

Abstract
Activated arginase has been implicated in many diseases including cancer, immune cell dysfunction, infections, and vascular disease. Enhanced arginase activity has been reported in lungs of patients with pulmonary artery hypertension. We used hypoxia as a model for pulmonary hypertension and studied the effect of exposure to hypoxia on arginase activity in human lung microvascular endothelial cells (HMVEC). Hypoxia induces upregulation of arginase activity as well as mRNA and protein levels of arginase II (Arg II), the only arginase isoform we were able to identify in HMVEC. In endothelial cells, arginase shares and competes for the substrate l-arginine with nitric oxide (NO) synthase (NOS). Through regulation of substrate availability for NOS, arginase is able to modulate NO production. To evaluate the role of Arg II in regulation of NO production under hypoxia, we compared NO output (RFL-6 reporter assay) in cells with normal and silenced Arg II. Exposure to hypoxia led to an increase in NO levels produced by HMVEC. Inhibition of Arg II by specific small interfering RNA or by the pharmacological inhibitor BEC additionally enhanced the levels of NO. Another possible role for activated arginase is involvement in regulation of cell proliferation. However, we showed that hypoxia decreased cell proliferation and upregulated Arg II did not have an effect on cell proliferation. Since hypoxia-inducible factors (HIF) are a family of transcriptional factors activated by hypoxia, we tested the possibility of involvement of HIF-1 and HIF-2 in regulation of Arg II under hypoxia. The silencing of HIF-2 but not HIF-1 prevented the activation of Arg II by hypoxia.
Keywords: nitric oxide, hypoxia-inducible factor-2α, cell proliferation
arginase is a binuclear manganese metalloenzyme that catalyzes the hydrolysis of l-arginine to l-ornithine and urea. The highest arginase activity has been found in the liver, where arginase is a final enzyme in the urea cycle. Liver is the only organ containing all enzymes in the urea cycle, and the arginase function in ureagenesis is well characterized; however, lesser levels of arginase activity were found in a number of other tissues that lack a complete urea cycle (18, 19). In addition, it has been shown that arginase activity is represented at least by two distinctive isofoms: arginase I (Arg I), the enzyme found primarily in liver, and arginase II, a second isoform (Arg II) (9, 15, 30, 48). Arg II is present in low levels or absent in the liver but is expressed in other organs, particularly kidney. Arg I and II are encoded by two different genes and localized in cytoplasm and in mitochondria, respectively (5, 19). The physiological roles of arginases beyond the urea cycle are not well understood; however, a number of recent publications have implicated the arginases in many disease processes, including vascular disease, infectious disease, immune cell dysfunction, and cancer (10, 31, 33).
In endothelial cells, arginase is able to modulate the level of nitric oxide (NO) production. NO synthase (NOS) and arginase share a common substrate and may compete for l-arginine. The Km for arginine is in the millimolar range for mammalian arginases, but it is in the micromolar range for the various NOS isoenzymes. On the other hand, the Vmax of arginase at physiological pH (approximately 1,400 μmol·min−1·mg−1; calculated for rat liver arginase) is >1,000 times that of the NOS enzymes (approximately 1 μmol·min−1·mg−1), indicating similar rates of substrate usage for NO synthesis at low arginine concentrations (50). Therefore arginase can limit the availability of arginine for NO synthesis by intact cells. The existence of the competitive use of l-arginine by NOS and arginases in the vascular system has been reported in systemic hypertension, erectile dysfunction, and atherosclerosis (10, 16, 28, 29, 52).
Another possible role of arginase in endothelial cells has been linked with the regulation of cell proliferation. The product of arginine hydrolysis by arginase, ornithine, serves as a precursor for polyamine synthesis. Polyamines are essential for proliferation of vascular endothelial cells (32), and upregulated arginase could lead to enhanced synthesis of polyamines (21, 22, 50).
Hypoxia is a critical component of many acute and chronic lung diseases including chronic obstructive pulmonary disease (COPD), acute respiratory distress syndrome (ARDS), and pulmonary arterial hypertension (PAH). It is known that hypoxic conditions such as living at high altitude or chronic hypoxia secondary to lung disease and sleep-disordered breathing could lead to the development of PAH (26, 38, 47). Exposure of animals and vascular cell cultures to hypoxic conditions is a commonly used model in the study of pulmonary hypertension and vascular disease (34, 43). The mechanisms and signal transduction processes involved in hypoxia sensing by cells of the pulmonary vasculature have yet to be fully elucidated, and new findings may offer new insights into the pathogenesis of pulmonary hypertension. It has been shown recently that Arg II is upregulated in endothelial cells of patients with PAH; however, the mechanism of this upregulation was not offered (51). To better understand the mechanisms and role of this arginase upregulation, we studied 1) the expression of arginase in human lung microvascular endothelial cells (HMVEC) exposed to hypoxia in context with the role of hypoxia-inducible factor 1 (HIF-1)/HIF-2α; and 2) the involvement of arginase in the regulation of NO production and proliferation by these cells.
MATERIALS AND METHODS
Reagents.
All cell culture reagents unless specified were purchased from Invitrogen (Carlsbad, CA). All chemicals not specified are from Sigma-Aldrich (St. Louis, MO). S-(2-boronoethyl)-l-cysteine, HCI (BEC), and protease inhibitors were purchased from EMD Chemicals (Gibbstown, NJ).
Cell culture and exposure to hypoxia.
HMVEC from nonsmoking donors without pulmonary and cardiovascular diseases were purchased from Lonza (Walkerville, MD). Monolayers were maintained in EBM-2 basal medium containing 5% fetal bovine serum, growth factors, and antibiotics (Bullet kit, Lonza). Arginase activity and expression were studied in confluent monolayers of HMVEC exposed at 37°C to 0% or 1% O2-5% CO2-balance N2 (hypoxia) or air-5% CO2 (normoxia) for 24 h. All other measurements were done in HMVEC exposed to 1% O2-5% CO2-balance N2 (hypoxia) or air-5% CO2 (normoxia) for 8–24 h.
Rat lung fibroblasts (RFL-6) were purchased from American Type Culture Collection (Manassas, VA) and maintained in DMEM containing 15% fetal bovine serum and antibiotics. For experiments, RFL-6 were seeded in six-multiwell plates and were used after they reached confluence.
Arginase assays.
Arginase activity was determined in HMVEC lysates as previously described (53). Cells were scraped into lysis buffer (50 mM Tris·HCl pH 7.5, 0.1 mM EDTA, 0.1 mM EGTA, and protease inhibitors). Cell lysates (100 μl) were added to 150 μl of 10 mM MnCl2 in 50 mM Tris·HCl, pH 7.5, and arginase was activated by heating this mixture to 55°C for 10 min. Reaction was initiated with 100 μl of l-arginine (0.5 M; pH 9.7), and after incubation at 37°C for 60 min, the reaction was stopped by addition of 750 μl of an acid solution mixture (H2SO4:H3PO4:H2O, 1:3:7). The mixtures were heated with 50 μl of α-isonitrosopropriophenone (9% in absolute ethanol) at 100°C for 45 min. The urea concentration was determined spectrophotometrically by the absorbance at 550 nm.
Western blot analysis.
HMVEC were scraped in RIPA buffer (50 mM Tric·HCl, pH 7.5, 150 mM NaCl, 0.1% SDS, 0.5% deoxycholate, and 1% Nonidet P-40) containing protease inhibitor cocktail and phophatase inhibitor cocktail. The lysates were clarified, and samples (10–20 μg of protein) were separated by SDS-PAGE and transferred to a nitrocellulose membrane. Membranes were blotted with specific antibodies to Arg II, HIF-1, HIF-2 (Santa Cruz Biotechnology, Santa Cruz, CA), or β-actin (Sigma).
Silencing by small intefering RNA.
Silencer predesigned small intefering RNA (siRNA) from Applied Biosystems/Ambion (Austin, TX) targeting HIF-1α (ID no. s6539), HIF-2α (ID no. s4699), and Arg II (ID no. 119113) were used in knockdown experiments. To demonstrate that the transfection does not induce nonspecific effects on gene expression, a negative control siRNA (no. 4390843), which has no homology to known sequences from mice, rats, or humans, was used. HMVEC were transfected with siRNAs (30 nM final concentration) using siPORT NeoFX transfection reagent (Ambion) according to the manufacturer's recommendations. After 24 h, cells were changed to fresh media and exposed to 1% O2-5% CO2 (hypoxia) or air-5% CO2 for 24 h posttransfection. To estimate cytotoxic effects induced by transfection, we compared control nontransfected cells with cells transfected with negative siRNA. We found that transfection reduces the number of cells by 25–30% but does not affect any other parameters we measured including cGMP levels, protein content, and mRNA levels (data not shown).
Detection of endothelium-derived NO using RFL-6 reporter cell assay.
To measure bioactive NO released by endothelial cells, a RFL-6 reporter cell assay was used as described by others (17). Briefly, HMVEC were exposed to hypoxia or normoxia for 24 h, and for the final 30 min, 20 U/ml superoxide dismutase (SOD) was added to the cells. For NO measurement, cells were incubated for 2 min in the solution supplemented with 20 U/ml SOD, 0.3 mmol/l 3-isobutyl-1-methylxanthine (IBMX), and 10 μmol/l calcium ionophore A23187. The supernatants were then transferred to the RFL-6 cells (that had been preincubated with 0.3 mmol/l IBMX for 10 min at 37°C). After a 2-min incubation at 37°C, the supernatants were removed from the RFL-6 cells, and the cells were rapidly frozen in liquid nitrogen. The cGMP content of the RFL-6 cells was determined using a cGMP assay kit (Cayman Chemical, Ann Arbor, MI).
Gene expression measured by RT-PCR.
Total cellular RNA was isolated with a microRNA isolation kit (Ambion), according to the manufacturer's instructions, and treated with DNAse I (Ambion). Purified RNA (0.5–1 μg) was converted to cDNA with SuperScript III reverse transcriptase (Invitrogen). Real-time PCR was conducted using Applied Biosystems (Foster City, CA) Power SYBR Green PCR master mix on an ABI Prism 7500 sequence detection system. PCR primers for human Arg II (accession no. NM_001172), HIF-1α (accession no. NM_001530), HIF-2α (accession no. NM_001430), ornithine decarboxylase 1 (ODC-1; accession no. NM_002539.1), and β-actin (accession no. NM_001101) were as follows: Arg II forward 5′-AAGCTGGCTTGATGAAAAGGC-3′, reverse 5′-GCGTGGATTCACTATCAGGTTGT-3′ (product size 119 bp); β-actin forward 5′-GCCAACCGCGAGAAGATGA-3′, reverse 5′-CATCACGATGCCAGTGGTA-3′ (product size 120 bp) HIF-1α forward 5′-TGCTCATCAGTTGCCACTTC-3′; reverse; 5′-TCCTCACACGCAAATAGCTG-3′(product size 92 bp); HIF-2α forward 5′-GGGCCAGGTGAAAGTCTACA-3′, reverse 5′-TGCTGGATTGGTTCACACAT-3′(product size 105 bp); ODC-1 forward 5′-CCCAGCGTTGGACAAATACT-3′, reverse 5′-TCCATAGACGCCATCATTCA-3′ (product size 205 bp). All samples were run in triplicate. Melting curve analyses for amplification products indicated one specific product for each gene and no primer-dimer formation. For negative controls, the same RNA preparations were used with the omission of the reverse-transcriptase step to confirm the absence of DNA contamination. Relative gene expression was estimated by ABI Prism 7500 software with β-actin mRNA used as an internal reference.
Effect of hypoxia on endothelial cell proliferation.
The rate of HMVEC proliferation was estimated by [3H]thymidine incorporation into newly synthesized DNA. HMVEC were transfected with Arg II or negative siRNA and seeded in 24-well plates. Twenty-four huors later, HMVEC were exposed to 1% O2 (hypoxia) or normoxia for an additional 48 h. For the last 6 h, [3H]thymidine (0.5 μCu/well) was added to the cell media. After exposure, cells were fixed in 10% trichloroacetic acid (TCA) at room temperature for 15 min and then washed twice in 5% TCA. The acid-insoluble material was dissolved in 0.2 N NaOH-0.2% SDS and counted for radioactivity in a liquid scintillation counter.
Statistical analysis.
All results are expressed as means ± SE. Statistical analysis was performed using the two-tailed Student's t-test, and P < 0.05 was considered statistically significant.
RESULTS
Hypoxia upregulates arginase II in HMVEC.
Exposure of HMVEC to hypoxic conditions significantly upregulated arginase activity, and it was oxygen-concentration dependent (Fig. 1A). Exposure to 1% O2 for 24 h led to two to three times higher arginase activity, and exposure to 0% O2 increased arginase activity up to five to six times. There was a slight difference in the degree of arginase upregulation between different HMVEC lots. These changes in activity correlated with changes in mRNA and protein levels of Arg II (Fig. 1, B–D), the only arginase isoform we were able to identify in HMVEC. We tested other human endothelial cells, e.g., human umbilical vein endothelial cells and human pulmonary arterial endothelial cells (HPAEC), and also observed upregulation of Arg II under hypoxic conditions (data not shown).
Fig. 1.
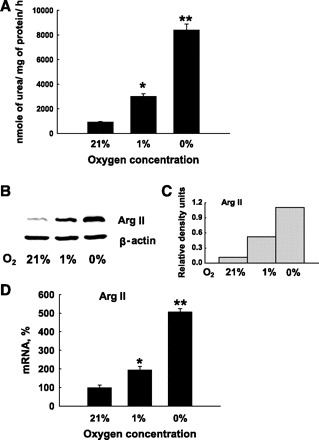
Expression and activity of arginase (Arg) II in human lung microvascular endothelial cells (HMVEC). HMVEC contain arginase activity that was higher in cell lysates of HMVEC exposed to reduced concentrations of oxygen for 24 h (A). This elevation in arginase activity corresponded to changes in protein level (B) and mRNA level (D) of Arg II. C: densitometric analysis of Arg II protein content shown in (B) normalized to β-actin protein level in the same lysate samples. The results shown are representative of at least three independent experiments. Each experiment (A and D) was run in triplicate. Each bar represents the mean ± SE. *P < 0.01 vs. normoxia and **P < 0.01 vs. normoxia and 1% O2 hypoxia.
Effect of Arg II on NO production in hypoxic conditions.
HMVEC have very low levels of NO production under basal conditions, so conventional techniques for measuring NO production such as the Griess reaction were not suitable. To measure the level of NO in HMVEC, we chose the more sensitive RFL-6 reporter assay in which the media from endothelial cells are transferred to RFL-6 fibroblasts that contain high levels of soluble guanylate cyclase. In this assay, the level of NO produced by endothelial cells corresponds to the level of cGMP produced by RFL-6 cells. The media from normal, nonactivated HMVEC produce cGMP levels in RFL-6 cells slightly higher than background levels. However, the activation of HMVEC with the Ca2+ ionophore A23187 dramatically increased the levels of cGMP (Fig. 2A). So in all experiments with hypoxia exposures, we made measurements on cells activated with A23187. Exposure of HMVEC to 1% O2 led to an increase in activated NO production which was completely abolished by NG-nitro-l-arginine methyl ester (l-NAME), an inhibitor of NOS (Fig. 2B).
Fig. 2.
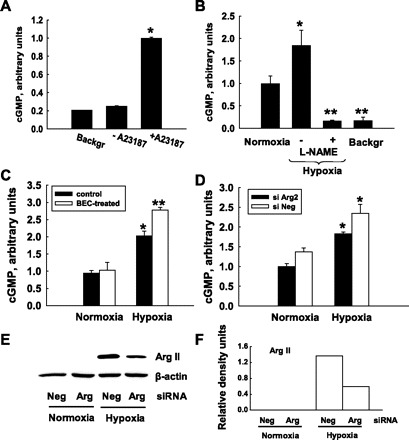
Indirect measurement of nitric oxide (NO) production in endothelial cells by cGMP levels in the RFL-6 reporter system. A: stimulation of NO production by A23187. Media from nonstimulated HMVEC produced an amount of cGMP close to background (Backgr) level. Stimulation of HMVEC with Ca2+ ionophore A23187 significantly increased NO production, allowing for the detection of cGMP over background levels. *P < 0.01 vs. background. B: HMVEC were exposed to normoxia (21% O2) or hypoxia (1% O2) for 24 h. Exposure to hypoxia increased NO production while treatment with NG-nitro-l-arginine methyl ester (l-NAME) completely abolished the hypoxia-induced increase in NO production. *P < 0.05 vs. normoxia; **P < 0.01 vs. normoxia and hypoxia alone. C and D: inhibition of Arg II by BEC (C) or by Arg II-specific small interfering RNA (siRNA; D) enhances NO production by HMVEC in hypoxic conditions. C: *P < 0.01 vs. normoxia control; **P < 0.01 vs. normoxia control and P < 0.05 vs. hypoxia control. D: *P < 0.05 vs. normoxia control. All measurements were done in duplicate; graphs are typical representatives of at least three separate experiments. Each bar represents the mean ± SE (for experiments in A–D). E: Western blotting demonstrates that siRNA to Arg II successfully suppressed Arg II protein expression in experiments in D while negative siRNA (siNeg) does not have effect on Arg II protein level. F: densitometric analysis of Arg II protein content shown in E normalized to β-actin protein level in the same lysate samples.
To study the involvement of Arg II in regulation of NO production under normal and hypoxic conditions, we measured NO production in control cells and in the cells with inhibited Arg II. The inhibition of Arg II was achieved either by the treatment of HMVEC with the nonspecific arginase inhibitor, BEC, or by silencing Arg II with specific siRNA. Inhibition of Arg II led to an increase in NO production in normoxic and hypoxic conditions, and the increase in hypoxic conditions was statistically significant (Fig. 2, C and D). The efficiency of inhibition with siRNA was monitored by Western blotting (Fig. 2, E and F).
Effect of Arg II on proliferation of HMVEC under hypoxic conditions.
The proliferation of HMVEC was estimated by the measurement of [3H]-thymidine incorporation. The exposure of HMVEC to 1% O2 significantly inhibited the proliferation rate of HMVEC. However, inhibition of Arg II expression by siRNA did not have an effect on proliferation of HMVEC in normoxic or hypoxic conditions (Fig. 3A). Polyamines are formed from ornithine in a sequence of enzymatic reactions. The rate-limiting step in polyamine biosynthesis is the decarboxylation of ornithine by ornithine decarboxylase (ODC) into putrescine, which is then converted to the higher polyamines spermidine and spermine (36). We measured ODC by real-time PCR and found that hypoxia exposure significantly reduced ODC mRNA levels in HMVEC (Fig. 3B).
Fig. 3.
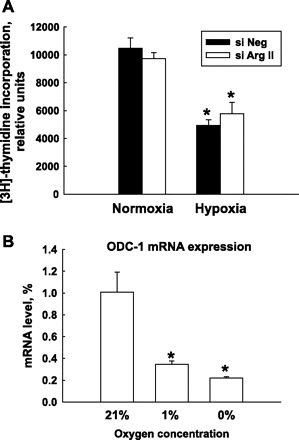
HMVEC proliferation under normoxia and hypoxia conditions. A: HMVEC were transfected with siNeg or Arg II siRNA (siArg II). Twenty-four hours after transfection, cells were exposed to 21% or 1% O2 for an additional 48 h. During the last 6 h of the exposure, [3H]thymidine was added to cells. Exposure of HMVEC to 1% O2 dramatically decreased the rate of HMVEC proliferation. Silencing of Arg II did not have an effect on HMVEC proliferation. B: expression of ornithine decarboxylase 1 (ODC-1) mRNA in HMVEC exposed to different oxygen concentrations for 24 h. The results shown are representative of three or more independent experiments made in triplicate. *P < 0.01 vs. normoxia.
HIF-2α but not HIF-1α is involved in the regulation of Arg II expression under hypoxic conditions.
The upregulation of Arg II mRNA under hypoxia suggests the involvement of transcriptional factors in this process. Since hypoxia-inducible factors (HIF) are a major family of transcriptional factors activated by hypoxia, we tested the possibility of involvement of HIF-1α and HIF-2α in regulation of Arg II under hypoxia. At first we were not able to identify both HIF-1α and HIF-2α after 24 h of exposure to hypoxia by Western blotting. Then we performed a time course study of the induction of HIF-1 and HIF-2 protein accumulation under hypoxia (Fig. 4, A and B). We found that both HIF-1 and HIF-2 proteins were induced in HMVEC after short exposures to hypoxia. For both isoforms, the highest level of protein accumulation was at 8 h exposure to hypoxia, followed by decline and complete disappearance by 24 h. Next we showed that we can effectively and specifically inactivate HIF-1α and HIF-2α in HMVEC by transfecting the cells with siRNA to each factor. As shown in Fig. 4, the HIF-α siRNAs led to a highly selective downregulation of the respective HIF-α mRNA. Quantitative reverse transcription-PCR analysis revealed that we were able to reduce mRNA levels for HIF-1 and HIF-2 down to 40% of their respective levels in control cells (Fig. 4C). Western blotting of the samples exposed to hypoxia confirmed the diminished levels of HIF-α proteins after transfection with siRNA (Fig. 4, D and E). Next we analyzed how the silencing of HIF-1 or HIF-2 affected hypoxia-induced upregulation of Arg II mRNA in HMVEC. The effect was compared with Arg II expression in HMVEC transfected with Arg II siRNA. We found that silencing of HIF-1α did not have an effect on Arg II expression, whereas the silencing of HIF-2α attenuated the upregulation of Arg II under hypoxia. On Western blotting the silencing of HIF-2α had the same effect on Arg II expression under hypoxia as silencing of Arg II itself (Fig. 5, A and B). Quantitative real-time PCR showed that transfection with HIF-2 siRNA significantly attenuated the hypoxic increase in Arg II mRNA expression but less effectively than the Arg II siRNA (Fig. 5C).
Fig. 4.
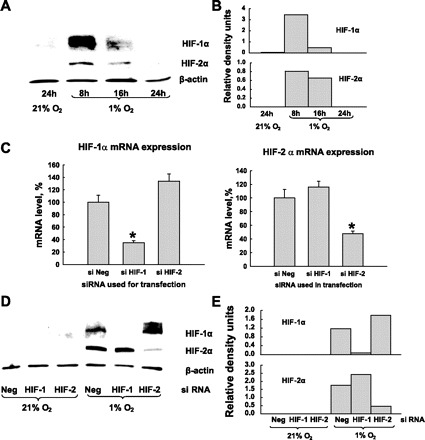
Expression of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in HMVEC. A: hypoxia activates both HIF-1 and HIF-2 in HMVEC. Cells were exposed to 21% or 1% O2 for 8, 16, or 24 h. B: densitometric analysis of HIF-1 and HIF-2 protein expression normalized to β-actin protein level in the same lysate samples. C and D: inhibition of HIF-1 and HIF-2 with gene-specific siRNA. Twenty-four hours after transfection, HMVEC were exposed to hypoxia for another 8 h. Inhibition of HIF-1 and HIF-2 was monitored by real-time PCR (C) and Western blotting (D). Protein expression was analyzed with densitometry analysis (E). The results shown are representative of two independent experiments. Real-time PCR was run in triplicate. *P < 0.01 vs. transfection with siNeg.
Fig. 5.
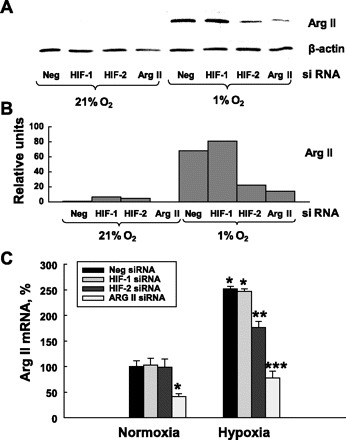
Effects of HIF-1 and HIF-2 silencing on Arg II protein expression. Silencing of HIF-2 but not HIF-1 partially abolished Arg II upregulation under hypoxic conditions. HIF-1, HIF-2, and Arg II were inhibited with specific siRNA. Twenty-four hours after transfection, HMVEC were exposed to 21% and 1% O2 for an additional 24 h. Arg II expression was measured by Western blotting (A), which was analyzed by densitometry (B), and real-time PCR (C). The results shown are representative of two independent experiments for PCR made in triplicate, and Western blots are representative of three independent experiments. *P < 0.01 vs. transfection with siNeg and exposure to normoxia; **P < 0.05 vs. transfection with siNeg and exposure to hypoxia; ***P < 0.01 vs. transfection with siNeg and exposure to hypoxia.
DISCUSSION
Reduced oxygen tensions have been reported to have different effects on arginase depending on isoform and species. It has been shown that hypoxia (1% O2) and anoxia (0% O2) increased Arg I mRNA expression in rat wound-derived macrophages 2.5- and 7.5-fold, respectively. Arg I mRNA expression was also induced by hypoxia and anoxia (4.5-fold) in mouse peritoneal macrophages. In marked contrast and demonstrating differences in the regulation of the two arginase genes, Arg II mRNA detected in murine cells was decreased by hypoxia and anoxia by 50% and 85%, respectively (1, 23). In agreement with these data we found that, in porcine pulmonary smooth muscle and endothelial cells, Arg II activity and expression were downregulated in response to hypoxia, while Arg I was upregulated (our unpublished data). In human endothelial cells we were able to detect only the expression of the Arg II mRNA and protein, and the same has been reported by others (39, 41, 46). To our surprise, in HMVEC, hypoxia activated Arg II expression and activity. This Arg II upregulation recently was reported by another group in HMVEC and in human pulmonary smooth muscle cells (6, 45). Hence, human Arg II has a different response to hypoxia compared with other species studied. It has been reported that at least five mRNA species of Arg II exist in human tissues, whereas only a single type of Arg II mRNA was found in the mouse (30, 40). This raises the possibility that the multiple types of Arg II mRNA in humans arise from the usage of different promoters and that hypoxia upregulates only one mRNA type from the promoter featured only in human cells.
Our results also demonstrate that HMVEC exposed to hypoxia are able to produce more NO in response to A23187 stimulation. Elevated NO production after hypoxia has been observed in other types of endothelial cells and has been explained by activation of endothelial NOS (eNOS) secondary to increased association of eNOS with heat shock protein 90 (3, 7, 13, 37, 42, 44). NO is an important regulator of vascular tone, and many studies have been devoted to identifying the role of NO in the normal and hypertensive pulmonary circulation (14, 27). The majority of these investigations have reported a decrease in lung NO production under hypoxia and in patients with PAH (8, 20, 24). However, recently, Bernal et al. (3) demonstrated an increase in NO levels in hypoxic-intact mouse lungs. Measurements of eNOS expression have also generated some controversial results; while one group reported reductions in eNOS gene expression in PAH patients (12), other studies did not reveal a decrease in eNOS on protein levels in lungs of patients with PAH (51). This citation probably represents the best point of view on this entangled problem: “Resolving the mechanism of the proposed hypoxia-induced increase in lung NO synthesis requires further experimentation, and the discrepancy in this field with regard to the question of whether NO is decreased or increased in hypoxia may be caused by different contributions of different pulmonary cell types (endothelial, alveolar epithelial cells, airway epithelial cells, etc), as well as the techniques used for detection of NO generation” (49). We would add that time of exposure to hypoxia, degree of hypoxia, and species used in these experiments are also important factors that contribute to the data discrepancies.
Our data clearly demonstrate that Arg II is able to modulate NO production in human endothelial cells under hypoxic conditions. Inhibition of Arg II with siRNA or with the pharmacological inhibitor BEC led to an increase in NO levels. It has been shown that arginase is responsible for reduced NO levels in many pathological conditions (10, 16, 28, 29, 52, 53). This is another example of competition between eNOS and Arg II for the substrate l-arginine. However, the increase in NO levels we observed was less dramatic than we expected. One possible explanation is that Arg II and eNOS have different subcellular distributions and Arg II competes for the substrate only with eNOS in close proximity. We speculate that since Arg II is localized in mitochondria, it could modulate NO production only in and around mitochondria. However, this hypothesis requires further investigation.
It is well known that chronic hypoxia in animal models increases endothelial cell proliferation (34). However, there is little information about the influence of acute hypoxia on pulmonary endothelial cell proliferation. We found just one study by Tucci et al. (47) that fully addressed endothelial cell proliferation and cycling under hypoxia. This study showed that acute hypoxic exposure slows endothelial cell division but does not arrest division. In our experimental conditions, hypoxia induced a significant decrease in HMVEC proliferation. We observed similar results for human pulmonary artery endothelial cells and human umbilical endothelial cells (data not shown). These data are in contrast to recently published results by another group (45), which demonstrated that the number of HMVEC increased under hypoxia and arginase was involved in this process. There are several possible reasons for these contradictory findings. One is variation in the source of the cells. In addition, the state of health, age, and medications of humans from whom cells are harvested and the methods of harvesting and processing of these cells may greatly affect the cell phenotype and their response to the stimulus. To decrease variations in cell responses in our studies, we used only cells from nonsmoking individuals without heart and lung disease. The serum concentration in the cell culture, seeding density of cells, and technique used to estimate cell proliferation may also add to data discrepancies. When proliferation is measured by counting cells, it raises the possibility that the increase in endothelial cell numbers under hypoxia observed, as reported by Toby et al. (45), may occur from proliferation as well as from better survival rate of cells under hypoxic conditions. Increases in arginase activity usually lead to an increase in polyamine synthesis from ornithine and increased cell proliferation. However, this is a sufficient condition only in the presence of active ODC, the rate-controlling enzyme in polyamine synthesis. Though in many situations ODC is coinduced with arginase (50), we observed a dramatic reduction in ODC expression under hypoxia. Decrease in ODC expression under hypoxia in HPAEC has been reported by others (25). This could explain why upregulated arginase failed to enhance cell proliferation in HMVEC under hypoxia in our study.
Very little is known about the transcriptional regulation of Arg II. Our data suggest that HIF-2α is involved in Arg II activation under hypoxia. Computer database analysis of the Arg II gene sequence revealed the presence of several potential hypoxia-responsive element-binding motifs (5′-RCGTG-3′). It has been shown that HIF-α subunits regulate both shared and unique target genes (35). We found that HIF-1α does not have any effect on Arg II, and Arg II gene is the target for HIF-2α only under hypoxic conditions in HMVEC. This is of interest since several recent publications establish a possible link between HIF-2α expression and pulmonary vascular diseases. It has been shown that rare HIF-2α-activating mutations in humans have been linked to the development of severe pulmonary hypertension (35). There is also experimental support for a critical role of HIF-2α in the pulmonary vasculature since hif2+/− mice are protected from hypoxia-induced right ventricular hypertrophy (4).
With transfection of HMVEC with HIF-2 siRNA we were able to silence HIF-2α down to 40% of the original level. This less-than-complete silencing may be the reason that we observed only partial attenuation of Arg II upregulation in hypoxia. Another possible explanation is that HIF-2α regulates Arg II in cooperation with other transcriptional factors. For example, it is known that many targets of HIF-2α are regulated as a result of physical and functional interactions with ETS-transcriptional factors family (2, 11, 35).
In conclusion, we found that hypoxia induces upregulation of Arg II in human endothelial cells. This upregulated arginase was able to modulate NO production but did not affect endothelial cell proliferation. We also showed for the first time that hypoxia-induced expression of Arg II is regulated by HIF-2α. The present study was specifically focused on examining the role of Arg II under hypoxia in context with the regulation of cell proliferation rate and the competition with eNOS for a common substrate. However, an additional role for Arg II in the regulation of physiological responses in the presence of hypoxia cannot be excluded.
GRANTS
This work was supported in part by National Institutes of Health Grants HL-68607 and HL-85133 and a VA Merit Review award (to J. M. Patel) as well as by Florida Department of Health Grants 08KN-08-17234 (to K. Krotova) and 04NIR-14 (to S. Zharikov).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
REFERENCES
- 1.Albina JE, Reichner JS. Oxygen and the regulation of gene expression in wounds. Wound Repair Regen 11: 445–451, 2003 [DOI] [PubMed] [Google Scholar]
- 2.Aprelikova O, Wood M, Tackett S, Chandramouli GVR, Barrett JC. Role of ETS transcription factors in the hypoxia-inducible factor-2 target gene selection. Cancer Res 66: 5641–5647, 2006 [DOI] [PubMed] [Google Scholar]
- 3.Bernal PJ, Leelavanichkul K, Bauer E, Cao R, Wilson A, Wasserloos KJ, Watkins SC, Pitt BR, St. Croix CM. Nitric oxide-mediated zinc release contributes to hypoxic regulation of pulmonary vascular tone. Circ Res 102: 1575–1583, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brusselmans K, Compernolle V, Tjwa M, Wiesener MS, Maxwell PH, Collen D, Carmeliet P. Heterozygous deficiency of hypoxia-inducible factor-2alpha protects mice against pulmonary hypertension and right ventricular dysfunction during prolonged hypoxia. J Clin Invest 111: 1519–1527, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cederbaum SD, Yu H, Grody WW, Kern RM, Yoo P, Iyer RK. Arginases I and II: do their functions overlap? Mol Genet Metab 81, Suppl 1: S38–S44, 2004 [DOI] [PubMed] [Google Scholar]
- 6.Chen B, Calvert AE, Cui H, Nelin LD. Hypoxia promotes human pulmonary artery smooth muscle cell proliferation through induction of arginase. Am J Physiol Lung Cell Mol Physiol 297: L1151–L1159, 2009 [DOI] [PubMed] [Google Scholar]
- 7.Chen Jx, Meyrick B. Hypoxia increases Hsp90 binding to eNOS via PI3K-Akt in porcine coronary artery endothelium. Lab Invest 84: 182–190, 2003 [DOI] [PubMed] [Google Scholar]
- 8.Demoncheaux EA, Higenbottam TW, Kiely DG, Wong JM, Wharton S, Varcoe R, Siddons T, Spivey AC, Hall K, Gize AP. Decreased whole body endogenous nitric oxide production in patients with primary pulmonary hypertension. J Vasc Res 42: 133–136, 2005 [DOI] [PubMed] [Google Scholar]
- 9.Dizikes GJ, Grody WW, Kern RM, Cederbaum SD. Isolation of human liver arginase cdna and demonstration of nonhomology between the two human arginase genes. Biochem Biophys Res Commun 141: 53–59, 1986 [DOI] [PubMed] [Google Scholar]
- 10.Durante W, Johnson FK, Johnson RA. Arginase: a critical regulator of nitric oxide synthesis and vascular function. Clin Exp Pharmacol Physiol 34: 906–911, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elvert G, Kappel A, Heidenreich R, Englmeier U, Lanz S, Acker T, Rauter M, Plate K, Sieweke M, Breier G, Flamme I. Cooperative interaction of hypoxia-inducible factor-2alpha (HIF-2alpha) and Ets-1 in the transcriptional activation of vascular endothelial growth factor receptor-2 (Flk-1). J Biol Chem 278: 7520–7530, 2003 [DOI] [PubMed] [Google Scholar]
- 12.Giaid A, Saleh D. Reduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N Engl J Med 333: 214–221, 1995 [DOI] [PubMed] [Google Scholar]
- 13.Hampl V, Cornfield D, Cowan N, Archer S. Hypoxia potentiates nitric oxide synthesis and transiently increases cytosolic calcium levels in pulmonary artery endothelial cells. Eur Respir J 8: 515–522, 1995 [PubMed] [Google Scholar]
- 14.Hampl V, Herget J. Role of nitric oxide in the pathogenesis of chronic pulmonary hypertension. Physiol Rev 80: 1337–1372, 2000 [DOI] [PubMed] [Google Scholar]
- 15.Haraguchi Y, Takiguchi M, Amaya Y, Kawamoto S, Matsuda I, Mori M. Molecular cloning and nucleotide sequence of cDNA for human liver arginase. Proc Natl Acad Sci USA 84: 412–415, 1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hsu LL, Champion HC, Campbell-Lee SA, Bivalacqua TJ, Manci EA, Diwan BA, Schimel DM, Cochard AE, Wang X, Schechter AN, Noguchi CT, Gladwin MT. Hemolysis in sickle cell mice causes pulmonary hypertension due to global impairment in nitric oxide bioavailability. Blood 109: 3088–3098, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ishii K, Sheng H, Warner TD, Forstermann U, Murad F. A simple and sensitive bioassay method for detection of EDRF with RFL-6 rat lung fibroblasts. Am J Physiol Heart Circ Physiol 261: H598–H603, 1991 [DOI] [PubMed] [Google Scholar]
- 18.Iyer R, Jenkinson CP, Vockley JG, Kern RM, Grody WW, Cederbaum S. The human arginases and arginase deficiency. J Inherit Metab Dis 21, Suppl 1: 86–100, 1998 [DOI] [PubMed] [Google Scholar]
- 19.Jenkinson CP, Grody WW, Cederbaum SD. Comparative properties of arginases. Comp Biochem Physiol B Biochem Mol Biol 114: 107–132, 1996 [DOI] [PubMed] [Google Scholar]
- 20.Kaneko FT, Arroliga AC, Dweik RA, Comhair SA, Laskowski D, Oppedisano R, Thomassen MJ, Erzurum SC. Biochemical reaction products of nitric oxide as quantitative markers of primary pulmonary hypertension. Am J Respir Crit Care Med 158: 917–923, 1998 [DOI] [PubMed] [Google Scholar]
- 21.Li H, Meininger CJ, Hawker JR, Jr, Haynes TE, Kepka-Lenhart D, Mistry SK, Morris SM, Jr, Wu G. Regulatory role of arginase I and II in nitric oxide, polyamine, and proline syntheses in endothelial cells. Am J Physiol Endocrinol Metab 280: E75–E82, 2001 [DOI] [PubMed] [Google Scholar]
- 22.Li H, Meininger CJ, Kelly KA, Hawker JR, Jr, Morris SM, Jr, Wu G. Activities of arginase I and II are limiting for endothelial cell proliferation. Am J Physiol Regul Integr Comp Physiol 282: R64–R69, 2002 [DOI] [PubMed] [Google Scholar]
- 23.Louis CA, Reichner JS, Henry WL, Jr, Mastrofrancesco B, Gotoh T, Mori M, Albina JE. Distinct arginase isoforms expressed in primary and transformed macrophages: regulation by oxygen tension. Am J Physiol Regul Integr Comp Physiol 274: R775–R782, 1998 [DOI] [PubMed] [Google Scholar]
- 24.Machado RF, Londhe Nerkar Mv, Dweik RA, Hammel J, Janocha A, Pyle J, Laskowski D, Jennings C, Arroliga AC, Erzurum SC. Nitric oxide and pulmonary arterial pressures in pulmonary hypertension. Free Radic Biol Med 37: 1010–1017, 2004 [DOI] [PubMed] [Google Scholar]
- 25.Manalo DJ, Rowan A, Lavoie T, Natarajan L, Kelly BD, Ye SQ, Garcia JGN, Semenza GL. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood 105: 659–669, 2005 [DOI] [PubMed] [Google Scholar]
- 26.McLaughlin VV, McGoon MD. Pulmonary arterial hypertension. Circulation 114: 1417–1431, 2006 [DOI] [PubMed] [Google Scholar]
- 27.Michelakis ED. The role of the NO axis and its therapeutic implications in pulmonary arterial hypertension. Heart Fail Rev 8: 5–21, 2003 [DOI] [PubMed] [Google Scholar]
- 28.Ming XF, Barandier C, Viswambharan H, Kwak BR, Mach F, Mazzolai L, Hayoz D, Ruffieux J, Rusconi S, Montani JP, Yang Z. Thrombin stimulates human endothelial arginase enzymatic activity via RhoA/ROCK pathway: implications for atherosclerotic endothelial dysfunction. Circulation 110: 3708–3714, 2004 [DOI] [PubMed] [Google Scholar]
- 29.Morris CR, Kuypers FA, Kato GJ, Lavrisha L, Larkin S, Singer T, Vichinsky EP. Hemolysis-associated pulmonary hypertension in thalassemia. Ann NY Acad Sci 1054: 481–485, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morris SM, Bhamidipati D, Kepka-Lenhart D. Human type II arginase: sequence analysis and tissue-specific expression. Gene 193: 157–161, 1997 [DOI] [PubMed] [Google Scholar]
- 31.Morris SM., Jr Recent advances in arginine metabolism: roles and regulation of the arginases. Br J Pharmacol 157: 922–930, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morrison RF, Seidel ER. Vascular endothelial cell proliferation: regulation of cellular polyamines. Cardiovasc Res 29: 841–847, 1995 [PubMed] [Google Scholar]
- 33.Munder M. Arginase: an emerging key player in the mammalian immune system. Br J Pharmacol 158: 638–651, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pak O, Aldashev A, Welsh D, Peacock A. The effects of hypoxia on the cells of the pulmonary vasculature. Eur Respir J 30: 364–372, 2007 [DOI] [PubMed] [Google Scholar]
- 35.Patel SA, Simon MC. Biology of hypoxia-inducible factor-2[alpha] in development and disease. Cell Death Differ 15: 628–634, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pegg AE, McCann PP. Polyamine metabolism and function. Am J Physiol Cell Physiol 243: C212–C221, 1982 [DOI] [PubMed] [Google Scholar]
- 37.Presley T, Vedam K, Velayutham M, Zweier JL, Ilangovan G. Activation of Hsp90-eNOS and increased NO generation attenuate respiration of hypoxia-treated endothelial cells. Am J Physiol Cell Physiol 295: C1281–C1291, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Preston IR. Clinical perspective of hypoxia-mediated pulmonary hypertension. Antioxid Redox Signal 9: 711–721, 2007 [DOI] [PubMed] [Google Scholar]
- 39.Ryoo S, Lemmon CA, Soucy KG, Gupta G, White AR, Nyhan D, Shoukas A, Romer LH, Berkowitz DE. Oxidized low-density lipoprotein-dependent endothelial arginase II activation contributes to impaired nitric oxide signaling. Circ Res 99: 951–960, 2006 [DOI] [PubMed] [Google Scholar]
- 40.Salimuddin Nagasaki A, Gotoh T, Isobe H, Mori M. Regulation of the genes for arginase isoforms and related enzymes in mouse macrophages by lipopolysaccharide. Am J Physiol Endocrinol Metab 277: E110–E117, 1999. [DOI] [PubMed] [Google Scholar]
- 41.Scalera F, Closs EI, Flick E, Martens-Lobenhoffer J, Boissel JP, Lendeckel U, Heimburg A, Bode-Böger SM. Paradoxical effect of l-arginine: acceleration of endothelial cell senescence. Biochem Biophys Res Commun 386: 650–655, 2009 [DOI] [PubMed] [Google Scholar]
- 42.Shi Y, Baker JE, Zhang C, Tweddell JS, Su J, Pritchard KA., Jr Chronic hypoxia increases endothelial nitric oxide synthase generation of nitric oxide by increasing heat shock protein 90 association and serine phosphorylation. Circ Res 91: 300–306, 2002 [DOI] [PubMed] [Google Scholar]
- 43.Stenmark KR, Meyrick B, Galie N, Mooi WJ, McMurtry IF. Animal models of pulmonary arterial hypertension: the hope for etiological discovery and pharmacological cure. Am J Physiol Lung Cell Mol Physiol 297: L1013–L1032, 2009 [DOI] [PubMed] [Google Scholar]
- 44.Strijdom H, Friedrich SO, Hattingh S, Chamane N, Lochner A. Hypoxia-induced regulation of nitric oxide synthase in cardiac endothelial cells and myocytes and the role of the PI3-K/PKB pathway. Mol Cell Biochem 321: 23–35, 2009 [DOI] [PubMed] [Google Scholar]
- 45.Toby IT, Chicoine LG, Cui H, Chen B, Nelin LD. Hypoxia-induced proliferation of human pulmonary microvascular endothelial cells depends on epidermal growth factor receptor tyrosine kinase activation. Am J Physiol Lung Cell Mol Physiol 298: L600–L606, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Topal G, Brunet A, Walch L, Boucher JL, David-Dufilho M. Mitochondrial arginase II modulates nitric-oxide synthesis through nonfreely exchangeable l-arginine pools in human endothelial cells. J Pharmacol Exp Ther 318: 1368–1374, 2006 [DOI] [PubMed] [Google Scholar]
- 47.Tuder RM, Yun JH, Bhunia A, Fijalkowska I. Hypoxia and chronic lung disease. J Mol Med 85: 1317–1324, 2007 [DOI] [PubMed] [Google Scholar]
- 48.Vockley JG, Jenkinson CP, Shukla H, Kern RM, Grody WW, Cederbaum SD. Cloning and characterization of the human yype II arginase gene. Genomics 38: 118–123, 1996 [DOI] [PubMed] [Google Scholar]
- 49.Weissmann N. Nitric oxide-mediated zinc release: a new (modulatory) pathway in hypoxic pulmonary vasoconstriction. Circ Res 102: 1451–1454, 2008 [DOI] [PubMed] [Google Scholar]
- 50.Wu GY, Morris SM. Arginine metabolism: nitric oxide and beyond. Biochem J 336: 1–17, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu W, Kaneko FT, Zheng S, Comhair SAA, Janocha AJ, Goggans T, Thunnissen FBJM, Farver C, Hazen SL, Jennings C, Dweik RA, Arroliga AC, Erzurum SC. Increased arginase II and decreased NO synthesis in endothelial cells of patients with pulmonary arterial hypertension. FASEB J 18: 1746–1748, 2004 [DOI] [PubMed] [Google Scholar]
- 52.Zhang C, Hein TW, Wang W, Miller MW, Fossum TW, McDonald MM, Humphrey JD, Kuo L. Upregulation of vascular arginase in hypertension decreases nitric oxide-mediated dilation of coronary arterioles. Hypertension 44: 935–943, 2004 [DOI] [PubMed] [Google Scholar]
- 53.Zharikov S, Krotova K, Hu H, Baylis C, Johnson RJ, Block ER, Patel J. Uric acid decreases NO production and increases arginase activity in cultured pulmonary artery endothelial cells. Am J Physiol Cell Physiol 295: C1183–C1190, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]