

Abstract
Insulin is believed to regulate glucose homeostasis mainly via direct effects on the liver, muscle, and adipose tissues. The contribution of insulin's central nervous system effects to disorders of glucose metabolism has received less attention. To evaluate whether postnatal reduction of insulin receptors (IRs) within the ventromedial hypothalamus (VMH), a brain region critical for glucose sensing, contributes to disorders of peripheral glucose metabolism, we microinjected a lentiviral vector expressing an antisense sequence to knockdown IRs or a control lentiviral vector into the VMH of nonobese nondiabetic rats. After 3–4 mo, we assessed 1) glucose tolerance, 2) hepatic insulin sensitivity, and 3) insulin and glucagon secretion, using the glucose clamp technique. Knockdown of IRs locally in the VMH caused glucose intolerance without altering body weight. Increments of plasma insulin during a euglycemic clamp study failed to suppress endogenous glucose production and produced a paradoxical rise in plasma glucagon in the VMH-IR knockdown rats. Unexpectedly, these animals also displayed a 40% reduction (P < 0.05) in insulin secretion in response to an identical hyperglycemic stimulus (∼220 mg/dl). Our data demonstrate that chronic suppression of VMH-IR gene expression is sufficient to impair glucose metabolism as well as α-cell and β-cell function in nondiabetic, nonobese rats. These data suggest that insulin resistance within the VMH may be a significant contributor to the development of type 2 diabetes.
the prevalence of type 2 diabetes, a disorder characterized by insulin resistance, inadequate insulin secretion, and inappropriate glucagon responses continues to rise at an alarming rate in the United States and around the world. An inadequate understanding of the basic pathophysiological mechanisms promoting the disease, and as a result optimal therapeutic approaches, has resulted in a substantial burden on healthcare costs. While treating peripheral insulin resistance in classic insulin-sensitive tissues such as the liver, adipose tissue, and muscle or enhancing insulin secretion has therapeutic benefit, in most cases it is insufficient to prevent disease progression (24). One potential target that has not generally been considered in the therapeutic equation is the central nervous system (CNS), although it has been elegantly shown by a number of studies to be insulin responsive (19, 20, 22, 26). Of particular interest in this regard is the ventromedial hypothalamus (VMH), a brain region that plays a vital role in monitoring energy and glucose homeostasis (17, 18, 23). The VMH contains glucose sensing neurons that are activated by either high or low glucose levels and for this purpose use mechanisms that are similar to those used by islets (12, 23, 25, 27, 28). A subset of VMH neurons also expresses insulin receptors (5, 18), and blockade of insulin action within this brain region promotes glucagon secretion under fasting and hypoglycemic conditions (20). Moreover, recent studies conducted in humans demonstrated that CNS insulin signaling is downregulated in patients with type 2 diabetes (16). Whether the postnatal development of impaired insulin signaling in the brain plays a significant role in the pathogenesis of diabetes independently of changes in body weight has not been determined.
It has been previously reported that targeted genetic manipulations that cause prenatal deletion of insulin receptors in POMC- and GLUT4-expressing neurons cause impaired glucose homeostasis (1, 9, 14). However, coexisting obesity and/or potential prenatal adaptations during brain development complicate the alterations in glucose metabolism produced in these knockout mice. The current study was undertaken to determine whether a local postnatal reduction in insulin signaling within the VMH, much like what is seen in peripheral tissues in type 2 diabetes, is sufficient to diminish insulin action and islet function in nonobese adult rats that are not prone to develop diabetes. For this purpose, we specifically knocked down insulin receptor gene expression in the VMH using an antisense lentiviral vector. This intervention reproduced many of the early pathophysiological changes associated with type 2 diabetes, including glucose intolerance and impaired islet function in the absence of obesity.
RESEARCH DESIGN AND METHODS
Animals.
Adult male Sprague-Dawley rats (250–275 g) were used for the study. All rats were individually housed in the Yale Animal Resources Center in temperature- (22–23°C) and humidity-controlled rooms. The animals were fed rat chow (Agway Prolab 3000, Syracuse, NY) and water ad libitum and were acclimatized to handling and a 12:12-h light-dark cycle (lights on between 0700 and 1900) before experimental manipulation. Principles of laboratory animal care were followed, and the Institutional Animal Care and Use Committee at Yale University approved experimental protocols.
Stereotaxic surgery.
Rats were placed into a stereotaxic frame, and stainless steel microinjection needles (from bregma: 2.6 mm posterior, 0.6 mm lateral and 8.0 mm ventral) were inserted down to the level of the VMH. A lentiviral vector (1.5 μl/side) containing the insulin receptor antisense (IR-LV) or a control vector was injected into the VMH over the course of 15 min at a rate of 0.1 μl/min. The needles were then left in place for an additional 5 min before being removed, and the incision was closed. This technique has been demonstrated to localize the injection to the VMH (predominantly to the ventromedial nucleus compared with the arcuate) and minimize spread to adjacent areas of the brain (3). Subsequently, the animals were returned to their home cages and 16 wk later underwent a series of metabolic studies before being euthanized. Insulin receptor knockdown was verified in VMH micropunches and accuracy of IR-LV delivery was determined histologically in coronal brain sections. This viral vector has been previously utilized for knockdown of insulin receptors (7, 8).
Oral glucose tolerance tests.
Six and twelve weeks after injection of the lentiviral vector, the animals were fasted overnight before undergoing an oral glucose tolerance test (OGTT)the next day. Glucose was gavaged (2 g/kg body wt), and tail vein glucose levels were measured at 0, 15, 30, 60, 90, and 120 min from a 0.3-μl tail vein sample for measurement of glucose. They were then allowed to recover for subsequent glucose clamp studies.
Glucose clamp studies.
At 15 wk postlentiviral injection, the animals underwent aseptic surgery to have vascular catheters implanted. Vascular catheters were prepared using a long piece of polyethylene tubing (PE-50) attached to a 3-cm piece of silicone tubing. The tubing was inserted into the left carotid artery for blood sampling and right jugular vein for infusion. Catheters were tunneled subcutaneously and exteriorized at the back of the neck (20). The animals were then allowed to recover for at least 1 wk in their home cages before receiving a hyperinsulinemic-euglycemic clamp or hyperglycemic clamp study.
Hyperinsulinemic-euglycemic clamp.
On the day of the study, overnight fasted rats (n = 6 with the control lentivirus and n = 6 with the IR siRNA lentiviral vector) were connected to infusion pumps ∼1–1.5 h prior to the start of the experiment and then left undisturbed to recover from handling stress. Following the recovery period, blood was withdrawn from the arterial line to assess baseline hormone concentrations, and a primed continuous infusion of [3H]glucose was initiated for 90 min before as well as during the clamp procedure. The hyperinsulinemic-euglycemic clamp consisted of a constant low-dose insulin infusion (2.5 mU·kg−1·min−1) and a variable-rate glucose infusion designed to maintain plasma glucose levels between 110 and 120 mg/dl for 90 min. Throughout the study, blood samples were collected at 30-min intervals to assess plasma glucagon and insulin concentrations and every 10 min for measurement of the glucose tracer. Following each sample collection, the blood was centrifuged, and plasma samples were aliquoted into appropriate tubes for storage. Plasma samples were kept at −20°C until analysis. The red blood cells were resuspended in an equivalent volume of artificial plasma and reinfused back into the animal to prevent volume depletion and anemia. At the end of the study, the animals were euthanized with an overdose of pentobarbital sodium, and the brains were rapidly removed and frozen in dry ice for assessment of the accuracy of viral vector delivery and quantitation of insulin receptor protein.
Hyperglycemic clamp.
Hyperglycemic clamps were performed in a separate series of animals (6 with the control lentivirus and 6 with the IR siRNA lentiviral vector) to assess β-cell function. The procedures used here were identical to those used for the hyperinsulinemic-euglycemic clamp (see above), except that neither tracer nor insulin was infused; instead, the animals were infused with a variable 35% dextrose solution to maintain plasma glucose at ∼220 mg/dl. Blood was collected at 0, 10, 20, 30, 60, 90, and 120 min to assess plasma insulin concentrations.
Immunoblot analysis.
Frozen tissue micropunches from the VMH were homogenized in 30 μl of lysis buffer (5 M urea, 2 M thiourea, 35 mM Tris, 65 mM CHAPS, and 65 mM DTT) using a plastic pestle and ultrasonicator, and protein contents were assessed with the Bradford protein assay. Equal amounts of protein were resolved under reducing conditions in a 4–20% gradient SDS-polyacrylamide gel. Protein samples were then transferred onto nitrocellulose membranes. Success of the protein transfer was assessed by exposure of the nitrocellulose membranes to Ponceau S solution, which was followed by a thorough washing prior to soaking the membranes in 5% blocking solution overnight. The next day, membranes were washed with 0.01 M PBS-Tween (PBS-T), incubated with primary antibodies directed against the insulin receptor and β-actin, washed with PBS-T, and incubated with appropriate secondary antibodies conjugated to peroxidase. Following incubation with the secondary antibody, the membranes were washed with PBS-T and immersed into chemiluminescence. The membranes were then exposed to Kodak X-OMAT film. Film analysis was undertaken using a computerized image analysis system running Kodak Digital Science One D software. Protein levels were expressed as a ratio between the protein of interest and β-actin to correct for differences in loading and transfer. Only animals that demonstrated at least a 50% reduction in insulin receptor protein compared with LV control rats were included in the study.
Hormone analysis.
Plasma catecholamine concentrations were analyzed by HPLC using electrochemical detection, and plasma hormone concentrations were determined using commercially available radioimmunoassay kits.
Statistical analysis.
Treatment effects were analyzed using one- or two-way analysis of variance (ANOVA) for independent or repeated measures as appropriate, followed by post hoc analysis using the Prism analytical software. P < 0.05 was set as the criterion for statistical significance.
RESULTS
Local microinjection of the lentiviral vector containing the siRNA against the insulin receptor (IR) gene was very effective in suppressing IR expression within the VMH compared with the control virus. As shown in Fig. 1A, Western blots for IR run on VMH micropunches demonstrated >50% IR knockdown (IR KD) within the VMH.
Fig. 1.
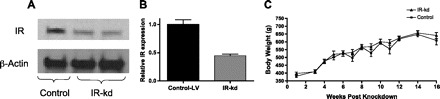
Local knockdown of insulin receptors (IR) within the VMH using a lentiviral vector (A and B) does not alter body weight (C). VMH-IR knockdown, ▴; control vector-treated animals, ■.
The knockdown of VMH-IR did not affect body weight (Fig. 1B) in rats fed normal chow. However, there was a gradual reduction in glucose tolerance. Six weeks after virus injection, the VMH-IR knockdown rats showed a slight but insignificant impairment of oral glucose tolerance, as observed in previously reported studies (8). However, by 12 wk out, VMH-IR knockdown animals exhibited significantly impaired glucose tolerance compared with their control counterparts (Fig. 2). One hour after oral gavage, plasma glucose levels were ∼30 mg/dl higher in the VMH-IR knockdown rats (P < 0.05 vs. control).
Fig. 2.
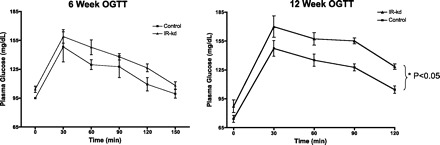
Effect of knockdown of VMH-IR on oral glucose tolerance (OGTT). Knockdown (KD) of VMH-IR (▴) leads to impaired glucose tolerance at 12 wk post-KD compared with control vector-treated animals (■). All values ± SE, *P < 0.05.
Subsequently (at 16 wk), the effect of VMH-IR knockdown on hepatic insulin sensitivity was evaluated (Fig. 3). Insulin levels during the clamp increased two- to threefold and were similar in both groups (80 ± 10 μU/ml in controls vs. 90 ± 11 μU/ml in IR KD). The rate of glucose infusion required to maintain euglycemia during the clamp was reduced by 30% in the VMH-IR knockdown rats (4.4 ± 0.2 Controls vs. 2.9 ± 0.2 mg·kg−1·min−1 IR KD, P < 0.05). The basal hepatic glucose production was not significantly different between control and VMH-IR KD groups (14.3 ± 3 mg·kg−1·min−1 controls vs. 16.2 ± 2 mg·kg−1·min−1 IR-kD). However, there was a significant impairment in the capacity of insulin to suppress hepatic glucose production (8.1 ± 2 mg·kg−1·min−1 Controls vs. 12.7 ± 1 mg·kg−1·min−1 VMH-IR-kD, P < 0.05; Fig. 3C). The rate of glucose utilization was not significantly affected by the knockdown of VMH insulin receptors (16.4 ± 2 control vs. 17.1 ± 2 IR KD). As expected with this relatively small dose of insulin, glucagon levels were unaltered in the control rats. In contrast, the VMH-IR knockdown showed a paradoxical twofold increase in plasma glucagon in response euglycemic hyperinsulinemia (P < 0.05; Fig. 3D).
Fig. 3.
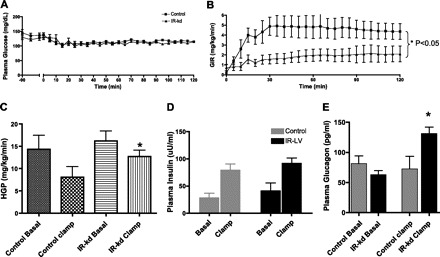
Local knockdown of VMH-IR leads to hepatic insulin resistance coupled with an inappropriate glucagon secretion. A: euglycemic clamp studies (A) were conducted in VMH-IR KD (▴) and control vector-treated animals (■). B: glucose infusion rate (GIR) was significantly lower in animals with VMH-IR knockdown. C: insulin-mediated suppression of hepatic glucose production (HGP) was significantly impaired in VMH-IR KD animals (filled bars) compared with controls (gray bars). D: plasma insulin levels were similar in both groups. E: also, an inappropriate elevation in glucagon levels was observed in VMH-IR KD animals after a euglycemic clamp. All values ± SE, *P < 0.05.
To assess whether the impairment in oral glucose tolerance after VMH-IR knockdown could be entirely accounted for by impaired insulin action, glucose-stimulated insulin secretion was examined in a separate group of animals using the hyperglycemic clamp (Fig. 4). VMH-IR knockdown animals required significantly less exogenous glucose infusion rates to maintain the standardized glucose stimulus (28.0 ± 3.8 mg·kg−1·min−1 Controls vs 19.0 ± 1.1 mg·kg−1·min−1 VMH-IR-kd, P < 0.05). Surprisingly, this was due to a nearly 50% reduction in insulin secretion in the VMH-IR knockdown rats (P < 0.05) (rather than insulin resistance and an attempt at compensatory hyperinsulinemia).
Fig. 4.
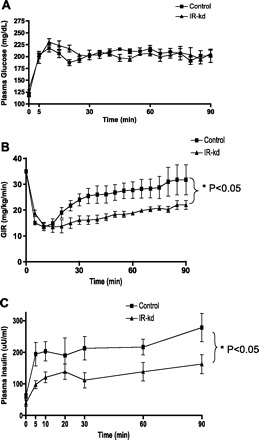
A: hyperglycemic clamp studies were conducted in VMH-IR KD (▴) and control vector-treated animals (■). B: GIR was significantly lower in animals with VMH-IR knockdown . C: This was accompanied by a significant attenuation of insulin secretory responses. All values ± SE, *P < 0.05.
DISCUSSION
Insulin resistance in classical insulin-responsive tissues such as liver, muscle, and adipocytes characteristically precedes the development of type 2 diabetes and is believed to account for the association between obesity and type 2 diabetes (24). Until recently, relatively little attention had been given to the potential role of impaired insulin signaling in brain in the pathogenesis of type 2 diabetes, as it was earlier thought to be insulin insensitive. More than three decades ago, Woods et al. (22) demonstrated that intracerebroventricular (icv) administration of insulin exerted an inhibitory effect on food intake and body weight. Subsequently, it was recognized that insulin receptors are widely expressed throughout the brain and that stimulation of insulin signaling via icv insulin delivery has the capacity to modulate the expression of neuropeptides that regulate food intake as well as glucose production (17). A recent study in humans demonstrated that insulin signaling pathways are attenuated in patients with type 2 diabetes (16). Moreover, mice lacking neuronal insulin receptors demonstrate obesity and insulin resistance (2, 21), and suppression of insulin signaling by third ventricle delivery of a PI 3-kinase inhibitor reduces, whereas increased insulin signaling in the hypothalamus increases, the plasma glucose-lowering effect of insulin in insulin-deficient diabetic rats (6). In the VMH, a key glucose-sensing region of the brain, many neurons possess insulin receptors as well as the insulin-responsive glucose transporter GLUT4 and respond to insulin application with changes in neuronal firing rate (5). In keeping with these findings, we (20) have previously shown that local blockade of insulin action within the VMH, using either an anti-insulin affibody or an insulin receptor antagonist, causes an immediate increase in both circulating glucose and glucagon in nondiabetic rats, suggesting that insulin acts on VMH neurons to tonically inhibit glucagon secretion. The importance of insulin's CNS effects in the regulation of glucose metabolism is also consistent with observations from a number of recent studies. It has been reported that icv delivery of insulin suppresses hepatic glucose production (13, 15, 17, 18, 22). In addition, models in which neuronal insulin receptors were specifically deleted support the role of central insulin signaling in the development of glucose intolerance and insulin resistance (15). For example, it has recently been demonstrated that the conditional ablation of insulin receptors in GLUT4, and not simply in peripheral GLUT4-expressing insulin-responsive tissues, is required for full progression to diabetes (15). It is also noteworthy in this regard that many VMH glucose-sensing neurons express both insulin receptors and GLUT4 (14).
Although the importance of CNS insulin receptors in the development of diabetes has been demonstrated using more specific genetic manipulations using knockout mice than those used here (10, 11, 21), and while it is likely that the regulation of peripheral glucose metabolism by the CNS is not restricted to a single brain region, the approach used here has several advantages. It allowed us 1) to specifically target the VMH insulin receptors in postnatal “adult” rats, thereby avoiding developmental or other adaptive effects associated with whole brain gene deletion, and 2) to avoid the confounding influence of altered body weight on peripheral insulin action as well as insulin secretion. The result was the recapitulation of the early stages of diabetes in rats not prone to develop the disease, specifically, glucose intolerance, hepatic insulin resistance, inappropriate glucagon secretion, and, surprisingly, impaired insulin secretion in the absence of changes in body weight. It should be noted that the VMH-IR knockdown animals gradually develop glucose intolerance over an extended period of time, much the same as is seen in humans who develop impaired glucose tolerance. It is intriguing to speculate that the aging process may have accentuated the impact of the reduction in VMH insulin signaling on glucose metabolism.
The demonstration of hepatic insulin resistance during the euglycemic insulin clamp experiment in rats with impaired VMH insulin receptor signaling may be due in part to the paradoxical rise in circulating glucagon observed during the study. Dysregulation of α-cell glucagon secretion is a characteristic feature of the diabetic state and is linked to hepatic insulin resistance (23). However, it is possible that neural pathways emanating from the VMH innervate the liver and are directly affected by VMH insulin receptor knockdown as well. Earlier studies had demonstrated a direct effect of intraventricular insulin infusion on hepatic glucose production when given with somatostatin to prevent changes in circulating glucagon and insulin (18). Thus, it is likely that insulin acts within the hypothalamus to regulate glucose production both directly and indirectly via changes in islet function. It should be noted that the doses of insulin used in the current studies were targeted to look at the effects of modest increases in peripheral insulin levels on hepatic glucose production and thus were insufficient to assess whether or not the suppression of VMH insulin receptor gene expression was capable of exerting an effect on insulin-stimulated peripheral glucose uptake as well.
Although our data indicating a role for insulin receptor signaling within the VMH in the regulation of hepatic glucose production is consistent with previous reports using less targeted approaches to this specific brain region, the finding that this intervention also suppressed β-cell insulin secretion in response to a standardized hyperglycemic stimulus was unexpected. The presence of hepatic insulin resistance would be anticipated to induce compensatory hyperinsulinemia, particularly in the absence of frank diabetes. It is intriguing to speculate that the appearance of suppressed glucose-stimulated insulin secretion in conjunction with the suppression of VMH insulin action might be the result of the disruption of a neural circuit connecting glucose-sensing neurons in the VMH with pancreatic -cells. Interestingly, mice with a deletion of insulin receptors in GLUT4-positive neurons appears to have a similar effect on insulin secretion (15). However, this alteration in β-cell function observed in these knockout mice occurs in the background in which there is concomitant deletion of insulin receptors in muscle and adipose tissue as well. Our study suggests that changes in hypothalamic insulin signaling may be the predominant mediator of the β-cell dysfunction seen in these mice.
The development of type 2 diabetes involves the convergence of many overlapping factors that contribute to the development of insulin resistance and eventually beta cell failure (24). A contributory factor is the coexistence of obesity that magnifies insulin resistance and acts to exacerbate the condition. The fact that VMH insulin receptor knockdown animals do not develop florid diabetes may in part reflect the absence of weight gain in these animals compared with controls. This might be a consequence of the specificity to the viral targeting strategy, which was directed at the ventromedial nucleus rather than the arcuate nucleus. However, we cannot exclude the possibility that there were undetected changes in body fat distribution that contributed to the appearance of glucose intolerance and insulin resistance. On the other hand, our data provide support for the intriguing possibility that the role of the brain extends beyond the pathogenesis of obesity, and that it may play a direct role in the dysregulation of islet function that leads to the development of type 2 diabetes, as well. Furthermore, these studies suggest that drugs designed to improve insulin sensitivity in type 2 diabetes may need to target key insulin-responsive brain glucose regulatory neurons to achieve maximal efficacy.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
REFERENCES
- 1. Banno R, Zimmer D, De Jonghe BC, Atienza M, Rak K, Yang W, Bence KK. PTP1B and SHP2 in POMC neurons reciprocally regulate energy balance in mice. J Clin Invest 120: 720–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bruning JC, Gautam D, Burks DJ, Gillette J, Schubert M, Orban PC, Klein R, Krone W, Muller-Wieland D, Kahn CR. Role of brain insulin receptor in control of body weight and reproduction. Science 289: 2122–2125, 2000 [DOI] [PubMed] [Google Scholar]
- 3. Cheng H, Zhou L, Zhu W, Wang A, Tang C, Chan O, Sherwin RS, McCrimmon RJ. Type 1 corticotropin-releasing factor receptors in the ventromedial hypothalamus promote hypoglycemia-induced hormonal counterregulation. Am J Physiol Endocrinol Metab 293: E705–E712, 2007 [DOI] [PubMed] [Google Scholar]
- 5. Cotero VE, Zhang BB, Routh VH. The response of glucose-excited neurones in the ventromedial hypothalamus to decreased glucose is enhanced in a murine model of type 2 diabetes mellitus. J Neuroendocrinol 22: 65–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gelling RW, Morton GJ, Morrison CD, Niswender KD, Myers MG, Jr, Rhodes CJ, Schwartz MW. Insulin action in the brain contributes to glucose lowering during insulin treatment of diabetes. Cell Metab 3: 67–73, 2006 [DOI] [PubMed] [Google Scholar]
- 7. Grillo CA, Piroli GG, Evans AN, Macht VA, Wilson SP, Scott KA, Sakai RR, Mott DD, Reagan LP. Obesity/hyperleptinemic phenotype adversely affects hippocampal plasticity: Effects of dietary restriction. Physiol Behav 2011. February 24 [Epub ahead of print] PMID: 21354191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Grillo CA, Tamashiro KL, Piroli GG, Melhorn S, Gass JT, Newsom RJ, Reznikov LR, Smith A, Wilson SP, Sakai RR, Reagan LP. Lentivirus-mediated downregulation of hypothalamic insulin receptor expression. Physiol Behav 92: 691–701, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hill JW, Xu Y, Preitner F, Fukuda M, Cho YR, Luo J, Balthasar N, Coppari R, Cantley LC, Kahn BB, Zhao JJ, Elmquist JK. Phosphatidyl inositol 3-kinase signaling in hypothalamic proopiomelanocortin neurons contributes to the regulation of glucose homeostasis. Endocrinology 150: 4874–4882, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Koch L, Wunderlich FT, Seibler J, Konner AC, Hampel B, Irlenbusch S, Brabant G, Kahn CR, Schwenk F, Bruning JC. Central insulin action regulates peripheral glucose and fat metabolism in mice. J Clin Invest 118: 2132–2147, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Konner AC, Janoschek R, Plum L, Jordan SD, Rother E, Ma X, Xu C, Enriori P, Hampel B, Barsh GS, Kahn CR, Cowley MA, Ashcroft FM, Bruning JC. Insulin action in AgRP-expressing neurons is required for suppression of hepatic glucose production. Cell Metab 5: 438–449, 2007 [DOI] [PubMed] [Google Scholar]
- 12. Levin BE, Routh VH, Kang L, Sanders NM, Dunn-Meynell AA. Neuronal glucosensing: what do we know after 50 years? Diabetes 53: 2521–2528, 2004 [DOI] [PubMed] [Google Scholar]
- 13. Lin HV, Plum L, Ono H, Gutierrez-Juarez R, Shanabrough M, Borok E, Horvath TL, Rossetti L, Accili D. Divergent regulation of energy expenditure and hepatic glucose production by insulin receptor in AgRP and POMC neurons. Diabetes 59: 337–346, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lin HV, Ren H, Samuel VT, Lee HY, Lu TY, Shulman GI, Accili D. Diabetes in mice with selective impairment of insulin action in Glut4-expressing tissues. Diabetes 60: 700–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lin HV, Ren H, Samuel VT, Lee HY, Lu TY, Shulman GI, Accili D. Diabetes in mice with selective impairment of insulin action in Glut4-expressing tissues. Diabetes 60: 700–709, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu Y, Liu F, Grundke-Iqbal I, Iqbal K, Gong CX. Deficient brain insulin signalling pathway in Alzheimer's disease and diabetes. J Pathol 225: 54–62, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Obici S, Feng Z, Karkanias G, Baskin DG, Rossetti L. Decreasing hypothalamic insulin receptors causes hyperphagia and insulin resistance in rats. Nat Neurosci 5: 566–572, 2002 [DOI] [PubMed] [Google Scholar]
- 18. Obici S, Zhang BB, Karkanias G, Rossetti L. Hypothalamic insulin signaling is required for inhibition of glucose production. Nat Med 8: 1376–1382, 2002 [DOI] [PubMed] [Google Scholar]
- 19. Pagotto U. Where does insulin resistance start? Brain Diabetes Care 32, Suppl 2: S174–S177, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Paranjape SA, Chan O, Zhu W, Horblitt AM, McNay EC, Cresswell JA, Bogan JS, McCrimmon RJ, Sherwin RS. Influence of insulin in the ventromedial hypothalamus on pancreatic glucagon secretion in vivo. Diabetes 59: 1521–1527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Plum L, Belgardt BF, Bruning JC. Central insulin action in energy and glucose homeostasis. J Clin Invest 116: 1761–1766, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schwartz MW, Figlewicz DP, Baskin DG, Woods SC, Porte D., Jr Insulin in the brain: a hormonal regulator of energy balance. Endocr Rev 13: 387–414, 1992 [DOI] [PubMed] [Google Scholar]
- 23. Sherwin RS. Bringing light to the dark side of insulin: a journey across the blood-brain barrier. Diabetes 57: 2259–2268, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shulman GI. Cellular mechanisms of insulin resistance. J Clin Invest 106: 171–176, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang S, Tulina N, Carlin DL, Rulifson EJ. The origin of islet-like cells in Drosophila identifies parallels to the vertebrate endocrine axis. Proc Natl Acad Sci USA 104: 19873–19878, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Woods SC, Lotter EC, McKay LD, Porte D., Jr Chronic intracerebroventricular infusion of insulin reduces food intake and body weight of baboons. Nature 282: 503–505, 1979 [DOI] [PubMed] [Google Scholar]
- 27. Xu E, Kumar M, Zhang Y, Ju W, Obata T, Zhang N, Liu S, Wendt A, Deng S, Ebina Y, Wheeler MB, Braun M, Wang Q. Intra-islet insulin suppresses glucagon release via GABA-GABAA receptor system. Cell Metab 3: 47–58, 2006 [DOI] [PubMed] [Google Scholar]
- 28. Yang XJ, Kow LM, Funabashi T, Mobbs CV. Hypothalamic glucose sensor: similarities to and differences from pancreatic beta-cell mechanisms. Diabetes 48: 1763–1772, 1999 [DOI] [PubMed] [Google Scholar]