

Abstract
While exercise training (ExT) appears to influence cerebrovascular function during type 1 diabetes (T1D), it is not clear whether this beneficial effect extends to protecting the brain from ischemia-induced brain injury. Thus our goal was to examine whether modest ExT could influence transient focal ischemia-induced brain injury along with nitric oxide synthase (NOS)-dependent dilation of cerebral (pial) arterioles during T1D. Sprague-Dawley rats were divided into four groups: nondiabetic sedentary, nondiabetic ExT, diabetic (streptozotocin; 50 mg/kg ip) sedentary, and diabetic ExT. In the first series of studies, we measured infarct volume in all groups of rats following right MCA occlusion for 2 h, followed by 24 h of reperfusion. In a second series of studies, a craniotomy was performed over the parietal cortex, and we measured responses of pial arterioles to an endothelial NOS (eNOS)-dependent, a neuronal NOS (nNOS)-dependent, and a NOS-independent agonist in all groups of rats. We found that sedentary diabetic rats had significantly larger total, cortical, and subcortical infarct volumes following ischemia-reperfusion than sedentary nondiabetic, nondiabetic ExT, and diabetic ExT rats. Infarct volumes were similar in sedentary nondiabetic, ExT nondiabetic, and ExT diabetic rats. In contrast, ExT did not alter infarct size in nondiabetic compared with sedentary nondiabetic rats. In addition, ExT diabetic rats had impaired eNOS- and nNOS-dependent, but not NOS-independent, vasodilation that was restored by ExT. Thus ExT of T1D rats lessened ischemic brain injury following middle cerebral artery occlusion and restored impaired eNOS- and nNOS-dependent vascular function. Since the incidence of ischemic stroke is increased during T1D, we suggest that our finding are significant in that modest ExT may be a viable preventative therapeutic approach to lessen ischemia-induced brain injury that may occur in T1D subjects.
Keywords: ADP, NMDA, nitroglycerin, pial arterioles, middle cerebral artery occlusion, ischemia-reperfusion
exercise training (ext) has been shown to be beneficial for reducing the risk of premature death and disease, including cardiovascular- and cerebrovascular-related diseases. Investigators have suggested that ExT may reduce the risk of these diseases by influencing many diverse cellular networks. ExT has been shown to increase the synthesis/release of a number of important growth factors (vascular endothelial growth factor, brain-derived neurotrophic factor, fibroblast growth factor, insulin-like growth factor-I) to promote neurogenesis, suppress inflammation and apoptosis, enhance endothelial nitric oxide synthase (eNOS) and eNOS-dependent vascular function, and/or increase the activity of antioxidant enzymes in blood and in tissues of several organ systems (7, 13–19, 21, 27, 35, 37–39, 44, 47, 49). Thus the protective effects of ExT on the peripheral and cerebral circulations may be based on maintaining an adequate blood supply, maintaining vascular function, suppressing inflammation, and enhancing repair processes.
Ischemic stroke is one of the leading causes of death and permanent disability in humans. Epidemiological studies have suggested that diabetes mellitus (type 1 and type 2) is a critical risk factor for ischemic stroke (2, 5, 22, 25) and worsens outcome following ischemic stroke (3, 22, 48). In previous studies, our laboratory reported that impaired eNOS- and neuronal nitric oxide synthase (nNOS)-dependent responses of cerebral arterioles and the basilar artery observed in type 1 diabetes (T1D) rats could be restored to that observed in nondiabetic rats by modest ExT (30, 31). In addition, we found that superoxide levels were increased in sedentary diabetic rats, and ExT inhibited this increase in superoxide (31). Although ExT appears to influence vascular function in large and small cerebral blood vessels during T1D via an influence on oxidative stress, it is not clear whether the beneficial effects of ExT during T1D extend to protecting the brain from ischemia-induced brain injury. Thus the goal of the present study was to determine whether ExT could affect brain injury following cerebral ischemia-reperfusion during T1D. In addition, we examined the potential relationship between ischemia-induced brain injury and cerebrovascular function during ExT in nondiabetic and diabetic rats by examining responses of cerebral arterioles to eNOS- and nNOS-dependent agonists.
METHODS
Induction of diabetes.
All procedures were reviewed and approved by the Institutional Animal Care and Use Committee at the Louisiana State University Health Sciences Center-Shreveport. Male Sprague-Dawley rats (Harlan Sprague-Dawley, Indianapolis, IN; 180–200 g body wt) were randomly divided into one of four groups: sedentary nondiabetic, ExT nondiabetic, sedentary diabetic, and ExT diabetic. All rats had access to food and water ad libitum. The diabetic groups of rats were injected with streptozotocin (50 mg/kg ip) to induce diabetes, and the nondiabetic groups of rats were injected with vehicle (sodium citrate buffer). Blood samples, for measurement of blood glucose concentration, were obtained 3 days after injection of streptozotocin or vehicle, and on the day of the experiment. Blood glucose concentration was determined by using an Accu-Chek II Blood Glucose Monitor (Boehringer Mannheim Diagnostics, Indianapolis, IN). An animal with a blood glucose concentration of >300 mg/dl on samples obtained 3 days after injection of streptozotocin and on the day of the experiment was considered to be diabetic.
ExT.
Rats were subjected to modest levels of ExT using standard techniques similar to that which our laboratory has described previously (31). Treadmill exercise was started 3 days following injection of streptozotocin or vehicle and was carried out 5 days/wk until the day before the experiment. Experiments were conducted 6–8 wk after starting the ExT. The length of time on the treadmill was initially 10–15 min/day for the first 5 days at 0% grade at a speed of 20–25 M/min. Then, over an 8- to 10-day period, the duration on the treadmill was gradually increased to 60 min at a speed of 20–25 M/min. This level of ExT produces a similar increase in citrate synthase activity in skeletal muscle of nondiabetic and T1D rats and is considered modest in duration and intensity (30).
Transient focal cerebral ischemia.
Transient focal cerebral ischemia was performed using methods our laboratory has described previously (45, 52, 53). On the day of the experiment, sedentary nondiabetic (n = 11), ExT nondiabetic (n = 6), sedentary diabetic (n = 6), and ExT diabetic (n = 6) rats were anesthetized with ketamine (100 mg/kg ip)/xylazine (15 mg/kg ip). A laser Doppler flow probe (PeriFlux System 5000; Perimed; Ardmore, PA) was attached to the right side of the dorsal surface of the skull (2 mm caudal and 6 mm lateral to the bregma). A 4–0 monofilament nylon suture was prepared by rounding its tip by heating and coating with silicon. The right common and external carotid arteries were exposed and ligated. The middle cerebral artery (MCA) was occluded by inserting the filament from the basal area of the external carotid artery and advancing it cranially into the internal carotid artery to the point where the MCA branched from the internal carotid artery. Onset of MCA occlusion (MCAO) was determined by a rapid drop in regional cerebral blood flow, as measured by the laser Doppler. After occluding the MCA for 2 h, reperfusion was initiated by removing the suture. The rats were allowed to recover for 24 h, when the evaluation of brain infarct volume was determined.
Assessment of brain injury following MCAO.
Following 24 h of reperfusion, the rats were euthanized with Inactin (thiobutabarbital sodium; 150 mg/kg ip). The brains were quickly removed and placed in ice-cold saline for 5 min. The brains were then cut into six 2-mm-thick coronal sections. The sections were stained with 2% 2,3,5-triphenyl-tetrazolium chloride (TTC) for 15 min at 37°C. Slice images were digitalized, and the infarct lesion was evaluated using Kodak Molecular Imaging Software (Eastman Kodak, Rochester, NY). Complete lack of staining with TTC was defined as the infarct lesion. The infarct volume was expressed as a percentage of the contralateral hemisphere. We measured the total infarct volume, cortical infarct volume, and subcortical infarct volume in all groups of rats. The investigator was blinded to the group of rat during the measurement of infarct volume. Our laboratory has used these methods in previous studies (45, 52).
In vivo reactivity of pial arterioles.
Rats were prepared for in vivo studies 6–8 wk after injection of streptozotocin or vehicle. Rats were anesthetized with Inactin (100 mg/kg IP). A tracheotomy was performed and the animals were ventilated mechanically with room air and supplemental oxygen. A catheter was inserted into a femoral vein for injection of supplemental anesthesia, and a femoral artery was cannulated for measurement of arterial pressure and to obtain a blood sample for the measurement of blood glucose concentration. After placement of all catheters, the animal was placed in a head holder, and a craniotomy was made over the left parietal cortex to expose the pial microcirculation (8). The cranial window was suffused with artificial cerebral spinal fluid, at a temperature of 37 ± 1°C, and bubbled with 95% nitrogen and 5% carbon dioxide. The cranial window was connected via a three-way valve to an infusion pump, which allowed for infusion of agonists into the suffusate. Arterial blood gases were monitored and maintained within normal limits.
The cranial window was suffused for 30–45 min before responses to the agonists were examined. Then we examined dilation of pial arterioles in sedentary nondiabetic (n = 5), ExT nondiabetic (n = 9), sedentary diabetic (n = 6), and ExT diabetic (n = 5) rats in response to adenosine 5′-diphosphate (ADP; 10 and 100 μM), N-methyl-d-aspartic acid (NMDA; 100 and 300 μM), and nitroglycerin (0.1 and 1.0 μM). Agonists were mixed in artificial cerebral spinal fluid and superfused over the cranial window preparation. Diameter of pial arterioles was measured using a video image-shearing monitor (Instrumentation for Physiology and Medicine, San Diego, CA). The diameter of pial arterioles was measured immediately before the application of agonists and every min for 5 min during application of the agonists. Steady-state responses were reached within 2–3 min after application, and the diameter of pial arterioles returned to baseline within 5 min after application of the agonists was stopped.
Statistical analysis.
Analysis of variance with Fisher's test for significance was used to compare infarct volumes, baseline diameter of pial arterioles, responses of pial arterioles to the agonists, body weight, blood glucose concentration, and mean arterial blood pressure between sedentary and ExT nondiabetic and diabetic rats. A P value of ≤0.05 was considered to be significant.
Drugs and chemicals.
Streptozotocin, ADP, ketamine, xylazine, sodium citrate, Inactin, and TTC were purchased from Sigma-Aldrich (St. Louis, MO). Nitroglycerin was purchased from American Reagent Laboratories (Shirley, NY).
RESULTS
Baseline conditions.
Baseline diameter of pial arterioles and mean arterial pressure were similar in all groups of rats (Table 1). However, blood glucose concentration was significantly higher in the diabetic groups of rats compared with the nondiabetic groups, regardless of ExT (Table 1). Furthermore, body weight was similar in sedentary and ExT nondiabetic rats, but was significantly less in sedentary and ExT diabetic rats compared with the nondiabetic rats (Table 1).
Table 1.
Baseline diameter of cerebral arterioles, mean arterial pressure, blood glucose concentration, and body weight in nondiabetic, nondiabetic-ExT, T1D, and T1D-ExT rats
| Nondiabetic | Nondiabetic-ExT | T1D | T1D-ExT | |
|---|---|---|---|---|
| Baseline diameter, μm | 43 ± 2 | 44 ± 2 | 44 ± 3 | 45 ± 2 |
| Mean arterial pressure, mmHg | 136 ± 5 | 134 ± 3 | 120 ± 5 | 140 ± 4 |
| Blood glucose, mg/dl | 106 ± 11 | 114 ± 6 | 426 ± 42*† | 410 ± 28*† |
| Body weight, g | 366 ± 15 | 393 ± 12 | 279 ± 5*† | 293 ± 12*† |
Values are means ± SE. ExT, exercise training; T1D, type 1 diabetes.
P < 0.05 vs. nondiabetic rats.
P < 0.05 vs. nondiabetic-ExT rats.
MCAO/reperfusion-induced brain injury.
Total infarct volume induced by a 2-h MCAO followed by 24 h of reperfusion was greater in sedentary diabetic than in sedentary nondiabetic rats (Fig. 1). In addition, total infarct volume was similar in sedentary nondiabetic rats and ExT nondiabetic rats (Fig. 1). In contrast, ExT decreased total infarct volume in diabetic rats compared with sedentary diabetic rats (Fig. 1). Identification of the location of the infarct revealed that both the cortical and subcortical infarct volumes were increased in sedentary diabetic rats, that ExT did not influence the distribution of the infarct volumes in sedentary or ExT nondiabetic rats, and that ExT prevented the increase in cortical and subcortical infarct volumes in diabetic rats (Fig. 2).
Fig. 1.
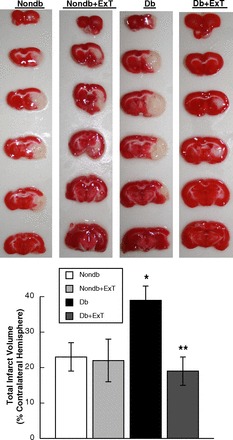
Top: representative 2-mm-thick TTC-stained coronal sections of the brains from sedentary and exercise training (ExT) nondiabetic (Nondb) and diabetic (Db) rats subjected to 2-h middle cerebral artery occlusion (MCAO) and 24 h of reperfusion. Dark stain indicates viable tissue, and complete lack of stain defines the infarct region. Bottom: mean data depicting total infarct volume in sedentary and ExT Nondb and Db rats. Values are means ± SE. *P < 0.05 vs. sedentary and ExT Nondb rats. **P < 0.05 vs. sedentary Db rats.
Fig. 2.
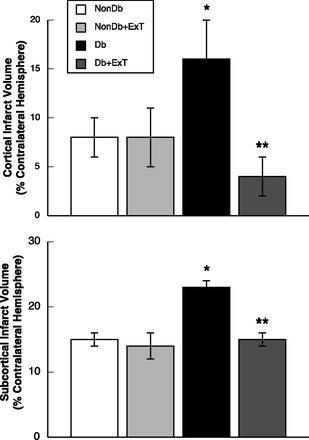
Cortical and subcortical infarct volumes in sedentary and ExT Nondb and Db rats. Values are means ± SE. *P < 0.05 vs. sedentary and ExT Nondb rats. **P < 0.05 vs. sedentary Db rats.
Responses of pial arterioles.
ADP- (Fig. 3) and NMDA-induced (Fig. 4) dilation of pial arterioles was similar in sedentary and ExT nondiabetic rats. In contrast, dilation of pial arterioles in response to ADP (Fig. 3) and NMDA (Fig. 4) was significantly less in sedentary diabetic rats compared with sedentary and ExT nondiabetic rats (P < 0.05). Furthermore, we found that ExT reversed impaired vasodilation to ADP and NMDA in diabetic rats to levels observed in sedentary and ExT nondiabetic rats. Thus, eNOS- and nNOS-induced dilation of pial arterioles is impaired in sedentary diabetic rats, and this is reversed by ExT. In contrast to that observed with ADP and NMDA, nitroglycerin produced similar dose-related dilation of pial arterioles in all groups of rats (Fig. 5). These findings, regarding the influence of ExT on vascular function, are similar to what our laboratory has shown in a previous study (31).
Fig. 3.
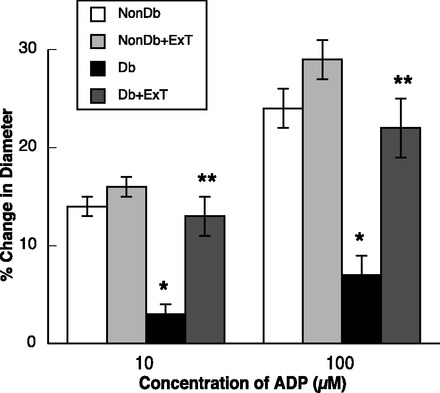
Response of cerebral arterioles to ADP in sedentary and ExT Nondb and Db rats. Values are means ± SE. *P < 0.05 vs. sedentary and ExT Nondb rats. **P < 0.05 vs. sedentary Db rats.
Fig. 4.
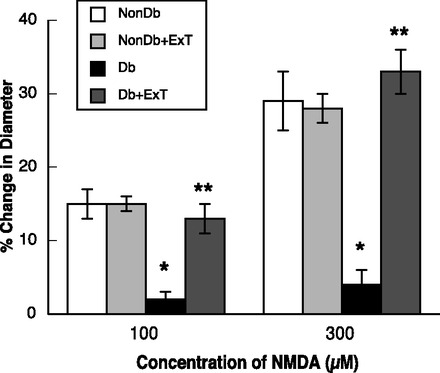
Response of cerebral arterioles to N-methyl-d-aspartic acid (NMDA) in sedentary ExT Nondb and Db rats. Values are means ± SE. *P < 0.05 vs. sedentary and ExT Nondb rats. **P < 0.05 vs. sedentary Db rats.
Fig. 5.
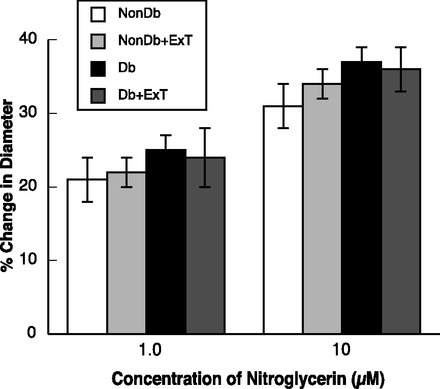
Response of cerebral arterioles to nitroglycerin in sedentary and ExT Nondb and Db rats. Values are means ± SE.
DISCUSSION
The major new finding of the present study is that modest ExT can limit brain injury following cerebral ischemia-reperfusion during T1D. In addition, this influence of ExT on infarct volume following ischemia-reperfusion in diabetic rats is accompanied by prevention of impaired eNOS- and nNOS-dependent cerebrovascular function. Based on these findings, we suggest that modest ExT has beneficial effects on the cerebral microcirculation and may have significant therapeutic potential for the prevention of diabetes-aggravated brain injury following ischemic stroke.
Consideration of methods.
Ischemic stroke accounts for ∼80–85% of all strokes. We used a model of transient focal ischemia to determine whether ExT could influence brain injury following ischemia-reperfusion in nondiabetic and diabetic rats (53). This methodology uses a silicon-coated suture to produce occlusion of the MCA that appears to be more consistent than the poly-l-lysine-coated suture. Our laboratory has shown previously that the poly-l-lysine-coated suture is adequate in diameter to produce occlusion of the MCA, but may lack suitable thickness in the trunk of the suture to allow the MCA area to be supplied by the internal carotid and posterior carotid arteries, thereby producing a more variable infarct (53). In contrast, the silicon-coated suture can obstruct all sources of blood flow from these arteries. Thus we suggest that the use of this modified suture yields a more consistent ischemic area/infarct volume, and we suggest that this model is appropriate for the study of cerebral ischemic events.
We used treadmill exercise in nondiabetic and diabetic rats to examine the influence of ExT on reactivity of cerebral arterioles and on infarct volume following MCAO. We and others have used the regimen for ExT described in the present study to examine the influence of modest ExT on various aspects of diabetes-induced diseases (30, 31, 41, 54). In previous studies, our laboratory found that this modest level of ExT prevented diabetes-induced alterations in vascular function of large and small cerebral arterioles, without influencing body weight or blood glucose concentration (30, 31). In addition, studies by Shao et al. (41) reported that a similar level of ExT, as used in the present study, produced a similar increase in citrate synthase activity in ExT nondiabetic and T1D rats (indicating that the degree of ExT was similar in nondiabetic and T1D rats) and could attenuate cardiac dysregulation observed in T1D rats, again without influencing body weight or blood glucose levels in either nondiabetic or T1D rats. In addition, these investigators (41) reported that there was no difference in cardiac function between sedentary nondiabetic and ExT nondiabetic rats, suggesting that the level of ExT was modest, but still adequate, to influence parameters in T1D. Thus we suggest that the regimen of ExT used in the present study may be modest, but is adequate to influence cerebrovascular dysfunction and brain injury following MCAO during T1D.
We used ADP and NMDA to examine eNOS- and nNOS-dependent responses of cerebral arterioles, respectively. We and others have shown that ADP dilates cerebral arterioles via activation of nitric oxide synthase (NOS), presumably eNOS (1, 9, 32). In addition, we and others have shown that NMDA dilates cerebral arterioles via the activation of nNOS (10–12, 46). Based on these previous findings, we suggest that ADP and NMDA are appropriate agonists to evaluate NOS-dependent dilatation of cerebral arterioles.
Influence of diabetes on the incidence of stroke.
Diabetes (type 1 and type 2) is an important risk factor for cerebral ischemia, and the risk for ischemic stroke is about twice as much in a diabetic individual as in an individual without diabetes (2, 22, 42). In addition, brain injury is increased, and functional outcomes are worse following cerebral ischemia during diabetes (3, 22, 34, 48). The mechanisms by which diabetes contributes to an increase in the risk for cerebral ischemia and brain injury following cerebral ischemia appear to be related to hyperglycemia (3, 5, 23) and the consequences of hyperglycemia, i.e., an increase in oxidative stress (26, 50). Kusaka et al. (26) found that brain injury following cerebral ischemia was increased in T1D rats due to the influence of angiotensin II (via AT1 receptors) activating NADPH oxidase. The findings of the present study are in agreement with and extend the findings of Kusaka et al. (26). We found that reactivity of cerebral arterioles was decreased in T1D rats, and brain injury following cerebral ischemia was increased in T1D rats. Furthermore, we found that ExT could reverse impaired cerebrovascular reactivity and limit brain injury following cerebral ischemia during T1D. Although we do not have definitive information regarding the precise mechanism by which ExT protects the brain during T1D, we speculate that ExT may decrease oxidative stress (31).
Influence of ExT on the risk of stroke.
A number of epidemiological studies using human subjects have examined the relationship between pre-ExT on the risk of ischemic stroke. The findings from these studies have been mixed. Some studies have reported that pre-ExT was associated with better functional outcomes after having a stroke, a less severe level of stroke, and/or a decrease in the occurrence of ischemic stroke (6, 20, 24, 28, 40, 43). The mechanisms that contributed to the protective effects of pre-ExT on functional outcomes/occurrence of stroke are not entirely clear from these epidemiological studies, but have been suggested to be related to effects of pre-ExT on metabolic pathways, blood pressure, blood cholesterol, glucose tolerance, and/or body weight. In contrast to studies that have shown a protective influence of pre-ExT, a recent prospective study has suggested that pre-ExT may decrease the occurrence of ischemic stroke, but does not improve functional outcomes following stroke (36). Thus, based on epidemiological studies, it is difficult to provide a global statement regarding the beneficial effects of pre-ExT on the pathogenesis of stroke, but it appears that the majority of evidence suggests that pre-ExT has a beneficial effect on both the occurrence and functional outcomes of ischemic stroke.
Investigators also have examined the influence of pre- and post-ExT on the pathogenesis of stroke using animal models. Endres et al. (7) examined the influence of pre-ExT on eNOS expression and ischemic injury in wild-type and eNOS−/− mice. These investigators found that 3 wk of pre-ExT before induction of MCAO could protect against cerebral ischemic injury in wild-type mice, but not in eNOS−/− mice, and concluded that this protection was related to an increase in eNOS activity. Others have examined the influence of post-ExT on brain injury following cerebral ischemia. Cechetti et al. (4) found that post-ExT following a cerebral ischemic event could protect against cognitive dysfunction. The mechanism for the protective effect of post-ExT was not precisely determined, but was speculated to be related to an adaptive effect of antioxidant pathways due to an increase in oxidative stress produced by treadmill exercise. In another study, Zheng et al. (55) found that post-ExT improved recovery of brain function in rats following MCAO by a mechanism that appeared to be related to an increase in the expression of angiopoietin-1 and/or Tie-2, which influence neovascularization. Finally, a study by Zhang et al. (51) found that post-ExT could improve brain injury following MCAO by a mechanism that appeared to promote mitochondrial biogenesis. Thus it appears that post-ExT following the induction of an ischemic stroke can protect against neurological dysfunction and brain injury. The present study extends the findings of previous studies by demonstrating a positive influence of pre-ExT on brain injury and cerebrovascular function during T1D. However, we did not find a significant beneficial effect of pre-ExT on cerebrovascular reactivity or brain injury following cerebral ischemia-reperfusion in normal (nondiabetic) rats in the present study or in our laboratory's previous studies (30, 31), even though we found an increase in eNOS protein in ExT nondiabetic rats (31). The explanation for the discrepancy between the present study and the study of Endres et al. (7) regarding the beneficial effects of pre-ExT on brain injury following cerebral ischemia-reperfusion in normal (nondiabetic) animals is not clear, but may be related to species differences (mice vs. rats) and/or differences in the intensity and duration of ExT. Future studies will need to examine the influence of intensity and duration of ExT on cerebrovascular reactivity and brain injury following cerebral ischemia.
Relationship between cerebrovascular reactivity and brain injury following cerebral ischemia.
We measured cerebrovascular function and infarct volume in sedentary and ExT nondiabetic and diabetic rats. We suggest that there may be a relationship between altered cerebrovascular function and brain infarct volume following cerebral ischemia-reperfusion. During a transient focal cerebral ischemic event, the reduction in blood flow from the arterial occlusion is greatest in the center of the ischemic territory (ischemic core) and less at the periphery of the ischemic core (ischemic penumbra) (53). Following an ischemic stroke, cerebrovascular reactivity is impaired (29, 33), thereby reducing the potential increases in blood flow to the ischemic area by functional activation of many components within brain tissue, i.e., a functional alteration in the neurovascular unit. During a disease state such as T1D, this condition may be amplified, given that vascular function in response to eNOS- and nNOS-dependent stimuli is compromised (31). In the present study, we found a significant increase in the infarct volume in both the cortical and subcortical regions of the brain in T1D rats that was restored to that observed in nondiabetic rats by ExT. Thus it is conceivable that, if there are preexisting alterations in cerebrovascular function (i.e., the neurovascular unit) during T1D, that this might influence the severity of the infarct volume following cerebral ischemia-reperfusion by limiting adequate cerebral blood flow to the ischemia area.
In summary, we found that transient focal cerebral ischemia produced a larger infarct volume in T1D rats compared with nondiabetic rats. In addition, we found that ExT limited brain injury following cerebral ischemia-reperfusion in T1D rats. Furthermore, we found that ExT restored impaired eNOS- and nNOS-dependent dilation of cerebral arterioles in T1D rats, but did not alter NOS-independent vasodilation. We speculate that ExT may have a potential therapeutic value for the treatment of T1D-induced cerebrovascular dysfunction and may be important in the prevention of T1D-induced cerebrovascular abnormalities, including the risk of ischemic stroke.
GRANTS
This study was supported by funds from National Heart, Lung, and Blood Institute Grant HL-090657 and funds from the LSU Health Science Center-Shreveport.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: D.M.A. and W.G.M. conception and design of research; D.M.A., H.S., and W.G.M. performed experiments; D.M.A. and W.G.M. analyzed data; D.M.A. and W.G.M. interpreted results of experiments; D.M.A. drafted manuscript; D.M.A., H.S., and W.G.M. approved final version of manuscript; H.S. and W.G.M. edited and revised manuscript.
REFERENCES
- 1. Ayajiki K, Okamura T, Toda N. Involvement of nitric oxide in endothelium-dependent, phasic relaxation caused by histamine in monkey cerebral arteries. Jpn J Pharmacol 60: 357–362, 1992 [DOI] [PubMed] [Google Scholar]
- 2. Baird TA, Parsons MW, Barber PA, Butcher KS, Desmond PM, Tress BM, Colman PG, Jerums G, Chambers BR, Davis SM. The influence of diabetes mellitus and hyperglycaemia on stroke incidence and outcome. J Clin Neurosci 9: 618–626, 2002 [DOI] [PubMed] [Google Scholar]
- 3. Bravata DM, Kim N, Concato J, Brass LM. Hyperglycaemia in patients with acute ischaemic stroke: how often do we screen for undiagnosed diabetes? QJM 96: 491–497, 2003 [DOI] [PubMed] [Google Scholar]
- 4. Cechetti F, Worm PV, Elsner VR, Bertoldi K, Sanches E, Ben J, Siqueira IR, Netto CA. Forced treadmill exercise prevents oxidative stress and memory deficits following chronic cerebral hypoperfusion in the rat. Neurobiol Learn Mem 97: 90–96, 2012 [DOI] [PubMed] [Google Scholar]
- 5. Chukwuma CS, Tuomilehto J. Diabetes and the risk of stroke. J Diabetes Complications 7: 250–262, 1993 [PubMed] [Google Scholar]
- 6. Deplanque D, Masse I, Lefebvre C, Libersa C, Leys D, Bordet R. Prior TIA, lipid-lowering drug use, and physical activity decrease ischemic stroke severity. Neurology 67: 1403–1410, 2006 [DOI] [PubMed] [Google Scholar]
- 7. Endres M, Gertz K, Lindauer U, Katchanov J, Schultze J, Schrock H, Nickenig G, Kuschinsky W, Dirnagl U, Laufs U. Mechanisms of stroke protection by physical activity. Ann Neurol 54: 582–590, 2003 [DOI] [PubMed] [Google Scholar]
- 8. Fang Q, Sun H, Mayhan WG. Impairment of nitric oxide synthase-dependent dilatation of cerebral arterioles during infusion of nicotine. Am J Physiol Heart Circ Physiol 284: H528–H534, 2003 [DOI] [PubMed] [Google Scholar]
- 9. Faraci FM. Role of endothelium-derived relaxing factor in cerebral circulation: large arteries vs. microcirculation. Am J Physiol Heart Circ Physiol 261: H1038–H1042, 1991 [DOI] [PubMed] [Google Scholar]
- 10. Faraci FM, Breese KR. Nitric oxide mediates vasodilatation in response to activation of N-methyl-d-aspartate receptors in brain. Circ Res 72: 476–480, 1993 [DOI] [PubMed] [Google Scholar]
- 11. Faraci FM, Breese KR, Heistad DD. Responses of cerebral arterioles to kainate. Stroke 25: 2080–2084, 1994 [DOI] [PubMed] [Google Scholar]
- 12. Faraci FM, Brian JE. 7-Nitroindazole inhibits brain nitric oxide synthase and cerebral vasodilatation in response to N-methyl-d-aspartate. Stroke 26: 2172–2176, 1995 [DOI] [PubMed] [Google Scholar]
- 13. Franke WD, Stephens GM, Schmid PG. Effects of intense exercise training on endothelium-dependent exercise-induced vasodilatation. Clin Physiol 18: 521–528, 1998 [DOI] [PubMed] [Google Scholar]
- 14. Fuchsjager-Mayrl G, Pleiner J, Wiesinger GF, Sieder AE, Quittan M, Nuhr MJ, Francesconi C, Seit HP, Francesconi M, Schmetterer L, Wolzt M. Exercise training improves vascular endothelial function in patients with type 1 diabetes. Diabetes Care 25: 1795–1801, 2002 [DOI] [PubMed] [Google Scholar]
- 15. Fukai T, Siegfried MR, Ushio-Fukai M, Cheng Y, Kojda G, Harrison DG. Regulation of the vascular extracellular superoxide dismutase by nitric oxide and exercise training. J Clin Invest 105: 1631–1639, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Green DJ, Maiorana A, O'Driscoll G, Taylor R. Effect of exercise training on endothelium-derived nitric oxide function in humans. J Physiol 561: 1–25, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Griffin KL, Woodman CR, Price EM, Laughlin MH, Parker JL. Endothelium-mediated relaxation of porcine collateral-dependent arterioles is improved by exercise training. Circulation 104: 1393–1398, 2001 [DOI] [PubMed] [Google Scholar]
- 18. Hambrecht R, Adams V, Erbs S, Linke A, Krankel N, Shu Y, Baither Y, Gielen S, Thiele H, Gummert JF, Mohr FW, Schuler G. Regular physical activity improves endothelial function in patients with coronary artery disease by increasing phosphorylation of endothelial nitric oxide synthase. Circulation 107: 3152–3158, 2003 [DOI] [PubMed] [Google Scholar]
- 19. Higashi Y, Yoshizumi M. Exercise and endothelial function: role of endothelium-derived nitric oxide and oxidative stress in healthy subjects and hypertensive patients. Pharmacol Ther 102: 87–96, 2004 [DOI] [PubMed] [Google Scholar]
- 20. Hu FB, Stampfer MJ, Solomon C, Liu S, Colditz GA, Speizer FE, Willett WC, Manson JE. Physical activity and risk for cardiovascular events in diabetic women. Ann Intern Med 134: 96–105, 2001 [DOI] [PubMed] [Google Scholar]
- 21. Johnson P. Antioxidant enzyme expression in health and disease: effects of exercise and hypertension. Comp Biochem Physiol C Toxicol Pharmacol 133: 493–505, 2002 [DOI] [PubMed] [Google Scholar]
- 22. Jorgensen HS, Nakayama H, Raaschou HO, Olsen TS. Effect of blood pressure and diabetes on stroke in progression. Lancet 344: 156–159, 1994 [DOI] [PubMed] [Google Scholar]
- 23. Kagansky N, Levy S, Knobler H. The role of hyperglycemia in acute stroke. Arch Neurol 58: 1209–1212, 2001 [DOI] [PubMed] [Google Scholar]
- 24. Krarup LH, Truelsen T, Gluud C, Andersen G, Zeng X, Korv J, Oskedra A, Boysen G. Prestroke physical activity is associated with severity and long-term outcome from first-ever stroke. Neurology 71: 1313–1318, 2008 [DOI] [PubMed] [Google Scholar]
- 25. Kurl S, Laukkanen JA, Niskanen L, Laaksonen D, Sivenius J, Nyyssonen K, Salonen JT. Metabolic syndrome and the risk of stroke in middle-aged men. Stroke 37: 806–811, 2006 [DOI] [PubMed] [Google Scholar]
- 26. Kusaka I, Kusaka G, Zhou C, Ishikawa M, Nanda A, Granger DN, Zhang JH, Tang J. Role of AT1 receptors and NAD(P)H oxidase in diabetes-aggravated ischemic brain injury. Am J Physiol Heart Circ Physiol 286: H2442–H2451, 2004 [DOI] [PubMed] [Google Scholar]
- 27. Lawson DL, Chen L, Mehta JL. Effects of exercise-induced oxidative stress on nitric oxide release and antioxidant activity. Am J Cardiol 80: 1640–1642, 1997 [DOI] [PubMed] [Google Scholar]
- 28. Lee IM, Hennekens CH, Berger K, Buring JE, Manson JE. Exercise and risk of stroke in male physicians. Stroke 30: 1–6, 1999 [DOI] [PubMed] [Google Scholar]
- 29. Leffler CW, Beasley DG, Busija DW. Cerebral ischemia alters cerebral microvascular reactivity in newborn pigs. Am J Physiol Heart Circ Physiol 257: H266–H271, 1989 [DOI] [PubMed] [Google Scholar]
- 30. Mayhan WG, Sun H, Mayhan JF, Patel KP. Influence of exercise on dilatation of the basilar artery during diabetes mellitus. J Appl Physiol 96: 1730–1737, 2004 [DOI] [PubMed] [Google Scholar]
- 31. Mayhan WG, Arrick DM, Patel KP, Sun H. Exercise training normalizes impaired NOS-dependent responses of cerebral arterioles in type 1 diabetic rats. Am J Physiol Heart Circ Physiol 300: H1013–H1020, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mayhan WG. Endothelium-dependent responses of cerebral arterioles to adenosine 5′-diphosphate. J Vasc Res 29: 353–358, 1992 [DOI] [PubMed] [Google Scholar]
- 33. Mayhan WG, Amundsen SM, Faraci FM, Heistad DD. Responses of cerebral arteries after ischemia and reperfusion in cats. Am J Physiol Heart Circ Physiol 255: H879–H884, 1988 [DOI] [PubMed] [Google Scholar]
- 34. Ntaios G, Egli M, Faouzi M, Michel P. J-shaped association between serum glucose and functional outcome in acute ischemic stroke. Stroke 41: 2366–2370, 2010 [DOI] [PubMed] [Google Scholar]
- 35. Powers SK, Ji LL, Leeuwenburgh C. Exercise training-induced alterations in skeletal muscle antioxidant capacity: a brief review. Med Sci Sports Exerc 107: 987–997, 1999 [DOI] [PubMed] [Google Scholar]
- 36. Rist PM, Lee IM, Kase CS, Gaziano JM, Kurth T. Physical activity and functional outcomes from cerebral vascular events in men. Stroke 42: 3352–3356, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Roberts CK, Barnard RJ, Jasman A, Balon TW. Acute exercise increases nitric oxide synthase activity in skeletal muscle. Am J Physiol Endocrinol Metab 277: E390–E394, 1999 [DOI] [PubMed] [Google Scholar]
- 38. Rush JW, Turk JR, Laughlin MH. Exercise training regulates SOD-1 and oxidative stress in porcine aortic endothelium. Am J Physiol Heart Circ Physiol 284: H1378–H1387, 2003 [DOI] [PubMed] [Google Scholar]
- 39. Rush JWE, Laughlin MH, Woodman CR, Price EM. SOD-1 expression in pig coronary arterioles is increased by exercise training. Am J Physiol Heart Circ Physiol 279: H2068–H2076, 2000 [DOI] [PubMed] [Google Scholar]
- 40. Sacco RL, Gan R, Boden-Albala B, Lin IF, Kargman DE, Hauser WA, Shea S, Paik MC. Leisure-time physical activity and ischemic stroke risk: the Northern Manhattan Stroke Study. Stroke 29: 380–387, 1998 [DOI] [PubMed] [Google Scholar]
- 41. Shao CH, Wehrens XH, Wyatt TA, Parbhu S, Rozanski GJ, Patel KP, Bidasee KR. Exercise training during diabetes attenuates cardiac ryanodine receptor dysregulation. J Appl Physiol 106: 1280–1292, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Stegmayr B, Asplund K. Diabetes as a risk factor for stroke. A population perspective. Diabetologia 38: 1061–1068, 1995 [DOI] [PubMed] [Google Scholar]
- 43. Stroud N, Mazwi TM, Case LD, Brown RDJ, Brott TG, Worrall BB, Meschia JF. Prestroke physical activity and early functional status after stroke. J Neurol Neurosurg Psychiatry 80: 1019–1022, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sun D, Huang A, Koller A, Kaley G. Enhanced NO-mediated dilations in skeletal muscle arterioles of chronically exercised rats. Microvasc Res 64: 491–496, 2002 [DOI] [PubMed] [Google Scholar]
- 45. Sun H, Zhao H, Sharpe GM, Arrick DM, Mayhan WG. Effect of chronic alcohol consumption on brain damage following transient focal ischemia. Brain Res 1194: 73–80, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sun H, Patel KP, Mayhan WG. Impairment of neuronal nitric oxide synthase-dependent dilatation of cerebral arterioles during chronic alcohol consumption. Alcohol Clin Ex Res 26: 663–670, 2002 [PubMed] [Google Scholar]
- 47. Tanabe T, Maeda S, Miyauchi T, Iemitsu M, Takanashi M, Irukayama-Tomobe Y, Yokota T, Ohmori H, Matsuda M. Exercise training improves ageing-induced decrease in eNOS expression of the aorta. Acta Physiol Scand 178: 3–10, 2003 [DOI] [PubMed] [Google Scholar]
- 48. Williams LS, Rotich J, Qi R, Fineberg N, Espay A, Bruno A, Fineberg SE, Tierney WR. Effects of admission hyperglycemia on mortality and costs in acute ischemic stroke. Neurology 59: 67–71, 2002 [DOI] [PubMed] [Google Scholar]
- 49. Xiang L, Naik J, Hester RL. Exercise-induced increase in skeletal muscle vasodilatory responses in obese Zucker rats. Am J Physiol Regul Integr Comp Physiol 288: R987–R991, 2005 [DOI] [PubMed] [Google Scholar]
- 50. Yorek MA. The role of oxidative stress in diabetic vascular and neural disease. Free Radic Res 37: 471–480, 2003 [DOI] [PubMed] [Google Scholar]
- 51. Zhang Q, Wu Y, Zhang P, Sha H, Jia J, Hu Y, Zhu J. Exercise induces mitochondrial biogenesis after brain ischemia in rats. Neuroscience 205: 10–17, 2012 [DOI] [PubMed] [Google Scholar]
- 52. Zhao H, Mayhan WG, Arrick DM, Xiong W, Sun H. Dose-related influence of chronic alcohol consumption on cerebral ischemia/reperfusion injury. Alcohol Clin Exp Res 35: 1265–1269, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhao H, Mayhan WG, Sun H. A modified suture technique produces consistent cerebral infarction in rats. Brain Res 1246: 158–166, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zheng H, Mayhan WG, Patel KP. Exercise training improves the defective centrally mediated erectile responses in rats with type I diabetes. J Sex Med 8: 3086–3097, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zheng Q, Zhu D, Bai Y, Wu Y, Jia J, Hu Y. Exercise improves recovery after ischemic brain injury by inducing the expression of angiopoietin-1 and Tie-2 in rats. Tohoku J Exp Med 224: 221–228, 2011 [DOI] [PubMed] [Google Scholar]