

Abstract
Mutagenesis of mice with N-ethyl-N-nitrosourea (ENU) is a phenotype-driven approach to unravel gene function and discover new biological pathways. Phenotype-driven approaches have the advantage of making no assumptions about the function of genes and their products and have been successfully applied to the discovery of novel gene-phenotype relationships in many physiological systems. ENU mutagenesis of mice is used in many large-scale and more focused projects to generate and identify novel mouse models for the study of gene functions and human disease. This review examines the strategies and tools used in ENU mutagenesis screens to efficiently generate and identify functional mutations.
Keywords: N-ethyl-N-nitrosourea, phenotype-driven, inflammatory bowel disease, mouse models
phenotype-driven and gene-driven mutagenic approaches are important tools for studying the normal and abnormal function of genes (47). Gene-driven or “reverse genetic” approaches involve mutating a gene of interest to see whether it causes an abnormal phenotype. This approach is direct but requires, and may be limited by, prior knowledge or assumptions of the function of the gene. Here we review a phenotype-driven or “forward genetic” approach. Phenotype-driven approaches involve identifying organisms with an abnormal phenotype, followed by identifying the mutant gene. This approach is useful for identifying novel, difficult-to-predict functions of genes but is time consuming, because identifying the mutant gene may not be straightforward.
Early phenotype-driven approaches were used to study gene function by firstly characterizing variant phenotypes in natural or induced mutants in lower organisms, such as Caenorhabditis elegans and Drosophila melanogaster, followed by identifying the underlying gene and its mutation (9, 23, 49). The function of many human genes has been inferred from these forward genetic studies but, because of their genetic diversity and evolutionary distance, these lower organisms are not the model of choice for the study of human gene function and disease. Mice, on the other hand, have a similar number of genes and share similar physiological systems to humans, making them a more useful model organism. With the completion of the mouse genome project and the availability of various tools for manipulating the mouse genome, mice have become the primary model for the study of human diseases.
One of the first tools available for genetic manipulation in the mouse was the use of N-ethyl-N-nitrosourea (ENU) to induce random mutations throughout the genome (57). These random mutations allowed large-scale phenotype-driven screens to be used as part of a forward genetic approach to generate and identify novel mouse models for the study of gene functions (5, 17, 42, 52). Since then, gene targeting technologies and the ability to manipulate mouse embryonic stem (ES) cells have also allowed a genotype-driven or reverse genetic approach for the study of gene function through the generation of transgenic and knockout mice. In this review we examine the strategies and tools used by ENU mutagenesis screens and how improvements in the technology of both forward and reverse genetics have allowed an integrated approach.
ENU Mutagenesis
ENU is a powerful synthetic alkylating compound that acts by transferring its ethyl group to any nucleophilic nitrogen or oxygen sites of nucleic acids. The transferred ethyl groups form DNA adducts that can cause mispairing and, if not repaired, result in mutations in spermatogonial stem cells during DNA replication. The vast majority of induced mutations are single base-pair substitutions in the form of A-T to T-A transversions (44%) or A-T to G-C transitions (38%) (48, 65). Most ENU-induced point mutations result in missense (64%) mutations followed by splicing (26%) and nonsense (10%) mutations. Missense mutations have the potential to generate a series of alleles where the gene product has no biological activity (null), reduced activity (hypomorphic), or increased activity (hypermorphic). Although the site of ENU action is seemingly random, analysis of recently published data on the spectrum of ENU-induced mutations has shown that there is a bias toward genes with higher G + C content or bases flanked by G or C (6). This bias may be a limiting factor since it causes a particular class of amino acid change more efficiently than others. Thus the effect of amino acid class changes relying on G-C to C-G transversions or G-C to T-A transitions will be underrepresented (2).
The mutation frequency of ENU was first estimated by using a specific locus test for recessive alleles of easily detectable phenotypes such as eye and coat color (57). These early studies estimated a mutation frequency of one mutation per gene in every 175 to 655 gametes. More recent sequence-based studies on ENU-induced mutations have revealed frequencies of one mutation every 1 to 2.7 Mb, depending on the strain of mouse used and the concentration of ENU administered. Optimized treatment regimens used in these studies consisted of three weekly intraperitoneal injections of 80–100 mg/kg (5, 15, 24). The haploid size of the mouse genome is estimated to be ∼2.6 Gb; hence even at the conservative mutation rate of one every 2.7 Mb, each gamete of an ENU-treated male (G0) would carry ∼1,000 mutations (5). Of these mutations, it is estimated that ∼20 would potentially give rise to functional mutations since only ∼1–3% of the mouse genome is coding or regulatory regions.
Apart from being a mutagen, ENU is also toxic and carcinogenic at the concentrations used in the optimized treatment regimens. Treated mice are sterile for ∼10 wk and, depending on the strain used, not all mice survive the treatment or regain fertility (28). Recent region-specific ENU screens have also suggested that a mutation in ∼14–19% of genes causes sterility or results in embryonic lethal mutants (32, 72). A comparison of mouse strains used in various ENU screens has shown that, in general, outbred or hybrid mice are more tolerant of ENU than inbred strains (28). However, the use of outbred or hybrid strains adds to the complexity of genotyping the resulting mutant phenotype owing to their mixed genetic background. Additionally, interstrain specific polymorphisms can potentially mask or alter the effect of the mutation (5). To avoid these complications, inbred strains are the preferred choice in genomewide ENU mutagenesis screens.
Advantages of Genomewide Screens Using ENU Mutagenesis
Using ENU mutagenesis to identify genes involved in biological processes and diseases has a number of advantages over other techniques. ENU-induced mutagenesis does not rely on prior knowledge or assumptions about the genes involved, so the likelihood of generating novel information is high (52). ENU mutagenesis screens are designed so that mutations in many genes can be analyzed in each mouse, resulting in an efficient screening strategy. By comparison, conventional insertional gene knockout requires considerable knowledge of a particular gene and the analysis of only one gene or closely linked group of genes at a time. The development of insertional mutagenesis techniques using gene traps and/or transposons has allowed random insertional mutagenesis screens to be implemented (1, 35, 51). Gene traps and transposons randomly insert a fragment of DNA, which can encode reporter genes or conditional elements to help identify the site of insertion and control gene expression, into the mouse genome. DNA fragments inserted in coding or regulatory regions result in functional mutants. However, unlike ENU mutagenesis, almost all mutations induced by insertional mutagenesis result in null alleles. ENU-induced alleles are similar to those that arise naturally. By inducing changes in amino acid residues in individual protein domains or splice products, the alleles produced by ENU mutagenesis reveal separate gene functions that are not apparent from the production of null alleles by insertional mutagenesis.
Genomewide Screening Strategies
Before embarking on a screen of ENU-induced variants it is important to have robust screening strategies in place. Large-scale genomewide projects typically rely on a series of noninvasive standardized tests to identify phenotypes that are variant from wild-type mice to maximize recovery. One of the first standardized screening protocols used was SHIRPA (Smith Kline Beecham, Harwell, MRC Mouse Genome Centre, Imperial College, Royal London Hospital, Phenotype Assessment), which described up to 40 tests to identify behavioral and neurological phenotypic mutants (56). Recent standardized protocols such as EMPReSS (European Mouse Phenotyping Resource for Standardized Screens) have a wider variety of protocols, including blood tests to identify disorders of other physiological systems such as hormonal, metabolic, and immunological systems (10). EMPReSS also uses imaging techniques such as magnetic resonance imaging and dual-energy X-ray to examine cardiovascular and respiratory function as well as body composition. Standardized protocols for necropsy and histopathology have also been developed to detect and reference tissue abnormalities in mice (10, 64). See Table 1 for examples of current large-scale ENU mutagenesis projects.
Table 1.
Current genomewide large-scale ENU mouse mutagenesis projects
| Project | Screen Type | Phenotype | Website | References |
|---|---|---|---|---|
| Australian Phenomics Facility | Recessive | Immunology Hearing Male fertility Gastrointestinal tract | http://www.apf.edu.au/about/projects/ | 22, 34, 66 |
| Genomics Institute of the Novartis Research Foundation | Dominant and Recessive | Immunology Neurobiology Vision Metabolic disorders Hearing | http://www.gnf.org/technology/organismal/mammalian-genetics-phenotyping.htm | 38 |
| MRC Harwell Mutagenesis Project | Dominant and Recessive | All Major Physiological Systems | http://www.har.mrc.ac.uk/research/mutagenesis/ | 20, 21, 74 |
| Munich ENU Project | Dominant and Recessive | Dysmorphology Allergy Metabolic disorders Aldosterone | http://www.helmholtz-muenchen.de/en/ieg/group-functional-genetics/enu-screen/index.html | 3, 4, 68 |
| The Scripps Research Institute | Dominant and Recessive | All major physiological systems | http://mutagenetix.scripps.edu/home.cfm | 7, 8, 50 |
| The Jackson Laboratory | Recessive | All major physiological systems | http://research.jax.org/collaboration/index.html | 12, 19, 36, 62 |
| RIKEN ENU Mutagenesis Project | Dominant and Recessive | All major physiological systems | http://www.brc.riken.jp/lab/gsc/mouse/ | 30, 59, 70 |
A summary of large-scale N-ethyl-N-nitrosourea (ENU) mutagenesis projects that have published mutations in the past 24 mo. This table does not claim to be complete.
As the males treated with ENU (G0 males, see Fig. 1A) regain their fertility they are mated to wild-type females to produce the first-generation (G1) pedigrees. These G1 mice are heterozygous for ENU-induced mutations carried by the gametes of the G0 male. ENU mutagenesis phenotype-driven screens for functional mutations can be broadly classed into two categories: screens for dominant mutations and screens for recessive mutations.
Fig. 1.
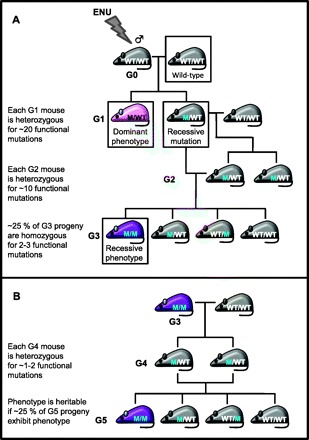
Breeding strategies for genomewide screening of dominant and recessive mutations. A: a G0 male is treated with N-ethyl-N-nitrosourea (ENU) and mated to a wild-type female. The G0 male will carry ∼1,000 ENU-induced mutations. The genotype for a particular ENU-mutated gene is shown with “WT” denoting a wild-type allele and “M” denoting a mutant allele that influences function. G1 progeny carrying a dominant functional mutation will exhibit a variant phenotype. G1 progeny not exhibiting a variant phenotype may carry recessive mutations and are crossed to a wild-type mouse. A G2 mouse is backcrossed to its G1 parent to generate G3 progeny. If ∼25% of G3 mice exhibit a common variant phenotype, they are homozygous for a recessive functional mutation. B: heritability of the variant phenotype is tested by crossing an affected G3 mouse to a wild-type mouse. G4 siblings are then intercrossed to generate G5 progeny. The variant phenotype is heritable if ∼25% of G5 mice exhibit the phenotype.
Screens for dominant functional mutations are relatively quick since G1 mice will exhibit a variant phenotype if they carry a dominant mutation. Heritability of this mutation is assessed by mating the affected G1 mouse or its siblings to a wild-type mouse. Assuming the phenotype segregates as a Mendelian trait, ∼50% of the resultant progeny (G2) should exhibit the abnormal phenotype.
A number of phenotypes and genes have been identified by screens for dominantly acting genes. As part of the Munich ENU project, a clinical chemical blood screen for dominant mutants identified a hyperglycemic pedigree (68). The ENU-induced mutation causing the phenotype was a missense mutation, a T to G transversion, causing an amino acid change from methionine to arginine at position 210 in the glucokinase (GCK) enzyme. Similar to conventional GCK-knockout mice, mice homozygous for GCKM210R die a few days after birth, whereas heterozygous mice were shown to have a 50% reduction in GCK enzyme activity. Since GCK activity is required for glycolysis the heterozygous mice are hyperglycemic by 10 days of age and eventually develop diabetes similar to the human disorder, maturity-onset diabetes of the young type 2 (MODY2). The MRC Harwell Mutagenesis Project also described an ENU-induced missense amino acid change in GCK from isoleucine to phenylalanine at position 366 (67). GCKI366F heterozygous mice have decreased GCK activity but unlike GCKM210R, homozygous mice were viable and not perinatally lethal. A third large-scale project, the RIKEN ENU Mutagenesis Project, identified two nonsense mutations, six missense mutations, and three splicing mutations of the GCK gene (27). Subsequent analysis revealed that the missense mutations had different effects on mRNA and protein expression as well as enzyme activity. Together, the different point mutations identified by the three screens provide an allelic series for the GCK gene, including null alleles and hypomorphic alleles, to better study its role in a complex disorder such as MODY2.
Although screening for dominant mutations in G1 mice is relatively fast, published data from large-scale dominant screens revealed that only ∼2% of functional mutations are dominant (26, 46). Therefore breeding strategies have been described to recover recessive mutations in the third generation (G3). These strategies involve mating G1 mice not exhibiting an abnormal phenotype to wild-type mice. The resultant G2 mice are then either backcrossed to their G1 parent or intercrossed with a G2 sibling to generate G3 mice, a proportion of which should be homozygous for multiple mutations (Fig. 1A). The former strategy has a greater probability of recovering recessive mutations, whereas the latter strategy has a lower probability of generating mice that are homozygous for more than one recessive mutation, which may confound any abnormal phenotype observed (29). Heritability of the recessive mutation can be tested by mating affected G3 mice to wild-type mice; littermates from the resultant progeny (G4) are subsequently intercrossed. If the recessive mutation is inherited in a Mendelian pattern, ∼25% of the G5 progeny should exhibit the abnormal phenotype (Fig. 1B). However, if the screen requires necroscopy or histopathology to identify the phenotype of interest, multiple pairs of G3 siblings of the affected mice are intercrossed to test heritability. If at least one pair produces affected G4 progeny at Mendelian ratios, the mutation is considered to be heritable.
Screens for recessive mutations have uncovered many interesting phenotypes, genes, and pathways. The RIKEN ENU mutagenesis project has recently generated a mouse model of infantile neuroaxonal dystrophy from a recessive screen (70). The point mutation was identified as a G to A transition causing an amino acid change from glycine to arginine at position 373 of the phospholipase A2 enzyme (iPLA2β) encoded by the Pla2G6 gene. The mutation abolishes the enzymes activity leading to disruption of cell membrane homeostasis, which results in axonal degeneration. The PLA2G6G373R mutant mice displayed severe motor dysfunction by 10 wk of age, unlike conventional PLA2G6-knockout mice, which did not display a significant neurodegenerative phenotype until after 1 yr of age (40, 60). A significant reduction in bone mass and strength after 6 mo of age was also described in PLA2G6-knockout mice (53). A similar phenotype was also seen in PLA2G6G373R mutant mice, only it was observed much earlier. Although the PLA2G6G373R mutation was in a gene already associated with neurodegeneration, the early onset of the phenotypes provides an improved mouse model for further studies in these disorders and an insight into the sequence-function relationship of the gene. In contrast, a recent recessive mutation identified by the ENU mutagenesis project at the Genomics Institute of the Novartis Research Foundation was in a gene that was previously not associated with diabetes (38). The mutation was identified as a point mutation with a corresponding amino acid change from tyrosine to histidine at position 344 of the Sec61α1 protein. Sec61α1T344H mutant mice exhibited diabetes and hepatosteatosis caused by an increase in apoptosis of pancreatic β-cells. Increased apoptosis was thought to be due to the Sec61α1T344H mutant protein disrupting endoplasmic reticulum (ER) function and protein processing, resulting in ER stress and apoptosis.
Screens to identify genes that affect the immune system have been particularly successful in identifying genes not previously associated with immune function or immune-mediated diseases. These screens also allowed a deeper understanding of the function of genes that were already known to have immunological function (17, 25). An excellent example of the former is the uncovering of the function of the Roquin gene (37, 69, 76). Mutations in roquin were found to be causal in a strain of mice with severe autoimmune disease. The function of roquin was previously unknown and subsequent work has shown that roquin acts to limit expression of a stimulatory protein on lymphocytes, inducible T cell costimulator, by modulating the rate of mRNA degradation. This work illuminated a novel immunoregulatory pathway.
Although large-scale screens have the potential to recover high numbers of functional mutations, they are quite laborious and time consuming, requiring large animal houses and high numbers of trained personnel. To reduce costs and increase feasibility many small-scale ENU mutagenesis projects have focused on screening phenotypes for specific physiological systems. Examples include muscular phenotypes (43), hair loss (33), hearing and vestibulary function (21), cardiovascular disease (19), T cell development (30), and anemia (34).
From Phenotype to Gene
Once inheritance of an abnormal phenotype has been established the strategy currently used to identify the causative gene is gene mapping to narrow the region containing the mutation to a manageable size, followed by DNA sequencing. Two important features of the ENU approach facilitate this process. Firstly, ENU variants are typically induced in the fully sequenced C57BL/6 genome, so causality can be established readily, even for genes of previously unknown function. Secondly, sequencing need only be limited to exons and the 50-bp flanking sequence, which contains the splice donor and acceptor sequences, since virtually all ENU-induced functional variants so far discovered are in these sequence elements.
To generate mice for mapping, the affected mice are outcrossed to wild-type mice of a different inbred strain. The progeny are then backcrossed or intercrossed. Since the region containing the mutated gene is inherited along with other polymorphisms, the outcrossing allows the strain-specific polymorphisms close to the mutation to be followed and identified by linkage analysis. With the availability of large databases of strain-specific single-nucleotide polymorphisms (SNPs), linkage analysis using these SNPs can, initially, assign the mutation to a chromosomal region of ∼20–60 Mb (73). During linkage analysis the search for candidate genes in the mapped region can be performed using genome browsers such as those hosted by Ensembl (http://www.ensembl.org/index.html) and RIKEN (http://omicspace.riken.jp/db/genome.html). RIKEN have also developed a data mining tool called Positional Medline (PosMed) that suggests and ranks candidate genes in the mapped region based on database searches using phenotypic keywords (75). Exons and flanking regions from candidate genes in the region can then be sequenced to identify the mutation. A mouse model for human retinal degeneration was recently identified with PosMed (59). The mutation causing the phenotype was mapped to the proximal region of chromosome 19. Although there were more than 400 genes in the mapped region, the PosMed system sorted and presented three candidate genes that could play a role in retinal degeneration: Rom1, Stx3, and Best1. Exons from the three genes were subsequently sequenced and a single missense mutation at position 1195 of the Rom1 gene was identified. If no mutations are found or there are no obvious candidate genes, further rounds of backcrossing and intercrossing are required to reduce the region to ∼1 Mb. This, of course, does not rule out the possibility that there are other phenotype-causing mutations in the mapped region.
Although the use of SNPs has accelerated the process of fine mapping and allowed a narrower region to be defined, the breeding required for this analysis is still the most time consuming process in identifying the ENU-induced mutation. This will, at some point in the not too distant future, be greatly reduced by the use of rapid and inexpensive next-generation sequencing technologies. Indeed, an ENU-induced mutation causing heterotaxy identified by next-generation sequencing has already been published (77). This mutation was identified as a missense mutation in the Megf8 gene, when a 2.2-Mb region of chromosome 7 was completely sequenced by using the massively parallel sequencing technique. As the technologies become more widely available, a dramatic increase in the number of novel mouse models for human diseases identified by genomewide ENU mutagenesis screens could be expected.
Site-Specific Screens
Linkage analysis to determine genetic causes for human disorders have only been moderately successful due to the genetic heterogeneity of humans. Although genetic loci can be identified and correlated with the development of a disease, the feasibility and usefulness of evaluating the role of each gene in the region by gene-driven or reverse genetic approaches can be limited since gene-targeting technologies usually produce null alleles. To better understand the role of these genetic loci in disease development, site- or region-specific ENU mutagenesis screens have been developed to maximize the discovery and generation of allelic series for candidate genes. Two main strategies have been used to generate DNA libraries for these site-specific screens, the cryopreservation of sperm or tissue from G1 progeny of ENU-treated mice (5, 42, 52, 58) or ES cells treated with ENU (13, 45). Mutations in the gene or region of interest from these libraries are rapidly detected by a number of techniques, including temperature gradient capillary electrophoresis, denaturing high-performance liquid chromatography, and direct sequencing. Once detected, the mutation can be recovered by in vitro fertilization or the generation of chimeric mice, to characterize any potential phenotype. A recent mouse model generated for the study of arterial tortuosity syndrome (ATS) was identified from a site-specific screen (14). Human ATS is caused by mutations in the glucose transporter 10 (GLUT10) encoded by the Slc2A10 gene. To generate a relevant GLUT10 mutant mouse model, exon 2 of the Slc2A10 gene was sequenced from sperm and DNA archives of G1 progeny from an ENU-treated male. Seven different missense mutations were identified from the screen, three of which were predicted to interfere with protein structure. Of the three mutations that were of interest only two, G128E and S150F, were successfully recovered by in vitro fertilization. Subsequent analysis of the two mutant mouse lines revealed a vascular phenotype characterized by thickening and irregular shape of blood vessels. Although the phenotypes were not as severe as those found in humans with ATS, the GLUT10G128E and GLUT10S150F mutant mice are still useful as mouse models for ATS.
A further application of site-specific screens is to quickly identify recessive mutations in genes of interest. The International Knockout Mouse Consortium (IKMC) was recently formed to systematically generate ES cell lines that have each gene of the mouse genome knocked out (16). At the time of writing, the IKMC has generated ∼14,000 mutant ES cell lines. By crossing knockout mice from these archives with mice generated from site-specific ENU mutagenesis screens, functional recessive mutations in the gene of interest on any chromosome, including sex-linked chromosomes, can be detected in the first generation. This breeding strategy is also useful for generating functional mutants for genes that are lethal when knocked out (42).
Modifier and Sensitized Screens
One factor that can limit the success of linkage analysis in humans is that many disorders are caused by a combination of environmental factors and genes. The same limitation affects the generation of mouse models for these disorders by gene-driven screens. Although genomewide ENU mutagenesis screens are capable of generating models for these multifactorial disorders, it is almost impossible to show heritability of the disorder if multiple genes are involved. To increase the efficiency of generating mouse models for multifactorial disorders, many recent screens use both phenotype-driven and gene-driven approaches (11, 18, 20, 54, 74). Unlike standard ENU mutagenesis screens, these modifier or sensitized screens use mice that have also been modified by genetic, environmental, or pharmacological manipulations to develop or predispose to develop a disorder of interest (29). Modifier screens search for ENU-induced mutations that suppress or enhance the development of the disorder. Mice used in this type of screen are usually genetically modified, such as knockout or transgenic mice, which are known to develop a disorder of interest. These mice can be treated directly with ENU or crossed to an ENU-treated wild-type mouse to identify mutations that modulate the development of the disorder (see Fig. 2). The latter strategy is used when transgenic mice or mice heterozygous for a knockout gene display the disorder of interest (55, 61). An example of a modifier screen is the use of conventional Mpl-knockout mice to identify genes that suppress thrombocytopenia (11, 31). Mpl is the receptor for thrombopoietin required for the normal production of platelets. Mice lacking either thrombopoietin or its receptor exhibit thrombocytopenia. Since Mpl-knockout mice were on a C57BL/6 background, they were treated directly with ENU before mating to an untreated isogenic C57BL/6 mouse. Mutations that suppressed thrombocytopenia were identified by screening the G1 progeny for normal levels of platelets. Mutations in two genes have been identified that suppress thrombocytopenia in two separate modifiers screens. The first was in the Plt4 gene (11) followed by the Plt6 gene (31). Plt4 encodes the c-Myb transcription factor whereas Plt6 encodes c-Myb's coregulator, p300. Together, these mouse models have identified a novel role for c-Myb/p300 in suppressing TPO-independent platelet production.
Fig. 2.
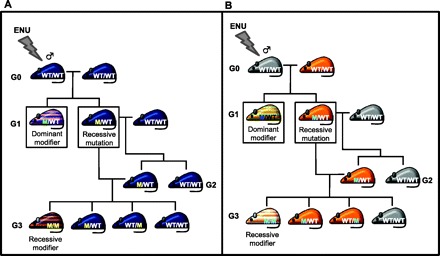
Breeding strategy for modifier screens in mice with an existing gene mutation. In modifier screens, mice that exhibit a disorder of interest are used to find ENU-induced mutations that suppress or enhance the development of the disorder. Two different strategies for modifier screens are shown. A: in this approach, homozygous knockout mice displaying a disorder of interest (blue-colored mice) are screened. A G0 knockout male treated with ENU is mated to an untreated knockout female. G1 progeny showing altered development or severity of the disorder carry a dominant functional mutation that modifies the phenotype (blue and pink striped mouse). G1 progeny not exhibiting any alterations in the disorder may carry recessive ENU-induced mutations and are mated to an untreated knockout mouse. G2 progeny are then backcrossed to the G1 parent carrying ENU-induced mutations. If ∼25% of G3 mice exhibit altered development or severity of the disorder, they are homozygous for a recessively acting mutation that modifies the disorder (blue and orange striped mouse). B: in this approach, mice displaying the disorder of interest are heterozygous for a gene-targeted allele (orange-colored mice). A G0 wild-type male is treated with ENU and mated to a heterozygous female exhibiting a disorder of interest. G1 progeny showing altered development or severity of the disorder carry a dominant modifier (orange and green striped mouse). Heterozygous G1 progeny not exhibiting any alterations in the disorder may carry recessive ENU-induced mutations and are mated to a wild-type mouse. Heterozygous G2 progeny are backcrossed to the G1 parent carrying ENU-induced mutations. If ∼25% of G3 mice exhibit altered development or severity of the disorder, they are homozygous for a recessive modifier (orange and white striped mouse). Note that some mice in the G3 generation may be homozygous for the predisposing mutation and are not shown here.
Unlike mice used in modifier screens, sensitized mice do not normally develop the phenotype of interest; they are rather predisposed to developing the phenotype with additional stimulus. Examples of this include ENU mutagenesis projects searching for mouse models of inflammatory bowel disease (IBD) by using subpathological concentrations of dextran sodium sulfate (DSS) to predispose mice to developing IBD-like phenotypes (see Fig. 3). Sensitized mice may also exhibit an abnormal condition that predisposes them to develop a phenotype of interest. Thus modifier screens and sensitizer screens can overlap. One such screen also uses Mpl-knockout mice; however, the phenotype of interest was not the lack of platelets but rather the lack of red blood cells (54). Since Mpl-knockout mice are thrombocytopenic, they are also predisposed to develop anemia. A pedigree exhibiting a significant decrease in red blood cells was shown to have a mutation in the Ank-1 gene, which is required for normal erythropoiesis. The mutation was identified as a point mutation in a splicing acceptor site of the gene causing incorrect splicing and a premature stop codon. This mutation was the first null allele for ankyrin-1 in the mouse. Other examples of sensitized screens include the use of mice heterozygous for the insulin receptor knockout mutation (20) or the spontaneous leptin mutation (18) in the search for new models of diabetes. These data show that the use of sensitized mice in ENU mutagenesis screens provides a more efficient strategy for identifying regulatory or susceptibility genes for particular complex human disorders.
Fig. 3.
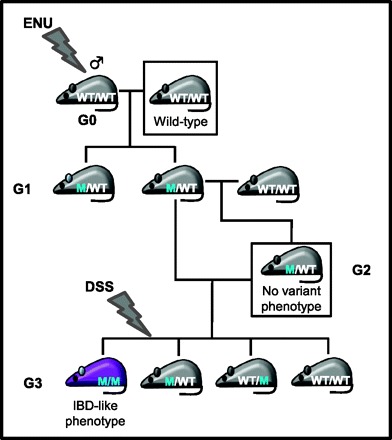
A breeding strategy for a dextran sodium sulfate (DSS) sensitizer screen. The DSS sensitizer screen shown here uses lethal histopathological analysis to assess inflammatory bowel disease (IBD)-like phenotypes, so the screening strategy involves treating G3 progeny to assess whether their parents are carriers of functional ENU-induced mutations causing IBD-like phenotypes. G2 progeny are backcrossed to their G1 parent to generate G3 progeny, which are treated with 1% DSS. If ∼25% of a G3 litter develop an IBD-like phenotype their parents are heterozygous for a recessive functional mutation. If more than 50% of a G3 litter develop an IBD-like phenotype then one or both parents carry a dominant functional mutation.
ENU Mutagenesis in IBD Research
Chronic IBD, which includes Crohn's disease and ulcerative colitis, is a common condition that afflicts people of all ages in most Western populations (39). Although the condition is rarely fatal in itself, it is responsible for considerable morbidity and predisposes individuals to an increased incidence of colorectal cancer. It is widely held to result from interactions between environmental factors and genes that lead to a chronic dysregulated response to nonpathogenic intestinal microflora. To further understand the basis for IBD and to produce new mouse models of disease, screens of ENU variants are underway and a number of gene variants that predispose to IBD have already been discovered.
Mutations in the gene encoding the major acidic mucin of the lower gastrointestinal tract Muc2, which encodes mucin 2, have been identified in ENU mouse strains with an ulcerative colitis-like disease, namely Eeyore, Winnie (22), and Kenny (see Fig. 4A; unpublished observations). Subsequent analysis of Eeyore and Winnie showed that the mutations disrupted mucin 2 production leading to an accumulation of misfolded protein in goblet cells. The accumulated protein triggers the cells ER stress response, which ultimately leads to goblet cell death. Additionally, the Woodrat mouse strain was generated in a DSS sensitizer screen (8). The Woodrat mutation is in the membrane-bound transcription factor peptidase site 1 (S1P) gene (Mbtps1). S1P has been shown to be required for the unfolded protein response (UPR) during ER stress. Thus, without this UPR, ER stress can result in cell death and increased mucosal permeability, which leads to the development of colitis. These findings demonstrate that compromising the mucosal barrier either by a lack of mucin production or abnormal cell death predisposes to colitis.
Fig. 4.
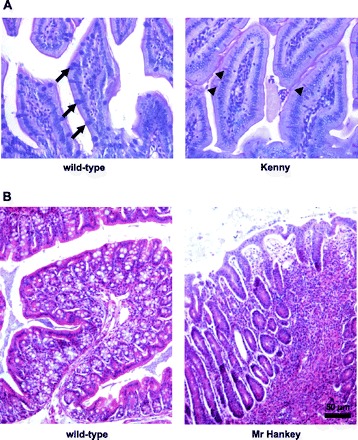
IBD-like phenotypes in Kenny and Mr Hankey mice. A: mucin production by goblet cells in the small intestine is disrupted in Kenny mice. Alcian blue/periodic acid-Schiff (PAS) stain of ileum sections show goblet cells stain dark blue with Alcian blue in wild-type ileum (arrows) whereas only smaller cells that stain pink with PAS can be observed in Kenny ileum (arrow heads). B: hematoxylin and eosin stain of colon sections showing wild-type colon with no inflammation and Mr Hankey colon with transmural colonic inflammation.
ENU mutagenesis has revealed that intestinal inflammation can also arise as a result of immune system deficiencies. The Sphinx mouse, identified from a flow cytometric screen for immunological phenotypes, has an ENU-induced mutation causing an amino acid change from glycine to cysteine at position 38 of the GTPase of immunity-associated protein 5 (Gimap5), which is required for T and B cell survival (7). The mutation was effectively a null allele since no detectable Gimap5 protein was detected in Sphinx mice leading to disrupted lymphocyte homeostasis and thus lymphopenia. A second pedigree, Mr Hankey (named after the “South Park” character), was identified when mice presented with persistent diarrhea and weight loss in an ENU screen searching for IBD phenotypes. Subsequent histological analysis revealed that Mr Hankey also presented with transmural colonic inflammation (see Fig. 4B; unpublished observations), a phenotype very similar to that of human ulcerative colitis, and is also immunodeficient. Gene mapping followed by sequencing of candidate genes in the region of genetic linkage revealed the presence of a mutation in Prkdc, a gene required for the development of lymphocytes and associated with immunodeficiency in mice and humans. The Mr Hankey Prkdc gene had a nonsense mutation at amino acid 545 of what is normally a protein of 4,128 residues. Thus the mutation is likely a null allele, which accounts for the observed lymphopenia. Immunocompromised animals such as Sphinx and Mr Hankey are prone to developing IBD, but usually only in the presence of some other extenuating factor such as infection, an imbalance in lymphocyte populations, or challenge with chemical sensitizers such as DSS. This is likely to be the case in these mouse lines as well since inflammation in Sphinx was ameliorated by antibiotic treatment and the onset of colitis in Mr Hankey was highly variable, suggesting the requirement for a stochastic factor.
These data validate ENU mutagenesis as an approach that can lead to the generation of mice with IBD-like phenotypes and that the underlying causal mutations can be discovered.
ENU Mutagenesis in Inflammation-Associated Cancer Research
The discovery of novel models for IBD will also benefit research onto inflammation-associated cancer as chronic inflammation has also been strongly associated with the development of cancer (see Ref. 71 for a comprehensive review). Numerous epidemiological and histological studies have shown that carcinogenesis is preceded and maintained by many conditions that display chronic inflammation, including IBD. The consequences of chronic inflammation, persistent tissue destruction with increased cellular proliferation in the presence of proinflammatory cytokines and prostaglandins, provide a microenvironment that increases the risk of malignant transformations leading to tumor formation. Although strongly associated, the molecular basis for the causal role of chronic inflammation in carcinogenesis has yet to be clarified (71). Thus ENU screens for IBD phenotypes could also potentially provide novel mouse models for inflammation-associated cancers of the lower gastrointestinal tract.
A mouse model, Min (multiple intestinal neoplasia), of the inherited human disease familial adenomatous polyposis (FAP), was discovered as part of an ENU screen for mutations causing increased tumor formation (44). Subsequent linkage analysis and sequencing revealed that the mutation was in the murine homolog (mApc) of a human tumor suppressor gene, adenomatous polyposis coli (APC) (63). An autosomal dominant mutation of the APC gene has been shown to cause FAP in humans, which can develop into colorectal cancer. The Min mouse has been extensively used to dissect the molecular pathways, and their modifiers, for the development of colorectal cancer (41). Numerous mutations in the APC gene have been subsequently generated, by site-directed mutagenesis, in an attempt to improve the model, since current models do not present with consistent metastasis as seen in humans. A possible direction for discovering a novel model of colorectal cancer with metastasis could be an ENU modifier screen using Min mice.
Conclusion
The diversity of functional mutations generated by ENU mutagenesis has provided numerous mouse models for the study of human disorders as well as the function and role of genes in different physiological systems. The variety of strategies and tools developed to combine phenotype-driven and gene-driven approaches has increased the efficiency in which useful mutations have been recovered. Together, the screens will eventually provide an allelic series for the study of every gene. As ENU-induced alleles are similar to those that arise naturally, the likelihood of generating novel mouse models for human disorders is high. These models will be invaluable tools as therapeutic targets and drug development.
GRANTS
This work was supported by National Institutes of Health award 1R21DK078946-01A1.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
ACKNOWLEDGMENTS
We thank the Australian Phenomics Facility, Australian National University for provision of the Mr. Hankey and Kenny Strains and in particular Prof. Chris Goodnow, Jessica Armstrong, and Michael Dobbie for advice and service.
REFERENCES
- 1. Abuin A, Hansen GM, Zambrowicz B. Gene trap mutagenesis. Handb Exp Pharmacol: 129–147, 2007 [DOI] [PubMed] [Google Scholar]
- 2. Acevedo-Arozena A, Wells S, Potter P, Kelly M, Cox RD, Brown SD. ENU mutagenesis, a way forward to understand gene function. Annu Rev Genomics Hum Genet 9: 49–69, 2008 [DOI] [PubMed] [Google Scholar]
- 3. Aigner B, Rathkolb B, Herbach N, Hrabe de Angelis M, Wanke R, Wolf E. Diabetes models by screen for hyperglycemia in phenotype-driven ENU mouse mutagenesis projects. Am J Physiol Endocrinol Metab 294: E232–E240, 2008 [DOI] [PubMed] [Google Scholar]
- 4. Aigner B, Rathkolb B, Klempt M, Wagner S, Michel D, de Angelis MH, Wolf E. N-ethyl-N-nitrosourea mutagenesis produced a small number of mice with altered plasma electrolyte levels. J Biomed Sci 16: 53–59, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Augustin M, Sedlmeier R, Peters T, Huffstadt U, Kochmann E, Simon D, Schoniger M, Garke-Mayerthaler S, Laufs J, Mayhaus M, Franke S, Klose M, Graupner A, Kurzmann M, Zinser C, Wolf A, Voelkel M, Kellner M, Kilian M, Seelig S, Koppius A, Teubner A, Korthaus D, Nehls M, Wattler S. Efficient and fast targeted production of murine models based on ENU mutagenesis. Mamm Genome 16: 405–413, 2005 [DOI] [PubMed] [Google Scholar]
- 6. Barbaric I, Wells S, Russ A, Dear TN. Spectrum of ENU-induced mutations in phenotype-driven and gene-driven screens in the mouse. Environ Mol Mutagen 48: 124–142, 2007 [DOI] [PubMed] [Google Scholar]
- 7. Barnes MJ, Aksoylar H, Krebs P, Bourdeau T, Arnold CN, Xia Y, Khovananth K, Engel I, Sovath S, Lampe K, Laws E, Saunders A, Butcher GW, Kronenberg M, Steinbrecher K, Hildeman D, Grimes HL, Beutler B, Hoebe K. Loss of T cell and B cell quiescence precedes the onset of microbial flora-dependent wasting disease and intestinal inflammation in Gimap5-deficient mice. J Immunol 184: 3743–3754, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brandl K, Rutschmann S, Li X, Du X, Xiao N, Schnabl B, Brenner DA, Beutler B. Enhanced sensitivity to DSS colitis caused by a hypomorphic Mbtps1 mutation disrupting the ATF6-driven unfolded protein response. Proc Natl Acad Sci USA 106: 3300–3305, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brenner S. The genetics of Caenorhabditis elegans. Genetics 77: 71–94, 1974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brown SD, Chambon P, de Angelis MH. EMPReSS: standardized phenotype screens for functional annotation of the mouse genome. Nat Genet 37: 1155, 2005. [DOI] [PubMed] [Google Scholar]
- 11. Carpinelli MR, Hilton DJ, Metcalf D, Antonchuk JL, Hyland CD, Mifsud SL, Di Rago L, Hilton AA, Willson TA, Roberts AW, Ramsay RG, Nicola NA, Alexander WS. Suppressor screen in Mpl−/− mice: c-Myb mutation causes supraphysiological production of platelets in the absence of thrombopoietin signaling. Proc Natl Acad Sci USA 101: 6553–6558, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chan ER, Lavender H, Li G, Haviernik P, Bunting KD, Adams MD. An ENU-induced recessive mutation in Mpl leads to thrombocytopenia with overdominance. Exp Hematol 37: 276–284, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen Y, Yee D, Dains K, Chatterjee A, Cavalcoli J, Schneider E, Om J, Woychik RP, Magnuson T. Genotype-based screen for ENU-induced mutations in mouse embryonic stem cells. Nat Genet 24: 314–317, 2000 [DOI] [PubMed] [Google Scholar]
- 14. Cheng CH, Kikuchi T, Chen YH, Sabbagha NG, Lee YC, Pan HJ, Chang C, Chen YT. Mutations in the SLC2A10 gene cause arterial abnormalities in mice. Cardiovasc Res 81: 381–388, 2009 [DOI] [PubMed] [Google Scholar]
- 15. Coghill EL, Hugill A, Parkinson N, Davison C, Glenister P, Clements S, Hunter J, Cox RD, Brown SD. A gene-driven approach to the identification of ENU mutants in the mouse. Nat Genet 30: 255–256, 2002 [DOI] [PubMed] [Google Scholar]
- 16. Collins FS, Rossant J, Wurst W. A mouse for all reasons. Cell 128: 9–13, 2007 [DOI] [PubMed] [Google Scholar]
- 17. Cook MC, Vinuesa CG, Goodnow CC. ENU-mutagenesis: insight into immune function and pathology. Curr Opin Immunol 18: 627–633, 2006 [DOI] [PubMed] [Google Scholar]
- 18. Dokmanovic-Chouinard M, Chung WK, Chevre JC, Watson E, Yonan J, Wiegand B, Bromberg Y, Wakae N, Wright CV, Overton J, Ghosh S, Sathe GM, Ammala CE, Brown KK, Ito R, LeDuc C, Solomon K, Fischer SG, Leibel RL. Positional cloning of “Lisch-Like”, a candidate modifier of susceptibility to type 2 diabetes in mice. PLoS Genet 4: e1000137, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fernandez L, Marchuk DA, Moran JL, Beier DR, Rockman HA. An N-ethyl-N-nitrosourea mutagenesis recessive screen identifies two candidate regions for murine cardiomyopathy that map to chromosomes 1 and 15. Mamm Genome 20: 296–304, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Goldsworthy M, Hugill A, Freeman H, Horner E, Shimomura K, Bogani D, Pieles G, Mijat V, Arkell R, Bhattacharya S, Ashcroft FM, Cox RD. Role of the transcription factor sox4 in insulin secretion and impaired glucose tolerance. Diabetes 57: 2234–2244, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hardisty-Hughes RE, Parker A, Brown SD. A hearing and vestibular phenotyping pipeline to identify mouse mutants with hearing impairment. Nat Protoc 5: 177–190, 2010 [DOI] [PubMed] [Google Scholar]
- 22. Heazlewood CK, Cook MC, Eri R, Price GR, Tauro SB, Taupin D, Thornton DJ, Png CW, Crockford TL, Cornall RJ, Adams R, Kato M, Nelms KA, Hong NA, Florin TH, Goodnow CC, McGuckin MA. Aberrant mucin assembly in mice causes endoplasmic reticulum stress and spontaneous inflammation resembling ulcerative colitis. PLoS Med 5: e54, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hirsh D, Vanderslice R. Temperature-sensitive developmental mutants of Caenorhabditis elegans. Dev Biol 49: 220–235, 1976 [DOI] [PubMed] [Google Scholar]
- 24. Hitotsumachi S, Carpenter DA, Russell WL. Dose-repetition increases the mutagenic effectiveness of N-ethyl-N-nitrosourea in mouse spermatogonia. Proc Natl Acad Sci USA 82: 6619–6621, 1985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hoebe K, Beutler B. Forward genetic analysis of TLR-signaling pathways: an evaluation. Adv Drug Deliv Rev 60: 824–829, 2008 [DOI] [PubMed] [Google Scholar]
- 26. Hrabe de Angelis MH, Flaswinkel H, Fuchs H, Rathkolb B, Soewarto D, Marschall S, Heffner S, Pargent W, Wuensch K, Jung M, Reis A, Richter T, Alessandrini F, Jakob T, Fuchs E, Kolb H, Kremmer E, Schaeble K, Rollinski B, Roscher A, Peters C, Meitinger T, Strom T, Steckler T, Holsboer F, Klopstock T, Gekeler F, Schindewolf C, Jung T, Avraham K, Behrendt H, Ring J, Zimmer A, Schughart K, Pfeffer K, Wolf E, Balling R. Genome-wide, large-scale production of mutant mice by ENU mutagenesis. Nat Genet 25: 444–447, 2000 [DOI] [PubMed] [Google Scholar]
- 27. Inoue M, Sakuraba Y, Motegi H, Kubota N, Toki H, Matsui J, Toyoda Y, Miwa I, Terauchi Y, Kadowaki T, Shigeyama Y, Kasuga M, Adachi T, Fujimoto N, Matsumoto R, Tsuchihashi K, Kagami T, Inoue A, Kaneda H, Ishijima J, Masuya H, Suzuki T, Wakana S, Gondo Y, Minowa O, Shiroishi T, Noda T. A series of maturity onset diabetes of the young, type 2 (MODY2) mouse models generated by a large-scale ENU mutagenesis program. Hum Mol Genet 13: 1147–1157, 2004 [DOI] [PubMed] [Google Scholar]
- 28. Justice MJ, Carpenter DA, Favor J, Neuhauser-Klaus A, Hrabe de Angelis M, Soewarto D, Moser A, Cordes S, Miller D, Chapman V, Weber JS, Rinchik EM, Hunsicker PR, Russell WL, Bode VC. Effects of ENU dosage on mouse strains. Mamm Genome 11: 484–488, 2000 [DOI] [PubMed] [Google Scholar]
- 29. Justice MJ, Noveroske JK, Weber JS, Zheng B, Bradley A. Mouse ENU mutagenesis. Hum Mol Genet 8: 1955–1963, 1999 [DOI] [PubMed] [Google Scholar]
- 30. Kakugawa K, Yasuda T, Miura I, Kobayashi A, Fukiage H, Satoh R, Matsuda M, Koseki H, Wakana S, Kawamoto H, Yoshida H. A novel gene essential for the development of single positive thymocytes. Mol Cell Biol 29: 5128–5135, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kauppi M, Murphy JM, de Graaf CA, Hyland CD, Greig KT, Metcalf D, Hilton AA, Nicola NA, Kile BT, Hilton DJ, Alexander WS. Point mutation in the gene encoding p300 suppresses thrombocytopenia in Mpl−/− mice. Blood 112: 3148–3153, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kile BT, Hentges KE, Clark AT, Nakamura H, Salinger AP, Liu B, Box N, Stockton DW, Johnson RL, Behringer RR, Bradley A, Justice MJ. Functional genetic analysis of mouse chromosome 11. Nature 425: 81–86, 2003 [DOI] [PubMed] [Google Scholar]
- 33. Kim JK, Kim E, Baek IC, Kim BK, Cho AR, Kim TY, Song CW, Seong JK, Yoon JB, Stenn KS, Parimoo S, Yoon SK. Overexpression of Hr links excessive induction of Wnt signaling to Marie Unna hereditary hypotrichosis. Hum Mol Genet 19: 445–453, 2010 [DOI] [PubMed] [Google Scholar]
- 34. Lambe T, Simpson RJ, Dawson S, Bouriez-Jones T, Crockford TL, Lepherd M, Latunde-Dada GO, Robinson H, Raja KB, Campagna DR, Villarreal G, Jr, Ellory JC, Goodnow CC, Fleming MD, McKie AT, Cornall RJ. Identification of a Steap3 endosomal targeting motif essential for normal iron metabolism. Blood 113: 1805–1808, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Largaespada DA. Transposon mutagenesis in mice. Methods Mol Biol 530: 379–390, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee B, Kano K, Young J, John SW, Nishina PM, Naggert JK, Naito K. A novel ENU-induced mutation, peewee, causes dwarfism in the mouse. Mamm Genome 20: 404–413, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Linterman MA, Rigby RJ, Wong R, Silva D, Withers D, Anderson G, Verma NK, Brink R, Hutloff A, Goodnow CC, Vinuesa CG. Roquin differentiates the specialized functions of duplicated T cell costimulatory receptor genes CD28 and ICOS. Immunity 30: 228–241, 2009 [DOI] [PubMed] [Google Scholar]
- 38. Lloyd DJ, Wheeler MC, Gekakis N. A point mutation in Sec61alpha1 leads to diabetes and hepatosteatosis in mice. Diabetes 59: 460–470, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Loftus EV., Jr Clinical epidemiology of inflammatory bowel disease: Incidence, prevalence, and environmental influences. Gastroenterology 126: 1504–1517, 2004 [DOI] [PubMed] [Google Scholar]
- 40. Malik I, Turk J, Mancuso DJ, Montier L, Wohltmann M, Wozniak DF, Schmidt RE, Gross RW, Kotzbauer PT. Disrupted membrane homeostasis and accumulation of ubiquitinated proteins in a mouse model of infantile neuroaxonal dystrophy caused by PLA2G6 mutations. Am J Pathol 172: 406–416, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. McCart AE, Vickaryous NK, Silver A. Apc mice: models, modifiers and mutants. Pathol Res Pract 204: 479–490, 2008 [DOI] [PubMed] [Google Scholar]
- 42. Michaud EJ, Culiat CT, Klebig ML, Barker PE, Cain KT, Carpenter DJ, Easter LL, Foster CM, Gardner AW, Guo ZY, Houser KJ, Hughes LA, Kerley MK, Liu Z, Olszewski RE, Pinn I, Shaw GD, Shinpock SG, Wymore AM, Rinchik EM, Johnson DK. Efficient gene-driven germ-line point mutagenesis of C57BL/6J mice. BMC Genomics 6: 164–181, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Miller G, Neilan M, Chia R, Gheryani N, Holt N, Charbit A, Wells S, Tucci V, Lalanne Z, Denny P, Fisher EM, Cheeseman M, Askew GN, Dear TN. ENU mutagenesis reveals a novel phenotype of reduced limb strength in mice lacking fibrillin 2. PLoS One 5: e9137, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Moser AR, Pitot HC, Dove WF. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science 247: 322–324, 1990 [DOI] [PubMed] [Google Scholar]
- 45. Munroe RJ, Bergstrom RA, Zheng QY, Libby B, Smith R, John SW, Schimenti KJ, Browning VL, Schimenti JC. Mouse mutants from chemically mutagenized embryonic stem cells. Nat Genet 24: 318–321, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nolan PM, Peters J, Strivens M, Rogers D, Hagan J, Spurr N, Gray IC, Vizor L, Brooker D, Whitehill E, Washbourne R, Hough T, Greenaway S, Hewitt M, Liu X, McCormack S, Pickford K, Selley R, Wells C, Tymowska-Lalanne Z, Roby P, Glenister P, Thornton C, Thaung C, Stevenson JA, Arkell R, Mburu P, Hardisty R, Kiernan A, Erven A, Steel KP, Voegeling S, Guenet JL, Nickols C, Sadri R, Nasse M, Isaacs A, Davies K, Browne M, Fisher EM, Martin J, Rastan S, Brown SD, Hunter J. A systematic, genome-wide, phenotype-driven mutagenesis programme for gene function studies in the mouse. Nat Genet 25: 440–443, 2000 [DOI] [PubMed] [Google Scholar]
- 47. Nolan PM, Peters J, Vizor L, Strivens M, Washbourne R, Hough T, Wells C, Glenister P, Thornton C, Martin J, Fisher E, Rogers D, Hagan J, Reavill C, Gray I, Wood J, Spurr N, Browne M, Rastan S, Hunter J, Brown SD. Implementation of a large-scale ENU mutagenesis program: towards increasing the mouse mutant resource. Mamm Genome 11: 500–506, 2000 [DOI] [PubMed] [Google Scholar]
- 48. Noveroske JK, Weber JS, Justice MJ. The mutagenic action of N-ethyl-N-nitrosourea in the mouse. Mamm Genome 11: 478–483, 2000 [DOI] [PubMed] [Google Scholar]
- 49. Nusslein-Volhard C, Wieschaus E. Mutations affecting segment number and polarity in Drosophila. Nature 287: 795–801, 1980 [DOI] [PubMed] [Google Scholar]
- 50. Osborn O, Sanchez-Alavez M, Brownell SE, Ross B, Klaus J, Dubins J, Beutler B, Conti B, Bartfai T. Metabolic characterization of a mouse deficient in all known leptin receptor isoforms. Cell Mol Neurobiol 30: 23–33, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ostertag EM, Madison BB, Kano H. Mutagenesis in rodents using the L1 retrotransposon. Genome Biol 8, Suppl 1: S16–S24, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Quwailid MM, Hugill A, Dear N, Vizor L, Wells S, Horner E, Fuller S, Weedon J, McMath H, Woodman P, Edwards D, Campbell D, Rodger S, Carey J, Roberts A, Glenister P, Lalanne Z, Parkinson N, Coghill EL, McKeone R, Cox S, Willan J, Greenfield A, Keays D, Brady S, Spurr N, Gray I, Hunter J, Brown SD, Cox RD. A gene-driven ENU-based approach to generating an allelic series in any gene. Mamm Genome 15: 585–591, 2004 [DOI] [PubMed] [Google Scholar]
- 53. Ramanadham S, Yarasheski KE, Silva MJ, Wohltmann M, Novack DV, Christiansen B, Tu X, Zhang S, Lei X, Turk J. Age-related changes in bone morphology are accelerated in group VIA phospholipase A2 (iPLA2beta)-null mice. Am J Pathol 172: 868–881, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Rank G, Sutton R, Marshall V, Lundie RJ, Caddy J, Romeo T, Fernandez K, McCormack MP, Cooke BM, Foote SJ, Crabb BS, Curtis DJ, Hilton DJ, Kile BT, Jane SM. Novel roles for erythroid Ankyrin-1 revealed through an ENU-induced null mouse mutant. Blood 113: 3352–3362, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rinchik EM, Carpenter DA, Selby PB. A strategy for fine-structure functional analysis of a 6- to 11-centimorgan region of mouse chromosome 7 by high-efficiency mutagenesis. Proc Natl Acad Sci USA 87: 896–900, 1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rogers DC, Fisher EM, Brown SD, Peters J, Hunter AJ, Martin JE. Behavioral and functional analysis of mouse phenotype: SHIRPA, a proposed protocol for comprehensive phenotype assessment. Mamm Genome 8: 711–713, 1997 [DOI] [PubMed] [Google Scholar]
- 57. Russell WL, Kelly EM, Hunsicker PR, Bangham JW, Maddux SC, Phipps EL. Specific-locus test shows ethylnitrosourea to be the most potent mutagen in the mouse. Proc Natl Acad Sci USA 76: 5818–5819, 1979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sakuraba Y, Sezutsu H, Takahasi KR, Tsuchihashi K, Ichikawa R, Fujimoto N, Kaneko S, Nakai Y, Uchiyama M, Goda N, Motoi R, Ikeda A, Karashima Y, Inoue M, Kaneda H, Masuya H, Minowa O, Noguchi H, Toyoda A, Sakaki Y, Wakana S, Noda T, Shiroishi T, Gondo Y. Molecular characterization of ENU mouse mutagenesis and archives. Biochem Biophys Res Commun 336: 609–616, 2005 [DOI] [PubMed] [Google Scholar]
- 59. Sato H, Suzuki T, Ikeda K, Masuya H, Sezutsu H, Kaneda H, Kobayashi K, Miura I, Kurihara Y, Yokokura S, Nishida K, Tamai M, Gondo Y, Noda T, Wakana S. A monogenic dominant mutation in Rom1 generated by N-ethyl-N-nitrosourea mutagenesis causes retinal degeneration in mice. Mol Vis 16: 378–391, 2010 [PMC free article] [PubMed] [Google Scholar]
- 60. Shinzawa K, Sumi H, Ikawa M, Matsuoka Y, Okabe M, Sakoda S, Tsujimoto Y. Neuroaxonal dystrophy caused by group VIA phospholipase A2 deficiency in mice: a model of human neurodegenerative disease. J Neurosci 28: 2212–2220, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Speca DJ, Rabbee N, Chihara D, Speed TP, Peterson AS. A genetic screen for behavioral mutations that perturb dopaminergic homeostasis in mice. Genes Brain Behav 5: 19–28, 2006 [DOI] [PubMed] [Google Scholar]
- 62. Stylianou IM, Svenson KL, VanOrman SK, Langle Y, Millar JS, Paigen B, Rader DJ. Novel ENU-induced point mutation in scavenger receptor class B, member 1, results in liver specific loss of SCARB1 protein. PLoS One 4: e6521, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Su LK, Kinzler KW, Vogelstein B, Preisinger AC, Moser AR, Luongo C, Gould KA, Dove WF. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science 256: 668–670, 1992 [DOI] [PubMed] [Google Scholar]
- 64. Sundberg JP, Sundberg BA, Schofield P. Integrating mouse anatomy and pathology ontologies into a phenotyping database: tools for data capture and training. Mamm Genome 19: 413–419, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Takahasi KR, Sakuraba Y, Gondo Y. Mutational pattern and frequency of induced nucleotide changes in mouse ENU mutagenesis. BMC Mol Biol 8: 52–61, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Theodoratos A, Whittle B, Enders A, Tscharke DC, Roots CM, Goodnow CC, Fahrer AM. Mouse strains with point mutations in TAP1 and TAP2. Immunol Cell Biol 88: 72–78, 2010 [DOI] [PubMed] [Google Scholar]
- 67. Toye AA, Moir L, Hugill A, Bentley L, Quarterman J, Mijat V, Hough T, Goldsworthy M, Haynes A, Hunter AJ, Browne M, Spurr N, Cox RD. A new mouse model of type 2 diabetes, produced by N-ethyl-nitrosourea mutagenesis, is the result of a missense mutation in the glucokinase gene. Diabetes 53: 1577–1583, 2004 [DOI] [PubMed] [Google Scholar]
- 68. Van Burck L, Blutke A, Kautz S, Rathkolb B, Klaften M, Wagner S, Kemter E, Hrabe de Angelis M, Wolf E, Aigner B, Wanke R, Herbach N. Phenotypic and pathomorphological characteristics of a novel mutant mouse model for maturity-onset diabetes of the young type 2 (MODY 2). Am J Physiol Endocrinol Metab 298: E512–E523, 2010 [DOI] [PubMed] [Google Scholar]
- 69. Vinuesa CG, Cook MC, Angelucci C, Athanasopoulos V, Rui L, Hill KM, Yu D, Domaschenz H, Whittle B, Lambe T, Roberts IS, Copley RR, Bell JI, Cornall RJ, Goodnow CC. A RING-type ubiquitin ligase family member required to repress follicular helper T cells and autoimmunity. Nature 435: 452–458, 2005 [DOI] [PubMed] [Google Scholar]
- 70. Wada H, Yasuda T, Miura I, Watabe K, Sawa C, Kamijuku H, Kojo S, Taniguchi M, Nishino I, Wakana S, Yoshida H, Seino K. Establishment of an improved mouse model for infantile neuroaxonal dystrophy that shows early disease onset and bears a point mutation in Pla2g6. Am J Pathol 175: 2257–2263, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Westbrook AM, Szakmary A, Schiestl RH. Mechanisms of intestinal inflammation and development of associated cancers: lessons learned from mouse models. Mutat Res 705: 40–59, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wilson L, Ching YH, Farias M, Hartford SA, Howell G, Shao H, Bucan M, Schimenti JC. Random mutagenesis of proximal mouse chromosome 5 uncovers predominantly embryonic lethal mutations. Genome Res 15: 1095–1105, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wiltshire T, Pletcher MT, Batalov S, Barnes SW, Tarantino LM, Cooke MP, Wu H, Smylie K, Santrosyan A, Copeland NG, Jenkins NA, Kalush F, Mural RJ, Glynne RJ, Kay SA, Adams MD, Fletcher CF. Genome-wide single-nucleotide polymorphism analysis defines haplotype patterns in mouse. Proc Natl Acad Sci USA 100: 3380–3385, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wong F, Fan L, Wells S, Hartley R, Mackenzie FE, Oyebode O, Brown R, Thomson D, Coleman MP, Blanco G, Ribchester RR. Axonal and neuromuscular synaptic phenotypes in Wld(S), SOD1(G93A) and ostes mutant mice identified by fiber-optic confocal microendoscopy. Mol Cell Neurosci 42: 296–307, 2009 [DOI] [PubMed] [Google Scholar]
- 75. Yoshida Y, Makita Y, Heida N, Asano S, Matsushima A, Ishii M, Mochizuki Y, Masuya H, Wakana S, Kobayashi N, Toyoda T. PosMed (Positional Medline): prioritizing genes with an artificial neural network comprising medical documents to accelerate positional cloning. Nucleic Acids Res 37: W147–W152, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Yu D, Tan AH, Hu X, Athanasopoulos V, Simpson N, Silva DG, Hutloff A, Giles KM, Leedman PJ, Lam KP, Goodnow CC, Vinuesa CG. Roquin represses autoimmunity by limiting inducible T-cell co-stimulator messenger RNA. Nature 450: 299–303, 2007 [DOI] [PubMed] [Google Scholar]
- 77. Zhang Z, Alpert D, Francis R, Chatterjee B, Yu Q, Tansey T, Sabol SL, Cui C, Bai Y, Koriabine M, Yoshinaga Y, Cheng JF, Chen F, Martin J, Schackwitz W, Gunn TM, Kramer KL, De Jong PJ, Pennacchio LA, Lo CW. Massively parallel sequencing identifies the gene Megf8 with ENU-induced mutation causing heterotaxy. Proc Natl Acad Sci USA 106: 3219–3224, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]