

Abstract
We have previously demonstrated that inhibition of vasodilator prostanoids, PGI2 and PGE2, and nitric oxide (NO) synthsesis by a selective cyclooxygenase-2 (COX-2) inhibitor, NS-398, restores blood pressure as a result of increased systemic and renal levels of 20-hydroxyeicosatetraenoic acid (20-HETE) in endotoxemic rats. The aim of this study was to further investigate the effects of NS-398 on the changes in expression and/or activity of COX-2, cyctochrome P450 4A1 (CYP4A1), inducible NO synthase (iNOS), and peroxynitrite formation in serum, renal, cardiac, and/or vascular tissues of lipopolysaacharide (LPS)-treated rats. LPS (10 mg/kg, i.p.)-induced decrease in blood pressure was associated with increased protein levels of COX-2, iNOS, and nitrotyrosine in kidney, heart, thoracic aorta, and superior mesenteric artery. The activities of COX-2 and iNOS as well as levels of PGI2, PGE2, and nitrotyrosine were also increased in the systemic circulation and renal, cardiac, and vascular tissues of LPS-treated rats. In contrast, renal, cardiac, and vascular CYP4A1 protein expression as well as systemic and tissue levels of 20-HETE were decreased in endotoxemic rats. These effects of LPS, except COX-2 protein expression, were prevented by NS-398 (10 mg/kg, i.p.), given 1 h after injection of LPS. These data suggest that COX-2-derived vasodilator prostanoids, PGI2 and PGE2, produced during endotoxemia increase iNOS protein expression and activity as well as peroxynitrite formation resulting in decreased CYP4A1 protein expression and 20-HETE synthesis. Taken together, we concluded that an increase in 20-HETE levels associated with a decrease in the production of vasodilator prostanoids and NO participates in the effect of NS-398 to prevent hypotension in the rat model of septic shock.
Keywords: Endotoxin, Blood Pressure, Cyclooxygenase-2, Prostaglandins, Nitric Oxide, Peroxynitrite, 20-HETE
1. Introduction
The expression of inducible nitric oxide (NO) synthase (iNOS) is enhanced in many tissues in response to mediators released by the lipid A part of lipopolysaccharide (LPS), also known as endotoxin, which is the most potent microbial mediator of the pathogenesis of sepsis and septic shock [1,2]. This leads to increased generation of NO, which contributes to a fall in blood pressure, vascular hyporeactivity, multiple organ failure, and the high mortality rate that are associated with septic shock [2–5]. Systemic blockade of iNOS opposes the fall in blood pressure in endotoxic shock [2,3,5]. This is not only due to withdrawal of the vasodilator effects of NO, but also is associated with increased activity of vasoconstrictor pathways including the sympathetic nervous, renin-angiotensin, endothelin, and 20-hydroxyeicosatetraenoic acid (20-HETE) systems [2,6].
20-HETE is an ω-hydroxylation product of arachidonic acid that is produced by cytochrome P450 (CYP) enzymes, mainly by the CYP4A and CYP4F isoforms in the kidney, heart, liver, brain, lung, and the vasculature [6–8]. In the vasculature, 20-HETE causes vasoconstriction in several vascular beds, including renal, cerebral, aortic, mesenteric, and coronary arteries [9–13]. 20-HETE activates protein kinase C [14–17], MAPK [18–20], Raf/Mitogen-activated protein kinase kinase (MEK)/ERK [21], and Rho-kinase [10] pathways, which all contribute to the regulation of vascular tone. Furthermore, 20-HETE phosphorylates and blocks KCa channel activity [13,14,17,22], thereby allowing for sustained depolarization of vascular smooth muscle and calcium entry through L-type calcium channels. 20-HETE also increases the conductance of L-type calcium channels through activation of PKC [23]. Recently, 20-HETE has been reported to enhance the activation of inward non-selective cation currents through transient receptor potential canonical six channels [24] which are implicated in the myogenic response [25]. It has also been reported that 20-HETE-induced constriction is also abolished by inhibition of COX with indomethacin [11,12], diclophenac [10], or the endoperoxide/thromboxane receptor antagonist, SQ-29548 [10,11]. Furthermore, prostaglandin analogs of 20-HETE, 20-OH-PGG2 and 20-OH-PGH2, produced by COX in vascular endothelial cells, have been shown to mediate the vasoconstrictor effects of 20-HETE [11,12]. As opposed to its vasoconstrictor effect, 20-HETE produces vasodilation in the renal, coronary, pulmonary, and basilar arteries [11,26–30]. These vasodilatory responses of 20-HETE have been attributed to NO release [29], conversion of 20-HETE to 20-OH-PGE2 and 20-OH-PGF2α by cyclooxygenase (COX) [10,30,27], and increased formation of PGE2 [27] and PGI2 [26–28,30]. CYP4A- and CYP4F-derived 20-HETE is also involved in LPS-induced acute systemic inflammation as a proinflammatory mediator [31,32].
It has been reported that NO inhibits renal CYP ω-hydroxylase activity and the production of 20-HETE [33,34]. Moreover, a NO-induced fall in the endogenous production of 20-HETE has also been found to contribute to the cyclic guanosine monophosphate (cyclic GMP)-independent vasodilator effects of NO in renal and cerebral microcirculations [33,35]. We have previously demonstrated that the fall in mean arterial pressure (MAP) in endotoxemic rats is also associated with a decrease in the expression of CYP4A1/A3 protein and CYP4A activity in kidney and increased production of NO in serum, kidney, heart, thoracic aorta, and superior mesenteric artery [36–46]. Moreover, administration of a synthetic analog of 20-HETE, N-[20-hydroxyeicosa-5(Z),14(Z)-dienoyl]glycine (5,14-HEDGE), prevented the vascular hyporeactivity, hypotension, and mortality associated with the changes in systemic and tissue NO production as well as iNOS protein expression in cardiac, renal, and vascular tissues of rats treated with LPS [41,42,46]. We have recently reported that 5,14-HEDGE also prevents the LPS-induced increase in iNOS-heat shock protein 90 (hsp90) complex formation and protein levels of phosphorylated vasodilator stimulated phosphoprotein (an index for protein kinase G activity), nicotinamide adenine dinucleotide phosphate (NADPH) subunits, and gp91phox and p47phox, and nitrotyrosine (a stable end-product of peroxynitrite), as well as the decrease in CYP4A1 protein expression in renal and cardiovascular tissues associated with elevated serum nitrotyrosine levels [46]. These findings support the hypothesis that NO-induced inhibition of 20-HETE production and removal of its influence on vascular tone contributes to the fall in blood pressure and vascular hyporeactivity in endotoxic shock.
Increased production of prostanoids by COX-2 has also been shown to contribute to systemic hypotension and related organ damage and decreased survival in animals and humans with sepsis [47–52]. Our previous studies with the use of the nonselective COX inhibitor, indomethacin, suggest that inhibition of renal iNOS protein expression as well as systemic nitrite and PGE2 levels by indomethacin restores MAP presumably due to increased production of 20-HETE derived from CYP4A in endotoxemic rats [40]. Our recent studies also suggest that a decrease in the expression and activity of COX-2 and iNOS associated with an increase in CYP4A1 expression and 20-HETE synthesis contributes to the effect of piroxicam, a preferential COX-1 inhibitor, helping to prevent the hypotension during rat endotoxemia [45]. More recently, we have demonstrated that inhibition of PGI2 and PGE2 synthesis and NO production by a selective COX-2 inhibitor, N-(2-cyclohexyloxy-4-nitrophenyl)-methansulphonamide (NS-398), restores blood pressure due to increased systemic and renal levels of 20-HETE [44]. The present study was conducted to further investigate the effects of NS-398 on the LPS-induced changes in the expression and activity of COX-2, CYP4A1, and iNOS as well as peroxynitrite formation in systemic circulation and renal, cardiac, and vascular tissues of rats. The results of this study have been presented in an abstract form [53].
2. Materials and Methods
2.1. Endotoxic shock model
Experiments were performed on male Wistar rats (n = 36) (Research Center of Experimental Animals, Mersin University, Mersin, Turkey) weighing 200 to 300 g that were fed with a standard chow. They were synchronized by maintenance of controlled environmental conditions throughout the experiments. The circadian rhythmicity of the animals were entrained by a standardized 12h light and 12 h dark. All experiments were carried out according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The protocol was approved by the Ethics Committee of Mersin University School of Medicine. Endotoxic shock was induced as previously described by Tunctan et al. [36]. Rats were randomly divided into saline (n = 10), LPS (n = 10), NS-398 (n = 8), and LPS+NS-398 (n= 8) groups. In the saline and NS-398 groups, rats received saline (4 ml/kg, i.p.) at time 0. Rats in the LPS and LPS+NS-398 groups were treated with LPS (Escherichia coli LPS, O111:B4; Sigma Chemical Co., St. Louis, MO, USA) (10 mg/kg, i.p.; sublethal dose) at time 0. In the NS-398 and LPS+NS-398 groups, rats were treated with a selective COX-2 inhibitor, NS-398 (Sigma Chemical, St. Louis, MO, USA) (10 mg/kg, i.p.) [44], 1 h after injection of saline or LPS, respectively. LPS and NS-398 were dissolved in saline and ethanol (20%, w/v), respectively. MAP and heart rate (HR) were measured using a tail-cuff device (MAY 9610 Indirect Blood Pressure Recorder System, Commat Ltd., Ankara, Turkey) during a control period at time 0 and 1, 2, 3, and 4 h. All animals survived in the experiments. Rats were euthanized 4 h after the administration of saline or LPS and blood samples, kidney, heart, thoracic aorta, and superior mesenteric artery were collected from all animals. Sera were obtained from blood samples by centrifugation at 23,910 × g for 15 min at 4°C and stored at −20°C. The tissues were rapidly frozen in liquid nitrogen and stored at −80°C. The frozen tissues were ground into powder in liquid nitrogen and homogenized in 1 ml of an ice-cold 20 mM HEPES buffer (pH 7.5) containing 20 mM β-glycerophosphate, 20 mM sodium pyrophosphate, 0.2 mM sodium orthovanadate, 2 mM ethylenediaminetetraacetic acid, 20 mM sodium fluoride, 10 mM benzamidine, 1 mM dithiothreitol, 20 mM leupeptin, and 10 mM aprotinin [37]. The samples were centrifuged at 23,910 × g for 15 min at 4°C and then supernatants were removed and stored at −80°C for measurement of COX-2, CYP4A1, iNOS, nitrotyrosine, and actin protein levels and/or activities. Total protein amount was determined by Coomassie blue method using bovine serum albumin (BSA) as standard [36].
2.2. Immunoblotting
Immunoblotting for COX-2, CYP4A1, iNOS, nitrotyrosine, β-actin, α-sarcomeric actin/α-skeletal muscle actin, and α-smooth muscle actin proteins were performed according to the method described previously [38,42,43,45]. Briefly, tissue homogenates (140–175 μg of protein) were subjected to a 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then proteins were transferred to a nitrocellulose membrane. The membranes were blocked with 5% non-fat dry milk in Tris-buffered saline (mmol/L: Tris-HCl 25 [pH 7.4], NaCl 137, KCl 27 and 0.05% Tween 20) and incubated overnight with monoclonal antibodies (1:500 in 5% BSA) of anti-COX-2 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-CYP4A1 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-iNOS (BD Transduction Laboratories, San Jose, CA, USA), and anti-nitrotyrosine (Santa Cruz Biotechnology, Santa Cruz, CA, USA) followed by incubation with sheep anti-mouse IgG-horseradish peroxidase (Amersham Life Sciences, Cleveland, OH, USA) (1:1,000 in 0.1% BSA) for 1 h. The blots were developed with enhanced chemiluminescence (ECL Plus Western Blotting Detection Reagents) (Amersham Life Sciences, Cleveland, OH) according to the manufacturer’s instructions. Immunoreactive proteins were visualized using a gel-imaging system (EC3-CHEMI HR imaging system; Ultra-Violet Products, UVP, Cambridge, UK). Densitometric analysis was performed with NIH image software (Image J 1.28, Rasband WS, Image J, National Institute of Health, Bethesda, MD, USA). The membranes were reprobed with monoclonal antibodies against β-actin (Sigma Chemical Co., St. Louis, MO, USA) for kidney, α-sarcomeric actin/α-skeletal muscle actin (Sigma Chemical Co., St. Louis, MO, USA) for heart, and α-smooth muscle actin (Sigma Chemical Co., St. Louis, MO, USA) for vessels (1:500 in 5% BSA) as a loading control following by incubation with sheep anti-mouse IgG-horseradish peroxidase (1:1,000 in 0.1% BSA). Relative densities of immunoreactive bands for COX-2, CYP4A1, iNOS, and nitrotyrosine proteins in each sample were normalized to the density of corresponding bands for β-actin, α-sarcomeric actin/α-skeletal muscle actin, or α-smooth muscle actin.
2.3. Measurement of COX-2 and NOS activities, and 6-keto-PGF1α, PGE2, 20-HETE, and nitrotyrosine levels
COX-2 and NOS activities, and 6-keto-PGF1α, PGE2, 20-HETE, and nitrotyrosine levels in the sera and tissue samples were measured by ELISA according to the manufacturer’s instructions in the COX Activity Assay Kit (Cayman Chemical Co., Ann Arbor, MI, USA), Ultrasensitive Colorimetric Assay for Nitric Oxide Synthase (Oxford Biomedical Research Inc., Oxford, MI, USA), 6-keto-PGF1α ELISA Kit (Cayman Chemical Co., Ann Arbor, MI, USA), PGE2 ELISA Kit (Cayman Chemical Co., Ann Arbor, MI, USA), Hypertension ELISA (20-HETE) Kit (Detroit R&D, Inc., Detroit, MI, USA), and nitrotyrosine ELISA Kit (Northwest Life Science Specialities, LSS, Vancouver, WA, USA) assay kits, respectively.
2.4. Statistical analysis
All data were expressed as means ± S.E.M. Data were analysed by one-way ANOVA followed by Student-Newman-Keuls test for multiple comparisons, Kruskal-Wallis test followed by Dunns test for multiple comparisons and Student’s t or Mann-Whitney U tests when appropriate. A P value < 0.05 was considered to be statistically significant.
3. Results
3.1. Effect of selective COX-2 inhibition on the cardiovascular response to LPS
LPS caused a gradual fall in MAP (Fig. 1A) and an increase in HR (Fig. 1B) over the 4 h course of the experiment (p < 0.05). The change in MAP and HR reached a maximum 4 h after the administration of LPS. MAP fell by 25 mmHg and HR rose by 69 bpm in rats treated with LPS. A selective COX-2 inhibitor, NS-398, prevented the fall in MAP and the increase in HR in rats given LPS (p < 0.05). NS-398 had no effect on MAP and HR when given to rats treated with vehicle (p > 0.05).
Fig. 1.
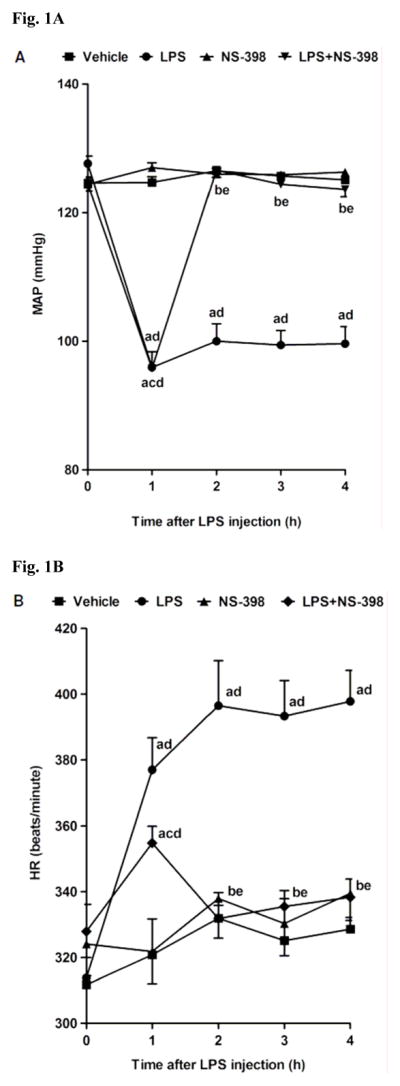
Time course of the effects of NS-398 on (A) MAP and (B) HR following administration of saline (vehicle) (4 ml/kg, i.p.) or LPS (10 mg/kg, i.p.) to conscious rats. NS-398 (10 mg/kg, i.p.) was given 1 h after administration of LPS. Data are expressed as means ± S.E.M. of 8–10 animals. aSignificant difference from the corresponding value seen in rats treated with saline (vehicle) (p < 0.05). bSignificant difference from the corresponding value seen in the rats treated with vehicle and LPS (p < 0.05). cSignificant difference from the corresponding value seen in the rats treated with vehicle and NS-398 (p < 0.05). dSignificant difference from the time 0 h value within a group (p < 0.05). eSignificant difference from the time 1 h value within a group (p < 0.05).
3.2. Effect of NS-398 on the LPS-induced increase in COX-2 protein expression and activity
LPS increased COX-2 protein expression in kidney (Fig. 2A), heart (Fig. 2B), thoracic aorta (Fig. 2C), and superior mesenteric artey (Fig. 2D) of rats (p < 0.05). The increase in COX-2 protein expression was not prevented in the tissues of rats treated with LPS and NS-398 (p > 0.05) (Fig. 2). NS-398 had no effect on the basal levels of COX-2 protein in vehicle-treated rats (p > 0.05). The LPS-induced increase in COX-2 protein expression was associated with an increase in COX-2 activity in serum (Fig. 3A), kidney (Fig. 3B), heart (Fig. 3C), thoracic aorta (Fig. 3D), and superior mesenteric artey (Fig. 3E) (p < 0.05). We have previously demonstrated that NS-398 prevented the increase in systemic (Figs. 4A and 5A) and renal levels (Figs. 4B and 5B) of 6-keto-PGF1α and PGE2 in LPS-treated rats (p < 0.05) [31]. In the present study, the LPS-induced increase in 6-keto-PGF1α and PGE2 levels in heart (Figs. 4C and 5C), thoracic aorta (Figs. 4D and 5D), and superior mesenteric artery (Figs. 4E and 5E) were also prevented by NS-398 (p < 0.05). In contrast to COX-2 protein expression, NS-398 prevented the LPS-induced increase in COX-2 activity and 6-keto-PGF1α and PGE2 levels in the sera and tissues (p < 0.05) (Figs. 3, 4 and 5). Basal levels of COX-2 activity and prostanoid levels were not changed by NS-398 in vehicle-treated rats (p > 0.05) (Figs. 3, 4, and 5).
Fig. 2.


Effect of NS-398 on changes in COX-2 protein expression in (A) kidney, (B) heart, (C) thoracic aorta, and (D) superior mesenteric artery measured 4 h after saline (vehicle) or LPS injection. Data are expressed as means±S.E.M of 4 animals. aSignificant difference from the corresponding value seen in rats treated with saline (p < 0.05).
Fig. 3.



Effect of NS-398 on changes in COX-2 activity in (A) serum, (B) kidney, (C) heart, (D) thoracic aorta, and (E) superior mesenteric artery measured 4 h after saline (vehicle) or LPS injection. COX-2 activity was measured using the COX Activity Assay Kit for the peroxidase activity of COX following the manufacturer’s instructions. The peroxidase activity was assayed colorimetrically by monitoring the appearance of oxidized N,N,N′,N′-tetramethyl-p-phenylenediamine (TMPD), a stable metabolite of PGH2, in the presence of arachidonic acid. The samples were treated with SC-560 (a COX-1 selective inhibitor) for distinguishing COX-2 activity from COX-1 activity. Data are expressed as means±S.E.M of 3–5 animals. aSignificant difference from the corresponding value seen in rats treated with saline (p < 0.05). bSignificant difference from the corresponding value seen in the rats treated with vehicle and LPS (p < 0.05).
Fig. 4.
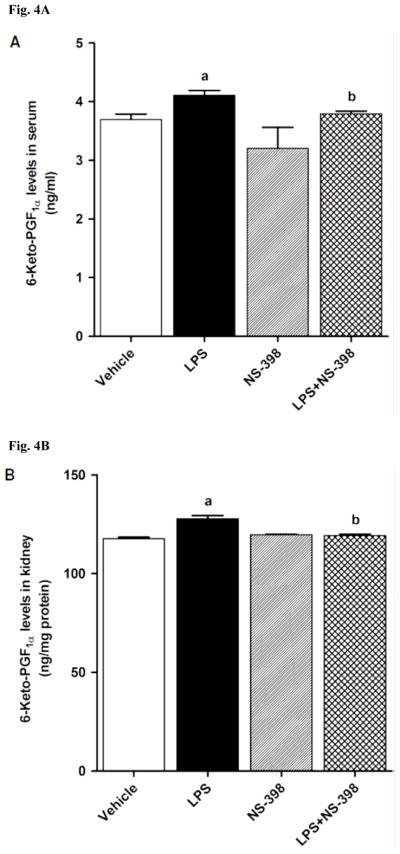
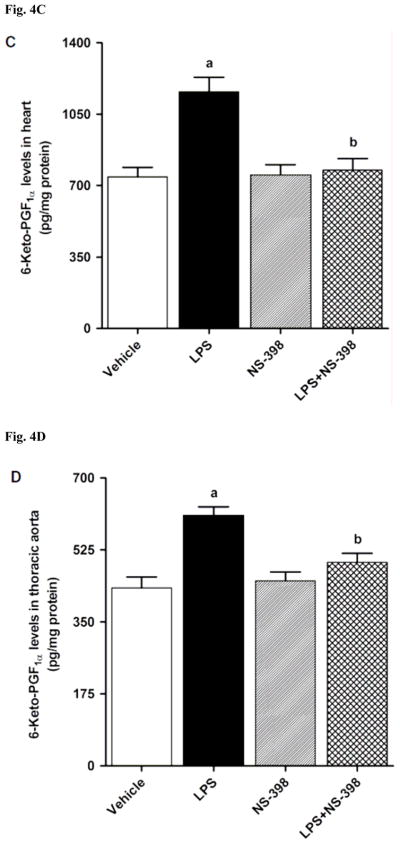

Effect of NS-398 on changes in 6-keto-PGF1α levels in (A) serum, (B) kidney, (C) heart, (D) thoracic aorta, and (E) superior mesenteric artery measured 4 h after saline (vehicle) or LPS injection. Data are expressed as means±S.E.M of 6 animals. aSignificant difference from the corresponding value seen in rats treated with saline (p < 0.05). bSignificant difference from the corresponding value seen in the rats treated with vehicle and LPS (p < 0.05).
Fig. 5.
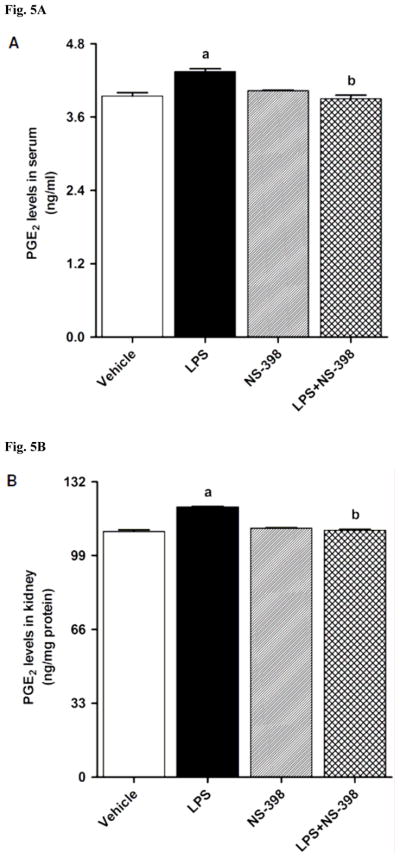
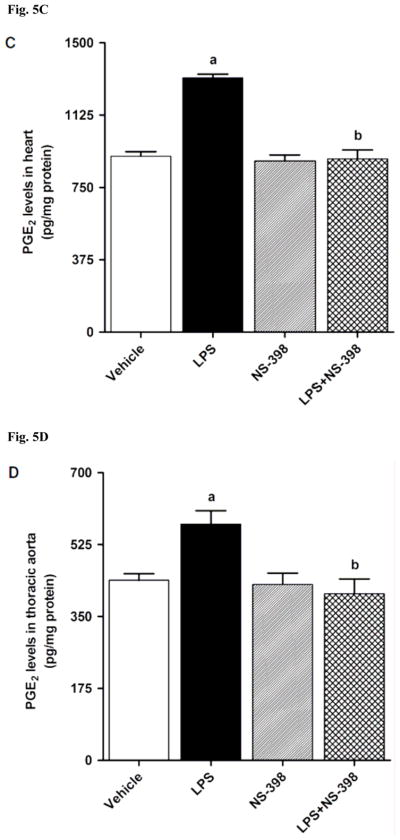
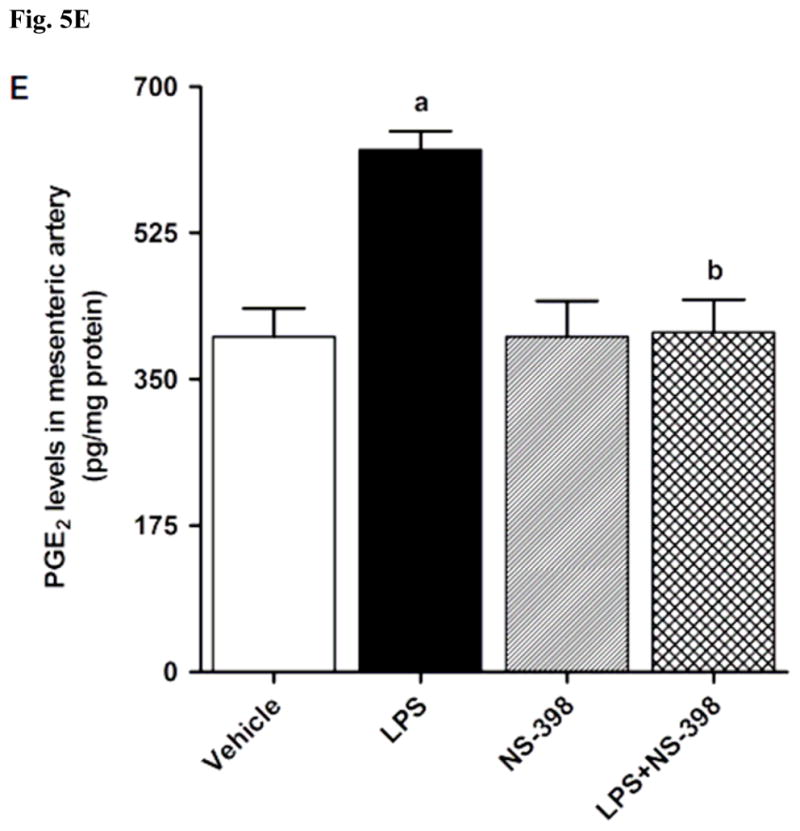
Effect of NS-398 on changes in PGE2 levels in (A) serum, (B) kidney, (C) heart, (D) thoracic aorta, and (E) superior mesenteric artery measured 4 h after saline (vehicle) or LPS injection. Data are expressed as means±S.E.M of 6 animals. aSignificant difference from the corresponding value seen in rats treated with saline (p < 0.05). bSignificant difference from the corresponding value seen in the rats treated with vehicle and LPS (p < 0.05).
3.3. Effect of NS-398 on the LPS-induced decrease in CYP4A1 protein expression and 20-HETE levels
CYP4A1 protein expression was decreased in the kidney (Fig. 6A), heart (Fig. 6B), thoracic aorta (Fig. 6C), and superior mesenteric artery (Fig. 6D) of endotoxemic rats (p < 0.05). We have previously demonstrated that NS-398 prevented the decrease in systemic (Fig. 7A) and renal (Fig. 7B) levels of 20-HETE in LPS-treated rats (p < 0.05) [44]. In the present study, the LPS-induced decrease in 20-HETE levels in heart (Fig. 7C), thoracic aorta (Fig. 7D), and superior mesenteric artery (Fig. 7E) were also prevented by NS-398 (p < 0.05). The LPS-induced decrease in CYP4A1 protein expression was associated with decreased 20-HETE levels in serum (Fig. 7A), kidney (Fig. 7B), heart (Fig. 7C), thoracic aorta (Fig. 7D), and superior mesenteric artery (Fig. 7E) (p < 0.05). The decrease in CYP4A1 protein expression and 20-HETE levels was prevented in the tissues of rats treated with LPS and NS-398 (p < 0.05) (Figs. 6 and 7). NS-398 had no effect on the basal levels of CYP4A1 protein and 20-HETE levels in vehicle-treated rats (p > 0.05) (Figs. 6 and 7).
Fig. 6.

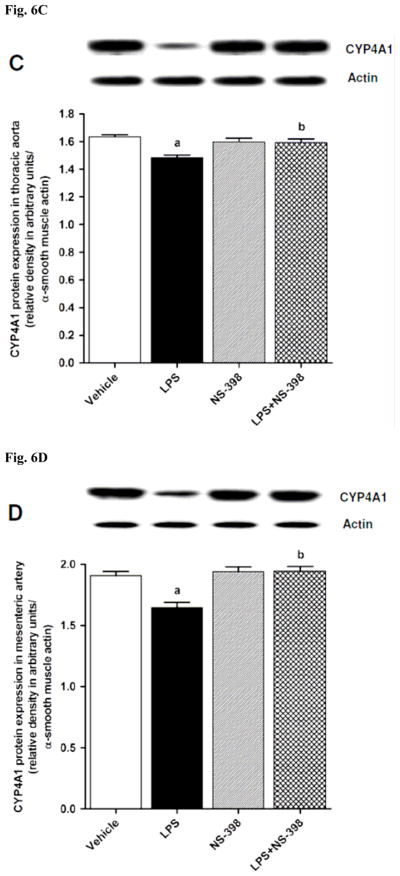
Effect of NS-398 on changes in CYP4A1 protein expression in (A) kidney, (B) heart, (C) thoracic aorta, and (D) superior mesenteric artery measured 4 h after saline (vehicle) or LPS injection. Data are expressed as means±S.E.M of 4 animals. aSignificant difference from the corresponding value seen in rats treated with saline (p < 0.05). bSignificant difference from the corresponding value seen in the rats treated with vehicle and LPS (p < 0.05).
Fig. 7.

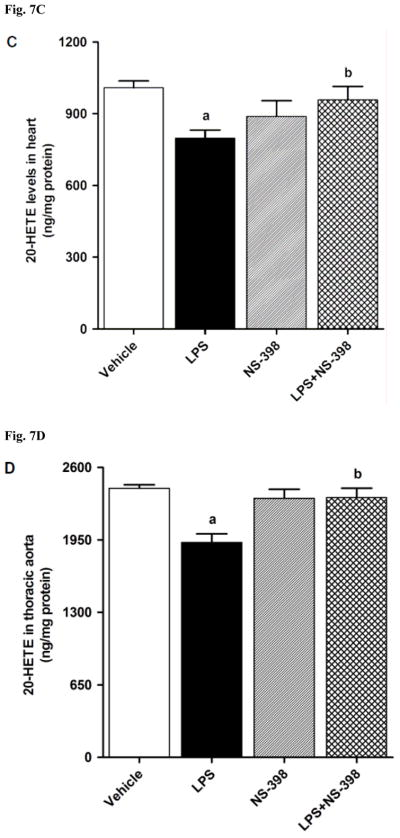
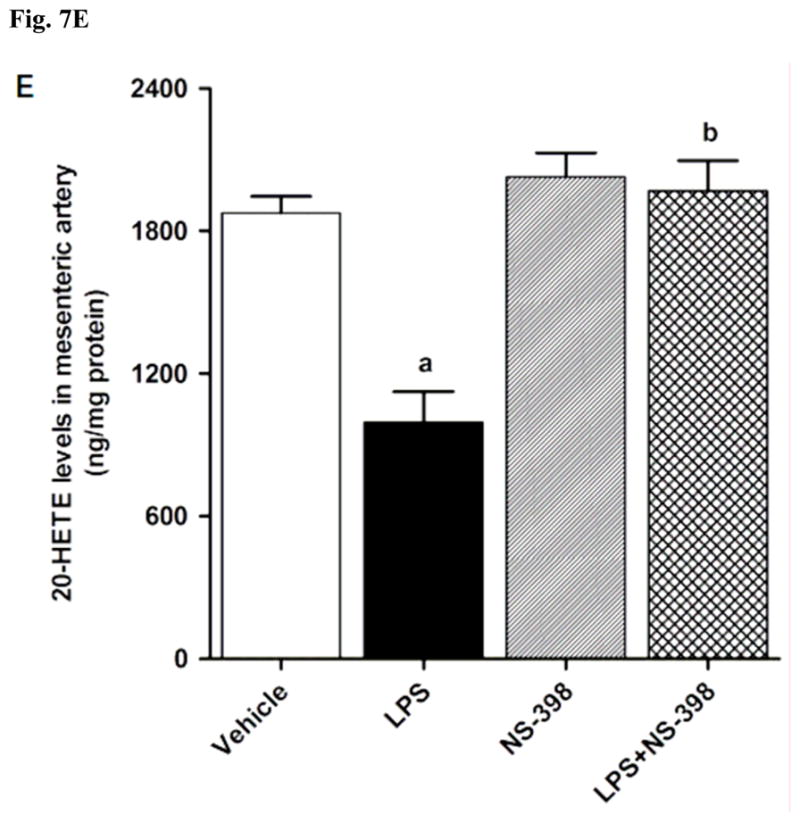
Effect of NS-398 on changes in 20-HETE levels in (A) serum, (B) kidney, (C) heart, (D) thoracic aorta, and (E) superior mesenteric artery measured 4 h after saline (vehicle) or LPS injection. Data are expressed as means±S.E.M of 5 animals. aSignificant difference from the corresponding value seen in rats treated with saline (p < 0.05). bSignificant difference from the corresponding value seen in the rats treated with vehicle and LPS (p < 0.05).
3.4. Effect of NS-398 on the LPS-induced increase in iNOS protein expression and activity
LPS increased iNOS protein expression in kidney (Fig. 8A), heart (Fig. 8B), thoracic aorta (Fig. 8C), and superior mesenteric artery (Fig. 8D) of rats (p < 0.05). The LPS-induced increase in iNOS protein expression was associated with an increase in NOS activity in serum (Fig. 9A), kidney (Fig. 9B), heart (Fig. 9C), thoracic aorta (Fig. 9D), and superior mesenteric artery (Fig. 9E) of rats (p < 0.05). The increase in iNOS protein expression and NOS activity was prevented in the serum and tissues of rats treated with LPS and NS-398 (p < 0.05) (Figs. 8 and 9). NS-398 had no effect on the basal levels of iNOS protein and NOS activity in vehicle-treated rats (p > 0.05) (Figs. 8 and 9).
Fig. 8.

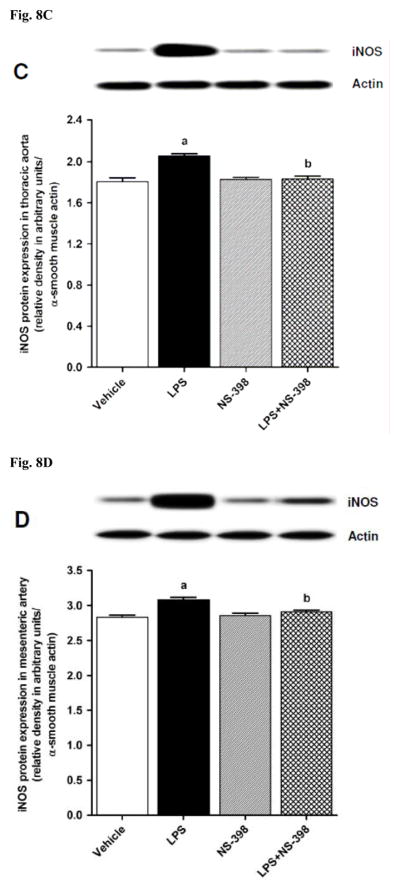
Effect of NS-398 on changes in iNOS protein expression in (A) kidney, (B) heart, (C) thoracic aorta, and (D) superior mesenteric artery measured 4 h after saline (vehicle) or LPS injection. Data are expressed as means±S.E.M of 4 animals. aSignificant difference from the corresponding value seen in rats treated with saline (p < 0.05). bSignificant difference from the corresponding value seen in the rats treated with vehicle and LPS (p < 0.05).
Fig. 9.

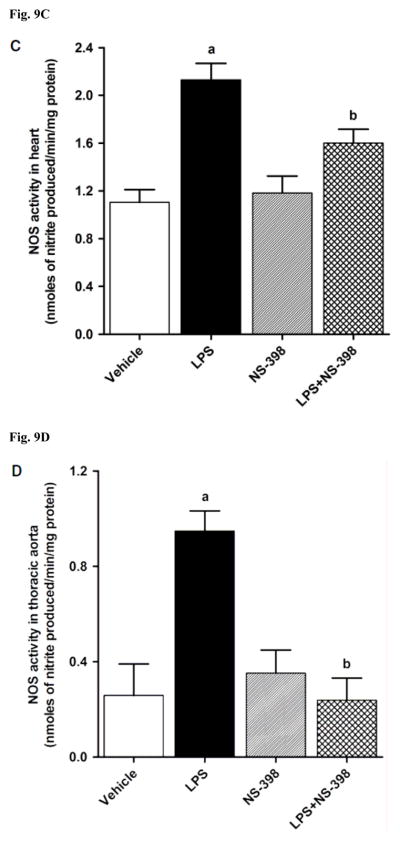
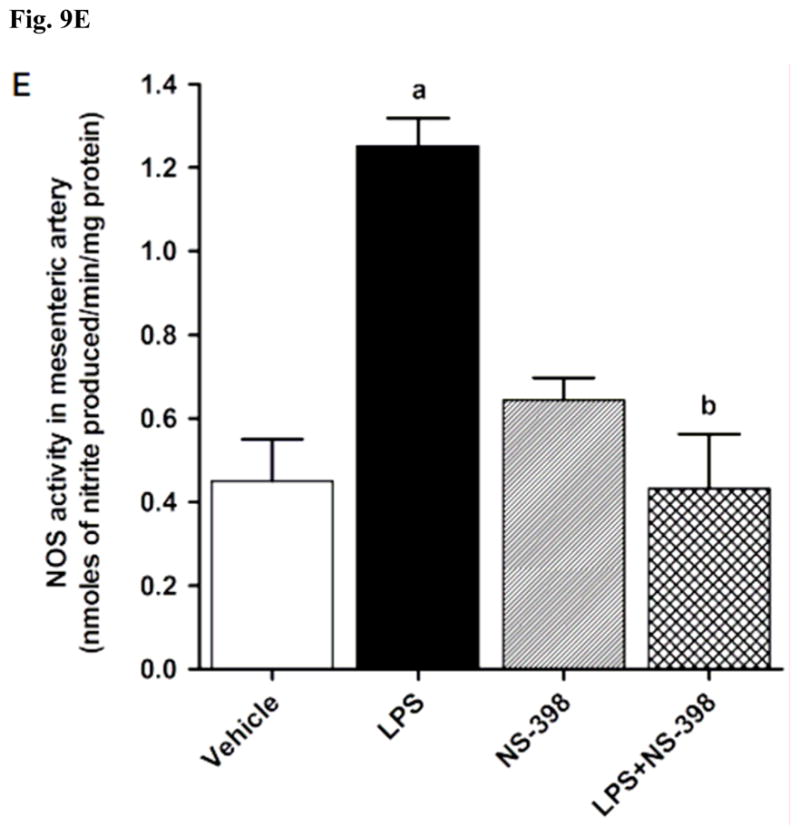
Effect of NS-398 on changes in NOS activity in (A) serum, (B) kidney, (C) heart, (D) thoracic aorta, and (E) superior mesenteric artery measured 4 h after saline (vehicle) or LPS injection. NOS activity was measured by the rate of L-arginine to nitrite/nitrate conversion using the Ultrasensitive Colorimetric Assay for Nitric Oxide Synthase following the manufacturer’s instructions. Data are expressed as means±S.E.M of 5 animals. aSignificant difference from the corresponding value seen in rats treated with saline (p < 0.05). bSignificant difference from the corresponding value seen in the rats treated with vehicle and LPS (p < 0.05).
3.5. Effect of NS-398 on the LPS-induced increase in nitrotyrosine levels
In order to investigate effects of COX-2 inhibition on the LPS-induced changes in peroxynitrite formation, nitrotyrosine (a stable end-product of peroxynitrite) levels were measured in the sera and tissue samples. Nitrotyrosine protein levels was increased in the kidney (Fig. 10A), heart (Fig. 10B), thoracic aorta (Fig. 10C), and superior mesenteric artey (Fig. 10D) of endotoxemic rats (p < 0.05). The LPS-induced increase in nitrotyrosine protein levels was associated with increased nitrotyrosine levels in serum (Fig. 12A), kidney (Fig. 12B), heart (Fig. 12C), thoracic aorta (Fig. 12D), and superior mesenteric artery (Fig. 12E) of rats (p < 0.05). The increase in nitrotyrosine protein levels and levels was prevented in the tissues of rats treated with LPS and NS-398 (p < 0.05) (Figs. 10 and 11). NS-398 had no effect on the basal levels of nitrotyrosine in vehicle-treated rats (p > 0.05) (Figs. 10 and 11).
Fig. 10.


Effect of NS-398 on changes in nitrotyrosine protein levels in (A) kidney, (B) heart, (C) thoracic aorta, and (D) superior mesenteric artery measured 4 h after saline (vehicle) or LPS injection. Data are expressed as means±S.E.M of 4 animals. aSignificant difference from the corresponding value seen in rats treated with saline (p < 0.05). bSignificant difference from the corresponding value seen in the rats treated with vehicle and LPS (p < 0.05).
Fig. 12.

Schematic diagram showing the involvement of iNOS, COX-2, CYP4A1, and NADPH oxidase in endotoxin-induced hypotension based on the current study. Endotoxin, the lipid A part of LPS which is the most potent microbial mediator of the pathogenesis of sepsis and septic shock, induces iNOS and COX-2 protein expression and activity associated with increased production of peroxynitrite. Conversely, endotoxin causes a decrease in CYP4A1 protein expression and 20-HETE production. The endotoxin-induced changes in protein expression and/or activity of these enzymes lead to a decrease in blood pressure. NS-398 prevents the effects of endotoxin on the increase in iNOS and COX-2 protein expression and activity, and nitrotyrosine levels as well as the decrease in CYP4A1 protein expression and 20-HETE levels, and thus, restores blood pressure during endotoxemia. It can be concluded that decreased expression and/or activity of COX-2 and iNOS and peroxynitrite formation associated with increased CYP4A1 expression and 20-HETE synthesis participate in the effect of NS-398 to prevent hypotension caused by endotoxemia in the rat. (
 ) stimulation; (
) stimulation; (
 ) inhibition.
) inhibition.
Fig. 11.
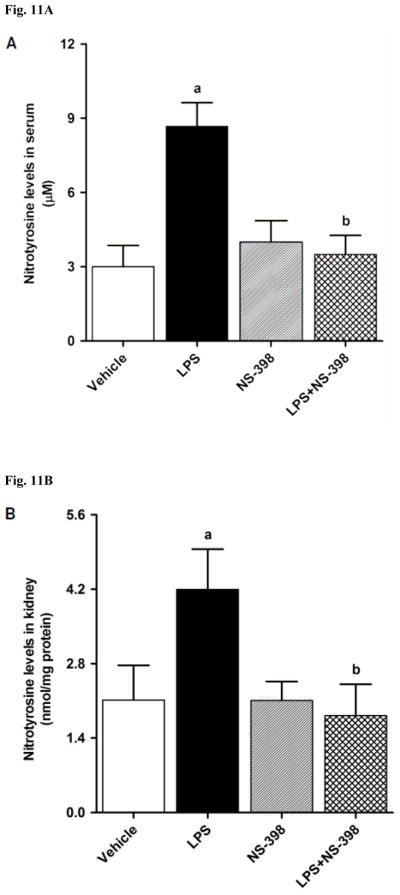


Effect of NS-398 on changes in nitrotyrosine levels in (A) serum, (B) kidney, (C) heart, (D) thoracic aorta, and (E) superior mesenteric artery measured 4 h after saline (vehicle) or LPS injection. Data are expressed as means±S.E.M of 6 animals. aSignificant difference from the corresponding value seen in rats treated with saline (p < 0.05). bSignificant difference from the corresponding value seen in the rats treated with vehicle and LPS (p < 0.05).
4. Discussion
The results of the present study indicate that an increase in the expression and activity of COX-2 and iNOS as well as peroxynitrite formation associated with decreased CYP4A1 expression and 20-HETE synthesis participates in the fall in blood pressure in rats treated with LPS. These data also demonstrate that decreased production of vasodilator prostanoids, NO, and peroxynitrite by NS-398, a selective COX-2 inhibitor, restores MAP and HR, which may be due to increased 20-HETE levels in systemic circulation as well as renal and cardiovascular tissues of endotoxemic rats.
There are several reports suggesting a direct link between arachidonic acid metabolites and NO under physiological and pathophysiological conditions [6–8,54,55]. The constitutive isoforms of COX and NOS enzymes play an important role in the regulation of several physiological states. On the other hand, under inflammatory conditions such as endotoxic shock, the inducible isoforms of these enzymes are expressed in a variety of cells and tissues resulting in the production of large amounts of prostanoids and NO. Our recent studies with a synthetic analog of 20-HETE, 5,14-HEDGE, suggest that an increase in systemic and renal 20-HETE levels associated with a decrease in iNOS and COX-2 protein expression and activity contributes to the effect of 5,14-HEDGE to prevent the vascular hyporeactivity, hypotension, and mortality during rat endotoxemia [41,42,46]. In contrast to the previous findings with 20-HETE [26–28], our results also suggest that 5,14-HEDGE is not to be converted to vasodilator prostanoids in the rat model of endotoxemia because the glycine substitution renders 5,14-HEDGE less susceptible to β-oxidation and the absence of the double bonds across the 8,9- and 11,12-carbons prevent its degradation by COX to arachidonic acid products [56]. More recently, we have demonstrated that inhibition of PGI2 and PGE2 synthesis, and NO production by a selective COX-2 inhibitor restores blood pressure due to increased systemic and renal levels of 20-HETE [44]. In the present study, the LPS-induced fall in MAP and rise in HR were associated with increased expression of COX-2 and iNOS in kidney, heart, thoracic aorta, and superior mesenteric artery. The activities of COX-2 and iNOS as well as PGI2 and PGE2 levels were also increased in the systemic circulation and renal, cardiac, and vascular tissues of LPS-treated rats. On the contrary, renal, cardiac, and vascular CYP4A1 protein expression as well as systemic and tissue levels of 20-HETE were decreased in endotoxemic rats. These effects of LPS, except COX-2 protein expression, were prevented by a selective COX-2 inhibitor, NS-398, given 1 h after injection of LPS. Although we did not investigate direct effect of NS-398 on the LPS-induced increase in iNOS expression and activity, it has been demonstrated that NS-398 inhibits LPS-induced increase in PGE2 formation associated with iNOS protein expression and nitrite production in a murine macrophage cell lines (RAW 264.7 and J774 cells) [57–59]. Based on the results from previous studies [36–46] and our present findings, it appears that decreased production of vasodilator prostanoids, PGI2 and PGE2, and NO as well as a vasoconstrictor eicosanoid, 20-HETE, participates in the effect of NS-398 to prevent the LPS-induced decrease in MAP and increase in HR in endotoxemic rats. It is also possible that inhibition of endogenously produced 20-HETE metabolism to vasodilator metabolites, 20-OH-PGE2 and 20-OH-PGF2α, by COX-2 might contribute to the effect of NS-398 in preventing the decrease in blood pressure in LPS-treated rats. However, additional experiments need to be conducted to demonstrate of the validity of the proposed hypothesis. Further characterization of the molecular mechanisms of interactions between COX, NOS, and CYP4A enzymes will provide the framework for extension of this work into understanding the role of arachidonic acid products and NO in the decrease in blood pressure during endotoxemia.
NO reacts with superoxide to form peroxynitrite, a powerful oxidant and nitrating molecule generated by several enzymatic reactions, including a variety of NADPH oxidases and uncoupled eNOS, and subsequent reaction of peroxynitrite with proteins results in nitrotyrosine formation. In vivo, peroxynitrite generation represents a NO-dependent pathogenic mechanism in conditions such as circulatory shock, stroke, myocardial infarction, chronic heart failure, and chronic inflammatory diseases [60–63]. It has been reported that NO-derived peroxynitrite formation plays an important role in vascular hyporeactivity to vasopressor agents in endotoxemic rats [64–66]. We have recently reported that the LPS-induced fall in blood pressure was associated with an increase in the protein levels of NADPH subunits, gp91phox and p47phox, and nitrotyrosine in the heart, thoracic aorta, kidney, and superior mesenteric artery, and elevated serum nitrotyrosine levels [46]. All of the LPS-induced changes were prevented by 20-HETE analog, 5,14-HEDGE [46]. In the present study, LPS caused an increase in the protein levels of nitrotyrosine in the renal and cardiovascular tissues of rats. Systemic and tissue levels of nitrotyrosine were also increased in endotoxemic rats. NS-398 also prevented the effects of LPS on the increase in nitrotyrosine levels. These results suggest that increased production of peroxynitrite also contributes to the LPS-induced hypotension during endotoxemia in rats.
In the present study we measured changes in the activity and expression of COX-2, iNOS, and CYP4A1 at 4 h after the administration of endotoxin. However, LPS produced a fall in MAP within 1 h and was sustained for 4 h. Administration of NS398 reversed the LPS-induced fall in blood pressure within 1 h. Therefore, one may argue that vasodilator prostanoids and NO and decreased levels of 20-HETE generated from an increased expression of COX-2 and iNOS, and decreased expression of CYP4A1, respectively, contribute to the sustained decrease in blood pressure. On the other hand, it is well established that iNOS and COX-2 expression are induced 1–2 h after LPS administration, and eNOS and COX-1 expression are downregulated [67–69]. It is also well known that increased NO production by eNOS is responsible for the fall in blood pressure within 1 h after LPS administration while iNOS-derived NO is responsible for the delayed hypotension and vascular hyporeactivity [69,70]. In addition, it has been shown that the fall in blood pressure associated with an increase in iNOS and eNOS expression, which is observed 2–3 h after endotoxin injection to rats, returns to baseline values after 12–24 h [71–74]. Gupta and Sharma [71] have also demonstrated that the fall in blood pressure, which occurs 2 h after LPS injection, is associated with an increase in aortic iNOS and eNOS protein expression, and systemic and tissue nitrate+nitrite (NOX) levels. Compensatory mechanisms, including activation of renin-angiotensin-aldosteron system, increased sensitivity of baroreceptor reflex mechanisms, and increased production of endothelin-1 and cathecolamines, which are known to activate phospholipase A2 and release arachidonic acid from tissue lipids, and results in prostaglandin synthesis, and also stimulate production of reactive nitrogen and oxygen species, have also been reported to be responsible for the changes in the formation of vasoregulatory molecules that could contribute to the fall in blood pressure during the early phase of LPS-induced endotoxemia [67–76]. Therefore, the vasodilator prostaglandins and NO could cause a decrease in blood pressure which is followed by increased expression of COX-2 and iNOS and decreased expression of CYP4A, resulting in sustained decrease in blood pressure. However, further studies on the measurement of expression of these enzyme systems at 1 h after LPS administration and the effect of NS398 are required to address this issue. In addition, we have previously demonstrated that the effects of a nonselective COX inhibitor, indomethacin, on the LPS-induced decrease in MAP, CYP4A1 protein expression, and CYP4A activity were minimized by the CYP4A inhibitor, aminobenzotriazole [40]. However, additional experiments need to be conducted using a selective inhibitor of 20-HETE synthesis, such as N-hydroxy-N′-(−4-butyl-2-methylphenyl)formamidine (HET0016), in order to demonstrate the effect of COX-2 inhibition on CYP4A- and CYP4F-derived 20-HETE production.
In conclusion, the present study provides an evidence that COX-2-derived vasodilator prostanoids, PGI2 and PGE2, produced during endotoxemia increase iNOS protein expression and activity as well as peroxynitrite formation resulting in decreased CYP4A1 protein expression and 20-HETE synthesis (Fig. 12). Our findings also suggest that increased 20-HETE levels associated with a decrease in the production of vasodilator prostanoids and NO participates in the effect of NS-398 to prevent hypotension in the rat model of septic shock. Impairment of cardiovascular and renal function is critically involved in the pathophysiological sequelae in septic shock finally resulting in multiorgan failure and death; restoration of these impaired functions should improve therapeutic benefit. In the light of the important role of prostanoids, NO, peroxynitrite, and 20-HETE in LPS-induced hypotension, multiple organ failure, and mortality, the interaction of COX, NOS, CYP4A, and NADPH pathways should be considered when developing new strategies for drug development in the treatment of endotoxic shock. More importantly, further studies with selective COX-2 inhibitors as well as arachidonic acid metabolites generated via CPY4A including stable analogs of 20-HETE in endotoxemia models could provide a novel approach to treat hypotension in septic shock.
Acknowledgments
Financial support was provided by grants from Novartis Turkey, TUBITAK (Project Code: SBAG-109S121), NIH 19134–37, NIH GM 31278, and the Robert A. Welch Foundation (GL625910).
Abbreviations
- 20-HETE
20-hydroxyeicosatetraenoic acid
- 5, 14-HEDGE
N-[20-hydroxyeicosa-5(Z),14(Z)-dienoyl]glycine
- BSA
bovine serum albumin
- COX
cyclooxygenase
- cyclic GMP
cyclic guanosine monophosphate
- CYP
cyctochrome P450
- eNOS
endothelial nitric oxide synthase
- HR
heart rate
- hsp90
heat shock protein 90
- iNOS
inducible nitric oxide synthase
- MAP
mean arterial pressure
- NADPH
nicotinamide adenine dinucleotide phosphate
- NO
nitric oxide
- NS-398
N-(2-cyclohexyloxy-4-nitrophenyl)-methansulphonamide
- PG
prostaglandin
- TxA2
thromboxane A2
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kleinert H, Schwarz PM, Förstermann U. Regulation of the expression of inducible nitric oxide synthase. Biol Chem. 2003;384:1343–64. doi: 10.1515/BC.2003.152. [DOI] [PubMed] [Google Scholar]
- 2.Tunctan B, Altug S. The use of nitric oxide synthase inhibitors in inflammatory diseases: a novel class of anti-inflammatory agents. Cur Med Chem Anti-Inflammatory & Anti-Allergy Agents. 2004;3:271–301. [Google Scholar]
- 3.Cauwels A. Nitric oxide in shock. Kidney Int. 2007;72:557–65. doi: 10.1038/sj.ki.5002340. [DOI] [PubMed] [Google Scholar]
- 4.Fernandes D, Assreuy J. Nitric oxide and vascular reactivity in sepsis. Shock. 2008;30:10–13. doi: 10.1097/SHK.0b013e3181818518. [DOI] [PubMed] [Google Scholar]
- 5.Hauser B, Bracht H, Matejovic M, Radermacher P, Venkatesh B. Nitric oxide synthase inhibition in sepsis? Lessons learned from large-animal studies. Anesth Analg. 2005;101:488–98. doi: 10.1213/01.ANE.0000177117.80058.4D. [DOI] [PubMed] [Google Scholar]
- 6.Miyata N, Roman RJ. Role of 20-hydroxyeicosatetraenoic acid (20-HETE) in vascular system. J Smooth Muscle Res. 2005;41:175–93. doi: 10.1540/jsmr.41.175. [DOI] [PubMed] [Google Scholar]
- 7.Roman RJ. P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol Rev. 2002;82:131–85. doi: 10.1152/physrev.00021.2001. [DOI] [PubMed] [Google Scholar]
- 8.Kroetz DL, Xu F. Regulation and inhibition of arachidonic acid omega-hydroxylases and 20-HETE formation. Annu Rev Pharmacol Toxicol. 2005;45:413–38. doi: 10.1146/annurev.pharmtox.45.120403.100045. [DOI] [PubMed] [Google Scholar]
- 9.Escalante B, Omata K, Sessa W, Lee SG, Falck JR, Schwartzman ML. 20-hydroxyeicosatetraenoic acid is an endothelium-dependent vasoconstrictor in rabbit arteries. Eur J Pharmacol. 1993;235:1–7. doi: 10.1016/0014-2999(93)90812-v. [DOI] [PubMed] [Google Scholar]
- 10.Randriamboavonjy V, Busse R, Fleming I. 20-HETE-induced contraction of small coronary arteries depends on the activation of Rho-kinase. Hypertension. 2003;41:801–6. doi: 10.1161/01.HYP.0000047240.33861.6B. [DOI] [PubMed] [Google Scholar]
- 11.Schwartzman ML, Falck JR, Yadagiri P, Escalante B. Metabolism of 20-hydroxyeicosatetraenoic acid by cyclooxygeanse: formation and identification of novel endothelium-dependent vasoconstrictor metabolites. J Biol Chem. 1989;264:11658–62. [PubMed] [Google Scholar]
- 12.Escalante B, Sessa WC, Falck JR, Yadagiri P, Schwartzman ML. Vasoactivity of 20-hydroxyeicosatetraenoic acid is dependent on metabolism by cyclooxygenase. J Pharmacol Exp Ther. 1989;248:229–32. [PubMed] [Google Scholar]
- 13.Zou AP, Fleming JT, Falck JR, Jacobs ER, Gebremedhin D, Harder DR, et al. 20-HETE is an endogenous inhibitor of the large-conductance Ca(2+)-activated K+ channel in renal arterioles. Am J Physiol. 1996;270:R228–37. doi: 10.1152/ajpregu.1996.270.1.R228. [DOI] [PubMed] [Google Scholar]
- 14.Lange A, Gebremedhin D, Narayanan J, Harder D. 20-Hydroxyeicosatetraenoic acid-induced vasoconstriction and inhibition of potassium current in cerebral vascular smooth muscle is dependent on activation of protein kinase C. J Biol Chem. 1997;272:27345–52. doi: 10.1074/jbc.272.43.27345. [DOI] [PubMed] [Google Scholar]
- 15.Nowicki S, Chen SL, Aizman O, Cheng XJ, Li D, Nowicki C, et al. 20-Hydroxyeicosa-tetraenoic acid (20 HETE) activates protein kinase C. Role in regulation of rat renal Na+,K+-ATPase. J Clin Invest. 1997;99(6):1224–30. doi: 10.1172/JCI119279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Obara K, Koide M, Nakayama K. 20-Hydroxyeicosatetraenoic acid potentiates stretch-induced contraction of canine basilar artery via PKC alpha-mediated inhibition of KCa channel. Br J Pharmacol. 2002;137:1362–70. doi: 10.1038/sj.bjp.0704960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun CW, Falck JR, Harder DR, Roman RJ. Role of tyrosine kinase and PKC in the vasoconstrictor response to 20-HETE in renal arterioles. Hypertension. 1999;33:414–8. doi: 10.1161/01.hyp.33.1.414. [DOI] [PubMed] [Google Scholar]
- 18.Muthalif MM, Benter IF, Karzoun N, Fatima S, Harper J, Uddin MR, et al. 20-Hydroxyeicosatetraenoic acid mediates calcium/calmodulin-dependent protein kinase II-induced mitogen-activated protein kinase activation in vascular smooth muscle cells. Proc Natl Acad Sci USA. 1998;95:12701–6. doi: 10.1073/pnas.95.21.12701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muthalif MM, Benter IF, Khandekar Z, Gaber L, Estes A, Malik S, et al. Contribution of Ras GTPase/MAP kinase and cytochrome P450 metabolites to deoxycorticosterone-salt-induced hypertension. Hypertension. 2000;35:457–63. doi: 10.1161/01.hyp.35.1.457. [DOI] [PubMed] [Google Scholar]
- 20.Muthalif MM, Uddin MR, Fatima S, Parmentier JH, Khandekar Z, Malik KU. Small GTP binding protein Ras contributes to norepinephrine-induced mitogenesis of vascular smooth muscle cells. Prostaglandins Other Lipid Mediat. 2001;65:33–43. doi: 10.1016/s0090-6980(01)00112-5. [DOI] [PubMed] [Google Scholar]
- 21.Akbulut T, Regner KR, Roman RJ, Avner ED, Falck JR, Park F. 20-HETE activates the Raf/MEK/ERK pathway in renal epithelial cells through an EGFR- and c-Src-dependent mechanism. Am J Physiol Renal Physiol. 2009;297:F662–70. doi: 10.1152/ajprenal.00146.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun CW, Alonso-Galicia M, Taheri MR, Falck JR, Harder DR, Roman RJ. Nitric oxide-20-hydroxyeicosatetraenoic acid interaction in the regulation of K+ channel activity and vascular tone in renal arterioles. Circ Res. 1998;83:1069–79. doi: 10.1161/01.res.83.11.1069. [DOI] [PubMed] [Google Scholar]
- 23.Gebremedhin D, Lange AR, Narayanan J, Aebly MR, Jacobs ER, Harder DR. Cat cerebral arterial smooth muscle cells express cytochrome P450 4A2 enzyme and produce the vasoconstrictor 20-HETE which enhances L-type Ca2+ current. J Physiol. 1998;507:771–81. doi: 10.1111/j.1469-7793.1998.771bs.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Basora N, Boulay G, Bilodeau L, Rousseau E, Payet MD. 20-hydroxyeicosatetraenoic acid (20-HETE) activates mouse TRPC6 channels expressed in HEK293 cells. J Biol Chem. 2003;278:31709–16. doi: 10.1074/jbc.M304437200. [DOI] [PubMed] [Google Scholar]
- 25.Brayden JE, Earley S, Nelson MT, Reading S. Transient receptor potential (TRP) channels, vascular tone and autoregulation of cerebral blood flow. Clin Exp Pharmacol Physiol. 2008;35:1116–20. doi: 10.1111/j.1440-1681.2007.04855.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carroll MA, Garcia MP, Falck JR, McGiff JC. Cyclooxygenase dependency of the renovascular actions of cytochrome P450-derived arachidonate metabolites. J Pharmacol Exp Ther. 1992;260:104–9. [PubMed] [Google Scholar]
- 27.Fang X, Faraci FM, Kaduce TL, Harmon S, Modrick ML, Hu S, et al. 20-Hydroxyeicosatetraenoic acid is a potent dilator of mouse basilar artery: role of cyclooxygenase. Am J Physiol Heart Circ Physiol. 2006;291:H2301–7. doi: 10.1152/ajpheart.00349.2006. [DOI] [PubMed] [Google Scholar]
- 28.Pratt PF, Falck JR, Reddy KM, Kurian JB, Campbell WB. 20-HETE relaxes bovine coronary arteries through the release of prostacyclin. Hypertension. 1998;31:237–41. doi: 10.1161/01.hyp.31.1.237. [DOI] [PubMed] [Google Scholar]
- 29.Yu M, McAndrew RP, Al-Saghir R, Maier KG, Medhora M, Roman RJ, et al. Nitric oxide contributes to 20-HETE-induced relaxation of pulmonary arteries. J Appl Physiol. 2002;93:1391–9. doi: 10.1152/japplphysiol.00247.2002. [DOI] [PubMed] [Google Scholar]
- 30.Carroll MA, Capparelli MF, Doumand AB, Cheng MK, Jiang H, McGiff JC. Renal vasoactive eicosanoids: interactions between cytochrome P450 and cyclooxygenase metabolites during salt depletion. Am J Hypertens. 2001;14:159A. [Google Scholar]
- 31.Theken KN, Deng Y, Kannon MA, Miller TM, Poloyac SM, Lee CR. Activation of the acute inflammatory response alters cytochrome P450 expression and eicosanoid metabolism. Drug Metab Dispos. 2011;39:22–9. doi: 10.1124/dmd.110.035287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anwar-mohamed A, Zordoky BN, Aboutabl ME, El-Kadi AO. Alteration of cardiac cytochrome P450-mediated arachidonic acid metabolism in response to lipopolysaccharide-induced acute systemic inflammation. Pharmacol Res. 2010;61:410–8. doi: 10.1016/j.phrs.2009.12.015. [DOI] [PubMed] [Google Scholar]
- 33.Alonso-Galicia M, Sun CW, Falck JR, Harder DR, Roman RJ. Contribution of 20-HETE to the vasodilator actions of nitric oxide in renal arteries. Am J Physiol. 1998;275:F370–8. doi: 10.1152/ajprenal.1998.275.3.F370. [DOI] [PubMed] [Google Scholar]
- 34.Wang MH, Wang J, Chang HH, Zand BA, Jiang M, Nasjletti A, et al. Regulation of renal CYP4A expression and 20-HETE synthesis by nitric oxide in pregnant rats. Am J Physiol. 2003;285:F295–302. doi: 10.1152/ajprenal.00065.2003. [DOI] [PubMed] [Google Scholar]
- 35.Alonso-Galicia M, Drummond HA, Reddy KK, Falck JR, Roman RJ. Inhibition of 20-HETE production contributes to the vascular responses to nitric oxide. Hypertension. 1997;29:320–5. doi: 10.1161/01.hyp.29.1.320. [DOI] [PubMed] [Google Scholar]
- 36.Tunctan B, Korkmaz B, Yildirim H, Tamer L, Atik U, Buharalioglu CK. Increased production of nitric oxide contributes to renal oxidative stress in endotoxemic rat. Am J Infect Dis. 2005;1:111–5. [Google Scholar]
- 37.Tunctan B, Korkmaz B, Yildirim H, Tamer L, Atik U, Buharalioglu CK. Reversal of endotoxin-induced hypotension by inhibition of inducible nitric oxide synthase activity is associated with improved oxidative status in rat heart, aorta and mesenteric artery. Turkish J Med Sci. 2006;36:71–80. [Google Scholar]
- 38.Tunctan B, Yaghini FA, Estes A, Malik KU. Inhibition by nitric oxide and cyclooxygenase of cytochrome P450 4A expression and activity contributes to endotoxin-induced hypotension in rats. Nitric Oxide: Biol Chem. 2006;14:51–7. doi: 10.1016/j.niox.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 39.Tunctan B, Korkmaz B, Dogruer ZN, Tamer L, Atik U, Buharalioglu CK. Inhibition of extracellular signal-regulated kinase (ERK1/2) activity reverses endotoxin-induced hypotension via decreased nitric oxide production in rats. Pharmacol Res. 2007;56:56–64. doi: 10.1016/j.phrs.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 40.Tunctan B, Yaghini FA, Estes A, Malik KU. Prostaglandins inhibit cytochrome P450 4A activity and contribute to endotoxin-induced hypotension in rats via nitric oxide production. Arch Pharm Res. 2008;31:856–65. doi: 10.1007/s12272-001-1238-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tunctan B, Korkmaz B, Buharalioglu CK, Sahan Firat S, Anjaiah S, Falck J, et al. A 20-HETE agonist, N-[20-hydroxyeicosa-5(Z),14(Z)-dienoyl]glycine, opposes the fall in blood pressure and vascular reactivity in endotoxin-treated rats. Shock. 2008;30:329–35. doi: 10.1097/SHK.0b013e31816471c6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cuez T, Korkmaz B, Buharalioglu CK, Sahan-Firat S, Falck J, Tunctan B, et al. A synthetic analogue of 20-HETE, 5,14-HEDGE, reverses endotoxin-induced hypotension via increased 20-HETE levels associated with decreased iNOS protein expression and vasodilator prostanoid production in rats. Basic Clin Pharmacol. 2010;106:378–88. doi: 10.1111/j.1742-7843.2009.00501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Korkmaz B, Buharalioglu K, Sahan-Firat S, Cuez T, Demiryurek TA, Tunctan B. Activation of MEK1/ERK1/2/iNOS/sGC/PKG pathway associated with peroxinitrite formation contributes to hypotension and vascular hyporeactivity in endotoxemic rats. Nitric Oxide: Biol Chem. 2011;24:160–72. doi: 10.1016/j.niox.2011.02.004. [DOI] [PubMed] [Google Scholar]
- 44.Tunctan B, Korkmaz B, Cuez T, Buharalioglu CK, Sahan-Firat S, Falck J, et al. Contribution of vasoactive eicosanoids and nitric oxide production to the effect of selective cyclooxygenase-2 inhibitor, NS-398, on endotoxin-induced hypotension in rats. Basic Clin Pharmacol Toxicol. 2010;107:877–82. doi: 10.1111/j.1742-7843.2010.00589.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Buharalioglu CK, Korkmaz B, Cuez T, Sahan-Firat S, Sari AN, Malik KU, et al. Piroxicam reverses endotoxin-induced hypotension in rats: contribution of vasoactive eicosanoids and nitric oxide. Basic Clin Pharmacol Toxicol. 2011;109:186–94. doi: 10.1111/j.1742-7843.2011.00708.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tunctan B, Korkmaz B, Cuez T, Sari AN, Kacan M, Gilik D, et al. Contribution of iNOS, COX-2 and CYP4A1 to the protective effect of a synthetic analog of 20-HETE, 5,14-HEDGE, against endotoxin-induced hypotension and mortality in experimental model of septic shock in rats and mice. Inflam Res. 2011;60:S55. [Google Scholar]
- 47.Tsiotou AG, Sakorafas GH, Anagnostopoulos G, Bramis J. Septic shock; current pathogenetic concepts from a clinical perspective. Med Sci Monit. 2005;11:RA76–85. [PubMed] [Google Scholar]
- 48.Ashorobi RB, Williams PA. Indomethacin and alpha-tocopherol enhanced survival in endotoxic rats. Cent Afr J Med. 1995;41:216–19. [PubMed] [Google Scholar]
- 49.Tunctan B, Altug S, Uludag O, Abacioglu N. Time-dependent variations in serum nitrite, 6-keto-prostaglandin F1α and thromboxane B2 levels induced by lipopolysaccharide in mice. Biol Rhythm Res. 2000;1:499–514. [Google Scholar]
- 50.Futaki N, Takahashi S, Kitagawa T, Yamakawa Y, Tanaka M, Higuchi S. Selective inhibition of cyclooxygenase-2 by NS-398 in endotoxin shock rats in vivo. Inflam Res. 1997;46:496–502. doi: 10.1007/s000110050232. [DOI] [PubMed] [Google Scholar]
- 51.Fatehi-Hassanabad Z, Muller M, Andriantsitohaina R, Furman BL, Parratt JR, Stoclet JC. Influence of indomethacin on the haemodynamic effects of lipopolysaccharide in rats. Fundam Clin Pharmacol. 1996;10:258–63. doi: 10.1111/j.1472-8206.1996.tb00304.x. [DOI] [PubMed] [Google Scholar]
- 52.Vayssettes-Courchay C, Bouysset F, Verbeuren TJ. Involvement of COX and NOS induction in the sympatho-activation during sepsis. Auton Neurosci. 2002;98:33–6. doi: 10.1016/s1566-0702(02)00027-9. [DOI] [PubMed] [Google Scholar]
- 53.Tunctan B, Sari AN, Kacan M, Gilik D, Buharalioglu CK, Sahan-Firat S, et al. NS-398, a selective COX-2 inhibitor, reverses endotoxin-induced hypotension via increased CYP4A1 expression and 20-HETE levels associated with decreased COX-2 and iNOS expression and/or activity in a rat model of septic shock. The 14th International Winter Eicosanoid Conference; Baltimore, Maryland, US. March 11–14; 2012. p. 29.p. S19-CVR. Abstract Book. [Google Scholar]
- 54.Mollace V, Muscoli C, Masini E, Cuzzocrea S, Salvemini D. Modulation of prostaglandin biosynthesis by nitric oxide and nitric oxide donors. Pharmacol Rev. 2005;57:217–52. doi: 10.1124/pr.57.2.1. [DOI] [PubMed] [Google Scholar]
- 55.Cuzzocrea S, Salvemini D. Molecular mechanisms involved in the reciprocal regulation of cyclooxygenase and nitric oxide synthase enzymes. Kidney Int. 2007;71:290–7. doi: 10.1038/sj.ki.5002058. [DOI] [PubMed] [Google Scholar]
- 56.Alonso-Galicia M, Falck JR, Reddy KM, Roman RJ. 20-HETE agonists and antagonists in the renal circulation. Am J Physiol. 1999;277:F790–6. doi: 10.1152/ajprenal.1999.277.5.F790. [DOI] [PubMed] [Google Scholar]
- 57.Chen BC, Lin WW. Pyrimidinoceptor potentiation of macrophage PGE(2) release involved in the induction of nitric oxide synthase. Br J Pharmacol. 2000;130:777–86. doi: 10.1038/sj.bjp.0703375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen CC, Chiu KT, Sun YT, Chen WC. Role of the cyclic AMP-protein kinase A pathway in lipopolysaccharide-induced nitric oxide synthase expression in RAW 264.7 macrophages. Involvement of cyclooxygenase-2. J Biol Chem. 1999;274:31559–64. doi: 10.1074/jbc.274.44.31559. [DOI] [PubMed] [Google Scholar]
- 59.Lin WW, Chen BC, Hsu YW, Lee CM, Shyue SK. Modulation of inducible nitric oxide synthase induction by prostaglandin E2 in macrophages: distinct susceptibility in murine J774 and RAW 264. 7 macrophages. Prostaglandins Other Lipid Mediat. 1999;58:87–101. doi: 10.1016/s0090-6980(99)00023-4. [DOI] [PubMed] [Google Scholar]
- 60.Esposito E, Cuzzocrea S. Role of nitroso radicals as drug targets in circulatory shock. Br J Pharmacol. 2009;157:494–508. doi: 10.1111/j.1476-5381.2009.00255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Szabó C, Módis K. Pathophysiological roles of peroxynitrite in circulatory shock. Shock. 2010;34:4–14. doi: 10.1097/SHK.0b013e3181e7e9ba. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen AF, Chen DD, Daiber A, Faraci FM, Li H, Rembold CM, et al. Free radical biology of the cardiovascular system. Clin Sci. 2012;123:73–91. doi: 10.1042/CS20110562. [DOI] [PubMed] [Google Scholar]
- 64.Cuzzocrea S, Mazzon E, Di Paola R, Esposito E, Macarthur H, Matuschak GM, et al. A role for nitric oxide-mediated peroxynitrite formation in a model of endotoxin-induced shock. J Pharmacol Exp Ther. 2006;319:73–81. doi: 10.1124/jpet.106.108100. [DOI] [PubMed] [Google Scholar]
- 65.d’Emmanuele di Villa Bianca R, Marzocco S, Di Paola R, Autore G, Pinto A, Cuzzocrea S, et al. Melatonin prevents lipopolysaccharide-induced hyporeactivity in rat. J Pineal Res. 2004;36:146–54. doi: 10.1046/j.1600-079x.2003.00111.x. [DOI] [PubMed] [Google Scholar]
- 66.Takakura K, Xiaohong W, Takeuchi K, Yasuda Y, Fukuda S. Deactivation of norepinephrine by peroxynitrite as a new pathogenesis in the hypotension of septic shock. Anesthesiology. 2003;98:928–34. doi: 10.1097/00000542-200304000-00020. [DOI] [PubMed] [Google Scholar]
- 67.Liu SF, Adcock IM, Old RW, Barnes PJ, Evans TW. Differential regulation of the constitutive and inducible nitric oxide synthase mRNA by lipopolysaccharide treatment in vivo in the rat. Crit Care Med. 1996;24:1219–25. doi: 10.1097/00003246-199607000-00026. [DOI] [PubMed] [Google Scholar]
- 68.Liu SF, Newton R, Evans TW, Barnes PJ. Differential regulation of cyclo-oxygenase-1 and cyclo-oxygenase-2 gene expression by lipopolysaccharide treatment in vivo in the rat. Clin Sci. 1996;90:301–6. doi: 10.1042/cs0900301. [DOI] [PubMed] [Google Scholar]
- 69.Yamaguchi N, Jesmin S, Zaedi S, Shimojo N, Maeda S, Gando S, Koyama A, Miyauchi T. Time-dependent expression of renal vaso-regulatory molecules in LPS-induced endotoxemia in rat. Peptides. 2006;27:2258–70. doi: 10.1016/j.peptides.2006.03.025. [DOI] [PubMed] [Google Scholar]
- 70.Szabo C, Mitchell JA, Thiemermann C, Vane JR. Nitric oxide-mediated hyporeactivity to noradrenaline precedes the induction of nitric oxide synthase in endotoxin shock. Br J Pharmacol. 1993;108:786–92. doi: 10.1111/j.1476-5381.1993.tb12879.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gupta A, Sharma AC. Despite minimal hemodynamic alterations endotoxemia modulates NOS and p38-MAPK phosphorylation via metalloendopeptidases. Mol Cell Biochem. 2004;265:47–56. doi: 10.1023/b:mcbi.0000044314.29395.fb. [DOI] [PubMed] [Google Scholar]
- 72.Gupta A, Sharma AC. Metalloendopeptidase inhibition regulates phosphorylation of p38-mitogen-activated protein kinase and nitric oxide synthase in heart after endotoxemia. Shock. 2003;20:375–81. doi: 10.1097/01.shk.0000087202.34916.0c. [DOI] [PubMed] [Google Scholar]
- 73.Mazzocchi G, Albertin G, Nussdorfer GG. Adrenomedullin (ADM), acting through ADM(22–52)-sensitive receptors, is involved in the endotoxin-induced hypotension in rats. Life Sci. 2000;66:1445–50. doi: 10.1016/s0024-3205(00)00455-0. [DOI] [PubMed] [Google Scholar]
- 74.Sharma AC, Sam AD, 2nd, Alden KJ, Moore SL, Law WR, Ferguson JL. Central versus peripheral mediation of naloxone’s perfusion effects in endotoxic rats. Shock. 2000;14:441–6. doi: 10.1097/00024382-200014040-00004. [DOI] [PubMed] [Google Scholar]
- 75.Mitaka C, Hirata Y, Yokoyama K, Nagura T, Tsunoda Y, Amaha K. Pathologic role of endothelin-1 in septic shock. J Cardiovasc Pharmacol. 1998;31:S233–5. doi: 10.1097/00005344-199800001-00065. [DOI] [PubMed] [Google Scholar]
- 76.Rogausch H, Vo NT, Del Rey A, Besedovsky HO. Increased sensitivity of the baroreceptor reflex after bacterial endotoxin. Ann N Y Acad Sci. 2000;917:165–8. doi: 10.1111/j.1749-6632.2000.tb05380.x. [DOI] [PubMed] [Google Scholar]