

Abstract
Congenital diaphragmatic hernia (CDH) is associated with significant mortality due to lung hypoplasia and pulmonary hypertension. The role of embryonic pulmonary innervation in normal lung development and lung maldevelopment in CDH has not been defined. We hypothesize that developmental defects of intrapulmonary innervation, in particular autonomic innervation, occur in CDH. This abnormal embryonic pulmonary innervation may contribute to lung developmental defects and postnatal physiological derangement in CDH. To define patterns of pulmonary innervation in CDH, human CDH and control lung autopsy specimens were stained with the pan-neural marker S-100. To further characterize patterns of overall and autonomic pulmonary innervation during lung development in CDH, the murine nitrofen model of CDH was utilized. Immunostaining for protein gene product 9.5 (a pan-neuronal marker), tyrosine hydroxylase (a sympathetic marker), vesicular acetylcholine transporter (a parasympathetic marker), or VIP (a parasympathetic marker) was performed on lung whole mounts and analyzed via confocal microscopy and three-dimensional reconstruction. Peribronchial and perivascular neuronal staining pattern is less complex in human CDH than control lung. In mice, protein gene product 9.5 staining reveals less complex neuronal branching and decreased neural tissue in nitrofen-treated lungs from embryonic day 12.5 to 16.5 compared with controls. Furthermore, nitrofen-treated embryonic lungs exhibited altered autonomic innervation, with a relative increase in sympathetic nerve staining and a decrease in parasympathetic nerve staining compared with controls. These results suggest a primary defect in pulmonary neural developmental in CDH, resulting in less complex neural innervation and autonomic imbalance. Defective embryonic pulmonary innervation may contribute to lung developmental defects and postnatal physiological derangement in CDH.
Keywords: lung development, nerve development, autonomic innervation
congenital diaphragmatic hernia (CDH) is a relatively common birth defect occurring in ∼1 of 2,500 live-born infants. The anatomic abnormalities in CDH include a defect in the diaphragm associated with persistence of abdominal viscera in the chest cavity, as well as lung developmental abnormalities, including lung hypoplasia and pulmonary vascular abnormalities. Lung hypoplasia in CDH is associated with decreased airway branching, decreased lung-to-body weight ratio, and decreased radial alveolar counts (3, 16, 22). The pulmonary vascular abnormalities include decreased pulmonary arterial branching, with increased muscularization of the intrapulmonary arteries (15, 17, 26, 30, 41). These abnormalities can lead to pulmonary hypertension and severe respiratory distress at birth. Despite recent advances in the care of infants with CDH, the reported mortality for infants born with CDH is 10–40% (1, 6, 10, 20, 25, 34). However, studies that included all cases with a prenatal diagnosis of CDH report mortality rates of 50–62%, thus revealing a significant hidden mortality due to fetal demise and postnatal death prior to definitive care (2, 12, 39). Specifically, persistent pulmonary hypertension is a major clinical factor contributing to early and delayed mortality in infants born with CDH (14).
The molecular mechanisms responsible for abnormal lung development in CDH remain incompletely understood. Furthermore, the contribution of defective pulmonary neural development to the characteristic developmental defects and postnatal physiological derangements in CDH has not been defined. Recent data using generic neural markers suggest decreased esophageal and tracheobronchial innervation in CDH (31–33). The development of lung innervation and the intrinsic pulmonary nervous system in normal lung remains an incompletely understood process. The embryonic lungs receive parasympathetic (vagal) and sympathetic extrinsic innervation, while intrinsic innervation of the lung is derived from neural crest cells (7, 8). Multiple studies have shown that functional innervation of the embryonic lung begins in the early stages of lung development. For example, branches from the vagus nerve to the developing trachea and lung buds are evident as early as embryonic (E) day 11.5 (E11.5) in mice (43). Subsequently, there is formation of a complex peribronchial neural plexus extending to the distal airways. Furthermore, neurotransmitters and neuropeptides are expressed in the early stages of lung development (5, 19, 44).
The effect of neural development on organogenesis is an emerging area of interest, and the specific role of embryonic pulmonary innervation in modulation of normal lung development (and, therefore, contribution to postnatal physiology) remains to be explored. In a system analogous to lung airway branching morphogenesis, parasympathetic innervation has recently been shown to be necessary for maintaining an epithelial progenitor cell population during salivary gland development (24). Removal of the parasympathetic submandibular neural ganglion from embryonic submandibular gland explants in culture, or chemical inhibition of acetylcholine signaling, results in decreased branching morphogenesis and decreased expression of epithelial progenitor cell markers (24). There is increasing evidence that the vasculature and embryonic blood flow play important roles in organogenesis of a variety of organs (13, 37). Furthermore, neural development during organogenesis closely parallels vascular development and is controlled by some of the same molecular factors (9). Therefore, defective neural development would be expected to be associated with defective vascular development and could potentially result in imbalanced autonomic innervation, which could contribute to altered embryonic pulmonary blood flow. Thus, improper nerve and blood vessel development could contribute to defects in lung organogenesis.
We hypothesized that defective pulmonary neural innervation occurs in CDH and results in an imbalance of pulmonary autonomic innervation. This altered embryonic pulmonary innervation and autonomic imbalance in CDH may contribute to specific lung defects in CDH through effects on airway or vascular development. Furthermore, defective pulmonary innervation, in particular autonomic imbalance, may lead to postnatal physiological derangements that contribute to pulmonary vascular hyperresponsiveness and pulmonary hypertension. To address this hypothesis, patterns of neural development in CDH were characterized in human autopsy lung specimens and in a nitrofen-induced mouse model of CDH.
MATERIALS AND METHODS
Human specimens.
Autopsy neonatal CDH or control lung specimens were obtained under approval of the University of Pittsburgh Committee for Oversight of Research Involving the Dead. Immunohistochemistry was performed with S-100 primary antibody (Santa Cruz Biotechnology, Santa Cruz, CA) and a biotinylated Vectastain ABC kit. Immunoperoxidase was detected using a diaminobenzidine kit (Dako, Carpinteria, CA).
Animals.
Time-mated CD-1 mice were obtained from Charles River. Experiments were performed in accordance with guidelines established by the University of Pittsburgh Institutional Animal Care and Use Committee.
Nitrofen model.
Timed-pregnant CD-1 mice were gavage-fed 25 mg of nitrofen dissolved in 1 ml of olive oil (nitrofen group) or 1 ml of olive oil alone (control) on day 8.5 of gestation. At various time points, pregnant mice were killed and embryonic lungs were dissected using a dissecting microscope.
Antibodies.
The following antibodies were used at the indicated dilutions for immunofluorescence: goat anti-E-cadherin (1:300 dilution; R & D Systems, Minneapolis, MN), rabbit anti-protein gene product 9.5 (PGP9.5, 1:300 dilution; AbD Serotec, Raleigh, NC), rabbit anti-tyrosine hydroxylase (1:500 dilution; Abcam, Cambridge, MA), goat anti-vesicular acetylcholine transporter (VAchT, 1:500 dilution; Abcam), and rabbit anti-VIP (1:1,000 dilution; Abcam).
Immunohistochemistry.
For whole mounts, embryonic lungs were fixed in 4% paraformaldehyde, washed in PBS, and fixed with graded methanol. Fixed whole mounts were washed in PBS and blocked with 10% normal donkey serum. Tissues were incubated with primary antibody overnight at 4°C, washed in 0.1% PBS-Tween 20 (PBST), and incubated overnight at 4°C with secondary antibody. After washes with 0.1% PBST, the tissue was mounted. For sections, embryonic lungs were fixed in 4% paraformaldehyde, cryoprotected in 30% sucrose overnight, embedded in Tissue-Tek optimal cutting temperature compound, and frozen in liquid nitrogen. Sections (6–8 μm) were cut by cryostat and mounted. Slides were washed in PBS and 0.1% PBST. Sections were incubated with primary antibody overnight at 4°C or at room temperature for 1 h. After washes with PBS and 0.1% PBST, the sections were incubated with secondary antibody for 1 h at room temperature. Slides were washed with PBS and mounted.
Western blot.
Total lung protein was extracted from whole embryonic lungs using extraction buffer and quantified using a bicinchoninic acid assay. Twenty micrograms of protein per sample were separated on a 12% SDS-polyacrylamide gel, transferred to a nitrocellulose membrane, blocked with 5% milk powder, and washed with Tris-buffered saline-Tween 20 (TBST). Membranes were incubated overnight at 4°C with primary rabbit anti-VIP (1:200 dilution; Abbiotec, San Diego, CA) and mouse anti-β-actin (1:20,000 dilution; Sigma, St. Louis, MO); then they were washed with TBST, incubated with goat anti-rabbit and goat anti-mouse horseradish peroxidase secondary antibodies, and washed with TBST. An enhanced chemiluminescence kit was used to visualize protein bands.
Quantitative and statistical analysis.
Immunofluorescent staining of embryonic lung whole mounts was imaged using confocal microscopy. z-Stack images were used for three-dimensional (3-D) reconstruction using Stereo InveStigator software (MBF Bioscience, Williston, VT). For quantification of neuronal staining, we utilized z-stacks of confocal images to calculate the volume of nerve staining as a percentage of total lung volume using Neurolucida software (MBF Bioscience). Neuronal staining in sections was quantified using ImageJ software (National Institutes of Health). Statistical analysis was performed using Student's t-test.
RESULTS
Pattern of pulmonary innervation in human CDH.
Previous data suggest decreased innervation of the lungs in CDH (31–33). We sought to further define the pattern of defective pulmonary innervation in CDH. Using immunostaining for the generic neural marker S-100, we compared human lung specimens obtained from autopsy of neonates who died from CDH (age at death 3 days-8.5 wk) with lung specimens from autopsy of neonates who died from nonpulmonary causes (age at death 5–6 days). To further characterize the pattern of defective pulmonary innervation in CDH, we further divided lung samples from each group (CDH vs. controls) into a central and a peripheral zone.
In the central zones, there was a subjectively less complex peribronchial neural plexus in CDH (Fig. 1, C and D) than control (Fig. 1, A and B) lung specimens. Qualitatively, the CDH plexuses were characterized by a decreased number of S-100-positive nerve branches and smaller nerve branches than controls. Quantification of S-100 staining revealed a significant decrease in central zone neural tissue in CDH compared with control lung specimens (Fig. 1E).
Fig. 1.
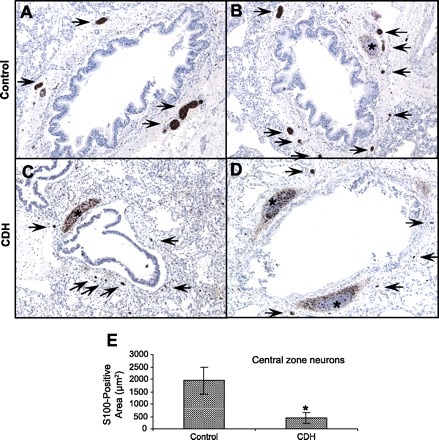
Comparison of central lung zone pulmonary innervation in control lungs and lung specimens from humans with congenital diaphragmatic hernia (CDH) using the pan-neural marker S-100. A and B: S-100 staining in the central zone of the control lung reveals a well-developed peribronchial neural plexus. Arrows, S-100-positive nerves; ∗, nonspecific S-100 cartilage staining. C and D: S-100 staining in the central zone of the CDH lung shows a less complex peribronchial neural plexus, with decreased number and size of nerve trunks (arrows). E: S-100-positive neurons in the central zone of control and CDH lungs expressed as total area of S-100-positive nerves on sections. There is a significant reduction in S-100-positive neural tissue in the central zone of CDH lungs compared with controls. S-100-positive cartilage cells were excluded from analysis. *P < 0.05.
Examination of S-100 immunostaining in peripheral lung zones similarly revealed decreased innervation of the peripheral airways and vasculature in CDH (Fig. 2, C and D) compared with control (Fig. 2, A and B) specimens. Defective innervation in peripheral lung zones was once again characterized by a decreased number of peribronchiolar and perivascular nerve branches in CDH compared with control specimens. Quantification once again showed a statistically significant decrease in S-100 immunostaining of peripheral lung zones in CDH compared with control specimens (Fig. 2E).
Fig. 2.

Comparison of peripheral lung zone pulmonary innervation in control and CDH human lung specimens using the pan-neural marker S-100. A and B: S-100 staining in the peripheral zone of the control lung reveals fine perivascular (V) and peribronchiolar (B) plexuses of nerves. Arrows, S-100-positive nerves. C and D: S-100 staining in the peripheral zone of the CDH lung shows a marked reduction in the number of S-100-positive perivascular and peribronchial nerves (arrows). E: S-100-positive neurons in the peripheral zone of control and CDH lungs expressed as total area of S-100-positive nerves on sections. There is a significant reduction in S-100-positive neural tissue in the peripheral zone of CDH lungs compared with controls. *P < 0.05.
Pattern of pulmonary innervation in nitrofen-treated murine embryonic lung.
We utilized the murine nitrofen model to study patterns of pulmonary innervation during lung development in CDH (23, 42). Lungs from nitrofen-exposed embryos exhibit histological findings that closely resemble the lung histology in human CDH (11). Using this technique, ∼70% of nitrofen-exposed embryos develop CDH, which becomes grossly evident after E14.5, but all nitrofen-treated embryonic lungs exhibit significant hypoplasia from early stages of lung development onward, with decreased airway branching and vascular defects characteristic of CDH even before closure of the diaphragm (data not shown). Therefore, because we are especially interested in the lung developmental defects during early stages of lung development, regardless of the presence of the diaphragmatic defect per se, for the following experiments, nitrofen-treated embryonic lungs were compared with controls without regard for whether a diaphragmatic defect was present.
Immunofluorescent staining of embryonic lung sections using the pan-neural marker PGP9.5 was performed. PGP9.5 staining of nitrofen-treated embryonic lungs compared with controls closely parallels the S-100 immunostaining pattern in postnatal human CDH lungs compared with controls (Fig. 3, A and B). Specifically, there was a reduction in the number of pulmonary peribronchial and perivascular nerve branches and smaller nerve branches in the nitrofen-treated embryonic lungs than in the controls.
Fig. 3.
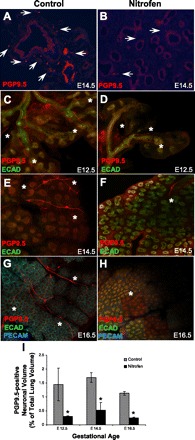
Comparison of pulmonary innervation in embryonic murine control and nitrofen-treated lungs using the pan-neural marker protein gene product 9.5 (PGP9.5). A and B: PGP9.5 staining in embryonic (E) day 14.5 (E14.5) control (A) and nitrofen-treated (B) mouse lung sections reveals a well-developed complex neural plexus in the control lung with a decreased number of nerve branches in the nitrofen-treated lung. Arrows, PGP9.5-positive nerves. Note similarity to S-100 staining pattern in Figs. 1 and 2. C–H: PGP9.5 immunofluorescent staining in confocal images of embryonic lung whole mounts from control and nitrofen-treated mice at E12.5, E14.5, and E16.5. In control lungs, there is a fine network of PGP9.5-positive nerves as early as E12.5, with increased complexity of branching into the periphery as gestation proceeds. In nitrofen-treated embryonic lungs, there is a less complex pattern of PGP9.5-positive innervation, with decreased neural branching from central nerve trunks at each gestational age. ∗, PGP9.5-positive nerve trunks. ECAD, E-cadherin; PECAM, platelet/endothelial cell adhesion molecule. Original magnification ×100. I: PGP9.5-positive neuronal volume in control and nitrofen-treated murine lung at E12.5, E14.5, and E16.5. Neuronal volume was calculated using z-stack images obtained from confocal microscopy of whole mounts and expressed as percentage of total lung volume. Quantification was performed using Stereo InveStigator software. *P < 0.05 vs. control.
To examine the pattern of pulmonary innervation in nitrofen-treated lungs in more detail and to eliminate selection bias of sections for quantification, we utilized confocal microscopy to analyze PGP9.5 immunofluorescent staining of embryonic lung whole mounts. Confocal images of PGP9.5 staining showed a fine complex pattern of neural branching into the lung periphery in control embryonic lungs from E12.5 onward (Fig. 3, C, E, and G). In nitrofen-treated embryonic lungs, there was a markedly less complex pattern of PGP9.5-positive nerve branching, especially in the lung periphery, at each embryonic stage from E12.5 onward (Fig. 3, D, F, and H). z-Stacks of confocal images of PGP9.5 staining were used to quantify PGP9.5-positive neural tissue in nitrofen-treated embryonic lungs compared with controls (Fig. 3I). At E12.5, E14.5, and E16.5, there was a statistically significant reduction in the volume of PGP9.5-positive neural tissue in nitrofen-treated embryonic lungs compared with controls, expressed as PGP9.5-positive neural volume as percentage of overall lung volume within the entire z-stack. This difference appeared to remain similar throughout gestation.
Autonomic imbalance in nitrofen-treated murine embryonic lung.
To further study the functional significance of the defective innervation of nitrofen-treated embryonic lungs, we used tyrosine hydroxylase as a sympathetic nerve marker and VAchT and VIP as parasympathetic markers to analyze patterns of autonomic innervation.
Immunofluorescent staining of embryonic lung whole mounts for tyrosine hydroxylase was analyzed using confocal microscopy. In control embryonic lungs at E12.5 and E14.5, there was a relatively less complex pattern of tyrosine hydroxylase-positive innervation than in control lungs for the pan-neural marker PGP9.5 (Fig. 4, A and C, compared with Fig. 3, C and E). Specifically, the fine branching network into the periphery of the lung that was seen with PGP9.5 was not seen with tyrosine hydroxylase. In contrast, in nitrofen-treated embryonic lungs at E12.5 and E14.5, the pattern of tyrosine hydroxylase-positive neural innervation was similar to the pattern of PGP9.5-positive neural innervation (compare Fig. 4, B and D, with Fig. 3, D and F). These results suggest a relative increase in sympathetic innervation in nitrofen-treated embryonic lungs compared with controls. Quantification of tyrosine hydroxylase staining in z-stacks of confocal images, expressed as the volume of tyrosine hydroxylase-positive nerves as a percentage of total lung volume, revealed a significant increase in relative tyrosine hydroxylase staining in nitrofen-treated embryonic lungs compared with controls (Fig. 4E).
Fig. 4.
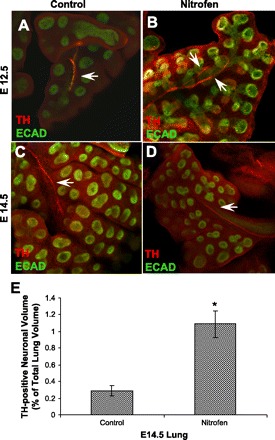
Comparison of pulmonary sympathetic innervation in embryonic murine control and nitrofen-treated lungs using the sympathetic marker tyrosine hydroxylase (TH). A–D: TH immunofluorescent staining in confocal images of embryonic lung whole mounts from control and nitrofen-treated mice at E12.5 and E14.5. In control lungs at E12.5 and E14.5, a central TH-positive nerve trunk, with little peripheral branching, is indentified. This is in contrast to the fine complex PGP9.5-positive branching pattern in control embryonic lungs (see Fig. 3). In nitrofen-treated lungs at E12.5 and E14.5, a central TH-positive nerve trunk, with little peripheral branching, is indentified. This pattern is similar to the truncated PGP9.5-positive branching pattern in nitrofen-treated embryonic lungs (see Fig. 3). These results suggest a relative increase in sympathetic innervation in nitrofen-treated embryonic lungs. Arrows, TH-positive nerves. Original magnification ×100. E: TH-positive neuronal volume in control and nitrofen-treated murine lung at E14.5. Neuronal volume was calculated using z-stack images obtained from confocal microscopy of whole mounts and expressed as percentage of total lung volume. *P < 0.05.
To examine parasympathetic innervation, we utilized immunofluorescent staining for VAchT and VIP. Immunofluorescent staining of embryonic lungs with VAchT revealed a pattern of innervation similar to that for the pan-neural marker PGP9.5. As shown at E12.5 and E16.5, there is complex VAchT-positive neural branching into the periphery of the lung in controls compared with a less complex truncated branching pattern of VAchT-positive nerves in nitrofen-treated embryonic lungs (Fig. 5, A–D). Quantification of VAchT staining in z-stacks of confocal images, expressed as the volume of VAchT-positive nerves as a percentage of total lung volume, confirmed a reduction of VAchT staining in nitrofen-treated embryonic lungs compared with controls (Fig. 5E).
Fig. 5.
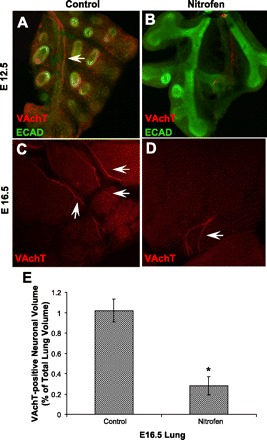
Comparison of pulmonary parasympathetic innervation in embryonic murine control and nitrofen-treated lungs using the parasympathetic marker vesicular acetylcholine transporter (VAchT). A–D: VAchT immunofluorescent staining in confocal images of embryonic lung whole mounts from control and nitrofen-treated mice at E12.5 and E16.5. In control lungs at E12.5 and E16.5, there is a fine network of VAchT-positive nerves as early as E12.5, with increased complexity of branching into the periphery as gestation proceeds. In nitrofen-treated embryonic lungs, there is a less complex pattern of VAchT-positive innervation, with decreased neural branching from central nerve trunks at each gestational age. These results suggest a relative decrease in parasympathetic innervation in nitrofen-treated embryonic lungs. Arrows, VAchT-positive nerves. Original magnification ×100. E: VAchT-positive neuronal volume in control and nitrofen-treated murine lung at E16.5. Neuronal volume was calculated using z-stack images obtained from confocal microscopy of whole mounts and expressed as percentage of total lung volume. *P < 0.05.
In addition to VAchT, VIP was used as another marker of parasympathetic innervation. In control lung sections, there was strong epithelial VIP staining mainly confined to epithelial nerve endings and/or neuroepithelial bodies within the distal airways and terminal buds (Fig. 6A). In nitrofen-treated embryonic lungs, there was a significant reduction in this distal airway VIP expression (Fig. 6B). In addition, VIP staining was also seen in a perivascular pattern, with a significant reduction in perivascular VIP staining in nitrofen-treated embryonic lungs compared with controls (Fig. 6, C and D). Western blot analysis confirmed a significant reduction in VIP protein expression in nitrofen-treated embryonic lungs compared with controls (Fig. 6E).
Fig. 6.
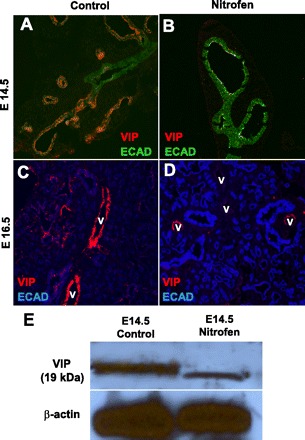
Immunofluorescent staining for VIP, a parasympathetic marker, in control and nitrofen-treated embryonic lungs. A and B: at E14.5, VIP is highly expressed within the distal airway epithelium, corresponding to nerve endings and/or neuroendocrine cells, in control embryonic lungs; this distal epithelial VIP expression is markedly reduced in nitrofen-treated embryonic lungs. C and D: at E16.5, VIP is also highly expressed within a perivascular pattern in control embryonic lungs; there is decreased perivascular VIP expression in nitrofen-treated embryonic lungs. V, blood vessels. Original magnification ×200. E: Western blot of VIP protein expression showing decreased VIP protein expression in nitrofen-treated embryonic lungs at E14.5 compared with control embryonic lungs at E14.5.
These results suggest an imbalance in pulmonary autonomic innervation in nitrofen-treated embryonic lungs compared with controls from the early stages of lung development onward. This imbalance is characterized by a relative increase in sympathetic innervation and a relative decrease in parasympathetic innervation. To further define differences in pulmonary innervation in nitrofen-treated lungs compared with controls, we created 3-D reconstructions of confocal images of PGP9.5, tyrosine hydroxylase, and VAchT staining in nitrofen-treated embryonic lungs compared with controls at E14.5. As shown in Fig. 7, A and B, 3-D reconstructions of confocal z-stacks of lung whole mounts stained for PGP9.5 revealed a complex pattern of pulmonary neural branching in control E14.5 embryonic lung and a markedly reduced pattern of branching from main nerve trunks into the periphery in nitrofen-treated lungs at E14.5. Compared with the pattern of pan-neural PGP9.5-positive nerve branching, the pattern of tyrosine hydroxylase-positive nerve branching is markedly reduced in control lungs at E14.5, while the pattern of tyrosine hydroxylase-positive nerve branching is similar to the pattern of PGP9.5-positive branching in nitrofen-treated lungs at E14.5, consistent with a relative increase of sympathetic (tyrosine hydroxylase-positive) innervation in nitrofen-treated embryonic lungs (Fig. 7, C and D). Finally, 3-D reconstructions of VAchT-positive nerves revealed a complex pattern of pulmonary VAchT-positive nerve branching in control embryonic lung at E14.5 and a markedly reduced pattern of VAchT-positive nerve branching in nitrofen-treated lungs at E14.5, consistent with a relative decrease in parasympathetic (VAchT-positive) innervation in nitrofen-treated embryonic lungs (Fig. 7, E and F).
Fig. 7.
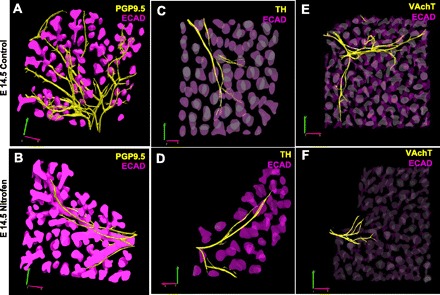
Three-dimensional reconstructions of PGP9.5-positive, TH-positive, and VAchT-positive neural branching in control and nitrofen-treated embryonic lungs at E14.5. Three-dimensional reconstructions were created from z-stacks of confocal images of whole mount lungs. A and B: pan-neural marker PGP9.5 reveals a complex neural branching pattern through the lung periphery in control lungs; neural branching pattern is significantly truncated in nitrofen-treated embryonic lungs. C and D: pattern of TH-positive nerves in control embryonic lungs is less complex, with reduced branching from the main central nerve trunks, than the overall neural branching pattern as defined by PGP9.5; pattern of TH-positive neural branching is similar in nitrofen-treated embryonic lungs, with minimal branching from the main central nerve trunks. In nitrofen-treated embryonic lungs, the TH-positive neural branching pattern closely resembles the overall PGP9.5-positive neural branching pattern (compare D with B). E and F: the parasympathetic marker VAchT reveals a complex neural branching pattern through the lung periphery in control lungs; the VAchT-positive neural branching pattern is significantly truncated in nitrofen-treated embryonic lungs. These results suggest an imbalance of autonomic innervation during embryonic lung development in the nitrofen-treated group compared with controls, with relatively increased sympathetic and decreased parasympathetic innervation. Original magnification ×100.
DISCUSSION
CDH is a relatively common and often devastating birth defect resulting in significant mortality and long-term morbidity in survivors (1, 6, 10, 20, 25, 34). The lungs in infants born with CDH exhibit specific developmental defects, including varying degrees of lung hypoplasia and decreased pulmonary arterial branching, with increased muscularization of the intrapulmonary arteries (3, 15–17, 22, 26, 30, 41). These abnormalities can lead to severe pulmonary hypertension and respiratory distress in the neonatal period. The failure of the pulmonary hypertension to resolve is a major factor contributing to poor outcomes in infants born with CDH (14). The underlying developmental processes contributing to the development of lung hypoplasia and pulmonary vascular defects and to the abnormal postnatal pulmonary vascular physiology in CDH remain undefined.
In human autopsy lung specimens and murine lungs treated with nitrofen as a model of CDH, we have shown defective pulmonary innervation in CDH. In the human specimens, staining with the generic neural marker S-100 reveals decreased neural innervation, with less complex branching of intrapulmonary nerves, in peribronchial and perivascular locations in specimens from infants with CDH compared with controls. This difference is especially marked in the lung periphery. Previous studies demonstrated decreased esophageal, tracheal, and bronchial neural crest-derived neural components in CDH (31–33). Our human data build on these earlier studies by further characterizing the pattern of defective neural innervation in CDH and demonstrating a marked decrease in pulmonary innervation of the airways and pulmonary vasculature in the lung periphery in CDH. Results utilizing PGP9.5 staining in nitrofen-treated mice closely resemble the human autopsy results. Specifically, fewer intrapulmonary nerves and a less complex branching pattern around airways and intrapulmonary arteries are observed in nitrofen-treated embryonic lungs than controls. In our mouse model, this decreased innervation in nitrofen-treated embryos is seen from E12.5 through the postnatal period, supporting the possibility that there is a primary developmental defect in intrapulmonary innervation beginning in the early stages of lung development (even before formation of the diaphragm) and persisting through birth.
To build on earlier findings and to further determine the significance of defective neural development in CDH, we used markers of sympathetic and parasympathetic nerves to determine the pattern of autonomic innervation in the developing lung. We hypothesize that an imbalance of sympathetic and parasympathetic autonomic innervation during lung development in CDH could contribute to lung developmental defects in CDH, in particular lung hypoplasia, and to postnatal physiological derangements resulting in persistent and severe pulmonary hypertension. Although, overall, PGP9.5 staining is decreased in nitrofen-treated embryonic lungs, neural staining for the sympathetic marker tyrosine hydroxylase is increased in nitrofen-treated lungs compared with controls from E12.5 to E16.5. In contrast, staining for the parasympathetic markers VAchT and VIP in nitrofen-treated lungs is decreased compared with controls. These novel results suggest an imbalance of autonomic innervation, with increased sympathetic and decreased parasympathetic innervation, within the developing lung in CDH initially in the early stages of lung development and throughout gestation. The pattern of VIP staining is especially interesting. In controls, there is marked VIP staining in what appear to be nerve endings or neuroendocrine bodies within the peripheral airway epithelium, with a lack of staining in more central larger airways. This peripheral VIP staining is markedly decreased in nitrofen-treated embryos.
An imbalance of autonomic innervation with increased sympathetic tone and decreased parasympathetic tone postnatally could be a major factor contributing to the pulmonary arterial hyperreactivity and pulmonary hypertension in infants with CDH (14). VIP is a known vasodilator and bronchodilator in lungs and is present in nerve endings of nonadrenergic noncholinergic neurons innervating the airways and the vasculature of the lung. VIP underexpression, in particular, has been associated with pulmonary hypertension and vascular changes such as increased muscularization of intrapulmonary arteries, as seen in CDH (35).
Beyond expected effects on postnatal physiology, an imbalance of autonomic innervation in nitrofen-treated lungs as early as E12.5 suggests that this imbalance may play a more interesting role developmentally, possibly contributing to the anatomic airway and vascular defects in CDH lungs. In a system analogous to lung airway branching morphogenesis, parasympathetic innervation has recently been demonstrated to regulate and maintain an epithelial progenitor cell population during salivary gland development. Removal of the parasympathetic submandibular ganglion from embryonic submandibular gland explants in culture or chemical inhibition of acetylcholine signaling results in decreased branching morphogenesis and decreased expression of epithelial progenitor cell markers (24). Thus, decreased parasympathetic innervation during development of the lungs in CDH may have a similar effect on airway branching morphogenesis, resulting in lung hypoplasia. In addition, defective innervation of pulmonary neuroendocrine cells or neuroepithelial bodies within the developing lung may impact airway branching morphogenesis. Pulmonary neuroendocrine cells are present at the early stages of lung development and seem to be key regulators of airway branching morphogenesis and epithelial proliferation through secretion of effectors such as calcitonin gene-related peptide, gastrin-releasing peptide, and bombesin (21, 27, 40, 45). Another way in which defective innervation may contribute to lung hypoplasia is via direct effects on airway smooth muscle contraction. Rhythmic contraction of the airway smooth muscle is known to promote lung growth by modulating embryonic airway pressure and, potentially, delivering growth factors within the amniotic fluid to the distal airways (36, 38). This process would presumably be influenced by early developmental alterations in autonomic innervation of the lung. Finally, defective autonomic innervation with increased sympathetic and decreased parasympathetic innervation of the embryonic lung may result in decreased embryonic pulmonary blood flow in CDH, which may have a direct impact on airway branching morphogenesis (13). Further examination of these various pathways is needed to prove the potential role of the imbalance of autonomic innervation in modulating branching morphogenesis and contributing to lung hypoplasia in CDH.
We have utilized the nitrofen model of CDH as our animal model for these experiments. The nitrofen model of CDH has been used extensively since it was first described in 1990 (23, 42). Lungs from nitrofen-exposed embryos exhibit histological findings that closely mimic the human disease. Importantly, the pulmonary vascular abnormalities in this model (decreased pulmonary arterial branching, increased muscularization of intrapulmonary arteries, and increased expression of α-smooth muscle actin) are similar to those in the human disease (11). Interestingly, nitrofen is thought to act through inhibition of retinoid signaling by inhibiting retinal dehydrogenase (18). Retinoid signaling has been shown to influence neural crest and peripheral neural development, at least suggesting that defective neural development may be a key early factor in the development of the lung defects in CDH (4, 28, 29).
In summary, using pan-neural markers in human tissue and a murine model of CDH-associated lung hypoplasia, we have confirmed the presence of defective pulmonary innervation in CDH and further defined its pattern. Furthermore, we have demonstrated, for the first time, an imbalance of autonomic innervation, with a relative increase in sympathetic innervation and a relative decrease in parasympathetic innervation, in nitrofen-exposed embryonic mouse lungs from the early stages of lung development onward. The lung developmental and physiological consequences of this autonomic imbalance remain to be fully explored.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
REFERENCES
- 1. Anonymous Does extracorporeal membrane oxygenation improve survival in neonates with congenital diaphragmatic hernia? The Congenital Diaphragmatic Hernia Study Group. J Pediatr Surg 34: 720–725, 1999 [DOI] [PubMed] [Google Scholar]
- 2. Anonymous Inhaled nitric oxide and hypoxic respiratory failure in infants with congenital diaphragmatic hernia. The Neonatal Inhaled Nitric Oxide Study Group (NINOS). Pediatrics 99: 838–845, 1997 [DOI] [PubMed] [Google Scholar]
- 3.Askenazi SS, Perlman M. Pulmonary hypoplasia: lung weight and radial alveolar count as criteria of diagnosis. Arch Dis Child 54: 614–618, 1979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Aybar MJ, Glavic A, Mayor R. Extracellular signals, cell interactions and transcription factors involved in the induction of the neural crest cells. Biol Res 35: 267–275, 2002 [DOI] [PubMed] [Google Scholar]
- 5. Balaguer L, Romano J, Ruiz-Pesini P. Serotonin immunoreactivity in the autonomic intrapulmonary ganglia of the fetal sheep. Neurosci Lett 133: 151–153, 1991 [DOI] [PubMed] [Google Scholar]
- 6. Boloker J, Bateman DA, Wung JT, Stolar CJ. Congenital diaphragmatic hernia in 120 infants treated consecutively with permissive hypercapnea/spontaneous respiration/elective repair. J Pediatr Surg 37: 357–366, 2002 [DOI] [PubMed] [Google Scholar]
- 7. Burns AJ, Delalande JM. Neural crest cell origin for intrinsic ganglia of the developing chicken lung. Dev Biol 277: 63–79, 2005 [DOI] [PubMed] [Google Scholar]
- 8. Burns AJ, Thapar N, Barlow AJ. Development of the neural crest-derived intrinsic innervation of the human lung. Am J Respir Cell Mol Biol 38: 269–275, 2008 [DOI] [PubMed] [Google Scholar]
- 9. Carmeliet P, Tessier-Lavigne M. Common mechanisms of nerve and blood vessel wiring. Nature 436: 193–200, 2005 [DOI] [PubMed] [Google Scholar]
- 10. Clark RH, Hardin WD, Jr, Hirschl RB, Jaksic T, Lally KP, Langham MR, Jr, Wilson JM. Current surgical management of congenital diaphragmatic hernia: a report from the Congenital Diaphragmatic Hernia Study Group. J Pediatr Surg 33: 1004–1009, 1998 [DOI] [PubMed] [Google Scholar]
- 11. Coleman C, Zhao J, Gupta M, Buckley S, Tefft JD, Wuenschell CW, Minoo P, Anderson KD, Warburton D. Inhibition of vascular and epithelial differentiation in murine nitrofen-induced diaphragmatic hernia. Am J Physiol Lung Cell Mol Physiol 274: L636–L646, 1998 [DOI] [PubMed] [Google Scholar]
- 12. Colvin J, Bower C, Dickinson JE, Sokol J. Outcomes of congenital diaphragmatic hernia: a population-based study in Western Australia. Pediatrics 116: e356–e363, 2005 [DOI] [PubMed] [Google Scholar]
- 13. Crivellato E, Nico B, Ribatti D. Contribution of endothelial cells to organogenesis: a modern reappraisal of an old Aristotelian concept. J Anat 211: 415–427, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dillon PW, Cilley RE, Mauger D, Zachary C, Meier A. The relationship of pulmonary artery pressure and survival in congenital diaphragmatic hernia. J Pediatr Surg 39: 307–312, 2004 [DOI] [PubMed] [Google Scholar]
- 15. Geggel RL, Murphy JD, Langleben D, Crone RK, Vacanti JP, Reid LM. Congenital diaphragmatic hernia: arterial structural changes and persistent pulmonary hypertension after surgical repair. J Pediatr 107: 457–464, 1985 [DOI] [PubMed] [Google Scholar]
- 16. George DK, Cooney TP, Chiu BK, Thurlbeck WM. Hypoplasia and immaturity of the terminal lung unit (acinus) in congenital diaphragmatic hernia. Am Rev Respir Dis 136: 947–950, 1987 [DOI] [PubMed] [Google Scholar]
- 17. Gosche JR, Islam S, Boulanger SC. Congenital diaphragmatic hernia: searching for answers. Am J Surg 190: 324–332, 2005 [DOI] [PubMed] [Google Scholar]
- 18. Greer JJ, Babiuk RP, Thebaud B. Etiology of congenital diaphragmatic hernia: the retinoid hypothesis. Pediatr Res 53: 726–730, 2003 [DOI] [PubMed] [Google Scholar]
- 19. Guembe L, Villaro AC. Histochemical demonstration of neuronal nitric oxide synthase during development of mouse respiratory tract. Am J Respir Cell Mol Biol 20: 342–351, 1999 [DOI] [PubMed] [Google Scholar]
- 20. Kays DW, Langham MR, Jr, Ledbetter DJ, Talbert JL. Detrimental effects of standard medical therapy in congenital diaphragmatic hernia. Ann Surg 230: 340–351, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. King KA, Torday JS, Sunday ME. Bombesin and [Leu8]phyllolitorin promote fetal mouse lung branching morphogenesis via a receptor-mediated mechanism. Proc Natl Acad Sci USA 92: 4357–4361, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kitagawa M, Hislop A, Boyden EA, Reid L. Lung hypoplasia in congenital diaphragmatic hernia. A quantitative study of airway, artery, and alveolar development. Br J Surg 58: 342–346, 1971 [DOI] [PubMed] [Google Scholar]
- 23. Kluth D, Kangah R, Reich P, Tenbrinck R, Tibboel D, Lambrecht W. Nitrofen-induced diaphragmatic hernias in rats: an animal model. J Pediatr Surg 25: 850–854, 1990 [DOI] [PubMed] [Google Scholar]
- 24. Knox SM, Lombaert IM, Reed X, Vitale-Cross L, Gutkind JS, Hoffman MP. Parasympathetic innervation maintains epithelial progenitor cells during salivary organogenesis. Science 329: 1645–1647, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lally KP, Lally PA, Van Meurs KP, Bohn DJ, Davis CF, Rodgers B, Bhatia J, Dudell G. Treatment evolution in high-risk congenital diaphragmatic hernia: ten years' experience with diaphragmatic agenesis. Ann Surg 244: 505–513, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Levin DL. Morphologic analysis of the pulmonary vascular bed in congenital left-sided diaphragmatic hernia. J Pediatr 92: 805–809, 1978 [DOI] [PubMed] [Google Scholar]
- 27. Li K, Nagalla SR, Spindel ER. A rhesus monkey model to characterize the role of gastrin-releasing peptide (GRP) in lung development. Evidence for stimulation of airway growth. J Clin Invest 94: 1605–1615, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Maden M. Retinoic acid in the development, regeneration and maintenance of the nervous system. Nat Rev Neurosci 8: 755–765, 2007 [DOI] [PubMed] [Google Scholar]
- 29. Maschhoff KL, Baldwin HS. Molecular determinants of neural crest migration. Am J Med Genet 97: 280–288, 2000 [DOI] [PubMed] [Google Scholar]
- 30. Naeye RL, Shochat SJ, Whitman V, Maisels MJ. Unsuspected pulmonary vascular abnormalities associated with diaphragmatic hernia. Pediatrics 58: 902–906, 1976 [PubMed] [Google Scholar]
- 31. Pederiva F, Lopez RA, Martinez L, Tovar JA. Tracheal innervation is abnormal in rats with experimental congenital diaphragmatic hernia. J Pediatr Surg 44: 1159–1164, 2009 [DOI] [PubMed] [Google Scholar]
- 32. Pederiva F, Lopez RA, Rodriguez JI, Martinez L, Tovar JA. Bronchopulmonary innervation defects in infants and rats with congenital diaphragmatic hernia. J Pediatr Surg 45: 360–365, 2010 [DOI] [PubMed] [Google Scholar]
- 33. Pederiva F, Rodriguez JI, Ruiz-Bravo E, Martinez L, Tovar JA. Abnormal intrinsic esophageal innervation in congenital diaphragmatic hernia: a likely cause of motor dysfunction. J Pediatr Surg 44: 496–499, 2009 [DOI] [PubMed] [Google Scholar]
- 34. Reickert CA, Hirschl RB, Atkinson JB, Dudell G, Georgeson K, Glick P, Greenspan J, Kays D, Klein M, Lally KP, Mahaffey S, Ryckman F, Sawin R, Short BL, Stolar CJ, Thompson A, Wilson JM. Congenital diaphragmatic hernia survival and use of extracorporeal life support at selected level III nurseries with multimodality support. Surgery 123: 305–310, 1998 [PubMed] [Google Scholar]
- 35. Said SI, Hamidi SA, Dickman KG, Szema AM, Lyubsky S, Lin RZ, Jiang YP, Chen JJ, Waschek JA, Kort S. Moderate pulmonary arterial hypertension in male mice lacking the vasoactive intestinal peptide gene. Circulation 115: 1260–1268, 2007 [DOI] [PubMed] [Google Scholar]
- 36. Schittny JC, Miserocchi G, Sparrow MP. Spontaneous peristaltic airway contractions propel lung liquid through the bronchial tree of intact and fetal lung explants. Am J Respir Cell Mol Biol 23: 11–18, 2000 [DOI] [PubMed] [Google Scholar]
- 37. Shah SR, Esni F, Jakub A, Paredes J, Lath N, Malek M, Potoka DA, Prasadan K, Mastroberardino PG, Shiota C, Guo P, Miller KA, Hackam DJ, Burns RC, Tulachan SS, Gittes GK. Embryonic mouse blood flow and oxygen correlate with early pancreatic differentiation. Dev Biol 349: 342–349, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sparrow MP, Lamb JP. Ontogeny of airway smooth muscle: structure, innervation, myogenesis and function in the fetal lung. Respir Physiol Neurobiol 137: 361–372, 2003 [DOI] [PubMed] [Google Scholar]
- 39. Stege G, Fenton A, Jaffray B. Nihilism in the 1990s: the true mortality of congenital diaphragmatic hernia. Pediatrics 112: 532–535, 2003 [DOI] [PubMed] [Google Scholar]
- 40. Sunday ME, Hua J, Dai HB, Nusrat A, Torday JS. Bombesin increases fetal lung growth and maturation in utero and in organ culture. Am J Respir Cell Mol Biol 3: 199–205, 1990 [DOI] [PubMed] [Google Scholar]
- 41. Taira Y, Yamataka T, Miyazaki E, Puri P. Comparison of the pulmonary vasculature in newborns and stillborns with congenital diaphragmatic hernia. Pediatr Surg Int 14: 30–35, 1998 [DOI] [PubMed] [Google Scholar]
- 42. Tenbrinck R, Tibboel D, Gaillard JL, Kluth D, Bos AP, Lachmann B, Molenaar JC. Experimentally induced congenital diaphragmatic hernia in rats. J Pediatr Surg 25: 426–429, 1990 [DOI] [PubMed] [Google Scholar]
- 43. Tollet J, Everett AW, Sparrow MP. Spatial and temporal distribution of nerves, ganglia, and smooth muscle during the early pseudoglandular stage of fetal mouse lung development. Dev Dyn 221: 48–60, 2001 [DOI] [PubMed] [Google Scholar]
- 44. Vaccaro R, Parisi Salvi E, Renda T. Early development of chick embryo respiratory nervous system: an immunohistochemical study. Anat Embryol (Berl) 211: 345–354, 2006 [DOI] [PubMed] [Google Scholar]
- 45. White SR, Hershenson MB, Sigrist KS, Zimmermann A, Solway J. Proliferation of guinea pig tracheal epithelial cells induced by calcitonin gene-related peptide. Am J Respir Cell Mol Biol 8: 592–596, 1993 [DOI] [PubMed] [Google Scholar]