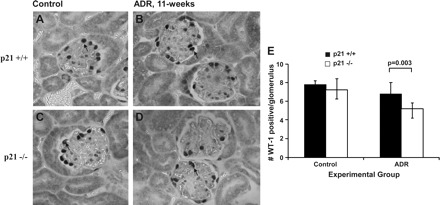
WT-1 immunostaining in p21+/+ and p21−/− mice following treatment with ADR. By immunohistochemistry (original magnification ×40), the number of WT-1-positive cells was decreased significantly in tissues from p21−/− mice 11 wk following disease induction compared with time-matched diseased p21+/+ mice (arrows indicate examples of positive cells; P < 0.05). A: representative immunostaining for WT-1, a surrogate marker for podocyte number, in tissue from p21+/+ mice that received vehicle only. B: representative immunostaining for WT-1 in tissue from p21+/+ mice 11 wk following disease induction. C: representative immunostaining for WT-1 in tissue from p21−/− mice that received vehicle only. D: representative immunostaining for WT-1 in tissue from p21−/− mice 11 wk following disease induction. E: quantification of WT-1-positive cells per glomerulus shown in graphic form (all glomeruli counted per section × 8 animals in each group).
Podocyte apoptosis in diseased p21−/− mice in ADR nephropathy.
Given the reduction in WT-1-positive cells in the p21−/− mice following disease induction by ADR treatment, the TUNEL assay was next performed to determine whether the decrease in WT-1-positive cells was secondary to apoptosis. At time points 1 and 2 wk, there was a statistically significant difference in the number of TUNEL-positive cells (per 200 glomeruli counted) in the p21−/− mice compared with the p21+/+ mice (week 1, means ± SE: 18 ± 1.7 for p21−/− mice vs. 9.9 ± 1.8 for p21+/+ mice, P = 0.0085; week 2, means ± SE: 37 ± 8.1 for p21−/− mice vs. 9.7 ± 2.8 for p21+/+ mice; P = 0.0130; Fig. 5). These data support the hypothesis that, in the course of disease induction following ADR treatment, apoptosis may account for decreased podocyte number in the p21−/− mice.
Fig. 5.

Apoptosis detection in p21+/+ and p21−/− mice following treatment with ADR. TdT-mediated dUTP nick end labeling (TUNEL) staining of tissue sections. Representative images of tissue sections from mice 1 wk following disease induction in p21+/+ group (A) vs. p21−/− group (B). C and D: quantification of TUNEL-positive cells per 200 glomeruli counted. Following disease induction, there was a statistically significantly greater number of TUNEL-positive cells in tissue from diseased p21−/− mice compared with tissue from diseased p21+/+ mice at 1 wk (P < 0.05; C) and at 2 wk (P < 0.05; D).
Reconstitution of p21 rescues p21−/− podocytes from ADR-induced apoptosis, in vitro.
To confirm the in vivo data, we next examined p21−/− and p21+/+ differentiated immortalized mouse podocytes in culture, previously characterized by our lab (64). Following exposure to ADR, p21+/+ podocytes exhibited apoptosis, measured by Hoechst 33342 staining, to statistically significant levels, compared with negative control and 6-h time point, at 18, 24, and 48 h (P < 0.05; Fig. 6, A, C, E). Similarly, following exposure to ADR, p21−/− podocytes also exhibited apoptosis by Hoechst 33342 staining to statistically significant levels, compared with negative control and 6-h time point, at 18, 24, and 48 h (P < 0.05; Fig. 6, B, D, E). By 48 h following injury induced by ADR exposure, there was a statistically significant difference in the number of apoptotic cells by Hoechst 33342 staining between p21−/− and p21+/+ podocytes, with a greater number of p21−/− apoptotic cells (94.89 ± SE 9.238 Hoechst-positive cells/300) compared with p21+/+ apoptotic cells (64.10 ± SE 5.009 Hoechst-positive cells/300; P = 0.010). Earlier time points did not show a statistically significant difference between p21−/− and p21+/+ podocytes. The results show that, over time, lack of p21 leads to a greater degree of apoptosis following ADR exposure (Fig. 6E).
Fig. 6.
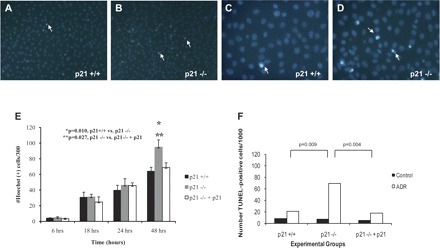
Apoptosis detection in p21+/+ and p21−/− podocytes in culture following treatment with ADR. A–D: representative images of podocytes stained with Hoechst 33342 following treatment with ADR (A and C: p21+/+ podocytes; B and D: p21−/− podocytes; A and B: original magnification ×10; C and D: original magnification ×20). E: quantification of Hoechst 33342-positive podocytes in culture per 300 cells counted. By 18 h, both p21+/+ and p21−/− podocytes in culture exhibited statistically significant levels of apoptosis compared with their respective baseline levels (P < 0.05). By 48 h, there was a statistically significant difference in the number of apoptotic cells by Hoechst staining between p21+/+ and p21−/− podocytes, with greater number of Hoechst-positive cells in the p21−/− podocytes (P < 0.05). Following reconstitution of p21 in p21−/− podocytes via retroviral transfection, there was a statistically significant decrease in Hoechst-positive cells 48 h after ADR treatment in the p21−/− podocytes with reconstituted p21 vs. the p21−/− podocytes (P < 0.05). F: quantification of TUNEL-positive podocytes in culture per 1,000 cells counted. At 48 h following ADR treatment, there was a statistically significantly greater number of TUNEL-positive cells in the p21−/− podocytes compared with the p21+/+ podocytes (P < 0.05). Following reconstitution of p21 via retroviral transfection, the number of TUNEL-positive cells was decreased significantly in the p21−/− podocytes with reconstituted p21 vs. the p21−/− podocytes (P < 0.05).
We next sought to determine whether reconstitution of p21 would rescue p21−/− podocytes from apoptosis. Following retroviral transfection of p21−/− podocytes with human p21 vector, by 48 h after injury induced by ADR treatment, there was a statistically significant decrease in apoptotic cells by Hoechst 33342 staining in p21−/− podocytes with reconstituted p21 (68.840 ± SE 5.980 Hoechst-positive cells/300) vs. p21−/− podocytes, approaching the level seen in the p21+/+ podocytes (P = 0.027; Fig. 6E). Earlier time points did not show a statistically significant difference in the number of apoptotic cells between p21−/− podocytes and p21−/− podocytes with reconstituted p21. These results demonstrate that reconstitution of p21 protects p21−/− podocytes from the increased susceptibility to apoptosis following ADR treatment.
To validate the results obtained by Hoechst 33342 staining, the TUNEL assay was utilized as another measure of apoptosis detection for the in vitro studies. Given that only the 48-h time point yielded a statistically significant difference in apoptosis between the groups by Hoechst 33342 staining, the TUNEL assay was performed at 48 h following ADR treatment. There was a statistically significant difference between the number of TUNEL-positive p21+/+ podocytes (21.438/1,000) compared with the number of TUNEL-positive p21−/− podocytes (69.125/1,000; P = 0.009; Fig. 6F). With reconstitution of p21 via retroviral transfection of p21−/− podocytes with human p21 vector, following ADR treatment for 48 h, there was a statistically significant decrease in the number of TUNEL-positive cells in p21−/− with reconstituted p21 podocytes (18/1,000) vs. the number of TUNEL-positive cells in p21−/− podocytes (P = 0.004). These studies validated the finding that reconstitution of p21 protected p21−/− podocytes from ADR-induced apoptosis.
Increased DNA damage in p21−/− podocytes following ADR exposure, in vitro.
To determine potential mechanisms for the protective effect of p21 in ADR-induced podocyte injury, we performed immunohistochemistry for proteins known to have important roles in the apoptosis cascade, including phospho-ERK and Bcl-2 (data not shown). There was no statistically significant difference in positive cells, when tissue was stained for the aforementioned apoptosis mediators, between p21+/+ and p21−/− mice. Although we and others identified these mediators as important in other models of glomerular injury, they do not appear to be the mechanism whereby p21 exerts its protective effect in murine ADR nephropathy (3, 64).
We previously found that DNA damage in podocytes leads to activation of cell cycle checkpoints, including the CDK inhibitor p21 (38, 48). These cell cycle checkpoints generate pauses in cell cycle progression when DNA damage is present, permitting the cell sufficient time for DNA repair or other cell fate decisions. Since ADR has been known to induce DNA damage in other cell culture models, we performed the CometAssay to determine whether DNA damage was present in podocytes following ADR exposure and whether the presence of p21 attenuated the extent of DNA damage. In Fig. 7, we show that DNA damage was present in podocytes following ADR-induced injury. The degree of DNA damage was significantly increased in p21−/− podocytes when compared with p21+/+ (113 positive cells per 200 cells counted ± SE 8.185 vs. 17.67 ± SE 7.055, P = 0.0009). With reconstitution of p21, the degree of DNA damage was significantly reduced (57 ± SE 5.859). The results provide supporting data that one mechanism whereby p21 is protective in the setting of ADR-induced podocyte injury may be in its capacity as a cell cycle checkpoint effector to maintain growth arrest in the setting of genotoxic stress.
Fig. 7.
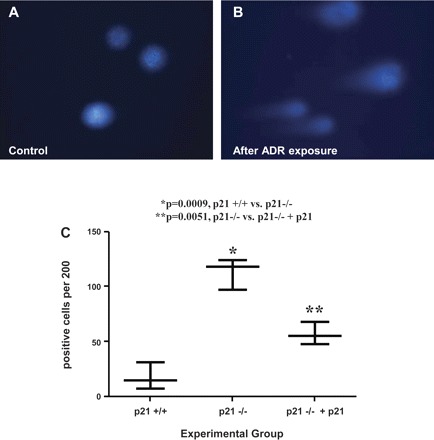
Increased DNA damage in p21−/− podocytes in culture following treatment with ADR. Damaged DNA was measured by the CometAssay in cultured podocytes following exposure to ADR at 48 h (original magnification ×20). A: representative micrograph of fluorescent DNA stain of control cells, showing undamaged and supercoiled DNA remaining within the nuclear cell membrane. B: representative micrograph of fluorescent DNA stain of ADR-treated podocytes, showing denatured DNA fragments migrating out from the cell in a long “comet tail.” C: quantification of podocytes with long comet tails. Following exposure to ADR, there was a statistically significant increase in the number of p21−/− podocytes with long comet tails compared with that seen in p21+/+ podocytes. Reconstitution of p21 significantly reduced the level of DNA damage (P < 0.05).
Actin cytoskeletal changes following ADR exposure, in vitro.
It is well-established that podocyte foot process effacement seen in FSGS is secondary to physical changes in the podocyte actin cytoskeleton, including disruption and reorganization. Because FSGS has been associated with cytoskeletal rearrangement, we next examined F-actin staining in our podocytes in culture. The results are shown in Fig. 8. Normal podocytes are characterized by a central group of thick well-defined actin stress fibers. Podocytes lose these stress fibers and form lamellipodia when injured. At baseline, before ADR exposure, p21−/− podocytes exhibited fewer stress fibers when compared with p21+/+ podocytes (P < 0.0001). With reconstitution of p21, the reduction in stress fibers was significantly less marked (P < 0.0001). Upon exposure to ADR, all groups exhibited a reduction in stress fibers to a significant degree (P < 0.0001) and the differences between groups remained significant, with p21−/− podocytes losing more stress fibers than the other groups (P < 0.0001). These results suggest that podocytes in culture have actin cytoskeletal changes that correlate with what is seen in vivo and that the presence of p21 protects the cell from marked actin reorganization and loss of stress fibers in response to ADR-induced injury.
Fig. 8.
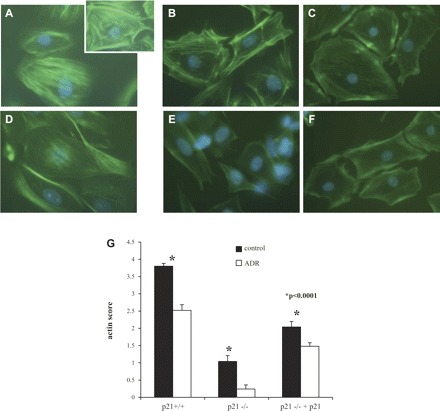
Diminished F-actin staining in p21−/− podocytes in culture at baseline and following treatment with ADR. The actin cytoskeleton was labeled with FITC-conjugated phalloidin to assess for actin cytoskeletal changes in podocytes following treatment with ADR. A: at baseline, before ADR exposure, the p21+/+ podocytes exhibit a central group of thick and well-defined actin stress fibers (inset shows close-up image; magnification ×20). B and C: before exposure, the p21−/− podocytes and p21−/− podocytes transfected with p21 exhibit decreased actin stress fibers compared with p21+/+ podocytes. D: following 48 h of treatment with ADR, there is decreased intensity of the actin stress fiber staining in p21+/+ podocytes. E: decrease in actin stress fibers is more marked in the p21−/− podocytes following ADR-induced injury. F: in p21−/− podocytes reconstituted with p21, there is less attenuation of actin stress fibers with injury. G: quantification of actin score, shown in graphic form. Within each group, upon treatment with ADR, the podocytes significantly lose actin stress fibers (P < 0.05). Compared with p21+/+ podocytes, p21−/− podocytes lose significantly more actin stress fibers. This loss is significantly reduced in the p21−/− podocytes with reconstituted p21 (P < 0.05).
DISCUSSION
FSGS is now the most common primary glomerular disease leading to ESRD, with the incidence increasing dramatically over the last 20 years (31). Emerging data support a strong correlation between injury to the glomerular visceral epithelial cell, or podocyte, and progressive kidney disease with consequent development of FSGS (53). The podocyte, with its highly complex cytoarchitecture that determines its elaborate function as a critical component of the glomerular filtration barrier, is a quiescent, terminally differentiated cell in the mature adult kidney under normal physiological conditions. Thus, the mechanisms that govern podocyte differentiation, and therefore proliferation, are essential to the normal function of this cell.
There is compelling evidence that a decline in podocyte number strongly correlates with, and likely underlies, development of FSGS in humans and animals (30, 41, 45, 66). There is an increasing body of literature showing that apoptosis (programmed cell death) of podocytes is a major cause of reduced podocyte number. Previous studies showed increased podocyte apoptosis in TGF-β transgenic mice (51), CD2AP null mice (52), and in rats exposed to puromycin aminonucleoside (16, 30, 50, 58). Indeed, several other potent stimuli for podocyte apoptosis have recently been described, including angiotensin II (13), mechanical strain (14), free radical generation (59), cyclosporine A (17), and the anthracycline antibiotic ADR (11). In vitro studies showed that during apoptosis, cells typically exit the cell cycle in late G1, although apoptotic cells can exit during any phase of the cell cycle (40).
Although, traditionally, cell cycle regulatory proteins have been considered to be involved solely in proliferation, there is mounting evidence that specific cell cycle proteins may also play important roles in the regulation of apoptosis (9, 19, 26, 34). Therefore, we sought to determine what role, if any, a specific cell cycle regulatory protein CDK inhibitor p21 has in the regulation of apoptosis in podocytes.
Several studies have highlighted CDK inhibitor p21 as a potentially important regulator of apoptosis (19). It is not entirely clear how p21 protects cells against apoptosis, but it has been proposed that either p21-mediated cell cycle arrest causes resistance to apoptosis or that p21 blocks apoptosis directly. However, paradoxically, in some cases, p21 can promote apoptosis (18, 35, 57, 64). Indeed, the function of p21 can vary substantially, based on its concentration and subcellular localization, the cellular context, and the cytotoxic agent. Therefore, an important question prompting this study was whether p21 was anti-apoptotic or proapoptotic in podocytes in an experimental model of FSGS and its in vitro correlate.
We previously showed that p21 levels are upregulated in podocytes in culture following exposure to ADR (11), a known podocyte toxin in rats and in susceptible murine strains (6, 47, 65, 67, 68). Because the role of p21 in podocyte apoptosis in the initiation and progression of FSGS was unknown, we utilized ADR to induce experimental FSGS. To validate the in vivo data, we then applied ADR to differentiated and immortalized podocytes in culture. A major finding in this study was that the presence of CDK inhibitor p21 was protective in the setting of ADR-induced podocyte injury, both in vivo and in vitro.
In ADR nephropathy, there was worse disease in the p21−/− mice compared with the p21+/+ mice by clinicopathologic parameters, including change in weight, plasma BUN, urinary albumin-to-creatinine ratio, and extent of glomerulosclerosis as semiquantitatively assessed by GSI. Worsened disease in the p21−/− mice correlated with early apoptosis and later reduction in podocyte number by WT-1 staining, underscoring the previously described relationship between a decline in podocyte number and progression to glomerulosclerosis. Upon ADR exposure of differentiated and immortalized podocytes in culture, at 18, 24, and 48 h, there was significant apoptosis in the p21−/− and p21+/+ cells when compared with the degree of apoptosis present for their respective negative control and 6-h time point. However, by 48 h following injury induction by ADR exposure, p21−/− podocytes exhibited significantly more apoptosis compared with that seen in p21+/+ podocytes.
To determine whether reconstitution of p21 would rescue the p21−/− podocytes from their increased susceptibility to ADR-induced apoptosis, p21 was transduced into p21−/− podocytes via retroviral transfection. By 48 h following injury induction by ADR exposure, p21−/− podocytes with reconstituted p21 demonstrated decreased apoptosis compared with p21−/− podocytes, with levels approaching those seen in the p21+/+ podocytes following ADR exposure. Thus, we demonstrated that reconstituted p21 protected the p21−/− podocytes from the increased susceptibility to apoptosis following ADR exposure. The protective effect of p21 also extends to the preservation of the normal pattern of the actin cytoskeleton following ADR-induced injury. In the absence of p21, there was a greater degree of loss of actin stress fibers and more distortion of the podocyte's normal architecture. In summary, we found that the presence of CDK inhibitor p21 is protective in the setting of ADR-induced podocyte injury, both in vitro and in vivo.
ADR has been known to induce DNA damage, thought to be secondary to DNA intercalation, free radical generation, and/or topoisomerase II inhibition (21). We previously found that DNA damage in podocytes leads to activation of cell cycle checkpoints (38, 48), molecular pathways that monitor passage through the cell cycle and generate pauses in cell cycle progression when DNA damage is present, permitting the cell sufficient time for DNA repair or other cell fate decisions (36). These observations support the hypothesis that p21, a downstream effector of p53 in the checkpoint pathway, plays an essential role in arresting the cell cycle in the presence of DNA damage. Others found that following ADR exposure, damaged cells that arrested their cell cycle did not undergo apoptosis. However, abnormal progression of the cell cycle, in the face of ADR-induced DNA damage, led to apoptosis (5). Therefore, one mechanism whereby p21 is protective may be in its capacity as a cell cycle checkpoint to maintain growth arrest in the setting of genotoxic stress. Indeed, we found that in podocytes lacking p21, there was a significantly increased level of DNA damage following ADR exposure when compared with p21+/+ podocytes. The extent of ADR-induced DNA damage was lessened when p21 was reconstituted. Therefore, it appears that without p21, cell cycle arrest cannot be maintained and the accumulation of ADR-induced damaged DNA leads to apoptosis. Taken together, p21 likely plays a critical role in the podocyte's cell cycle arrest in response to ADR-induced DNA damage and in the regulation of DNA damage response effectors.
Another potential mechanism whereby p21 is protective may be in its capacity to interact with proapoptotic molecules such as procaspase-3 (60, 61, 69) and apoptosis signal-regulating kinase-1 (2), inhibiting their activities. Several studies showed that p21 is potentially an important regulator of apoptosis by blocking apoptosis directly. The examination of these pathways has been ongoing in the cancer literature in recent years. Whether p21 may be blocking proapoptotic pathways in podocytes is unknown and will be a focus of future research efforts.
Defining the causal mechanisms of reduced podocyte number is fundamental to developing therapies for proteinuria and FSGS. In this study, we showed that p21 is prosurvival in the podocyte's response to ADR-induced injury and that one potential mechanism for this protective effect is the role of p21 in the DNA damage response, leading to cell cycle arrest in the presence of genotoxic stress. Ongoing studies are further defining the mechanisms of this protective effect as it relates to DNA damage and apoptosis.
GRANTS
This work was supported by a Robert Wood Johnson Foundation Grant 58826 to C. B. Marshall, by National Institutes of Health (NIH) Grant 3 RO1-DK-051096-06S1 to C. B. Marshall, and by the NIH Grants DK-60525, DK-56799, and DK-51096 to S. J. Shankland. S. J. Shankland is also an Established Investigator of the American Heart Association.
DISCLOSURES
No conflicts of interest are declared by the authors.
REFERENCES
- 1. Al-Douahji M, Brugarolas J, Brown PA, Stehman-Breen CO, Alpers CE, Shankland SJ. The cyclin kinase inhibitor p21WAF1/CIP1 is required for glomerular hypertrophy in experimental diabetic nephropathy. Kidney Int 56: 1691–1699, 1999 [DOI] [PubMed] [Google Scholar]
- 2. Asada M, Yamada T, Ichijo H, Delia D, Miyazono K, Fukumuro K, Mizutani S. Apoptosis inhibitory activity of cytoplasmic p21(Cip1/WAF1) in monocytic differentiation. EMBO J 18: 1223–1234, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Brinkkoetter PT, Olivier P, Wu JS, Henderson S, Krofft RD, Pippin JW, Hockenbery D, Roberts JM, Shankland SJ. Cyclin I activates Cdk5 and regulates expression of Bcl-2 and Bcl-XL in postmitotic mouse cells. J Clin Invest 119: 3089–3101, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brugarolas J, Chandrasekaran C, Gordon JI, Beach D, Jacks T, Hannon GJ. Radiation-induced cell cycle arrest compromised by p21 deficiency. Nature 377: 552–557, 1995 [DOI] [PubMed] [Google Scholar]
- 5. Cao W, Chi WH, Wang J, Tang JJ, Lu YJ. TNF-alpha promotes Doxorubicin-induced cell apoptosis and anti-cancer effect through downregulation of p21 in p53-deficient tumor cells. Biochem Biophys Res Commun 330: 1034–1040, 2005 [DOI] [PubMed] [Google Scholar]
- 6. Chen A, Sheu LF, Ho YS, Lin YF, Chou WY, Chou TC, Lee WH. Experimental focal segmental glomerulosclerosis in mice. Nephron 78: 440–452, 1998 [DOI] [PubMed] [Google Scholar]
- 7. Chen X, Moeckel G, Morrow JD, Cosgrove D, Harris RC, Fogo AB, Zent R, Pozzi A. Lack of integrin alpha1beta1 leads to severe glomerulosclerosis after glomerular injury. Am J Pathol 165: 617–630, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Clurman BE, Sheaff RJ, Thress K, Groudine M, Roberts JM. Turnover of cyclin E by the ubiquitin-proteasome pathway is regulated by cdk2 binding and cyclin phosphorylation. Genes Dev 10: 1979–1990, 1996 [DOI] [PubMed] [Google Scholar]
- 9. Coqueret O. New roles for p21 and p27 cell-cycle inhibitors: a function for each cell compartment? Trends Cell Biol 13: 65–70, 2003 [DOI] [PubMed] [Google Scholar]
- 10. Coresh J, Selvin E, Stevens LA, Manzi J, Kusek JW, Eggers P, Van Lente F, Levey AS. Prevalence of chronic kidney disease in the United States. JAMA 298: 2038–2047, 2007 [DOI] [PubMed] [Google Scholar]
- 11. Cormack-Aboud FC, Brinkkoetter PT, Pippin JW, Shankland SJ, Durvasula RV. Rosuvastatin protects against podocyte apoptosis in vitro. Nephrol Dial Transplant 24: 404–412, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Deman A, Ceyssens B, Pauwels M, Zhang J, Houte KV, Verbeelen D, Van den Branden C. Altered antioxidant defence in a mouse adriamycin model of glomerulosclerosis. Nephrol Dial Transplant 16: 147–150, 2001 [DOI] [PubMed] [Google Scholar]
- 13. Ding G, Reddy K, Kapasi AA, Franki N, Gibbons N, Kasinath BS, Singhal PC. Angiotensin II induces apoptosis in rat glomerular epithelial cells. Am J Physiol Renal Physiol 283: F173–F180, 2002 [DOI] [PubMed] [Google Scholar]
- 14. Durvasula RV, Petermann AT, Hiromura K, Blonski M, Pippin J, Mundel P, Pichler R, Griffin S, Couser WG, Shankland SJ. Activation of a local tissue angiotensin system in podocytes by mechanical strain. Kidney Int 65: 30–39, 2004 [DOI] [PubMed] [Google Scholar]
- 15. Fan YP, Weiss RH. Exogenous attenuation of p21(Waf1/Cip1) decreases mesangial cell hypertrophy as a result of hyperglycemia and IGF-1. J Am Soc Nephrol 15: 575–584, 2004 [DOI] [PubMed] [Google Scholar]
- 16. Fernandez L, Romero M, Soto H, Mosquera J. Increased apoptosis in acute puromycin aminonucleoside nephrosis. Exp Nephrol 9: 99–108, 2001 [DOI] [PubMed] [Google Scholar]
- 17. Fornoni A, Li H, Foschi A, Striker GE, Striker LJ. Hepatocyte growth factor, but not insulin-like growth factor I, protects podocytes against cyclosporin A-induced apoptosis. Am J Pathol 158: 275–280, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fotedar R, Brickner H, Saadatmandi N, Rousselle T, Diederich L, Munshi A, Jung B, Reed JC, Fotedar A. Effect of p21waf1/cip1 transgene on radiation induced apoptosis in T cells. Oncogene 18: 3652–3658, 1999 [DOI] [PubMed] [Google Scholar]
- 19. Gartel AL, Tyner AL. The role of the cyclin-dependent kinase inhibitor p21 in apoptosis. Mol Cancer Ther 1: 639–649, 2002 [PubMed] [Google Scholar]
- 20. Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol 119: 493–501, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gewirtz DA. A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochem Pharmacol 57: 727–741, 1999 [DOI] [PubMed] [Google Scholar]
- 22. Gilbertson DT, Liu J, Xue JL, Louis TA, Solid CA, Ebben JP, Collins AJ. Projecting the number of patients with end-stage renal disease in the United States to the year 2015. J Am Soc Nephrol 16: 3736–3741, 2005 [DOI] [PubMed] [Google Scholar]
- 23. Griffin SV, Hiromura K, Pippin J, Petermann AT, Blonski MJ, Krofft R, Takahashi S, Kulkarni AB, Shankland SJ. Cyclin-dependent kinase 5 is a regulator of podocyte differentiation, proliferation, and morphology. Am J Pathol 165: 1175–1185, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Griffin SV, Olivier JP, Pippin JW, Roberts JM, Shankland SJ. Cyclin I protects podocytes from apoptosis. J Biol Chem 281: 28048–28057, 2006 [DOI] [PubMed] [Google Scholar]
- 25. Hiromura K, Pippin JW, Blonski MJ, Roberts JM, Shankland SJ. The subcellular localization of cyclin dependent kinase 2 determines the fate of mesangial cells: role in apoptosis and proliferation. Oncogene 21: 1750–1758, 2002 [DOI] [PubMed] [Google Scholar]
- 26. Hiromura K, Pippin JW, Fero ML, Roberts JM, Shankland SJ. Modulation of apoptosis by the cyclin-dependent kinase inhibitor p27(Kip1). J Clin Invest 103: 597–604, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hoshi S, Shu Y, Yoshida F, Inagaki T, Sonoda J, Watanabe T, Nomoto K, Nagata M. Podocyte injury promotes progressive nephropathy in zucker diabetic fatty rats. Lab Invest 82: 25–35, 2002 [DOI] [PubMed] [Google Scholar]
- 28. Jat PS, Noble MD, Ataliotis P, Tanaka Y, Yannoutsos N, Larsen L, Kioussis D. Direct derivation of conditionally immortal cell lines from an H-2Kb-tsA58 transgenic mouse. Proc Natl Acad Sci USA 88: 5096–5100, 1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kim YG, Alpers CE, Brugarolas J, Johnson RJ, Couser WG, Shankland SJ. The cyclin kinase inhibitor p21CIP1/WAF1 limits glomerular epithelial cell proliferation in experimental glomerulonephritis. Kidney Int 55: 2349–2361, 1999 [DOI] [PubMed] [Google Scholar]
- 30. Kim YH, Goyal M, Kurnit D, Wharram B, Wiggins J, Holzman L, Kershaw D, Wiggins R. Podocyte depletion and glomerulosclerosis have a direct relationship in the PAN-treated rat. Kidney Int 60: 957–968, 2001 [DOI] [PubMed] [Google Scholar]
- 31. Kitiyakara C, Eggers P, Kopp JB. Twenty-one-year trend in ESRD due to focal segmental glomerulosclerosis in the United States. Am J Kidney Dis 44: 815–825, 2004 [PubMed] [Google Scholar]
- 32. Kuan CJ, al-Douahji M, Shankland SJ. The cyclin kinase inhibitor p21WAF1, CIP1 is increased in experimental diabetic nephropathy: potential role in glomerular hypertrophy. J Am Soc Nephrol 9: 986–993, 1998 [DOI] [PubMed] [Google Scholar]
- 33. Lennon R, Singh A, Welsh GI, Coward RJ, Satchell S, Ni L, Mathieson PW, Bakker WW, Saleem MA. Hemopexin induces nephrin-dependent reorganization of the actin cytoskeleton in podocytes. J Am Soc Nephrol 19: 2140–2149, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Levkau B, Koyama H, Raines EW, Clurman BE, Herren B, Orth K, Roberts JM, Ross R. Cleavage of p21Cip1/Waf1 and p27Kip1 mediates apoptosis in endothelial cells through activation of Cdk2: role of a caspase cascade. Mol Cell 1: 553–563, 1998 [DOI] [PubMed] [Google Scholar]
- 35. Lincet H, Poulain L, Remy JS, Deslandes E, Duigou F, Gauduchon P, Staedel C. The p21(cip1/waf1) cyclin-dependent kinase inhibitor enhances the cytotoxic effect of cisplatin in human ovarian carcinoma cells. Cancer Lett 161: 17–26, 2000 [DOI] [PubMed] [Google Scholar]
- 36. Lukas J, Lukas C, Bartek J. Mammalian cell cycle checkpoints: signalling pathways and their organization in space and time. DNA Repair (Amst) 3: 997–1007, 2004 [DOI] [PubMed] [Google Scholar]
- 37. Mancini G, Carbonara AO, Heremans JF. Immunochemical quantitation of antigens by single radial immunodiffusion. Immunochemistry 2: 235–254, 1965 [DOI] [PubMed] [Google Scholar]
- 38. Marshall CB, Pippin JW, Krofft RD, Shankland SJ. Puromycin aminonucleoside induces oxidant-dependent DNA damage in podocytes in vitro and in vivo. Kidney Int 70: 1962–1973, 2006 [DOI] [PubMed] [Google Scholar]
- 39. Marshall CB, Shankland SJ. Cell cycle regulatory proteins in podocyte health and disease. Nephron Exp Nephrol 106: e51–e59, 2007 [DOI] [PubMed] [Google Scholar]
- 40. Meikrantz W, Schlegel R. Apoptosis and the cell cycle. J Cell Biochem 58: 160–174, 1995 [DOI] [PubMed] [Google Scholar]
- 41. Meyer TW, Bennett PH, Nelson RG. Podocyte number predicts long-term urinary albumin excretion in Pima Indians with Type II diabetes and microalbuminuria. Diabetologia 42: 1341–1344, 1999 [DOI] [PubMed] [Google Scholar]
- 42. Mundel P, Reiser J, Kriz W. Induction of differentiation in cultured rat and human podocytes. J Am Soc Nephrol 8: 697–705, 1997 [DOI] [PubMed] [Google Scholar]
- 43. Mundel P, Reiser J, Zuniga Mejia Borja A, Pavenstadt H, Davidson GR, Kriz W, Zeller R. Rearrangements of the cytoskeleton and cell contacts induce process formation during differentiation of conditionally immortalized mouse podocyte cell lines. Exp Cell Res 236: 248–258, 1997 [DOI] [PubMed] [Google Scholar]
- 44. Nakamura T, Obata J, Kimura H, Ohno S, Yoshida Y, Kawachi H, Shimizu F. Blocking angiotensin II ameliorates proteinuria and glomerular lesions in progressive mesangioproliferative glomerulonephritis. Kidney Int 55: 877–889, 1999 [DOI] [PubMed] [Google Scholar]
- 45. Pagtalunan ME, Miller PL, Jumping-Eagle S, Nelson RG, Myers BD, Rennke HG, Coplon NS, Sun L, Meyer TW. Podocyte loss and progressive glomerular injury in type II diabetes. J Clin Invest 99: 342–348, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pavenstadt H, Kriz W, Kretzler M. Cell biology of the glomerular podocyte. Physiol Rev 83: 253–307, 2003 [DOI] [PubMed] [Google Scholar]
- 47. Pippin JW, Brinkkoetter PT, Cormack-Aboud FC, Durvasula RV, Hauser PV, Kowalewska J, Krofft RD, Logar CM, Marshall CB, Ohse T, Shankland SJ. Inducible rodent models of acquired podocyte diseases. Am J Physiol Renal Physiol 296: F213–F229, 2009 [DOI] [PubMed] [Google Scholar]
- 48. Pippin JW, Durvasula R, Petermann A, Hiromura K, Couser WG, Shankland SJ. DNA damage is a novel response to sublytic complement C5b-9-induced injury in podocytes. J Clin Invest 111: 877–885, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rincon J, Romero M, Viera N, Pedreanez A, Mosquera J. Increased oxidative stress and apoptosis in acute puromycin aminonucleoside nephrosis. Int J Exp Pathol 85: 25–33, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sanwal V, Pandya M, Bhaskaran M, Franki N, Reddy K, Ding G, Kapasi A, Valderrama E, Singhal PC. Puromycin aminonucleoside induces glomerular epithelial cell apoptosis. Exp Mol Pathol 70: 54–64, 2001 [DOI] [PubMed] [Google Scholar]
- 51. Schiffer M, Bitzer M, Roberts IS, Kopp JB, ten Dijke P, Mundel P, Bottinger EP. Apoptosis in podocytes induced by TGF-beta and Smad7. J Clin Invest 108: 807–816, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Schiffer M, Mundel P, Shaw AS, Bottinger EP. A novel role for the adaptor molecule CD2-associated protein in transforming growth factor-beta-induced apoptosis. J Biol Chem 279: 37004–37012, 2004 [DOI] [PubMed] [Google Scholar]
- 53. Shankland SJ. The podocyte's response to injury: role in proteinuria and glomerulosclerosis. Kidney Int 69: 2131–2147, 2006 [DOI] [PubMed] [Google Scholar]
- 54. Shankland SJ, Floege J, Thomas SE, Nangaku M, Hugo C, Pippin J, Henne K, Hockenberry DM, Johnson RJ, Couser WG. Cyclin kinase inhibitors are increased during experimental membranous nephropathy: potential role in limiting glomerular epithelial cell proliferation in vivo. Kidney Int 52: 404–413, 1997 [DOI] [PubMed] [Google Scholar]
- 55. Shankland SJ, Pippin JW, Reiser J, Mundel P. Podocytes in culture: past, present, and future. Kidney Int 72: 26–36, 2007 [DOI] [PubMed] [Google Scholar]
- 56. Sheaff RJ, Singer JD, Swanger J, Smitherman M, Roberts JM, Clurman BE. Proteasomal turnover of p21Cip1 does not require p21Cip1 ubiquitination. Mol Cell 5: 403–410, 2000 [DOI] [PubMed] [Google Scholar]
- 57. Shibata MA, Yoshidome K, Shibata E, Jorcyk CL, Green JE. Suppression of mammary carcinoma growth in vitro and in vivo by inducible expression of the Cdk inhibitor p21. Cancer Gene Ther 8: 23–35, 2001 [DOI] [PubMed] [Google Scholar]
- 58. Shiiki H, Sasaki Y, Nishino T, Kimura T, Kurioka H, Fujimoto S, Dohi K. Cell proliferation and apoptosis of the glomerular epithelial cells in rats with puromycin aminonucleoside nephrosis. Pathobiology 66: 221–229, 1998 [DOI] [PubMed] [Google Scholar]
- 59. Susztak K, Raff AC, Schiffer M, Bottinger EP. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes 55: 225–233, 2006 [PubMed] [Google Scholar]
- 60. Suzuki A, Tsutomi Y, Akahane K, Araki T, Miura M. Resistance to Fas-mediated apoptosis: activation of caspase 3 is regulated by cell cycle regulator p21WAF1 and IAP gene family ILP. Oncogene 17: 931–939, 1998 [DOI] [PubMed] [Google Scholar]
- 61. Suzuki A, Tsutomi Y, Miura M, Akahane K. Caspase 3 inactivation to suppress Fas-mediated apoptosis: identification of binding domain with p21 and ILP and inactivation machinery by p21. Oncogene 18: 1239–1244, 1999 [DOI] [PubMed] [Google Scholar]
- 62. Tucker JK. Focal segmental glomerulosclerosis in African Americans. Am J Med Sci 323: 90–93, 2002 [DOI] [PubMed] [Google Scholar]
- 63. Vielhauer V, Berning E, Eis V, Kretzler M, Segerer S, Strutz F, Horuk R, Grone HJ, Schlondorff D, Anders HJ. CCR1 blockade reduces interstitial inflammation and fibrosis in mice with glomerulosclerosis and nephrotic syndrome. Kidney Int 66: 2264–2278, 2004 [DOI] [PubMed] [Google Scholar]
- 64. Wada T, Pippin JW, Terada Y, Shankland SJ. The cyclin-dependent kinase inhibitor p21 is required for TGF-beta1-induced podocyte apoptosis. Kidney Int 68: 1618–1629, 2005 [DOI] [PubMed] [Google Scholar]
- 65. Wang Y, Wang YP, Tay YC, Harris DC. Progressive adriamycin nephropathy in mice: sequence of histologic and immunohistochemical events. Kidney Int 58: 1797–1804, 2000 [DOI] [PubMed] [Google Scholar]
- 66. Wharram BL, Goyal M, Wiggins JE, Sanden SK, Hussain S, Filipiak WE, Saunders TL, Dysko RC, Kohno K, Holzman LB, Wiggins RC. Podocyte depletion causes glomerulosclerosis: diphtheria toxin-induced podocyte depletion in rats expressing human diphtheria toxin receptor transgene. J Am Soc Nephrol 16: 2941–2952, 2005 [DOI] [PubMed] [Google Scholar]
- 67. Zheng Z, Pavlidis P, Chua S, D′Agati VD, Gharavi AG. An ancestral haplotype defines susceptibility to doxorubicin nephropathy in the laboratory mouse. J Am Soc Nephrol 17: 1796–1800, 2006 [DOI] [PubMed] [Google Scholar]
- 68. Zheng Z, Schmidt-Ott KM, Chua S, Foster KA, Frankel RZ, Pavlidis P, Barasch J, D'Agati VD, Gharavi AG. A Mendelian locus on chromosome 16 determines susceptibility to doxorubicin nephropathy in the mouse. Proc Natl Acad Sci USA 102: 2502–2507, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zhou BP, Liao Y, Xia W, Spohn B, Lee MH, Hung MC. Cytoplasmic localization of p21Cip1/WAF1 by Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nat Cell Biol 3: 245–252, 2001 [DOI] [PubMed] [Google Scholar]