

Abstract
Recently, covalent drugs have attracted great interest in the drug discovery community, with successful examples that have demonstrated their therapeutic effects. Here, we focus on the covalent inhibition of the fatty acid amide hydrolase (FAAH), which is a promising strategy in the treatment of pain and inflammation. Among the most recent and potent FAAH inhibitors (FAAHi), there are the cyclic piperidine/piperazine aryl ureas. FAAH hydrolyzes efficiently the amide bond of these compounds, forming a covalent enzyme-inhibitor adduct. To rationalize this experimental evidence, we performed an extensive computational analysis centered on the piperidine-based PF750 (1) and the piperazine-based JNJ1661010 (2), two potent lead compounds used to generating covalent inhibitors as clinical candidates. We found that FAAH induces a distortion of the amide bond of the piperidine/piperazine aryl ureas. QM/MM ΔELUMO-HOMO energies indicate that the observed enzyme-induced distortion of the amide bond favors the formation of a covalent FAAH- inhibitor adduct. These findings could help in the rational structure-based design of novel covalent FAAHi.
Introduction
The drug discovery community is looking at covalent drugs with a renewed interest.1 Indeed, covalent drugs have been approved for several clinical indications and have made a major positive impact on human health.2 In this scenario, fatty acid amide hydrolase (FAAH) represents a promising target for the discovery of covalent drugs to treat pain, inflammation and other diseases.3 FAAH is an integral membrane protein belonging to the amidase signature family of enzymes (Figure 1)4, and it is responsible for the deactivating hydrolysis of several endogenous fatty acid amides such as anandamide (AEA), palmytoylethanolamide (PEA) and oleoylethanolamide.5 Therefore, the inhibition of FAAH can be used to modulate the level of endogenous fatty acid ethanolamides,6 which in turn has been proven to induce anti-inflammatory, antidepressant, analgesic and anxiolytic effects.7 In addition, these actions occur in the absence of alteration in motility, sleep, weight gain or other side effects typically observed with direct CB1 agonists.8
Figure 1.

X-ray structure of the h/r FAAH protein in complex with the PF750 inhibitor (PDB code: 2VYA).10b The FAAH protein consists of two monomers: Mnr A is represented with blue ribbons and Mnr B is shown in yellow. The catalytic triad (Ser241-Ser217-Lys142) and the oxyanion hole (Ile238, Gly239, Gly240) residues are depicted in blue (Mnr A) and yellow sticks (Mnr B). PF750 is shown with green sticks.
FAAH is a serine hydrolase characterized by an unusual Ser-Ser-Lys catalytic triad (Ser241, Ser217, Lys142) that cleaves amides and esters with similar rates.9 Over the last few years, a number of crystal structures of FAAH bound to covalent inhibitors have been resolved.10 This experimental evidence, coupled with computational studies,11 has elucidated the enzymatic mechanism of FAAH; First, substrate hydrolysis is initiated by activation of the nucleophile Ser241 residue. This occurs through a proton transfer event that leads the side chain proton of Ser241 to Lys142, shuttled via Ser217. Then, the activated Ser241 attacks the carbonyl group of the substrate, leading to the formation of a tetrahedral intermediate. Finally, the protonation of the leaving group by Lys142, through a proton shuttle that again involves Ser217, facilitates the leaving group departure and formation of the acylated enzyme (Scheme 1a).
Scheme 1.

(a)Mechanism of substrate hydrolysis by FAAH (shown for a generic amide substrate). (b) Proposed mechanism of FAAH inhibition by the piperidine/piperazine ureas (shown for 1).18 FAAH residues are depicted in green, the leaving group is depicted in orange. The enzyme-induced conformational change in the piperidine/piperazine urea diminishes the conjugation of the nitrogen lone pair with the carbonyl and allows the nucleophilic attack by the Ser241. This leads to a covalent enzyme-inhibitor adduct.
Several classes of FAAH inhibitors12 (FAAHi) react and form a covalent bond with Ser241. Among these molecules are potent electrophilic compounds characterized by the presence of an activated carbonyl group. These include trifluoromethyl ketones, α-keto esters, α-keto amides, and α-ketoheterocycles, such as OL-135. However, most of these compounds have low target selectivity and limited efficacy in vivo.12
A class of FAAHi with a promising drug-like profile are potent and irreversible FAAHi based on a N-cyclohexylcarbamic acid O-aryl ester template, which include the compound URB524 (IC50 = 63 nM).8a, 13 Optimization of URB524 led to URB597 (the 3′-CONH2 derivative of URB524),14 a highly potent FAAHi in vitro (IC50 = 4.6 nM) and in vivo (ED50 = 0.15 mg/kg, in rat).8a, 15 Interestingly, a brain-impenetrant member of this class of compounds was recently disclosed (URB937, IC50 = 26.8 ± 4.9 nM) and shown to produce substantial analgesic effects in animal models, which is suggestive that inhibition of peripheral FAAH activity might represent a novel approach for the treatment of pain. 16
The unique ability of FAAH to cleave amides and esters at similar rates suggests, however, that not only carbamates but also ureas could act as good carbamoylating agents. Even though the substitution of the carbamate functionality with an acyclic urea led to mostly inactive compounds,13, 17 cyclic piperidine/piperazine-based molecules were identified by Pfizer and Cravatt’s lab as a novel class of potent FAAHi.18 Among them is the irreversible N-phenyl-4-(quinolin-3-ylmethyl)piperidine-1-carboxamide (PF750, 1) (IC50 = 16.2 nM)18 and the reversible piperazine-based N-Phenyl-4-(3-phenyl-1,2,4-thiadiaz ol-5-yl)-1-piperazinecarboxamide (JNJ1661010, 2) (IC50 = 33 ± 2.1 nM).19 The piperidine/piperazine moiety of these compounds seems to favor, somehow, the covalent interaction of the inhibitor with Ser241. In this respect, Cravatt and coworkers have proposed that the limited flexibility of piperidine/piperazine-based inhibitors could favor the conformational distortion of their urea functionality at the FAAH active site, favoring the formation of a covalent bond between the inhibitor and Ser241 (Scheme 1).18 This hypothesis, however, has not been investigated yet, and the mechanism of piperidine/piperazine FAAHi remains poorly understood at the atomic level.
Here, we report on a comparative study based on molecular dynamics (MD) simulations and quantum mechanics/molecular mechanics (QM/MM) computations that aims at characterizing the difference between cyclic and inactive acyclic ureas. Three structurally different compounds were considered: the two potent lead compounds piperidine-based 1 and the piperazine-based 2, along with an inactive acyclic 1-Cyclohexyl-3-naphthalen-2-ylurea, referred to here as Cpd3 (3) (IC50 = < 30,000 nM) (Figure 2).13 We have characterized the conformational flexibility of these compounds in water solution and in complex with FAAH. Our results support the hypothesis18 that FAAH is able to induce a distortion of the amide bond of the piperidine/piperazine compounds. The twist of the amide bond likely facilitates the amide bond hydrolysis, and formation of the covalent inhibitor-enzyme adduct.20 On the other hand, the rigidity of the planar urea moiety in the acyclic derivative seems to prevent its good fit into the catalytic site, which might partially explain its lack of inhibitory activity.
Figure 2.

The piperidine urea 1 (IC50 = 16.2 nM),18 the piperazine urea 2 (IC50 = 33 ± 2.1 nM),19 the acyclic urea 3 (IC50 = < 30,000 nM).13
Computational materials and methods
Structural models
Three model systems formed by FAAH in complex with either 1, 2 and with the inactive derivative 3 were considered for calculations (see Figure 1 and 2). All model systems were based on the crystallographic structure of the humanized rat FAAH protein ((h/r) FAAH) in complex with 1, solved at 2.75 Å resolution (PDB code: 2VYA).10b The X-ray structure of the FAAH/1 complex consists of monomer A (Mnr A) and monomer B (Mnr B) formed by 574 residues in total, one Cl− ion, 84 co-crystallized water molecules and two 1 residues (one per monomer) covalently attached to the Ser241 of FAAH through a carbamate bond. Importantly, this structure contains active site residues of the human protein, within the parent rat protein, including the key residues of the catalytic triad (Ser241, Ser217, Lys142) and those of the oxyanion hole (Ile238, Gly239, Gly240, Ser241). The binding mode of 1 was constructed by adding the leaving group to the crystallographic pose. The piperazinic 2 binding pose was generated via computational docking of the ligand into the FAAH binding site. Docking calculations were carried out using Autodock 4.221 (see Supplementary Information, SI). The final docked structure of 2 is in agreement with its binding pose proposed by Keith et al.19 The comparison between the binding mode of the co-crystallized ligand 1 and the docked 2 compound shows that the urea occupies the same region of the pocket (see SI). Due to the structural similarity between 1 and 3, the binding pose of the latter was modeled by manually modifying 1 within the binding site (see SI).
Molecular dynamics (MD) simulations
A standard MD setup was adopted to all the three complexes, namely FAAH/1, FAAH/2, and FAAH/3. Hydrogen atoms were added assuming standard bond lengths and angles. The simulations were performed with deprotonated Lys142, as proposed for the catalytic mechanism of FAAH.10a Each of the three complexes were immersed in a rectangular TIP3P water box.22 The starting size of the solvated system was approximately 100 Å × 100 Å × 125 Å for all the three complexes, containing about 36,550 water molecules and a total number of about 126,500 atoms. The charge of the systems was neutralized adding an exact number of counterions (8 Cl− ions were added in each model) whose optimal positions were located using the xLEAP program of the AMBER10 package.23 The AMBER99SB force field24 was adopted for the protein. The ligands 1, 2, and 3 were treated with the General Amber Force Field25 together with ad hoc charges calculated following the RESP procedure (see SI).26 The SHAKE27 algorithm was used for all the covalent bonds that contain an H atom, thus allowing a timestep of 2 fs for the integration of the equations of motion. All the simulations were performed by using the NAMD 2.7 package.28 Periodic boundary conditions were applied and the Particle-Mesh Ewald method was used to evaluate long-range electrostatic interactions with a direct cutoff of 10 Å. All the simulations were carried out with the following protocol. First, the systems were optimized and thermalized up to 300 K in the NVT ensemble using a Langevin bath29, in three consecutive steps: (1) the solvent was first equilibrated, slowly increasing the temperature from 0 to 100 K and maintaining the protein fixed, (2) the temperature was further increased up to 200 K while keeping fixed only the coordinates of backbone atoms of the protein, (3) constraints were released and the systems were simulated to reach the temperature of 300 K. Subsequently, 100 ns of MD were performed for each system in the NPT ensemble in standard condition using a Langevin Piston. Coordinates of the systems were collected every 2 ps, for a total of ~50,000 frames for each run. Statistics was accumulated during the last 98 ns of each run. The structural analysis here reported is obtained using data from the two FAAH monomers that form each complex, considered as independent, for a total of ~100,000 structures.
Unbound systems
The 1, 2, and 3 compounds were also simulated via classical MD in a pure water-box. 1 was immersed in a 43.1 × 31.0 × 29.4 Å3 box, with a number of water molecules equal to 1,314 and a total number of atoms of 3,991. The piperazine 2 was immersed in a 43.3 × 31.3 × 29.8 Å3 box of 1,348 water molecules, for a total of 4,089 atoms. The water box for the acyclic 3 was 40.3 × 33.7 × 29.2 Å3, including 1,202 water molecules and a total number of 3,646 atoms. Same protocol, as for the above-described simulations, was adopted for optimization and thermalization.
Analysis of structural data
The amide bond distortion was characterized by analyzing the following structural parameters: 1) the R2N–C–N–H dihedral angle (referred to as δ, scheme 2a); 2) the improper torsion centered on the secondary amine nitrogen (referred to as θ, scheme 2b).
Scheme 2.
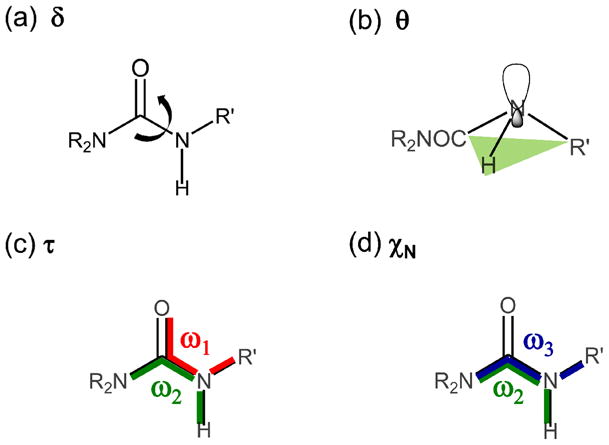
Descriptors used for the analysis of the amide bond distortion. (a) Dihedral angle δ = R2N-C-N-H, (b) improper torsion θ centered on the secondary amine nitrogen, (c) mean twisting angle τ = (ω1 + ω2)/2 and (d) χN = ω2 - ω3 + π(mod2π).30b
For completeness, the planarity of the amino group was also analyzed using the internal coordinates τ [3] and χN [4], originally defined by Dunitz.30 The τ and χN parameters are function of the torsion angles defined by the amide C–N bond, as follows:
| [3] |
| [4] |
Where ω1 = O=C–N–R′; ω2 = R2N–C–N–H; and ω3 = R2N–C–N–R′, (Scheme 2). The angle τ is a measure of the mean twisting angle between the C–N bond and it ranges from 0° (planar amide group) to 90° (when the two planes defined by the O=C–NR2 and the R′–N–H atoms are perpendicular). The χN parameter determines the degree of pyramidalization at the N atom, and it may take values approximately between 0° (planar sp2 geometry) to 60° (tetrahedral sp3 geometry). The geometrical meaning of τ and χN is shown in Scheme 2. We consider here the absolute values for these angles reported on the 0°–90° quadrant.
Quantum mechanics/molecular mechanics (QM/MM) calculations
QM/MM calculations were performed to obtain the HOMO-LUMO energy gap (ΔELUMO-HOMO) of the reactive complex formed by the inhibitor 1 and 2 and the enzyme. Due to the fact that 3 is neither experimentally active nor stable during the simulations, it was not considered for these calculations. Calculations were carried out using the two-layer ONIOM method,31 as implemented in Gaussian09.32 This approach allows treating the system at different levels of theory. The entire system (real) is computed with a low level of theory (MM), while the inner layer (model) is treated at a high level of theory (QM). The expression of the ONIOM energy, EONIOM, is given via a subtractive QM/MM scheme:
| [1] |
The electrostatic interaction between the two layers was taken into account via electrostatic embedding.33
For the structures extracted from each of the MD runs, we built a QM/MM subsystem by retaining the ligand and the 79 residues that were within 8 Å from the ligand. The QM region consisted of the ligand and the following residues of FAAH active site: the catalytic triad and oxyanion hole residues Ile238, Gly239, Gly240, as well as the Met191, Phe192, and the Asp237 residues. The N- and C-termini of the residues were capped with the standard Ace (acetyl) and Nme (N-metyl) residues parameterized with the Amber force field.24 A total number of 1,525 and 1,521 atoms were considered in the QM/MM calculations of the FAAH/1 and the FAAH/2 complexes, respectively (complete list available in SI). The QM part resulted in 178 atoms for the FAAH/1 system and 174 atoms for the FAAH/2 system. The remaining part of the systems was treated with the AMBER99SB force field.24 As a result, 500 snapshots were used to calculate the ΔELUMO-HOMO by means of single point energy calculations at the ONIOM (HF/6-311G**:Amber) level of theory, which was shown to give a good description of the orbital energies.34
In detail, we first calculated the ΔELUMO-HOMO for 50 structural snapshots equally distributed over the δ range (−40° to +35°). These calculations were performed for both FAAH/1 and FAAH/2 complexes, for a total of 100 structures. Additionally, to better address the difference between the most distorted and the planar structures, the average ΔELUMO-HOMO was determined using 100 structural snapshots randomly extracted within a range of 3 degrees centered on the most populated state of δ and θconformers of both complexes, for a total of additional 200 systems that showed the twist of the amide bond as detected during the dynamics. Namely, for the FAAH/1 complex, configurations were selected within −7° < δ < −4°, and 4° < θ< 7°. In the case of the FAAH/2 complex, the selected states were taken in −8° < δ < −5° and 4° < θ< 7° range (see Scheme 2 and Figure 3–4). For comparison, the average ΔELUMO-HOMO was also calculated for the less populated configurations characterized by the planarity of the ligand amide bond (δ and θ around ~0°). Also in this case, a set of structures from classical MD simulations of both FAAH/1 and FAAH/2 complexes were collected within a 3 degrees window, for a total of additional 200 model systems.
Figure 3.
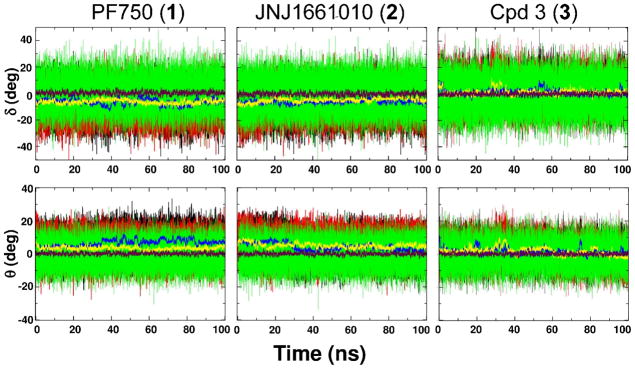
Dihedral angle δ (first row) and improper torsion θ (second row) value fluctuation during the 100 ns of MD simulation of 1 (first column), 2 (second column) and 3 (third column). Black and red thin lines are instantaneous values in Mnr A and Mnr B, respectively, while green thin lines are those in water. The running average values are calculated over 200 ps for δ and θ (blue and yellow thick lines for Mnr A and Mnr B, respectively). The maroon thick line represents the average value measured in water solution.
Figure 4.

Probability distributions of the δ (first column) and θ (second column) angles shown for 1 (first row), 2 (second row) and 3 (third row) in complex with FAAH and in water solution. The δ and θ frequency distributions are indicated with blue and red colors for the selected compounds bound to FAAH Mnr A and FAAH Mnr B respectively. The δ and θ distributions in water are shown in green. The population of the δ and θ angles was evaluated by using a bin size of 1°.
Results
MD simulations
(h/r) FAAH/PF750 (1) complex
During the NPT run, the structural stability was evaluated calculating the root mean square deviation (RMS-d) respect to the minimized X-ray structure. After an transient period of ~2 ns, the RMS-d for the all atoms of the protein settled at 2.42 ± 0.16 Å, while the RMS-d for the heavy atoms was about 1.56 ± 0.20 Å. The RMS-d of the 1 ligand remained below ~2 Å, while the 1 binding group (i.e., piperidine-urea group) was very stable (RMS-d of the N2–C=O atoms = 0.04 Å).
The 1 binding pose closely resembles the one proposed by Mileni et al.10b The carbonyl carbon of the ligand remained in proximity of Ser241 (C-O distance), at 2.95 ± 0.12 Å. Also, the H-bond pattern formed by the catalytic triad Ser241-Ser217-Lys142 was very stable; the distance between the side chain nitrogen of Lys142 and the hydroxyl oxygen of Ser217 ranged around 1.83 ± 0.27 Å, while the Ser217-Ser241 H-bonds was 2.04 ± 0.23 Å. Throughout the simulations, 1 conserved an ordered H-bond network with the oxyanion hole residues (Ser241, Gly240, Gly239, Ile238) and the main chain carbonyl oxygen of Met191. Overall, we found that the main interactions between FAAH and 1 were conserved during the simulations, which further evidenced the structural stability of the complex (see SI for details).
Amide Bond in 1
During the simulations, 1 underwent small conformational changes within FAAH binding site that involved primarily the piperidine moiety and a moderate twist of the hydrolytic amide bond. This evidence prompted us to compare the conformational flexibility of 1 within the enzyme with that in solution. We focused on the geometrical distortion of the amide bond during the simulations in terms of distribution of the dihedral angle δ and improper torsion θ, as described in the methods section. The time evolution of δ and θ during the MD is shown in Figure 3. There was a significant difference between the structural features of 1 within the enzyme and in solution. The average δ value θ within FAAH was equal to −6.35 (± 0.03°, standard error of the mean), while δ average was 0.41 ± 0.03° when simulated in a water box. The θvalues confirmed the out of the plane deformation of the 1′s urea functionality within the enzyme. Indeed, the average θvalue was equal to 5.00 ± 0.02° in complex with FAAH and 0.21 ± 0.02° in solution.
The probability distributions of δ and θ angles along the MD simulations are reported in Figure 4. The population of 1′s conformers in FAAH was centered on a δ value of around ~−5° while in solution δ was ~0° showing that the planarity of the C–N bond in water is strongly influenced by the enzymatic environment. Accordingly, the distribution curve of the θ angle of the inhibitor was centered on θ = ~6° when simulated in complex with FAAH, while θ was ~0° when simulated in solution (see Table 1).
Table 1.
Average over the last ~98 ns of MD simulation for the 1 inhibitor within FAAH and in water.
| δ | θ | τ | χN | |
|---|---|---|---|---|
| Mnr A | −6.42 (±0.03) | 6.63 (±0.03) | 13.01 (±0.03) | 23.15 (±0.05) |
| Mnr B | −6.29 (±0.03) | 3.38 (±0.02) | 11.01 (±0.03) | 22.34 (±0.05) |
| FAAH | −6.35 (±0.03) | 5.00 (±0.02) | 12.01 (±0.02) | 22.75 (±0.04) |
| Water box | 0.41 (±0.04) | 0.21 (±0.02) | 8.51 (±0.02) | 16.81 (±0.04) |
δ: R2N–C–N–H dihedral angle.θ: improper torsion centered on the secondary amine nitrogen. τ: mean twisting angle between the C–N bond. χN: out-of plane bending angle, measure of the degree of pyramidalization at the N atom. The standard error of the mean is reported in brackets. Angles are expressed in degrees.
For completeness, we here describe the amide bond distortion during dynamics according to the Dunitz internal coordinates τ and χN (Figure 5),30b as well. The average twist angle τ was 12.01 ± 0.02° in FAAH and 8.51 ± 0.02° in water, while the pyramidalization at the nitrogen, χN, was equal to 22.75 ± 0.04° in FAAH and 16.81 ± 0.04° in solution. Overall, these data further indicate the evident distortion of the hydrolytic amide bond of 1 when in complex with FAAH.
Figure 5.
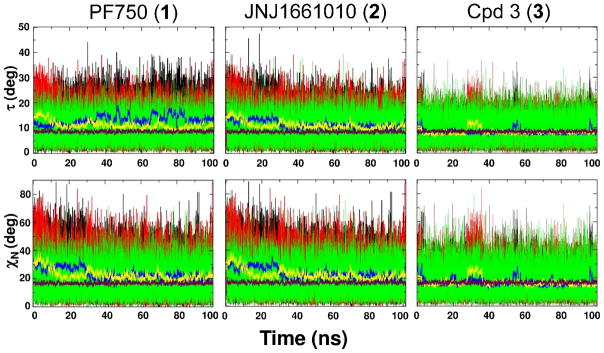
Twisting angle τ (first row), and the out-of-plane bending angle χN (second row) value fluctuation during the 100 ns of MD simulation by 1 (first column), 2 (second column) and 3 (third column). Black and red thin lines instantaneous values in Mnr A and Mnr B, respectively, while green thin lines are those in water. The running average values are calculated over 200 ps for τ and χN (blue and yellow thick lines for Mnr A and Mnr B, respectively). The average τ and χN angles in water are shown in maroon.
(h/r) FAAH/JNJ1661010 (2) complex
After the equilibration, during the ~100 ns the RMS-d of the overall FAAH protein atoms oscillated around 2.38 ± 0.13 Å. The fluctuation of the protein heavy atoms confirmed that FAAH conformation remained consistently stable throughout the simulations (RMS-d = 1.49 ± 0.18 Å). The RMS-d of 2 was around 0.5 – 1 Å, while the urea functional group was highly stable (RMS-d of the N2–C=O atoms = 0.04 ± 0.01 Å).
The key binding distances were well conserved in the ligand-protein complex. The C-O distance remained 2.90 ± 0.11 Å. The H-bond network of the catalytic triad was also well conserved: the Ser241-Ser217 average distance was 1.98 ± 0.20 Å and the Ser217-Lys142 H-bond oscillated around ~1.80 ± 0.11 Å. The interactions between the carbonyl oxygen of the 2 compound and the NH groups of the FAAH oxyanion hole were very stable, as well as the H-bond between the aniline hydrogen of 2 and the main chain carbonyl oxygen of the Met191. Data of key binding distances are reported in the SI. It is worth noting as 2 and 1 compounds showed an analogous network of binding interactions. Indeed, the orientation within FAAH active site of the piperazine 2 is similar to the experimental pose of the co-crystalized piperidine FAAHi 110b and PF384510c. Both compounds 1 and 2 place the heterocyclic moiety in the FAAH acyl chain binding channel, while the leaving group occupies the cytoplasmic access of FAAH active site. This binding orientation of the cyclic urea-based FAAHi has been recently explored by Pfizer and Cravatt’s lab through the modeling of benzotiophene piperidine and piperazine ureas as FAAHi.35 Their results resemble the existing crystallographic data and further support the proposed mechanism of FAAH inhibition.18
Amide Bond in 2
As for 1, the amide bond of 2 showed a distorted geometry when simulated in complex with FAAH. The average δ and θ values within FAAH were −5.22 ± 0.03° and 4.61 ± 0.02°, respectively. These values differ from those obtained in solution, where the amide bond remained planar (δ = −0.05 ± 0.03° and θ= −0.01 ± 0.02°). Values of δ and θ during the dynamics are shown in Figure 3.
The population of δ angle was peaked around −6°, while in solution was centered on 0°. The different behavior of the ligand within the enzyme and in solution can be appreciated by looking at the population of δ and θ in Figure 4. The distribution for the 2 in complex with FAAH was centered around θvalues of 5°/6°. In solution, the maximum of the distribution for θ values was centered upon zero, which indicated that the most abundant population of conformers was characterized by the planarity of the amide bond. Average values obtained for the Dunitz τ and χN angles are here reported for completeness. Within the enzyme, τ value was 11.16 ± 0.02°, and that the nitrogen was partially pyramidalized with an average χN value of 22.74 ± 0.04°. In solution, the average τ and χN angles were 8.48 ± 0.02° and 16.78 ± 0.04°, respectively. This further indicated that the amide functional group was more distorted when simulated in complex with the enzyme, compared to its behavior in solution. Structural data are reported in Table 2.
Table 2.
Average over the last ~98 ns of MD simulation for the 2 inhibitor within FAAH and in water.
| δ | θ | τ | χN | |
|---|---|---|---|---|
| Mnr A | −5.87 (±0.03) | 4.24 (±0.03) | 11.32 (±0.03) | 23.15 (±0.05) |
| Mnr B | −4.57 (±0.03) | 4.97 (±0.02) | 11.01 (±0.02) | 22.35 (±0.05) |
| FAAH | −5.22 (±0.03) | 4.61 (±0.02) | 11.16 (±0.02) | 22.74 (±0.04) |
| Water box | −0.05 (±0.03) | −0.01 (±0.02) | 8.48 (±0.02) | 16.78 (±0.04) |
δ: R2N–C–N–H dihedral angle.θ: torsion improper centered on the secondary amine nitrogen. τ: mean twisting angle between the C–N bond. χN: out-of plane bending angle, measure of the degree of pyramidalization at the N atom. The standard error of the mean is reported in brackets. Angles are expressed in degrees.
(h/r) FAAH/Cpd3 (3) complex
The MD simulations carried out on the inactive compound displayed a RMS-d for the whole protein of 2.33 ± 0.19 Å, while RMS-d was 1.45 ± 0.12 Å for the only heavy atoms. The RMS-d of the 3 was 1.36 ± 0.41 Å while the urea functional group shown to be very stable (RMS-d = 0.09 ± 0.03 Å). The carbamoylation distance was equal to 3.72 ± 0.19 Å, which was remarkably larger than that found for the active compounds. In addition, we observed the breaking of the catalytic triad H-bond network. In fact, the Ser241 – Ser217 distance was about 3.78 ± 0.36 Å, while the H-bond between the Ser217 and the Lys142 increased to a value of 3.14 ± 0.91 Å, during the simulations. Actually, along the simulations 3 moved away from Ser241, inducing the increase of the carbamoylation distance. This occurred at different times in the two monomers: after ~2 – 3 ns in Mnr B, and within the first picoseconds of dynamics in Mnr A (see the SI for further details). Overall, 3 did not retain the starting interactions between its carbonyl oxygen and the FAAH oxyanion hole, during the simulations, thus making useless any further investigation via QM/MM calculations. Moreover, in contrast to what observed with the active compounds, it was not possible to detect a stable H-bond interaction between the hydrogen atoms of the urea functional group and the main chain carbonyl oxygen of the enzymatic Met191.
Amide Bond in 3
The urea functionality conserved a planar geometry both in FAAH and in solution. Indeed, the δ angle displayed similar values within FAAH and in solution (δFAAH = −1.31 ± 0.03°; δwater box = −0.05 ± 0.03°). Likewise, θ assumed values equal to 1.59 ± 0.02° within the enzyme and 0.03 ± 0.02° in water. In fact, no correlation was observed between the δ and θ angles (see the δ vs. θ correlation plot is reported in Figure 6).
Figure 6.

Correlation diagrams between δ and θ angles for 1, 2 and 3 in complex with FAAH and in water. The δ vs θ correlation line is shown with blue (Mnr A) and red (Mnr B). In solution, the δ vs θ correlation line is in green. 1: the δ and θangles are linearly correlated both in FAAH and solution (R2FAAH = 0.60; R2H2O = 0.60). The δ vs θ regression lines are distinctly shifted with respect to those in solution: within FAAH the y-intercept is ~ −9.7°, while in solution the y-intercept crosses the axis origin (~ 0.3°). This further indicates the structural deformation of the amide bond of the inhibitor in complex with FAAH. 2: the δ and θ angles are linearly correlated (R2FAAH = 0.59; R2H2O = 0.62). The y-intercept is ~ −13.1°, while is about ~ −0.1° in water. 3: there is no correlation between the δ and θ angles.
δ and θ distributions resulted to be centered on zero, both when simulated in FAAH and in solution (Figure 4). As expected, the τ and χN angles showed comparable values in FAAH and in water. The average τ angle was around 8.20 ± 0.01° in FAAH and 8.41 ± 0.02° and in solution. The average χN was 16.68 ± 0.08° in FAAH and 16.64 ± 0.04° in water. Other relevant geometrical parameters are reported in Table 3. Overall, the urea geometry was quite rigid and planar, both within FAAH and in solution, indicating that FAAH was not able to induce the distortion of the amide bond in 3.
Table 3.
Average over the last ~98 ns of MD simulation for 3 within FAAH and in solution.
| δ | θ | τ | χN | |
|---|---|---|---|---|
| Mnr A | 1.31 (±0.03) | 1.53 (±0.03) | 8.18 (±0.02) | 16.62 (±0.04) |
| Mnr B | 1.30 (±0.03) | 1.63 (±0.02) | 8.22 (±0.02) | 16.74 (±0.04) |
| FAAH | 1.31 (±0.03) | 1.59 (±0.02) | 8.20 (±0.01) | 16.68 (±0.03) |
| Water box | −0.05 (±0.03) | 0.03 (±0.02) | 8.41 (±0.02) | 16.64 (±0.04) |
δ: R2N–C–N–H dihedral angle.θ: torsion improper centered on the secondary amine nitrogen. τ: mean twisting angle between the C–N bond. χN: out-of plane bending angle, measure of the degree of pyramidalization at the N atom. The standard error of the mean is reported in brackets. Angles are expressed in degrees.
QM/MM calculations
The nucleophilic attack on a carbonyl carbon can be described by the Fukui frontier molecular orbital theory.36 Accordingly, the reaction occurs when the highest occupied bonding molecular orbital (HOMO) of the nuclephile overlaps the lowest unoccupied antibonding molecular orbital (LUMO) of the electrophilic species. Along the reaction coordinate, the HOMO-LUMO gap becomes narrower and favors the reactivity. In this study, the hydroxyl oxygen of the catalytic Ser241 is the nucleophile that transfers electrons from its HOMO to the LUMO of the substrate (i.e. the carbonyl carbon of the ligand). We calculated the ΔELUMO-HOMO over the range of conformations of the amide bond and the average ΔELUMO-HOMO obtained for distorted and planar conformations, for both FAAH/1 and FAAH/2 complexes, as described in the methods section.
The most distorted conformations (−40° < δ < −35°) showed a ΔELUMO-HOMO of ~ 8.50 eV for the FAAH/1 complex, while ~ 9.50 eV for FAAH/2 (Figure 7). When the δ angle was close to 0° (i.e., planar conformations), the ΔELUMO-HOMO became gradually larger and it reached a value of 10.27 eV for FAAH/1 complex and 10.25 eV for FAAH/2. The ΔELUMO-HOMO trend, from distorted to planar conformations, is well visible in Figure 7, which highlights a smaller gap for distorted conformations (i.e., high reactivity towards amide bond hydrolysis).
Figure 7.

ΔELUMO-HOMO plotted against the dihedral angle δ for the FAAH/1 (upper graph) and the FAAH/2 (lower graph) complexes. 50 structures equally distributed over the δ range (−40° to +35°) were considered for each complex.
Centering the analysis on complexes showing a distorted C–N bond (see methods section), the average ΔELUMO-HOMO was 9.33 ± 0.18 eV for 1, while the analogue complexes characterized by a planar amide bond had the ΔELUMO-HOMO of 9.73 ± 0.18 eV, resulting in a ΔΔELUMO-HOMO of 0.40 eV. For complexes where 2 showed a distorted amide bond, the average ΔELUMO-HOMO was equal to 9.65 ± 0.24 eV. In this case, the planar analogues showed average ΔELUMO-HOMO of 10.02 ± 0.25 eV, resulting in a ΔΔELUMO-HOMO of 0.37 eV.
The localization of the frontier orbitals was also analyzed. For the FAAH/1 complex, LUMO of the distorted amide was well localized on the 1 carbonyl, being turned toward the HOMO of the Ser241 nucleophile. On the other hand, LUMO of the planar analogue was more delocalized and thus less prone to receive electrons from the HOMO electron-donor of the Ser241 (Figure 8). The FAAH/2 complex showed a similar topology of the frontier orbitals shape (Figure 8).
Figure 8.

Shape of the frontier orbitals for the FAAH/1 (first row) and the FAAH/2 (second row) complexes. Two representative snapshots characterized by the distorted (a and a′) and planar (b and b′) amide bond are shown for 1 and 2, respectively. The FAAH residues Ile238, Gly239, Gly240, Ser241, Ser217 are indicated in cyan, 1 is green and 2 is orange. The HOMO orbital of the Ser241 nucleophile is depicted in blue, while the LUMO orbital of the electrophile (i.e. the 1 and 2 carbonyl) is shown in red.
Overall, the QM/MM calculations showed a decreased ΔELUMO-HOMO for the protein/ligand complexes with distorted amide bond with respect to the planar analogues. These data reflect a greater reactivity of the distorted amides toward nucleophilic attack.
Discussion
Our results indicate that, within FAAH’s binding site, the piperidine/piperazine inhibitors assume a specific conformation, which is characterized by a twist of the amide bond and partial pyramidalization at the amide bond nitrogen. Importantly, this result can be used as a simple indication of the propensity of new urea-based compounds to act as a covalent inhibitor of FAAH.
The enzyme-induced distortion of the amide bond is evidenced by the δ and θ distribution curves of the piperidine/piperazine compounds bound to FAAH (Figure 4). Indeed, the amide bond of the inhibitors is planar when simulated in solution, as expected for force field-based simulations of this functional group, while it is distorted when simulated in complex with the enzyme FAAH (Figure 3 and 5).
As suggested by Greenberg,20 the amide bond distortion increases the sp3 character and the basicity of the amide nitrogen, making it available for protonation and coordination. As a consequence, distorted amides undergo nucleophilic attack more easily, compared to their planar analogs, as shown also by several informative computational studies.37 Moreover, recent DFT studies38 have shown that twisted amides, under different pH and solvent conditions, show a considerable acceleration of the hydrolysis compared to planar analogues. Also, kinetic experiments have demonstrated a significant acceleration for the hydrolysis of strained cyclic amides with distorted amide bond,39 such as the alkaline hydrolysis of the benzoquinuclidine-2-one, which is 1017 times faster than its strain-free counterpart.40 A final example is the highly twisted amide, 1-aza-2-adamantanone, which is exceedingly reactive toward hydrolysis in neutral conditions: at least 1010 times faster than that of a typical amide.41 These arguments support the hypothesis that FAAH-induced distortion of the amide bond of the piperidine/piperazine FAAH inhibitors, evidenced by our simulations, could make the amide functional group more susceptible to nucleophilic attack by Ser241.18 In other words, the amide distortion, which diminishes the conjugation of the nitrogen lone pair with the carbonyl, could lead to a more efficient hydrolysis of the amide bond. This would explain the inhibitory activity of piperidine/piperazine compounds via their prompt formation of a covalent bond with Ser241 of FAAH. Furthermore, the increased basicity of the distorted amide could promote an easy protonation and release of the aniline leaving group during the reaction. QM/MM simulations are in progress to quantify the effect of the amide bond distortion in facilitating the reaction and formation of a covalent enzyme-inhibitor adduct in FAAH’s catalytic site.
We calculated the free energy difference between the planar and most populated distorted amide conformations, according to the Boltzmann law of distribution. This difference turned out to be as small as ~1 kcal/mol, in agreement with the force field potential for such a slight amide bond distortion (See SI). Such a small gap between the planar and distorted conformations of the amide bond indicates an easy interchange between the two states. However, we observed a statistically relevant shift of the populated states of the ligand conformations toward distorted ones when in complex with FAAH, which favors the inhibitor hydrolysis (Figure 4). Our QM/MM calculations suggested indeed a higher reactivity of the distorted amides toward nucleophilic attack, relative to their planar analogues. We detected a lower ΔELUMO-HOMO (9.33 ± 0.18 eV for 1 and 9.65 ± 0.24 eV for 2) for distorted conformations, with respect to the planar analogues in which we found a larger Δ ELUMO-HOMO (9.73 ± 0.18 eV for 1 and 10.02 ± 0.25 eV for 2).
We speculate that the amide bond distortion could be primarily caused by the FAAH oxyanion hole residues (Ile238, Gly239, Gly240, Ser241), which chelate the carbonyl oxygen of the cyclic inhibitors, while the Met191 interacts with the ligand amide hydrogen (for detailed distances see SI). These simultaneous interactions that involve the urea functionality seem to be the major contribution to the twist of the amide bond in the piperidine/piperazine inhibitors. The peptide NH groups of the FAAH oxyanion hole draw electron density away from the urea functionality, while the Met191 H-bonds to the hydrogen of the ligand leaving group, weakening the amide bond. Furthermore, the heterocyclic moiety of the piperidine/piperazine inhibitors is closely surrounded by the Phe244, Tyr194, Ser193, Phe192, Val491, Ser193, Gly239, Gly216, Ser241 and Met191 residues. In this way, the enzyme generates a steric tension on the cyclic moiety of the inhibitor that enforces the deformation of the amide bond. It follows that the conformational flexibility of the piperidine/piperazine cycle appears to be critical for the activation of the urea-based FAAHi toward the nucleophilic attack. Noteworthy, the recent discovery of the piperidine and piperazine carbamates as efficacious inhibitors of the endocannabinoid hydrolases42 emphasizes the effectiveness of the piperidine/piperazine structural motif for generating new covalent inhibitors of FAAH. Conversely, the inactive 3 is unable to establish favorable interactions with the FAAH oxyanion hole and shows a loss of binding. This, coupled to lack of distortion of the amide bond, might explain the inability of 3 to inhibit FAAH.
Interestingly, the evidence of an enzyme-induced amide distortion reported herein can be related to other enzymes that are hypothesized to exploit the same mechanism to favor amide bond hydrolysis. For example, class A β-lactamases, which possess an oxyanion hole, are thought to induce a deformation of the sp2 carbon of the endocyclic amide bond of the substrateβ-lactam ring. This seems the major cause for β-lactam hydrolysis.43 Also, in the case of carboxypeptidase Y, it was suggested that distortion of the peptide bond within the substrate may be crucial for hydrolytic activity.44 In the case of the aspartic proteases, it was suggested that enzyme-induced distortion of the substrate amide could favor the attack
of water on the polarized carbonyl.45 Later, x-ray data of the aspartic proteinase endothiapepsin in complex with a potent difluorostatone-containing tripeptide renin confirmed this hypothesis.46 In the process of protein splicing, x-ray and NMR experiments have suggested that the loss of amide planarity could facilitate the catalytic mechanism, by destabilizing the scissile amide bond.47 Finally, a recent x-ray structure of the matrix metalloproteinase MMP-8 in complex with an N-hydroxylurea inhibitor showed a highly out-of-plane distortion of the amide bond, which undergoes hydrolysis.48 In this scenario, our findings further clarify, with unprecedented detail, how the enzyme can induce specific conformations of the amide bond to facilitate its hydrolysis.
Conclusions
We have presented a comparative MD and QM/MM study to rationalizing the mechanism used by the piperidine/piperazine urea-based compounds to inhibit FAAH. Three compounds were considered: two potent lead molecules, namely the piperidine 1 and the piperazine 2, and the inactive 3. The structural flexibility of these compounds was analyzed either when in complex with FAAH or when in water solution
MD simulations revealed a distortion of the amide bond of the piperidine/piperazine urea-based inhibitors when in complex with FAAH. This enzyme-induced conformational change may be a key step for FAAH inhibition. In fact, the linear inactive urea-based 3 does not show persistent deformations of the amide bond.
QM/MM calculations were used to evaluate the energy difference between the HOMO and LUMO orbitals (ΔELUMO-HOMO). Results confirmed that distorted amides are more reactive toward the nucleophilic attack, in comparison to planar conformation. This further indicates that the enzyme-induced distortion facilitates the amide hydrolysis.
Overall, our work corroborates the hypothesis18 that the inhibitory activity of the piperidine/piperazine aryl ureas originates from their flexibility. The results show indeed that FAAH is able to induce a distortion of the amide bond of piperidine/piperazine inhibitors, which prompts the formation a covalent enzyme-inhibitor adduct. In other words, here we provide a simple receipt to evaluate the propensity of new urea-based compounds to act as a covalent inhibitor. Therefore, we believe these findings could be used for the rational design of more potent inhibitors bearing a cyclic urea-based moiety more prone to distortion of the amide bond.
Supplementary Material
Acknowledgments
We thank CINECA – centre for high performance computing, Bologna (IT) – and the Italian Institute of Technology computational platform initiative for providing computer time.
Abbreviations
- AEA
anandamide
- Mnr A
monomer A
- Mnr B
monomer B
- ΔELUMO-HOMO
HOMO-LUMO energy gap
- FAAH
fatty acid amide hydrolase
- (h/r) FAAH
humanized rat FAAH protein
- FAAHi
FAAH inhibitors
- PEA
palmytoylethanolamide
Footnotes
Supporting information available
Further MD analysis and RESP charges, docking details for 2 and further QM characterization of the amide bond distortion.
References
- 1.Singh J, Petter RC, Baillie TA, Whitty A. The resurgence of covalent drugs. Nat Rev Drug Discov. 2011;10(4):307–317. doi: 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- 2.Potashman MH, Duggan ME. Covalent Modifiers: An Orthogonal Approach to Drug Design. Journal of Medicinal Chemistry. 2009;52(5):1231–1246. doi: 10.1021/jm8008597. [DOI] [PubMed] [Google Scholar]
- 3.Ahn K, Johnson DS, Cravatt BF. Fatty acid amide hydrolase as a potential therapeutic target for the treatment of pain and CNS disorders. Expert Opin Drug Discov. 2009;4(7):763–784. doi: 10.1517/17460440903018857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Desarnaud F, Cadas H, Piomelli D. Anandamide amidohydrolase activity in rat brain microsomes. Identification and partial characterization. J Biol Chem. 1995;270(11):6030–6035. doi: 10.1074/jbc.270.11.6030. [DOI] [PubMed] [Google Scholar]; (b) Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature. 1996;384(6604):83–87. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- 5.(a) Lambert DM, Vandevoorde S, Jonsson KO, Fowler CJ. The palmitoylethanolamide family: a new class of anti-inflammatory agents? Curr Med Chem. 2002;9(6):663–674. doi: 10.2174/0929867023370707. [DOI] [PubMed] [Google Scholar]; (b) Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258(5090):1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]; (c) Rodriguez de Fonseca F, Navarro M, Gomez R, Escuredo L, Nava F, Fu J, Murillo-Rodriguez E, Giuffrida A, LoVerme J, Gaetani S, Kathuria S, Gall C, Piomelli D. An anorexic lipid mediator regulated by feeding. Nature. 2001;414(6860):209–212. doi: 10.1038/35102582. [DOI] [PubMed] [Google Scholar]
- 6.Cravatt BF, Demarest K, Patricelli MP, Bracey MH, Giang DK, Martin BR, Lichtman AH. Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc Natl Acad Sci U S A. 2001;98(16):9371–9376. doi: 10.1073/pnas.161191698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Lichtman AH, Shelton CC, Advani T, Cravatt BF. Mice lacking fatty acid amide hydrolase exhibit a cannabinoid receptor-mediated phenotypic hypoalgesia. Pain. 2004;109(3):319–327. doi: 10.1016/j.pain.2004.01.022. [DOI] [PubMed] [Google Scholar]; (b) Gaetani S, Dipasquale P, Romano A, Righetti L, Cassano T, Piomelli D, Cuomo V. The endocannabinoid system as a target for novel anxiolytic and antidepressant drugs. Int Rev Neurobiol. 2009;85:57–72. doi: 10.1016/S0074-7742(09)85005-8. [DOI] [PubMed] [Google Scholar]; (c) Gobbi G, Bambico FR, Mangieri R, Bortolato M, Campolongo P, Solinas M, Cassano T, Morgese MG, Debonnel G, Duranti A, Tontini A, Tarzia G, Mor M, Trezza V, Goldberg SR, Cuomo V, Piomelli D. Antidepressant-like activity and modulation of brain monoaminergic transmission by blockade of anandamide hydrolysis. Proc Natl Acad Sci U S A. 2005;102(51):18620–18625. doi: 10.1073/pnas.0509591102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Kathuria S, Gaetani S, Fegley D, Valino F, Duranti A, Tontini A, Mor M, Tarzia G, La Rana G, Calignano A, Giustino A, Tattoli M, Palmery M, Cuomo V, Piomelli D. Modulation of anxiety through blockade of anandamide hydrolysis. Nat Med. 2003;9(1):76–81. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]; (b) Piomelli D. The molecular logic of endocannabinoid signalling. Nat Rev Neurosci. 2003;4(11):873–884. doi: 10.1038/nrn1247. [DOI] [PubMed] [Google Scholar]
- 9.(a) Ahn K, McKinney MK, Cravatt BF. Enzymatic pathways that regulate endocannabinoid signaling in the nervous system. Chem Rev. 2008;108(5):1687–1707. doi: 10.1021/cr0782067. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) McKinney MK, Cravatt BF. Structure and function of fatty acid amide hydrolase. Annu Rev Biochem. 2005;74:411–432. doi: 10.1146/annurev.biochem.74.082803.133450. [DOI] [PubMed] [Google Scholar]
- 10.(a) Bracey MH, Hanson MA, Masuda KR, Stevens RC, Cravatt BF. Structural adaptations in a membrane enzyme that terminates endocannabinoid signaling. Science. 2002;298(5599):1793–1796. doi: 10.1126/science.1076535. [DOI] [PubMed] [Google Scholar]; (b) Mileni M, Johnson DS, Wang Z, Everdeen DS, Liimatta M, Pabst B, Bhattacharya K, Nugent RA, Kamtekar S, Cravatt BF, Ahn K, Stevens RC. Structure-guided inhibitor design for human FAAH by interspecies active site conversion. Proc Natl Acad Sci U S A. 2008;105(35):12820–12824. doi: 10.1073/pnas.0806121105. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mileni M, Kamtekar S, Wood DC, Benson TE, Cravatt BF, Stevens RC. Crystal structure of fatty acid amide hydrolase bound to the carbamate inhibitor URB597: discovery of a deacylating water molecule and insight into enzyme inactivation. J Mol Biol. 2010;400(4):743–754. doi: 10.1016/j.jmb.2010.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Guimaraes CR, Boger DL, Jorgensen WL. Elucidation of fatty acid amide hydrolase inhibition by potent alpha-ketoheterocycle derivatives from Monte Carlo simulations. J Am Chem Soc. 2005;127(49):17377–17384. doi: 10.1021/ja055438j. [DOI] [PubMed] [Google Scholar]; (b) Lodola A, Mor M, Hermann JC, Tarzia G, Piomelli D, Mulholland AJ. QM/MM modelling of oleamide hydrolysis in fatty acid amide hydrolase (FAAH) reveals a new mechanism of nucleophile activation. Chem Commun (Camb) 2005;(35):4399–4401. doi: 10.1039/b503887a. [DOI] [PubMed] [Google Scholar]; (c) Lodola A, Mor M, Rivara S, Christov C, Tarzia G, Piomelli D, Mulholland AJ. Identification of productive inhibitor binding orientation in fatty acid amide hydrolase (FAAH) by QM/MM mechanistic modelling. Chem Commun (Camb) 2008;(2):214–216. doi: 10.1039/b714136j. [DOI] [PubMed] [Google Scholar]; (d) Lodola A, Mor M, Sirirak J, Mulholland AJ. Insights into the mechanism and inhibition of fatty acid amide hydrolase from quantum mechanics/molecular mechanics (QM/MM) modelling. Biochem Soc Trans. 2009;37(Pt 2):363–367. doi: 10.1042/BST0370363. [DOI] [PubMed] [Google Scholar]; (e) Tubert-Brohman I, Acevedo O, Jorgensen WL. Elucidation of hydrolysis mechanisms for fatty acid amide hydrolase and its Lys142Ala variant via QM/MM simulations. J Am Chem Soc. 2006;128(51):16904–16913. doi: 10.1021/ja065863s. [DOI] [PubMed] [Google Scholar]
- 12.Seierstad M, Breitenbucher JG. Discovery and development of fatty acid amide hydrolase (FAAH) inhibitors. J Med Chem. 2008;51(23):7327–7343. doi: 10.1021/jm800311k. [DOI] [PubMed] [Google Scholar]
- 13.Tarzia G, Duranti A, Tontini A, Piersanti G, Mor M, Rivara S, Plazzi PV, Park C, Kathuria S, Piomelli D. Design, synthesis, and structure-activity relationships of alkylcarbamic acid aryl esters, a new class of fatty acid amide hydrolase inhibitors. J Med Chem. 2003;46(12):2352–2360. doi: 10.1021/jm021119g. [DOI] [PubMed] [Google Scholar]
- 14.Mor M, Rivara S, Lodola A, Plazzi PV, Tarzia G, Duranti A, Tontini A, Piersanti G, Kathuria S, Piomelli D. Cyclohexylcarbamic acid 3′- or 4′-substituted biphenyl-3-yl esters as fatty acid amide hydrolase inhibitors: synthesis, quantitative structure-activity relationships, and molecular modeling studies. J Med Chem. 2004;47(21):4998–5008. doi: 10.1021/jm031140x. [DOI] [PubMed] [Google Scholar]
- 15.(a) Clapper JR, Duranti A, Tontini A, Mor M, Tarzia G, Piomelli D. The fatty-acid amide hydrolase inhibitor URB597 does not affect triacylglycerol hydrolysis in rat tissues. Pharmacol Res. 2006;54(5):341–344. doi: 10.1016/j.phrs.2006.06.008. [DOI] [PubMed] [Google Scholar]; (b) Piomelli D, Tarzia G, Duranti A, Tontini A, Mor M, Compton TR, Dasse O, Monaghan EP, Parrott JA, Putman D. Pharmacological profile of the selective FAAH inhibitor KDS-4103 (URB597) CNS Drug Rev. 2006;12(1):21–38. doi: 10.1111/j.1527-3458.2006.00021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clapper JR, Moreno-Sanz G, Russo R, Guijarro A, Vacondio F, Duranti A, Tontini A, Sanchini S, Sciolino NR, Spradley JM, Hohmann AG, Calignano A, Mor M, Tarzia G, Piomelli D. Anandamide suppresses pain initiation through a peripheral endocannabinoid mechanism. Nat Neurosci. 2010;13(10):1265–1270. doi: 10.1038/nn.2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sit SY, Conway C, Bertekap R, Xie K, Bourin C, Burris K, Deng H. Novel inhibitors of fatty acid amide hydrolase. Bioorg Med Chem Lett. 2007;17(12):3287–3291. doi: 10.1016/j.bmcl.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 18.Ahn K, Johnson DS, Fitzgerald LR, Liimatta M, Arendse A, Stevenson T, Lund ET, Nugent RA, Nomanbhoy TK, Alexander JP, Cravatt BF. Novel mechanistic class of fatty acid amide hydrolase inhibitors with remarkable selectivity. Biochemistry. 2007;46(45):13019–13030. doi: 10.1021/bi701378g. [DOI] [PubMed] [Google Scholar]
- 19.Keith JM, Apodaca R, Xiao W, Seierstad M, Pattabiraman K, Wu J, Webb M, Karbarz MJ, Brown S, Wilson S, Scott B, Tham CS, Luo L, Palmer J, Wennerholm M, Chaplan S, Breitenbucher JG. Thiadiazolopiperazinyl ureas as inhibitors of fatty acid amide hydrolase. Bioorg Med Chem Lett. 2008;18(17):4838–4843. doi: 10.1016/j.bmcl.2008.07.081. [DOI] [PubMed] [Google Scholar]
- 20.Greenberg A, Breneman CM, Liebman JF. The Amide Linkage: Structural Significance in Chemistry, Biochemistry, and Materials Science. Wiley; 2000. [Google Scholar]
- 21.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. Automated Docking Using a Lamarckian Genetic Algorithm and and Empirical Binding Free Energy Function. J Comput Chem. 1998;19:1639–1662. [Google Scholar]
- 22.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 23.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J Am Chem Soc. 1995;117:5179–5197. [Google Scholar]
- 24.Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins. 2006;65(3):712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem. 2004;25(9):1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 26.Bayly CI, Cieplak P, Cornell WD, Kollman PA. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J Phys Chem. 1993;97:10269–10280. [Google Scholar]
- 27.Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical-Integration of Cartesian Equations of Motion of a System with Constraints - Molecular-Dynamics of N-Alkanes. J Comput Phys. 1977;23(3):327–341. [Google Scholar]
- 28.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26(16):1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Feller SE, Zhang Y, Pastor RW, Brooks RW. Constant pressure molecular dynamics simulation: The Langevin piston method. J Chem Phys. 1995;103:4613–4621. [Google Scholar]
- 30.(a) Gilli G, Bertolasi V, Bellucci F, Ferretti V. Stereochemistry of the R,(X=)C(sp2)– N(sp3)R2R3 Fragment. Mapping of the Cis-Trans Isomerization Path by Rotation Around the C-N Bond from Crystallographic Structural Data. J Am Chem Soc. 1986;108:2420–2424. doi: 10.1021/ja00269a047. [DOI] [PubMed] [Google Scholar]; (b) Winkler FK, Dunitz JD. The non-planar amide group. J Mol Biol. 1971;59(1):169–182. doi: 10.1016/0022-2836(71)90419-0. [DOI] [PubMed] [Google Scholar]
- 31.(a) Humbel S, Sieber S, Morokuma K. The IMOMO method: Integration of different levels of molecular orbital approximations for geometry optimization of large systems: Test for n-butane conformation and SN2 reaction: RCL + Cl- J Chem Phys. 1996;105:1959–1967. [Google Scholar]; (b) Svensson M, Humbel S, Froese RDJ, Matsubara T, Sieber S, Morokuma K. ONIOM: A Multilayered Integrated MO + MM Method for Geometry Optimizations and Single Point Energy Predictions. A Test for Diels-Alder Reactions and Pt(P(t-Bu)3)2 + H2 Oxidative Addition. J Phys Chem. 1996;100:19357–19363. [Google Scholar]
- 32.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JJA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam NJ, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09, Revision A.1. Gaussian, Inc; Wallingford CT: 2009. [Google Scholar]
- 33.Vreven T, Byun KS, Komaromi I, Dapprich S, Montgomery JA, Morokuma K, Frisch MJ. Combining Quantum Mechanics Methods with Molecular Mechanics Methods in ONIOM. J Chem Theory Comput. 2006;2:815–826. doi: 10.1021/ct050289g. [DOI] [PubMed] [Google Scholar]
- 34.Morrell C, Grand A, Toro-Labbé A. New Dual Descriptor for Chemical Reactivity. J Phis Chem A. 2005;109:205–212. doi: 10.1021/jp046577a. [DOI] [PubMed] [Google Scholar]
- 35.Johnson DS, Ahn K, Kesten S, Lazerwith SE, Song Y, Morris M, Fay L, Gregory T, Stiff C, Dunbar JB, Jr, Liimatta M, Beidler D, Smith S, Nomanbhoy TK, Cravatt BF. Benzothiophene piperazine and piperidine urea inhibitors of fatty acid amide hydrolase (FAAH) Bioorg Med Chem Lett. 2009;19(10):2865–2869. doi: 10.1016/j.bmcl.2009.03.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fukui K. Theory of Orientation and Stereoselection. Springer-Verlag; New York: 1975. [Google Scholar]
- 37.(a) Alexandrova AN, Jorgensen WL. Why urea eliminates ammonia rather than hydrolyzes in aqueous solution. Journal of Physical Chemistry B. 2007;111(4):720–730. doi: 10.1021/jp066478s. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Blumberger J, Ensing B, Klein ML. Formamide hydrolysis in alkaline aqueous solution: Insight from ab initio metadynamics calculations. Angew Chem Int Edit. 2006;45(18):2893–2897. doi: 10.1002/anie.200600283. [DOI] [PubMed] [Google Scholar]; (c) Bryantsev VS, Firman TK, Hay BP. Conformational analysis and rotational barriers of alkyl- and phenyl-substituted urea derivatives. Journal of Physical Chemistry A. 2005;109(5):832–842. doi: 10.1021/jp0457287. [DOI] [PubMed] [Google Scholar]; (d) Cascella M, Raugei S, Carloni P. Formamide Hydrolysis Investigated by Multiple-Steering ab Initio Molecular Dynamics. J Phis Chem B. 2004;108:369–375. [Google Scholar]
- 38.(a) Lopez X, Mujika JI, Blackburn GM, Karplus M. Alkaline Hydrolysis of Amide Bonds: Effect of Bond Twist and Nitrogen Pyramidalization. J Phys Chem A. 2003;107:2304–2315. [Google Scholar]; (b) Mujika JI, Formoso E, Mercero JM, Lopez X. Reaction Mechanism of the Acidic Hydrolysis of Highly Twisted Amides: Rate Acceleration Caused by the Twist of the Amide Bond. J Phys Chem B. 2006;110:15000–15011. doi: 10.1021/jp0604037. [DOI] [PubMed] [Google Scholar]; (c) Mujika JI, Mercero JM, Lopez X. A Theoretical Evaluation of the pKa for Twisted Amides Using Density Functional Theory and Dielectric Continuum Methods. J Phys Chem A. 2003;107:6099–6107. [Google Scholar]; (d) Mujika JI, Mercero JM, Lopez X. Water-Promoted Hydrolysis of a Highly Twisted Amide: Rate Acceleration Caused by the Twist of the Amide Bond. J Am Chem Soc. 2005;127:445–4453. doi: 10.1021/ja044873v. [DOI] [PubMed] [Google Scholar]
- 39.Wang QP, Bennet AJ, Brown RS, Santarsiero BD. Distorted amides as models for activated peptide N-C(O) units. 3. Synthesis, hydrolytic profile, and molecular structure of 2,3,4,5-tetrahydro-2-oxo-1,5-propanobenzazepine. J Am Chem Soc. 1991;113:5757–5765. [Google Scholar]
- 40.Blackburn GM, Skaife CJ, Kay IT. Strain effects in acyl transfer reactions. Part 5. The kinetics of hydrolysis of benzoquinuclidin-2-one: a torsionally-distorted amide. J Chem Res. 1980:3650–3669. [Google Scholar]
- 41.Kirby AJ, Komarov IV, Feeder N. Synthesis, structure and reactions of the most twisted amide. J Chem Soc, Perkin Trans. 2001;2:522–529. [Google Scholar]
- 42.(a) Long JZ, Jin X, Adibekian A, Li W, Cravatt BF. Characterization of tunable piperidine and piperazine carbamates as inhibitors of endocannabinoid hydrolases. J Med Chem. 2010;53(4):1830–1842. doi: 10.1021/jm9016976. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Long JZ, Nomura DK, Vann RE, Walentiny DM, Booker L, Jin X, Burston JJ, Sim-Selley LJ, Lichtman AH, Wiley JL, Cravatt BF. Dual blockade of FAAH and MAGL identifies behavioral processes regulated by endocannabinoid crosstalk in vivo. Proc Natl Acad Sci U S A. 2009;106(48):20270–20275. doi: 10.1073/pnas.0909411106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.(a) Dal Peraro M, Vila AJ, Carloni P, Klein ML. Role of zinc content on the catalytic efficiency of B1 metallo beta-lactamases. J Am Chem Soc. 2007;129(10):2808–16. doi: 10.1021/ja0657556. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Park H, Brothers EN, Merz KM., Jr Hybrid QM/MM and DFT investigations of the catalytic mechanism and inhibition of the dinuclear zinc metallo-beta-lactamase CcrA from Bacteroides fragilis. J Am Chem Soc. 2005;127(12):4232–4241. doi: 10.1021/ja042607b. [DOI] [PubMed] [Google Scholar]
- 44.Stennicke HR, Mortensen UH, Breddam K. Studies on the hydrolytic properties of (serine) carboxypeptidase Y. Biochemistry. 1996;35(22):7131–7141. doi: 10.1021/bi952758e. [DOI] [PubMed] [Google Scholar]
- 45.Pearl LH. The catalytic mechanism of aspartic proteinases. FEBS Lett. 1987;214(1):8–12. doi: 10.1016/0014-5793(87)80003-0. [DOI] [PubMed] [Google Scholar]
- 46.Veerapandian B, Cooper JB, Sali A, Blundell TL, Rosati RL, Dominy BW, Damon DB, Hoover DJ. Direct observation by X-ray analysis of the tetrahedral “intermediate” of aspartic proteinases. Protein Sci. 1992;1(3):322–328. doi: 10.1002/pro.5560010303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.(a) Poland BW, Xu MQ, Quiocho FA. Structural insights into the protein splicing mechanism of PI-SceI. J Biol Chem. 2000;275(22):16408–13. doi: 10.1074/jbc.275.22.16408. [DOI] [PubMed] [Google Scholar]; (b) Romanelli A, Shekhtman A, Cowburn D, Muir TW. Semisynthesis of a segmental isotopically labeled protein splicing precursor: NMR evidence for an unusual peptide bond at the N-extein-intein junction. Proc Natl Acad Sci U S A. 2004;101(17):6397–6402. doi: 10.1073/pnas.0306616101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Campestre C, Agamennone M, Tortorella P, Preziuso S, Biasone A, Gavuzzo E, Pochetti G, Mazza F, Hiller O, Tschesche H, Consalvi V, Gallina C. N-Hydroxyurea as zinc binding group in matrix metalloproteinase inhibition: mode of binding in a complex with MMP-8. Bioorg Med Chem Lett. 2006;16(1):20–24. doi: 10.1016/j.bmcl.2005.09.057. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.