

Abstract
Chronic inflammation is a characteristic of obesity and is associated with accompanying insulin resistance, a hallmark of type 2 diabetes mellitus (T2DM). Although proinflammatory cytokines are known for their detrimental effects on adipose tissue function and insulin sensitivity, their beneficial effects in the regulation of metabolism have not drawn sufficient attention. In obesity, inflammation is initiated by a local hypoxia to augment angiogenesis and improve adipose tissue blood supply. A growing body of evidence suggests that macrophages and proinflammatory cytokines are essential for adipose remodeling and adipocyte differentiation. Phenotypes of multiple lines of transgenic mice consistently suggest that proinflammatory cytokines increase energy expenditure and act to prevent obesity. Removal of proinflammatory cytokines by gene knockout decreases energy expenditure and induces adult-onset obesity. In contrast, elevation of proinflammatory cytokines augments energy expenditure and decreases the risk for obesity. Anti-inflammatory therapies have been tested in more than a dozen clinical trials to improve insulin sensitivity and glucose homeostasis in patients with T2DM, and the results are not encouraging. One possible explanation is that anti-inflammatory therapies also attenuate the beneficial effects of inflammation in stimulating energy expenditure, which may have limited the efficacy of the treatment by promoting energy accumulation. Thus, the positive effects of proinflammatory events should be considered in evaluating the impact of inflammation in obesity and type 2 diabetes.
Keywords: inflammation, energy expenditure, insulin resistance, anti-inflammatory therapy, type 2 diabetes
obesity has reached epidemic proportions in many developed countries, including the United States. It induces marked insulin resistance and increases the risk of developing type 2 diabetes mellitus (T2DM) and other diseases (cardiovascular disease, some cancers, and asthma). One feature of obesity is a low-grade inflammation that likely originates in the expanding adipose tissue, and it is associated with infiltration of immune cells, including macrophages, lymphocytes, and leukocytes (22, 95, 131). The cytokines released from the inflammatory tissue affect the metabolic functions of several organs, including the liver, heart, muscle, and brain (46, 122). Proinflammatory cytokines released by the activated immune cells can impair insulin signaling in insulin-responsive organs and cause systemic insulin resistance, which increases the risk of developing hyperglycemia and T2DM. Insulin sensitizers such as thiazolidinediones (TZDs) have anti-inflammatory activity, which provides an additional rationale to pursue clinical studies to evaluate the efficacy of targeted anti-inflammatory therapeutics on T2DM (46, 95). In fact, anti-inflammatory therapies have been tested in many clinical trials to improve insulin sensitivity and restore glucose homeostasis.
However, the anti-inflammation therapies (anti-TNF, anti-IL-1, anti-IL-6, and salsalate) that will be discussed below have shown very limited efficacy with regard to improving insulin action and glucose homeostasis. While there could be many explanations (such as less efficacious than standard diabetes therapy, off-target effects, wrong patient population, etc.) for the limited benefit of anti-inflammatory therapies, one possibility is that our assumption that inflammation has only negative effects on insulin action and glucose homeostasis may not be correct. There might be some beneficial effects of inflammation in obesity. For example, inflammation plays an essential role in maintaining healthy adipose tissue through stimulating tissue remodeling (101). Inflammation increases energy expenditure and suppresses food intake, which would favor insulin sensitivity by limiting obesity (discussed below). Thus, the net outcomes of anti-inflammatory therapy on insulin sensitivity could depend on the extent to which the benefits outweigh the negatives in a given individual. In this review, we discuss the role of inflammation in adipose tissue remodeling and its impact on energy balance. We will use the beneficial effects of inflammation to explain the mixed results of the anti-inflammatory clinical trials that have been reported.
Why Does Obesity Increase Inflammation in Adipose Tissue?
White adipose tissue is a primary site of chronic inflammation in obesity and is characterized by expression of proinflammatory cytokines and infiltration of a variety of immune cells, including macrophages, T lymphocytes, B lymphocytes, natural killer cells, and neutrophils. (22, 95, 131). These immune cells, together with adipocytes and stromal vascular cells, constitute a cellular network that produces both proinflammatory and anti-inflammatory cytokines (40, 101, 132). While the proinflammatory cytokines are well known to impair insulin action in adipocytes, they are required for maintenance of angiogenesis, extracellular matrix remodeling, and clearance of dead cells in adipose tissue (15, 18, 40, 101, 129). These activities of inflammation are required to maintain a “healthy” microenvironment to sustain adipose tissue expansion. Angiogenesis is required for adipocyte differentiation and adipocyte function (17, 41). Inhibition of angiogenesis leads to suppression of adipose tissue growth, which has been shown to prevent obesity in mice (14). Recent studies suggest that proangiogenic factor (VEGF, vascular endothelial growth factor) and antiangiogenic factor (PEDF, pigment epithelium-derived factor) are elevated in plasma of obese humans and mice (20, 43, 116). In transgenic mice, adipocyte-specific overexpression of VEGF enhanced angiogenesis in adipose tissue and stimulated energy expenditure through fat “browning,” which is associated with improved insulin sensitivity (128). In contrast, infusion of PEDF inhibited angiogenesis and impaired insulin sensitivity in mice, whereas neutralizing PEDF improved insulin action (20). Macrophages, the major inflammatory cells in adipose tissue, have been reported to facilitate adipocyte differentiation (90), control lipolysis (65), and augment brown fat function in adipose tissue in mice (88). In obesity, adipocyte expansion in size disrupts the interaction between adipocytes and the extracellular matrix in adipose tissue. Inflammation facilitates restoration of the balance by stimulating adipocyte lipolysis to attenuate cell expansion.
Adipose tissue hypoxia is considered the initiator of chronic inflammation in obese states (134, 146). A reduction in interstitial oxygen was first observed in adipose tissue of obese mice (147) and then confirmed in human and mouse obesity models (104, 146). In growing tissues, a transient hypoxia is a common signal in the stimulation of new blood vessel formation (angiogenesis). If the angiogenic response is insufficient to resolve the hypoxia, a chronic inflammatory response will be activated (146). Inflammation is one of a number of alterations that have been found in adipose tissue in obesity (Fig. 1). The others include endoplasmic reticulum (ER) stress, decreased adiponectin, elevated leptin, increased lipolysis, decreased adipogenesis, adipocyte death, and insulin resistance. There is no unifying mechanism for all of these alterations. Adipose hypoxia may explain most, if not all, of the alterations in the adipose tissue (Fig. 1). Insufficient blood supply is the underlying mechanism for the adipose hypoxia (see review in Ref. 145).
Fig. 1.
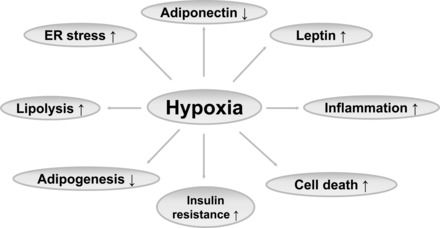
Hypoxia as a common root for various changes in adipose tissue in obesity. ER, endoplasmic reticulum. Hypoxia can induce complex changes in the endocrine and metabolic phenotype of the adipose tissue.
Hypoxia alters the balance between pro- and anti-inflammatory activities in adipose tissue. In cells, hypoxia induces expression of proinflammatory cytokines by activating transcription factors such as NF-κB (nuclear factor-κB) and HIF-1α (hypoxia-inducible factor 1α) in adipocytes and resident macrophages (147). Additionally, the local hypoxia may promote M2/M1 macrophage switching and induce leptin expression or adipocyte death to enhance the inflammatory response. M2 macrophages suppress the inflammatory response by secreting IL-10 and stimulate angiogenesis by producing angiogenic factors (37). In obesity, the differentiation of M2 into M1 macrophages is considered a major event that sustains chronic inflammation (76). In addition to the hypoxia hypothesis, there are other hypotheses for the origin of the inflammation in the adipose tissue (130). Yet, irrespective of the mechanism, most studies suggest that certain levels of inflammation are required for the maintenance of adipose tissue function by regulating extracellular matrix remodeling and adipocyte differentiation.
Inflammation: Impact on Energy Expenditure and Glucose Homeostasis
The primary cause of adiposity is a mismatch between energy intake and energy expenditure. Leptin and insulin play central roles in the regulation of energy balance in the body. Leptin induces satiety and augments energy expenditure to limit weight gain. In opposition, insulin induces energy accumulation and promotes weight gain. Yet, despite the marked increase in leptin in obesity, animals gain weight, presumably from central leptin resistance that is responsible for impaired control of food intake and energy expenditure. The energy surplus or weight gain is a major risk factor for insulin resistance, which triggers hyperinsulinemia. Adipose tissue inflammation is generally believed to contribute to the pathogenesis of insulin resistance through proinflammatory cytokines (see reviews, Refs. 46, 95, 122). However, the same cytokines stimulate energy expenditure and induce satiety and thus limit adiposity and improve glucose homeostasis. A role of inflammation in the control of energy expenditure will be discussed below.
We will examine the evidence from genetic and pharmacological studies in which both proinflammatory and anti-inflammatory cytokines have been tested in the regulation of energy balance. We recognize that the regulation of energy expenditure is complex and involves central and peripheral tissues (133). The proinflammatory cytokines to be discussed are interleukin-1 (IL-1), IL-6, tumor necrosis factor-α (TNF-α), leptin, and IL-18. The anti-inflammatory cytokines and signaling molecules are IL-1 receptor antagonist (IL-1Ra), mitogen-activated protein kinase phosphatase-1 (MPK1), adiponectin, and IL-10. In addition, we will discuss the studies in which inflammatory signaling pathways (NF-κB and JNK) have been manipulated. The major points about inflammation in energy balance are outlined in Fig. 2. Since the negative effects of proinflammatory cytokines on insulin action and adipose tissue function have been extensively documented in several outstanding review articles (46, 95, 122), we will not reiterate the negative effects in this article.
Fig. 2.
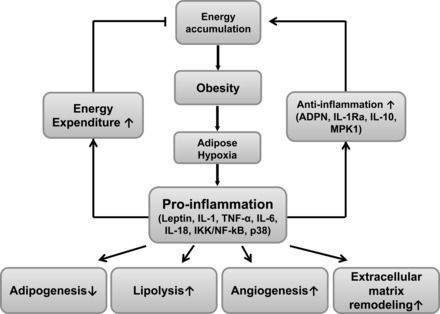
Impact of proinflammation and anti-inflammation events in adipose tissue on glucose homeostasis in peripheral tissues. Obesity and adipose tissue expansion and remodeling activate both proinflammation and anti-inflammation events. Expression of proinflammatory cytokines (leptin, IL-1, TNF-α, IL-6, IL-18, etc.) is enhanced in adipocytes, macrophages, and lymphocytes. To control chronic inflammation, the anti-inflammation molecules (ADPN, IL-1Ra, IL-10, etc.) are activated to balance the inflammatory impact. Both the pro- and anti-inflammatory events occurring in adipose tissue spill over onto peripheral tissues to alter insulin resistance and energy expenditure. The anti-inflammatory molecules tend to promote energy (fat and glucose) storage and improve insulin action, whereas the proinflammatory activities facilitate weight loss yet impair insulin action. A: on balance, the impact of glucose homeostasis is an integration of the pro- and anti-inflammatory activities. B: upon removal of proinflammation, the balance of homeostasis can be compromised due to loss of the beneficial increase in energy expenditure.
Before discussing the cytokines, or signaling molecules, we will briefly discuss the impact of endotoxemia on glucose homeostasis. Recent studies suggest that endotoxemia, secondarily to increased gut permeability and dietary-induced alterations of the intestinal microbiome, may contribute to the chronic inflammation associated with obesity (12, 56). As in obesity, endotoxemia induces similar inflammatory events and, on the surface, has overlapping metabolic features with inflammation. Moreover, a number of clinical trials have tested the impact of anti-inflammatory therapy on mortality in septic individuals. Although the patient population of endotoxemia is significantly different from individuals with T2DM, similar strategies have been used to test the role of inflammation.
There is rich literature regarding the acute impact of endotoxin (LPS) on inflammation and metabolism. The inflammatory response to LPS includes expression of many cytokines that are also upregulated by obesity, although the kinetics and origin of the cytokines are different. Following LPS exposure, the primary source of circulating cytokines is resident macrophages in the liver and other highly vascular tissues. LPS is a potent activator of the hypothalamo-pituitary-adrenal axis and is known to induce leptin secretion (even in the absence of obesity). The glucose response to LPS is dependent on the dose and duration of exposure to LPS and the species involved (80, 126). In general, high doses of LPS cause profound hypoglycemia and insulin resistance; low doses of LPS generally cause hyperglycemia and insulin resistance. At first, a combination of hypoglycemia and insulin resistance seems like a contradiction. However, the hypoglycemia is a result of impaired hormone-stimulated hepatic glucose production and increased insulin-independent glucose uptake in multiple tissues in both humans and rodents (2, 80, 81, 144). The insulin resistance is related to impaired insulin action in the liver or muscle through both direct and indirect effects of LPS. Proinflammatory cytokines are believed to impair insulin signaling directly in multiple tissues. The indirect effects include low blood flow in insulin-sensitive tissues (85, 135), increased glucocorticoid secretion (2, 81), and concomitant activation of the autonomic nervous system. LPS inhibits food intake and increases energy expenditure. However, in rodents, energy expenditure may decrease and hypothermia can develop if the animal is not in a thermoneutral environment or in response to a high dose of LPS (67).
Endotoxin concentrations can transiently rise after a normal meal, and high-fat diets can induce a low-grade endotoxemia (12, 56), which is proposed as a risk factor for the chronic inflammation in obesity. The impact of chronic low-grade endotoxemia on metabolism has been reported in two studies. Chronic (28 days) treatment with a low dose of LPS (300 μg·kg−1·day−1) induced obesity in mice (12). The adiposity was observed in the absence of detectable changes in energy intake. Energy expenditure (and/or core body temperature) was not assessed in the study but was likely reduced by LPS. Interestingly, despite the presence of obesity and inflammation in LPS-treated mice, there was no systemic insulin resistance. In a cat study, LPS was infused for 10 days (96). Initially LPS induced insulin resistance (increased basal insulin). However, by day 10, despite evidence of persistent inflammation, insulin concentration returned to normal. Thus the effects of sustained LPS delivery on hepatic and peripheral glucose metabolism can differ markedly from the acute effect of LPS. The mechanism of weight gain in the two LPS models remains to be identified. The anti-inflammatory response that is triggered by the chronic proinflammatory response may play a role in the inhibition of energy expenditure in LPS-treated mice.
Clinical trials of anti-TNF therapies have been conducted but yielded limited benefit in septic patients. Recently, the only immunomodulatory drug (drotrecogin-α) approved specifically for the treatment of severe sepsis was withdrawn from the market for its poor efficacy (8, 110–112). The reason for the limited efficacy of the drug is unknown. Several factors likely play a role, such as the timing and duration of therapy, patient heterogeneity, the therapeutic ability to access the site of inflammation, and the possibility of beneficial effect of TNF-α. Interestingly, timing of intervention is important. In rodents, if anti-TNF therapy was started before LPS administration, it was very effective. However, if it was given a few hours after LPS exposure, the anti-TNF therapy was not effective (7). Thus, anti-inflammatory approaches have not been very successful in improving outcomes in sepsis.
What we have learned from studies examining the impact of targeting inflammatory events in shaping the metabolic response to LPS or sepsis is that the net effect of inflammatory events on glucose homeostasis are dependent on both the duration of exposure and the magnitude of the inflammatory response. Moreover, while inflammatory mediators can impair insulin action, they also directly reduce blood glucose by impairing hepatic glucose production and augmenting peripheral glucose disposal independently of insulin. This complex interaction may explain the very dynamic and time-dependent effect of chronic inflammation on glucose homeostasis. If one considers that low-grade endotoxemia may contribute to obesity-associated inflammation, anti-inflammatory therapies should be able to attenuate the metabolic disorders in obesity. However, anti-inflammatory therapies have not been very effective. If the beneficial effect of targeting inflammation in sepsis is offset by the side effects of the therapies or time window of the intervention, similar challenges will present in the anti-inflammatory therapies that are designed to improve glucose homeostasis without negative consequences in obese and diabetic individuals.
Targeted modulation of pro- and anti-inflammatory cytokines and hormones.
TNF-α.
As the first proinflammatory cytokine reported in adipose tissue, TNF-α has been extensively documented as a participant in the pathogenesis of insulin resistance (47, 48). In cellular models, TNF-α inhibits insulin signaling through activation of several serine kinases including JNK (c-JUN NH2-terminal kinase), IKK, S6K (ribosomal protein S6 kinase 1), etc. (32, 115, 150). In adipose tissue, TNF-α is primarily produced by resident macrophages. The metabolic activity of TNF-α is complex, as indicated by comparing the phenotype of global TNF-α KO and TNF-receptor KO mice. While both mice have little or no phenotype on a chow diet, on a high-fat diet (HFD) TNF-α KO mice have normal weight gain. In contrast, the TNF receptor KO mice have a blunted weight gain when the receptor 2 (R2) but not R1 receptor is removed (120, 143). Diet-induced hyperinsulinemia was exacerbated when both receptors were removed. This suggests that TNF-α may maintain, rather than aggravate, insulin action in vivo. A more careful analysis of the TNF receptor KO mice (100) using littermate controls revealed that they are prone to obesity and have a low metabolic rate. Interestingly, when TNF-α is reconstituted only in the adipose tissue using the aP2 promoter in TNF-α KO mice, the transgenic mice exhibit less adiposity at 20 wk of age (142), suggesting a role for TNF-α in the maintenance of energy balance. Although ex vivo insulin action was impaired in adipose tissue, systemic insulin sensitivity was improved in the mice, as indicated by a low level of fasting insulin. In a more severe insulin resistance model, such as the db/db mouse, loss of TNF-α receptor has no phenotype (120). In summary, TNF-α has profound effects on adipose tissue biology and insulin signaling in adipocytes, but its contribution to systemic insulin resistance during obesity is limited.
IL-1.
The IL-1 superfamily includes more than 30 members. Of these, IL-1α, IL-1β, and IL-1 receptor antagonist (IL-1Ra) have been reported to regulate energy metabolism and glucose homeostasis. IL-1α and IL-1β share the same cell membrane receptor (type I receptor) and trigger similar intracellular signaling pathways. IL-1 activity is regulated both transcriptionally and posttranscriptionally. The latter occurs via proteolytic cleavage of pro-IL-1β by the inflammasome (55). In addition, IL-1 competes for receptor occupancy with the receptor antagonist IL-1Ra.
IL-1 modulates glucose homeostasis by impairing insulin signaling, modulating insulin secretion, augmenting insulin-independent glucose uptake, increasing energy expenditure, and stimulating autonomic nervous system activity (58, 70, 97, 107). Depending on the dose, IL-1 can induce hypoglycemia or hyperglycemia (70, 138). Moreover, IL-1β can inhibit insulin secretion in β-cells and induce islet failure (4). Thus, even though IL-1 can directly impair insulin action in vivo, an assortment of responses can be manifested, some of which actually improve rather than aggravate glucose homeostasis. IL-1 also can limit obesity by augmenting energy expenditure (78, 83). When superphysiological doses of IL-1 (IL-1α or IL-1β) are given directly (intracerebroventricularly) into the central nervous system, energy expenditure and body temperature increase, and food intake decreases (83), inducing a negative energy balance. IL-1 administration activates the hypothalamic-pituitary-adrenal axis, increasing plasma corticosterone and adrenocorticotropin, and decreases spontaneous physical activity in rats. Continuous injection of IL-1 into the central nervous system for 5 days reduces body weight secondarily to anorexia and an increase in energy expenditure (83). Although those observations were made with nonphysiological levels of IL-1, the studies consistently support the body's physiological response to IL-1. Thus, in conditions such as obesity, where IL-1 activity is chronically elevated, interventions that inhibit IL-1 activity may, in fact, induce positive energy balance and aggravate glucose homeostasis.
The importance of IL-1 in regulating energy metabolism is supported by the phenotype of mice lacking either IL-1Ra or the IL-1 receptor (33, 78). IL-1Ra is an anti-inflammatory protein secreted by adipose tissue and other tissues (51). IL-1Ra acts to block IL-1 interaction with its receptor. Plasma IL-1Ra is elevated in obesity (82). IL-1Ra KO mice have enhanced IL-1 activity, and the mice are resistant to obesity (78). Their energy expenditure (normalized for body weight) is enhanced, and they are protected from HFD-induced insulin resistance (124, 125). Given that the IL-1Ra KO mice are smaller than the wild-type (WT) controls, more advanced statistical analysis is required to normalize the energy expenditure data (53, 54). Inhibition of IL-1 activity by injection of recombinant IL-1Ra leads to insulin resistance in mice (124). In IL-1R KO mice, IL-1 is unable to stimulate energy expenditure, and the mice have increased (1.5- to 2.5-fold) visceral and subcutaneous fat (33). This was accompanied by hyperglycemia and insulin resistance. Thus, some of the IL-1 activity seen during obesity may serve to limit adiposity and reduce the risk of developing T2DM.
IL-6.
IL-6 is elevated in plasma and in adipose tissue in obesity (29, 57). The impact of IL-6 activity on energy and glucose metabolism is suggested by the phenotypes of both the IL-6 KO (137) and STAT3 (signal transducer and activator of transcription 3) KO mice (49). STAT3 is a transcription factor that is activated by IL-6. Global ablation of IL-6 leads to adult-onset obesity and insulin resistance (137). In transgenic mice, IL-6 overexpression in skeletal muscle stimulates energy expenditure and reduces food intake, which limits adiposity (28). Exercise is an important lifestyle intervention to control weight gain. Interestingly, as a myokine, IL-6 is secreted by contracting muscle and is elevated two- to threefold in the circulation during exercise (105, 106). The increase in IL-6 may help amplify exercise-induced lipolysis in adipose tissue; the mobilized fatty acids are then oxidized by the working muscle (106). The impact of IL-6 on glucose metabolism is controversial. IL-6 infusion does not reduce insulin sensitivity in human subjects (68), yet it does so in mice (59, 63, 64). In mice, suppression of IL-6 signal transduction by liver-specific STAT3 ablation leads to hepatic insulin resistance (49). Basal IL-6 may be required in the maintenance of glucose homeostasis by restraining gluconeogenesis. As IL-6 has beneficial effects on glucose metabolism and on energy balance during exercise, anti-IL-6 therapy may have unfavorable consequences in obesity and T2DM.
LEPTIN.
Leptin secretion increases as adipose tissue expands. Leptin limits weight gain by reducing food intake and increasing energy expenditure (84). However, obesity is associated with leptin resistance, which is manifested as hyperleptinemia. It is unclear whether the resistance is a primary contributing factor to the obesity and/or is secondary to the higher leptin tone. Moreover, it is not known whether the leptin resistance extends to tissues other than the brain. Leptin resistance is thought to primarily occur in the brain. Leptin can be proinflammatory. Leptin induces the expression of proinflammatory cytokines in macrophages and T cells (74, 75, 79) and stimulates macrophage phagocytosis and monocyte proliferation (30, 74). Leptin also activates other signaling pathways used by proinflammatory cytokine receptors, including MAPKs (mitogen-activated protein kinases p38 and ERK) (77, 86), JAK/STAT3 (6, 136) and phosphatidylinositol 3-kinase (PI3K) (77, 141). Leptin expression is induced by inflammatory mediators (39, 119, 155). The signaling pathway and expression pattern suggest that leptin may mediate proinflammatory responses.
The protein structure of leptin is similar to that of other proinflammatory cytokines including IL-6, IL-11, IL-12, LIF, G-CSF, etc. (19), which may permit cross-talk of those cytokines with the leptin receptor. Leptin stimulates T cell proliferation and Th1 differentiation (5, 79), controls regulatory T lymphocytes (Treg) in adipose tissue (24), inhibits anti-inflammatory cytokine expression (IL-4 and IL-10) (79), and promotes Th1-mediated inflammatory responses such as experimental colitis and collagen-induced arthritis (9, 123). These inflammatory responses are reduced in ob/ob or db/db mice. This may be due, in part, to the elevated corticosterone secretion in these mice (44). However, leptin is not absolutely required for the chronic inflammation in adipose tissue in obesity. ob/ob mice exhibit comparable adipose inflammation to WT obese mice (147). Low-energy expenditure, hypothermia, and cold intolerance are characteristics of leptin deficiency in ob/ob mice and leptin receptor-deficient (db/db) mice. Leptin controls food intake and energy expenditure through its actions in the brain. Leptin also acts on peripheral tissues (such as pancreatic islets) to regulate energy balance and feeding behavior. In obesity, leptin resistance in the brain and islets contributes to sustained weight gain and is involved in development of hyperleptinemia. A high level of leptin may promote chronic inflammation by inhibiting expression of anti-inflammatory cytokines and inducing proinflammatory cytokines in T cells and macrophages. We are unaware of any study reporting leptin resistance in immune cells in obesity.
IL-18.
IL-18 is another member of the IL-1 superfamily. In obese and T2DM subjects, the plasma concentration and adipose tissue expression of IL-18 are elevated (25). A major function of IL-18 is to induce IFNγ expression by T cells; however, IL-18 also has effects on adipose tissue and brain. IL-18 suppresses angiogenesis through IFNγ-dependent expression of interferon-γ-inducible protein 10 (IP-10). The specific role of IP-10 in adipose tissue is unclear (13). Global IL-18 inactivation in mice increases the risk for obesity, hyperphagia, and insulin resistance (87). Administration of recombinant IL-18 (rIL-18) intracerebroventricularly inhibited food intake, reversed hyperglycemia, and corrected T2DM subjects, IL-18 resistance may be present (154). Thus, elevated IL-18 may serve a protective role in obesity to limit adiposity.
ADIPONECTIN AND IL-10.
Adiponectin is an adipokine that has anti-inflammatory activity. Adiponectin concentration is reduced in plasma of obese subjects, and the reduction is associated with an elevation in chronic inflammation (23). Adiponectin expression in adipocytes is reduced by hypoxia and proinflammatory cytokines (146). In ob/ob mice, fat-specific overexpression of adiponectin makes the transgenic mice more obese, but the mice are healthy compared with their ob/ob littermates (61). Adiponectin has been reported to inhibit lipolysis in adipocytes (109). Despite the exaggerated adiposity in adiponectin-overexpressing mice, adipose tissue inflammation is not elevated. This is presumably due to the increased adiponectin expression in the mice (61). In vitro, adiponectin inhibited the inflammatory response in macrophages via increasing anti-inflammatory cytokines such as IL-10 (103). In cells, adiponectin induces activities of several anti-inflammatory signaling molecules, including SOCS3 (suppressor of cytokine signaling 3) (27), A20 (TNF-induced protein-3) (27), and AMPK (AMP-activated protein kinase) (98). These cellular activities likely explain the anti-inflammatory effects of adiponectin. Thus, the anti-inflammatory activity of adiponectin is closely associated with facilitation of energy accumulation and inhibition of energy expenditure.
IL-10 is a classical anti-inflammatory cytokine that suppresses the signal transduction of proinflammatory cytokines. The plasma concentration of IL-10 is reduced in obesity (23); the mechanism is not known. IL-10 expression is induced by proinflammatory cytokines such as TNF-α. IL-10 is mainly produced by M2 macrophages and Th2 lymphocytes. In obese mice, IL-10 is increased when M2 macrophages are stimulated by TZD treatment (76, 92). In physiological settings, IL-10 is required to resolve inflammation. In mice, global inactivation of IL-10 increases the risk of inflammation; KO mice suffer from chronic enterocolitis (69). The KO mice have less fat tissue and lower body weight relative to the control mice (69). Whether IL-10 alters food intake and/or energy expenditure has not been evaluated. Interestingly, hematopoietic inactivation of IL-10 did not alter the response of the mice to an HFD (66), suggesting that the low adiposity of IL-10 KO mice is not due to loss of IL-10 in blood cells or macrophages. IL-10 infusion improved insulin sensitivity in rodents (59). In transgenic mice, overexpression of IL-10 in skeletal muscle decreased local inflammation and improved insulin sensitivity in response to an HFD despite similar adiposity (45). In summary, reduction of plasma IL-10 may contribute to the elevation in chronic inflammation in obesity. Its impact on glucose homeostasis and energy balance is less clear in obesity where IL-10 is decreased.
Targeted modulation of inflammatory signaling pathways.
The reality is that multiple cytokines or adipokines are altered in obesity, and metabolic responses to obesity involve more than one cytokine. The cytokines discussed above do not act in isolation. One proinflammatory cytokine may induce expression of other cytokines during an inflammatory response. At the same time, the proinflammatory cytokines induce expression of anti-inflammatory cytokines, which tends to dampen the overall inflammatory response. Based on the metabolic phenotypes of the mouse models discussed above, anti-inflammatory cytokines inhibit energy expenditure and promote weight gain by eliminating the metabolic activities of proinflammatory cytokines. Thus, we propose that, during weight gain, the anti-inflammatory factors play a dominant role. We refer to this physiological state as “inflammation resistance” (148). This condition likely occurs in the early stage of obesity and limits energy expenditure, thereby facilitating weight gain. In this section, we focus on intracellular signaling molecules that integrate pro- and anti-inflammatory cytokine activity to control energy balance.
MPK1.
Mitogen-activated protein kinase-1 (MKP-1) is an anti-inflammatory intracellular kinase that inhibits MAPK (p38, JNK, and ERK). In rodents, MKP-1 expression is increased in response to HFD. In global MKP-1 KO mice, MAPK activation (p38, JNK, and ERK) is amplified in response to various stresses such as LPS, serum growth factors, JNK activator (anisomycin), and osmotic stress (16, 89, 139, 152). The mice exhibit a strong proinflammatory response with elevations in TNF-α and IL-6 (16). As a result, the KO mice are more susceptible to endotoxin shock. The heightened inflammatory status in MKP-1 KO mice is associated with higher energy expenditure and resistance to diet-induced obesity (140). MPK1 induces p38 activation, and this mechanism may inhibit hepatic glucose production. In obese mice, p38 MAPK is reported to inhibit gluconeogenesis through activation of X-box binding proteins (XBPs) (72). p38 phosphorylates the spliced form of X-box binding protein-1 (XBP-1s), leading to their nuclear translocation. In the nucleus, XBP-1s inhibits gluconeogenesis by targeting Forkhead box O1 (FoxO1) (153). Thus, loss of anti-inflammatory signaling molecules results in a net increase in proinflammatory tone that increases energy expenditure and makes mice resistant to obesity.
NF-κB AND IKKβ.
The IKKβ/NF-κB signaling pathway controls the expression of the major proinflammatory cytokines. It is activated by many obesity-associated factors such as inflammation, hypoxia, ER stress, diacylglycerol, ceramide, etc. (146). This signaling pathway has been investigated in several studies using transgenic mice to understand the relationship between inflammation and insulin resistance (3, 10, 11, 113, 149). Those studies suggest that an increase in IKKβ/NF-κB activity may induce systemic insulin resistance or hepatic insulin resistance. However, the relationship of insulin resistance and energy expenditure was not carefully examined in the majority of those studies. This issue was addressed in our studies of two lines of NF-κB transgenic mice. We enhanced NF-κB activity by fat-specific overexpression of the NF-κB p65 subunit (aP2-p65 mice) or by (globally) deleting the NF-κB p50 subunit in p50 KO mice (31, 132). In both models, NF-κB tone was enhanced, and inflammation was increased in the mice on either a regular chow diet or HFD. The resultant inflammation increased energy expenditure and reduced adiposity in mice on HFD (31, 132, 151). Insulin sensitivity was not impaired in the mice on a chow diet and was improved in the mice on HFD. The observations were extended into fat-specific IKKβ-overexpressing mice that have been recently reported by another group (50). The fat-specific IKKβ mice exhibited an increase in energy expenditure and resistance to diet-induced obesity. Inflammation in adipose tissue failed to induce systemic insulin resistance in the mice on the regular chow diet or an HFD. Thus, inflammation in those fat-specific IKKβ/NF-κB mice provides beneficial effects in the control of adiposity and insulin resistance.
The beneficial activity of inflammation is supported by studies examining the impact of IKKβ in knockout mice (3, 38). Insulin sensitivity was assessed using the hyperinsulinemic-euglycemic clamp in mice in which IKKβ was inactivated in myeloid cells (including macrophages and myeloid cells) (3). After 7 wk on an HFD, insulin sensitivity was improved in KO mice, and the change was associated with low adiposity. In a later study, the mice were found to have a heightened inflammatory response with elevated plasma IL-1β and greater lethality in response to endotoxin (38). Thus, IKKβ inactivation in myeloid cells generates a proinflammatory mouse model, in which insulin sensitivity was improved. This proinflammatory status might have played a role to attenuate weight gain. Although not discussed by the authors, the inflammation likely contributed to the improved insulin sensitivity in the mice on an HFD (3). Thus, the studies suggest that, if a proinflammatory state is created in vivo, the inflammation may enhance energy expenditure to protect the animals from diet-induced obesity and insulin resistance.
JNK.
The serine kinase JNK1 that is activated by ER stress, inflammation, and lipotoxicity is believed to contribute to obesity-induced insulin resistance. JNK1 phosphorylates IRS-1 (Ser307 in rodent and Ser312 in human IRS-1) in cellular models, and global JNK1 KO mice are more sensitive to insulin (1, 99). However, a recent study of the liver-specific JNK KO mice demonstrates that loss of JNK1 impairs hepatic insulin sensitivity and predisposes the liver to steatosis when mice are placed on an HFD (117). In a separate study, activation of JNK1 by inactivating XBP1 improves insulin sensitivity in the liver (52). In contrast, JNK1 activation in other tissues (adipose tissue, skeletal muscle, and brain) impairs insulin action (118). These results suggest that JNK-dependent inflammation may regulate insulin sensitivity in a tissue-specific manner.
Anti-Inflammatory Therapy in Clinical Trials
The association of inflammation with insulin resistance in obesity suggests that inflammation may contribute to the pathogenesis of T2DM. This is supported by the fact that insulin sensitizer thiazolidinediones (TZDs) have anti-inflammatory effects, and inflammatory cytokines can induce insulin resistance in cellular and animal models (46, 95). In the past two decades, anti-inflammatory therapies have been used to improve insulin sensitivity in more than a dozen small clinical trials (Table 1). Most of the anti-inflammatory drugs are those that are routinely used in the treatment of rheumatoid arthritis, such as TNF-α antibodies (Etanercept, Infliximab, Humira, etc.) (62, 73, 93, 114), an IL-1 antibody (XOMA052) or inhibitor (Anakinra) (71), an IL-6 antibody (Tocilizumab) (94, 121), and an inflammation suppressor (salsalate, a dimer of salicylic acid) (26, 91). The therapeutics reduced inflammation in the patients in all of the clinical trials, as indicated by a decrease in the inflammatory markers C-reactive protein and IL-6. However, the therapeutic efficacy of improving insulin sensitivity remains uncertain.
Table 1.
Clinical trials for anti-inflammatory therapy
| Target | Drug | Subjects | Treatment | Insulin Sensitivity | Major Change | References |
|---|---|---|---|---|---|---|
| TNF-α | Antibody CDP571 | 21 | 4 wk | No change | No change in FBG and insulin | 93 |
| Antibody Ro 45–2081 | 7 | Once | No change | No change in FBG and insulin | 102 | |
| Etanercept | 20 | 4 wk | No change | CRP↓, IL-6↓ | Dominguez H et al. J Vasc Res 42: 517–5525, 2005 | |
| Etanercept | 56 | 4 wk | No change | CRP↓, IL-6↓ | Bernstein LE et al. Arch Intern Med 166: 902–908, 2006 | |
| Etanercept | 56 | 4 wk | No change | ADPN↑ | 73 | |
| Etanercept | 40 | 6 mo | No change | FBG↓, ADPN↑ | 127 | |
| Etanercept | 1 | 20 mo | No change | FBG↓ | Cheung D et al. J Am Acad Dermatol 60: 1032–1036, 2009 | |
| Infliximab | Improved | Yazdani-Biuki B et al. Eur J Clin Invest 34: 641–642, 2004 | ||||
| Infliximab | 45* | 24 wk | Improved | FBG↓, Insulin↓ | 62 | |
| Infliximab | 27* | Once | Improved | Insulin↓ | 36 | |
| Humira | 9* | 8 wk | No change | CRP↓, IL-6↓ | 114 | |
| IL-1 | Anakinra | 69 | 13 wk | No change | FBG↓, β-cell function ↑ | 71 |
| XOMA 052 | 98 | Once | No change | Hb A1c↓, β-cell function ↑ | Donath MY et al. Diabetologia 51: S7, 2008 | |
| XOMA 052 | 420 | 24 wk | No change | http://asweetlife.org/a-sweet-life-staff/in-the-news/type-2-in-the-news/xoma-type-2-diabetes-drug-fails-to-meet-end-point-in-phase-2-trials/14912/ | ||
| Salsalate | 16 | 2–4 wk | Improved | FFA↓, ADPN↑ | 35 | |
| IKKβ | Salsalate | 20 | 4 wk | No change | FBG↓, GTT↑ | 26 |
| JNKS6K | Salsalate | 40 | 1 wk | No change | FBG↓, GTT↑, β-cell function ↑ | Koska J et al. Diabetologia 52: 385–393, 2009 |
| Salsalate | 27 | 14 wk | Not reported | Hb A1c ↓ | 34 | |
| Salsalate | 17 | 2 wk | No change | 11β-HSD1↓ | 91 | |
| IKKβ | Statins | 32,752 | 1.9–4.9 yr | Diabetes incidence ↑ | Cardiovascular events↓ | Preiss D et al. JAMA 305: 2556–2564, 2011 |
| JNK | ||||||
| HIF-1a | Statins | 90,000 | Years | Diabetes incidence↑ | Cardiovascular events↓ | 108 |
| IL-6 | Tocilizumab | 11* | 12 wk | Improved | Insulin↓, CRP↓, ADPN↑, | 121 |
| Tocilizumab | 10* | 1–6 mo | Not reported | Hb A1c↓ | 94 |
FBG, fasting blood glucose; CRP, C-reactive protein; Hb A1c, hemoglobin A1c; FFA, free fatty acid; ADPN, adiponectin; 11β-HSD1, 11β-hydroxysteroid dehydrogenase type 1.
Anti-TNF-α therapy was the first among those tested in clinical trials. There were more than 10 published studies for anti-TNF-α therapies in diabetic patients between 1996 and 2011 (Table 1). The drugs include Infliximab, Etanercept, and Humira (62, 73, 93, 114). Unfortunately, the therapy never entered the stage of large, multicenter trials due to inconsistent or negative results (93, 102). The therapy improved insulin sensitivity in patients with rheumatoid arthritis (36, 62, 114), but it was ineffective in diabetic patients without rheumatoid arthritis (73, 93, 114, 127). The discrepancy is likely related to the cortisone that was used in the management of rheumatoid arthritis in the diabetic patients. These studies suggest that TNF-α is not a good target in the improvement of insulin sensitivity or glucose disorder in T2DM patients.
IL-1 inhibitors improved glucose homeostasis in two of three small trials (Table 1). However, β-cell function, but not insulin action, was improved in the two trials. In the third trial, the IL-1 blocker (MOXA 052) failed to give the expected effects in the improvement of insulin resistance and glucose disorder (Table 1). Currently, the IL-1 antibody ACZ885 is being tested in a large clinical trial (600 subjects, NCT00900146*), and the final results are not yet known (22).
The impact of an IL-6 signaling inhibitor, Tocilizumab, on glucose homeostasis was evaluated in rheumatoid arthritis patients (94, 121). Tocilizumab improved glucose homeostasis (decreased Hb A1c) in diabetic patients and improved insulin action (decreased HOMA-IR) in nondiabetic subjects (94, 121). In the studies, the patients were receiving prednisolone for the management of rheumatoid arthritis. This makes the data interpretation complex, as prednisolone induces insulin resistance. The studies are not sufficient to suggest that IL-6 is a good target in the control of insulin resistance hyperglycemia in T2DM patients.
Salsalate, a dimer of salicylic acid, is used in the treatment of rheumatoid arthritis and osteoarthritis. Sodium salicylate (aspirin) at high doses improves insulin sensitivity in humans and mice (60, 149). Aspirin inhibits multiple serine kinases that phosphorylate IRS-1 in the suppression of the insulin signaling pathway (32). A recent study suggests that salicylate activates AMPK (AMP-activated protein kinase) through a direct interaction with AMPK protein, which is observed in the control of blood lipids (42). Aspirin also inhibits the expression of multiple inflammatory cytokines including IL-1 and TNF-α. The therapeutic value of aspirin is limited by the increased bleeding risk at the high dosages required to generate the metabolic effects. To overcome this weakness, salsalate (related to aspirin in structure) was chosen in diabetic clinical trials for its safety record in the treatment of rheumatoid arthritis (26, 34, 91). There have been reports from five small clinical trials using salsalate (Table 1). Insulin sensitivity (35) and glucose control (34) were improved in two small studies in T2DM subjects. Insulin concentration also was improved in young, obese, nondiabetic subjects (26). Glucose homeostasis was not evaluated in another study (91). A large trial for salsalate (284 subjects, NCT00799643*) is ongoing, but the preliminary results presented at a recent national meeting (American Diabetes Association) were that salsalate improved Hb A1c but did not change insulin sensitivity as assessed by fasting insulin and glucose. The improved Hb A1c was associated with an elevation in plasma insulin in the subjects. The results do not provide support to the role of inflammation in the pathogenesis of insulin resistance.
Statins are widely used in the control of blood cholesterol. In addition to the reduction of cholesterol, they also downregulate activities of several transcription factors, including NF-κB, AP-1, and HIF-1α, in the inflammatory pathway (21). The impact of statins on glucose metabolism and insulin sensitivity has been examined in a meta-analysis of more than 90,000 patients (108) who participated in major cardiovascular clinical trials (Table 1). The analysis reveals that statin therapy is associated with a 9% increase in the risk for developing T2DM. Compared with a moderate dose, high-dose statins exacerbate the risk. In these trials, participants did not have diabetes prior to therapy. Although statins are not the classical anti-inflammatory medicines, and the mechanism of statin-associated risk for diabetes is unknown, the anti-inflammatory activities of statins may contribute to the increased risk of T2DM in the study.
Our conclusion is that anti-inflammatory therapies are not very efficacious in improving insulin sensitivity in clinical trials. The therapies exhibit modest activities in improving glucose metabolism in some studies. The reason for the low efficacy is unknown. We propose that accounting for the beneficial effects of inflammation may be the key to understanding why obesity-associated inflammation is not as bad as we originally believed.
Conclusion
Many studies suggest that inflammation is a compensatory response in obesity. It has both positive and negative metabolic effects that impact energy expenditure, glucose homeostasis, and insulin action. Theoretically, the metabolic impact of inflammation is determined by the tissue location, the profiles of inflammatory mediators, and the cell types involved. Inflammation can increase energy expenditure and limit food intake as well as improve insulin sensitivity (148). This possibility has been supported by observations in many animal studies, which help us to gain a balanced view of inflammation. We recognize that those animal studies have limitations in terms of clinical relevance. However, the studies do help us to gain insight into the nature of obesity-associated inflammation. The negative impact of inflammation has been well documented in the regulation of glucose and lipid metabolism in obesity, and it has been the foundation of rationale for anti-inflammatory therapies in T2DM patients. Unfortunately, the therapies, in general, have done little to improve insulin sensitivity and glucose homeostasis, especially compared with the standard of care for individuals with diabetes. Currently, there is no consensus as to why the anti-inflammatory therapies have largely been unsuccessful. We propose that the positive activity of inflammation may be the key. Although the relative significance of the positive and negative effects of inflammation remains to be determined in obesity and T2DM, the evidence does not suggest that inflammation is a good therapeutic target to improve insulin sensitivity. Currently, the anti-inflammatory approaches cannot distinguish between the positive and negative effects of inflammation.
Several potential areas have been indicated in this review concerning future directions in obesity-associated inflammation. These areas deserve to be explored further. If inflammation prevents energy accumulation in obesity, why does obesity occur in the presence of inflammation? In the course of weight gain, does the anti-inflammatory activity become a dominant player in controlling energy balance and glucose homeostasis? Does the inhibition of islet function by inflammation have negative or positive effects on energy balance in obesity? As physical exercise is associated with chronic inflammation (e.g., elevated IL-6), should proinflammatory approaches be considered a part of the antiobesity strategy? Answers to these questions may lead to development of new approaches in the treatment of obesity and T2DM.
GRANTS
J. Ye is supported by National Institute of Diabetes and Digestive and Kidney Diseases grants (DK-085495 and DK-068036). O. P. McGuinness is supported by projects including the Vanderbilt Diabetes Research and Training Center (DK-020593), the Vanderbilt Mouse Metabolic Phenotyping Center (DK-059637), and RO1 projects (DK-043748 and DK-078188).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: J.Y. conception and design of research; J.Y. prepared figures; J.Y. and O.P.M. drafted manuscript; J.Y. and O.P.M. edited and revised manuscript; J.Y. and O.P.M. approved final version of manuscript; O.P.M. analyzed data.
ACKNOWLEDGMENTS
We highly appreciate comments from Dr. George Bray, Dr. Barbara Kahn, and Dr. Frank Greenway. J. Ye and O. P. McGuinness are the guarantors of this work, had full access to all the data, and take full responsibility for the integrity of the data and accuracy of data analysis.
REFERENCES
- 1. Aguirre V, Uchida T, Yenush L, Davis R, White MF. The c-Jun NH(2)-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser(307). J Biol Chem 275: 9047–9054, 2000 [DOI] [PubMed] [Google Scholar]
- 2. Agwunobi AO, Reid C, Maycock P, Little RA, Carlson GL. Insulin resistance and substrate utilization in human endotoxemia. J Clin Endocrinol Metab 85: 3770–3778, 2000 [DOI] [PubMed] [Google Scholar]
- 3. Arkan MC, Hevener AL, Greten FR, Maeda S, Li ZW, Long JM, Wynshaw-Boris A, Poli G, Olefsky J, Karin M. IKK-beta links inflammation to obesity-induced insulin resistance. Nat Med 11: 191–198, 2005 [DOI] [PubMed] [Google Scholar]
- 4. Arnush M, Heitmeier MR, Scarim AL, Marino MH, Manning PT, Corbett JA. IL-1 produced and released endogenously within human islets inhibits beta cell function. J Clin Invest 102: 516–526, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Batra A, Okur B, Glauben R, Erben U, Ihbe J, Stroh T, Fedke I, Chang HD, Zeitz M, Siegmund B. Leptin: a critical regulator of CD4+ T-cell polarization in vitro and in vivo. Endocrinology 151: 56–62, 2010 [DOI] [PubMed] [Google Scholar]
- 6. Baumann H, Morella KK, White DW, Dembski M, Bailon PS, Kim H, Lai CF, Tartaglia LA. The full-length leptin receptor has signaling capabilities of interleukin 6-type cytokine receptors. Proc Natl Acad Sci USA 93: 8374–8378, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Beutler B, Milsark IW, Cerami AC. Passive immunization against cachectin/tumor necrosis factor protects mice from lethal effect of endotoxin. Science 229: 869–871, 1985 [DOI] [PubMed] [Google Scholar]
- 8. Blackwell TS, Christman JW. Sepsis and cytokines: current status. Br J Anaesthesia 77: 110–117, 1996 [DOI] [PubMed] [Google Scholar]
- 9. Busso N, So A, Chobaz-Péclat V, Morard C, Martinez-Soria E, Talabot-Ayer D, Gabay C. Leptin signaling deficiency impairs humoral and cellular immune responses and attenuates experimental arthritis. J Immunol 168: 875–882, 2002 [DOI] [PubMed] [Google Scholar]
- 10. Cai D, Frantz JD, Tawa NE, Jr, Melendez PA, Oh BC, Lidov HG, Hasselgren PO, Frontera WR, Lee J, Glass DJ, Shoelson SE. IKKbeta/NF-kappaB activation causes severe muscle wasting in mice. Cell 119: 285–298, 2004 [DOI] [PubMed] [Google Scholar]
- 11. Cai D, Yuan M, Frantz DF, Melendez PA, Hansen L, Lee J, Shoelson SE. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat Med 11: 183–190, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, Neyrinck AM, Fava F, Tuohy KM, Chabo C, Waget A, Delmee E, Cousin B, Sulpice T, Chamontin B, Ferrieres J, Tanti JF, Gibson GR, Casteilla L, Delzenne NM, Alessi MC, Burcelin R. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 56: 1761–1772, 2007 [DOI] [PubMed] [Google Scholar]
- 13. Cao R, Farnebo J, Kurimoto M, Cao Y. Interleukin-18 acts as an angiogenesis and tumor suppressor. FASEB J 13: 2195–2202, 1999 [DOI] [PubMed] [Google Scholar]
- 14. Cao Y. Adipose tissue angiogenesis as a therapeutic target for obesity and metabolic diseases. Nat Rev Drug Discov 9: 107–115, 2010 [DOI] [PubMed] [Google Scholar]
- 15. Cao Y. Angiogenesis modulates adipogenesis and obesity. J Clin Invest 117: 2362–2368, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chi H, Barry SP, Roth RJ, Wu JJ, Jones EA, Bennett AM, Flavell RA. Dynamic regulation of pro- and anti-inflammatory cytokines by MAPK phosphatase 1 (MKP-1) in innate immune responses. Proc Natl Acad Sci USA 103: 2274–2279, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Christiaens V, Lijnen HR. Angiogenesis and development of adipose tissue. Mol Cell Endocrinol 318: 2–9, 2010 [DOI] [PubMed] [Google Scholar]
- 18. Cinti S, Mitchell G, Barbatelli G, Murano I, Ceresi E, Faloia E, Wang S, Fortier M, Greenberg AS, Obin MS. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J Lipid Res 46: 2347–2355, 2005 [DOI] [PubMed] [Google Scholar]
- 19. Cohen SL, Halaas JL, Friedman JM, Chait BT, Bennett L, Chang D, Hecht R, Collins F. Human leptin characterization. Nature 382: 589, 1996 [DOI] [PubMed] [Google Scholar]
- 20. Crowe S, Wu LE, Economou C, Turpin SM, Matzaris M, Hoehn KL, Hevener AL, James DE, Duh EJ, Watt MJ. Pigment epithelium-derived factor contributes to insulin resistance in obesity. Cell Metab 10: 40–47, 2009 [DOI] [PubMed] [Google Scholar]
- 21. Dichtl W, Dulak J, Frick M, Alber HF, Schwarzacher SP, Ares MP, Nilsson J, Pachinger O, Weidinger F. HMG-CoA reductase inhibitors regulate inflammatory transcription factors in human endothelial and vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 23: 58–63, 2003 [DOI] [PubMed] [Google Scholar]
- 22. Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol 11: 98–107, 2011 [DOI] [PubMed] [Google Scholar]
- 23. Esposito K, Pontillo A, Giugliano F, Giugliano G, Marfella R, Nicoletti G, Giugliano D. Association of low interleukin-10 levels with the metabolic syndrome in obese women. J Clin Endocrinol Metab 88: 1055–1058, 2003 [DOI] [PubMed] [Google Scholar]
- 24. Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, Lee J, Goldfine AB, Benoist C, Shoelson S, Mathis D. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med 15: 930–939, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fischer CP, Perstrup LB, Berntsen A, Eskildsen P, Pedersen BK. Elevated plasma interleukin-18 is a marker of insulin-resistance in type 2 diabetic and non-diabetic humans. Clin Immunol 117: 152–160, 2005 [DOI] [PubMed] [Google Scholar]
- 26. Fleischman A, Shoelson SE, Bernier R, Goldfine AB. Salsalate improves glycemia and inflammatory parameters in obese young adults. Diabetes Care 31: 289–294, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Folco EJ, Rocha VZ, Lopez-Ilasaca M, Libby P. Adiponectin inhibits proinflammatory signaling in human macrophages independent of interleukin-10. J Biol Chem 284: 25569–25575, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Franckhauser S, Elias I, Rotter Sopasakis V, Ferre T, Nagaev I, Andersson CX, Agudo J, Ruberte J, Bosch F, Smith U. Overexpression of Il6 leads to hyperinsulinaemia, liver inflammation and reduced body weight in mice. Diabetologia 51: 1306–1316, 2008 [DOI] [PubMed] [Google Scholar]
- 29. Fried SK, Bunkin DA, Greenberg AS. Omental and subcutaneous adipose tissues of obese subjects release interleukin-6: depot difference and regulation by glucocorticoid. J Clin Endocrinol Metab 83: 847–850, 1998 [DOI] [PubMed] [Google Scholar]
- 30. Gainsford T, Willson TA, Metcalf D, Handman E, McFarlane C, Ng A, Nicola NA, Alexander WS, Hilton DJ. Leptin can induce proliferation, differentiation, and functional activation of hemopoietic cells. Proc Natl Acad Sci USA 93: 14564–14568, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gao Z, Yin JJZ, He Q, McGuinness OP, Ye J. Inactivation of NF-κB p50 leads to insulin sensitization in liver through post-translational inhibition of p70S6K. J Biol Chem 284: 18368–18376 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gao Z, Zuberi A, Quon M, Dong Z, Ye J. Aspirin inhibits serine phosphorylation of insulin receptor substrate 1 in tumor necrosis factor-treated cells through targeting multiple serine kinases. J Biol Chem 278: 24944–24950, 2003 [DOI] [PubMed] [Google Scholar]
- 33. Garcia MC, Wernstedt I, Berndtsson A, Enge M, Bell M, Hultgren O, Horn M, Ahren B, Enerback S, Ohlsson C, Wallenius V, Jansson JO. Mature-onset obesity in interleukin-1 receptor I knockout mice. Diabetes 55: 1205–1213, 2006 [DOI] [PubMed] [Google Scholar]
- 34. Goldfine AB, Fonseca V, Jablonski KA, Pyle L, Staten MA, Shoelson SE. The effects of salsalate on glycemic control in patients with type 2 diabetes: a randomized trial. Ann Int Med 152: 346–357, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Goldfine AB, Silver R, Aldhahi W, Cai D, Tatro E, Lee J, Shoelson SE. Use of salsalate to target inflammation in the treatment of insulin resistance and type 2 diabetes. Clin Transl Sci 1: 36–43, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gonzalez-Gay MA, De Matias JM, Gonzalez-Juanatey C, Garcia-Porrua C, Sanchez-Andrade A, Martin J, Llorca J. Anti-tumor necrosis factor-alpha blockade improves insulin resistance in patients with rheumatoid arthritis. Clin Exper Rheumatol 24: 83–86, 2006 [PubMed] [Google Scholar]
- 37. Gordon S. Alternative activation of macrophages. Nat Rev Immunol 3: 23–35, 2003 [DOI] [PubMed] [Google Scholar]
- 38. Greten FR, Arkan MC, Bollrath J, Hsu LC, Goode J, Miething C, Goktuna SI, Neuenhahn M, Fierer J, Paxian S, Van Rooijen N, Xu Y, O'Cain T, Jaffee BB, Busch DH, Duyster J, Schmid RM, Eckmann L, Karin M. NF-κB is a negative regulator of IL-1β secretion as revealed by genetic and pharmacological inhibition of IKKβ. Cell 130: 918–931, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Grunfeld C, Feingold KR. Regulation of lipid metabolism by cytokines during host defense. Nutrition 12: S24–S26, 1996 [DOI] [PubMed] [Google Scholar]
- 40. Halberg N, Khan T, Trujillo ME, Wernstedt-Asterholm I, Attie AD, Sherwani S, Wang ZV, Landskroner-Eiger S, Dineen S, Magalang UJ, Brekken RA, Scherer PE. HIF 1 alpha induces fibrosis and insulin resistance in white adipose tissue. Mol Cell Biol 29: 4467–4483, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hausman GJ, Richardson RL. Adipose tissue angiogenesis. J Anim Sci 82: 925–934, 2004 [DOI] [PubMed] [Google Scholar]
- 42. Hawley SA, Fullerton MD, Ross FA, Schertzer JD, Chevtzoff C, Walker KJ, Peggie MW, Zibrova D, Green KA, Mustard KJ, Kemp BE, Sakamoto K, Steinberg GR, Hardie DG. The ancient drug salicylate directly activates AMP-activated protein kinase. Science 336: 918–922, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. He Q, Gao Z, Yin J, Zhang J, Yun Z, Ye J. Regulation of HIF-1a activity in adipose tissue by obesity-associated factors: adipogenesis, insulin and hypoxia. Am J Physiol Endocrinol Metab 300: E877–E885, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Heiman ML, Ahima RS, Craft LS, Schoner B, Stephens TW, Flier JS. Leptin inhibition of the hypothalamic-pituitary-adrenal axis in response to stress. Endocrinology 138: 3859–3863, 1997 [DOI] [PubMed] [Google Scholar]
- 45. Hong EG, Ko HJ, Cho YR, Kim HJ, Ma Z, Yu TY, Friedline RH, Kurt-Jones E, Finberg R, Fischer MA, Granger EL, Norbury CC, Hauschka SD, Philbrick WM, Lee CG, Elias JA, Kim JK. Interleukin-10 prevents diet-induced insulin resistance by attenuating macrophage and cytokine response in skeletal muscle. Diabetes 58: 2525–2535, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hotamisligil GS. Inflammation and metabolic disorders. Nature 444: 860–867, 2006 [DOI] [PubMed] [Google Scholar]
- 47. Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman BM. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J Clin Invest 95: 2409–2415, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science 259: 87–91, 1993 [DOI] [PubMed] [Google Scholar]
- 49. Inoue H, Ogawa W, Ozaki M, Haga S, Matsumoto M, Furukawa K, Hashimoto N, Kido Y, Mori T, Sakaue H, Teshigawara K, Jin S, Iguchi H, Hiramatsu R, LeRoith D, Takeda K, Akira S, Kasuga M. Role of STAT-3 in regulation of hepatic gluconeogenic genes and carbohydrate metabolism in vivo. Nat Med 10: 168–174, 2004 [DOI] [PubMed] [Google Scholar]
- 50. Jiao P, Feng B, Ma J, Nie Y, Paul E, Li Y, Xu H. Constitutive activation of IKKβ in adipose tissue prevents diet-induced obesity in mice. Endocrinology 153: 154–165, 2012 [DOI] [PubMed] [Google Scholar]
- 51. Juge-Aubry CE, Somm E, Giusti V, Pernin A, Chicheportiche R, Verdumo C, Rohner-Jeanrenaud F, Burger D, Dayer JM, Meier CA. Adipose tissue is a major source of interleukin-1 receptor antagonist: upregulation in obesity and inflammation. Diabetes 52: 1104–1110, 2003 [DOI] [PubMed] [Google Scholar]
- 52. Jurczak MJ, Lee AH, Jornayvaz FR, Lee HY, Birkenfeld AL, Guigni BA, Kahn M, Samuel VT, Glimcher LH, Shulman GI. Dissociation of inositol-requiring enzyme (IRE1α)-mediated c-Jun N-terminal kinase activation from hepatic insulin resistance in conditional X-box-binding protein-1 (XBP1) knock-out mice. J Biol Chem 287: 2558–2567, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kaiyala KJ, Morton GJ, Leroux BG, Ogimoto K, Wisse B, Schwartz MW. Identification of body fat mass as a major determinant of metabolic rate in mice. Diabetes 59: 1657–1666, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kaiyala KJ, Schwartz MW. Toward a more complete (and less controversial) understanding of energy expenditure and its role in obesity pathogenesis. Diabetes 60: 17–23, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kanneganti TD, Lamkanfi M, Nunez G. Intracellular NOD-like receptors in host defense and disease. Immunity 27: 549–559, 2007 [DOI] [PubMed] [Google Scholar]
- 56. Kelly CJ, Colgan SP, Frank DN. Of Microbes and meals: the health consequences of dietary endotoxemia. Nutr Clin Pract 27: 215–225, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kern PA, Ranganathan S, Li C, Wood L, Ranganathan G. Adipose tissue tumor necrosis factor and interleukin-6 expression in human obesity and insulin resistance. Am J Physiol Endocrinol Metab 280: E745–E751, 2001 [DOI] [PubMed] [Google Scholar]
- 58. Khalkhal A, Haddar A, Semiane N, Mallek A, Abdelmalek A, Castex F, Gross R, Dahmani Y. Obesity, insulin resistance and diabetes in the sand rat exposed to a hypercaloric diet; possible protective effect for IL1-β. Comptes Rendus Biologies 335: 271–278, 2012 [DOI] [PubMed] [Google Scholar]
- 59. Kim HJ, Higashimori T, Park SY, Choi H, Dong J, Kim YJ, Noh HL, Cho YR, Cline G, Kim YB, Kim JK. Differential effects of interleukin-6 and -10 on skeletal muscle and liver insulin action in vivo. Diabetes 53: 1060–1067, 2004 [DOI] [PubMed] [Google Scholar]
- 60. Kim JK, Kim YJ, Fillmore JJ, Chen Y, Moore I, Lee J, Yuan M, Li ZW, Karin M, Perret P, Shoelson SE, Shulman GI. Prevention of fat-induced insulin resistance by salicylate. J Clin Invest 108: 437–446, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kim JY, van de Wall E, Laplante M, Azzara A, Trujillo ME, Hofmann SM, Schraw T, Durand JL, Li H, Li G, Jelicks LA, Mehler MF, Hui DY, Deshaies Y, Shulman GI, Schwartz GJ, Scherer PE. Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J Clin Invest 117: 2621–2637, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kiortsis DN, Mavridis AK, Vasakos S, Nikas SN, Drosos AA. Effects of infliximab treatment on insulin resistance in patients with rheumatoid arthritis and ankylosing spondylitis. Ann Rheum Dis 64: 765–766, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Klover PJ, Clementi AH, Mooney RA. Interleukin-6 depletion selectively improves hepatic insulin action in obesity. Endocrinology 146: 3417–3427, 2005 [DOI] [PubMed] [Google Scholar]
- 64. Klover PJ, Zimmers TA, Koniaris LG, Mooney RA. Chronic exposure to interleukin-6 causes hepatic insulin resistance in mice. Diabetes 52: 2784–2789, 2003 [DOI] [PubMed] [Google Scholar]
- 65. Kosteli A, Sugaru E, Haemmerle G, Martin JF, Lei J, Zechner R, Ferrante AW., Jr Weight loss and lipolysis promote a dynamic immune response in murine adipose tissue. J Clin Invest 120: 3466–3479, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kowalski GM, Nicholls HT, Risis S, Watson NK, Kanellakis P, Bruce CR, Bobik A, Lancaster GI, Febbraio MA. Deficiency of haematopoietic-cell-derived IL-10 does not exacerbate high-fat-diet-induced inflammation or insulin resistance in mice. Diabetologia 54: 888–899, 2011 [DOI] [PubMed] [Google Scholar]
- 67. Kozak W, Conn CA, Kluger MJ. Lipopolysaccharide induces fever and depresses locomotor activity in unrestrained mice. Am J Physiol Regul Integr Comp Physiol 266: R125–R135, 1994 [DOI] [PubMed] [Google Scholar]
- 68. Krogh-Madsen R, Plomgaard P, Moller K, Mittendorfer B, Pedersen BK. Influence of TNF-α and IL-6 infusions on insulin sensitivity and expression of IL-18 in humans. Am J Physiol Endocrinol Metab 291: E108–E114, 2006 [DOI] [PubMed] [Google Scholar]
- 69. Kuhn R, Lohler J, Rennick D, Rajewsky K, Muller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell 75: 263–274, 1993 [DOI] [PubMed] [Google Scholar]
- 70. Lang CH, Dobrescu C. Interleukin-1 induced increases in glucose utilization are insulin mediated. Life Sci 45: 2127–2134, 1989 [DOI] [PubMed] [Google Scholar]
- 71. Larsen CM, Faulenbach M, Vaag A, Volund A, Ehses JA, Seifert B, Mandrup-Poulsen T, Donath MY. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med 356: 1517–1526, 2007 [DOI] [PubMed] [Google Scholar]
- 72. Lee J, Sun C, Zhou Y, Gokalp D, Herrema H, Park SW, Davis RJ, Ozcan U. p38 MAPK-mediated regulation of Xbp1s is crucial for glucose homeostasis. Nat Med 17: 1251–1260, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lo J, Bernstein LE, Canavan B, Torriani M, Jackson MB, Ahima RS, Grinspoon SK. Effects of TNF-α neutralization on adipocytokines and skeletal muscle adiposity in the metabolic syndrome. Am J Physiol Endocrinol Metab 293: E102–E109, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Loffreda S, Yang SQ, Lin HZ, Karp CL, Brengman ML, Wang DJ, Klein AS, Bulkley GB, Bao C, Noble PW, Lane MD, Diehl AM. Leptin regulates proinflammatory immune responses. FASEB J 12: 57–65, 1998 [PubMed] [Google Scholar]
- 75. Lord GM, Matarese G, Howard JK, Baker RJ, Bloom SR, Lechler RI. Leptin modulates the T-cell immune response and reverses starvation-induced immunosuppression. Nature 394: 897–901, 1998 [DOI] [PubMed] [Google Scholar]
- 76. Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest 117: 175–184, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Martin-Romero C, Sanchez-Margalet V. Human leptin activates PI3K and MAPK pathways in human peripheral blood mononuclear cells: possible role of Sam68. Cell Immunol 212: 83–91, 2001 [DOI] [PubMed] [Google Scholar]
- 78. Matsuki T, Horai R, Sudo K, Iwakura Y. IL-1 plays an important role in lipid metabolism by regulating insulin levels under physiological conditions. J Exp Med 198: 877–888, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Mattioli B, Straface E, Quaranta MG, Giordani L, Viora M. Leptin promotes differentiation and survival of human dendritic cells and licenses them for Th1 priming. J Immunol 174: 6820–6828, 2005 [DOI] [PubMed] [Google Scholar]
- 80. McGuinness OP. Defective glucose homeostasis during infection. Annu Rev Nutr 25: 9–35, 2005 [DOI] [PubMed] [Google Scholar]
- 81. Mehta NN, McGillicuddy FC, Anderson PD, Hinkle CC, Shah R, Pruscino L, Tabita-Martinez J, Sellers KF, Rickels MR, Reilly MP. Experimental endotoxemia induces adipose inflammation and insulin resistance in humans. Diabetes 59: 172–181, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Meier CA, Bobbioni E, Gabay C, Assimacopoulos-Jeannet F, Golay A, Dayer JM. IL-1 receptor antagonist serum levels are increased in human obesity: a possible link to the resistance to leptin? J Clin Endocrinol Metab 87: 1184–1188, 2002 [DOI] [PubMed] [Google Scholar]
- 83. MohanKumar SM, Smith CL, MohanKumar PS. Central adaptation to chronic administration of interleukin-1beta (IL-1beta) in rats. Brain Res Bull 62: 71–76, 2003 [DOI] [PubMed] [Google Scholar]
- 84. Morton GJ, Schwartz MW. Leptin and the central nervous system control of glucose metabolism. Physiol Rev 91: 389–411, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Mulligan KX, Morris RT, Otero YF, Wasserman DH, McGuinness OP. Disassociation of muscle insulin signaling and insulin-stimulated glucose uptake during endotoxemia. PLoS ONE 7: e30160, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Najib S, Sanchez-Margalet V. Human leptin promotes survival of human circulating blood monocytes prone to apoptosis by activation of p42/44 MAPK pathway. Cell Immunol 220: 143–149, 2002 [DOI] [PubMed] [Google Scholar]
- 87. Netea MG, Joosten LA, Lewis E, Jensen DR, Voshol PJ, Kullberg BJ, Tack CJ, van Krieken H, Kim SH, Stalenhoef AF, van de Loo FA, Verschueren I, Pulawa L, Akira S, Eckel RH, Dinarello CA, van den Berg W, van der Meer JW. Deficiency of interleukin-18 in mice leads to hyperphagia, obesity and insulin resistance. Nat Med 12: 650–656, 2006 [DOI] [PubMed] [Google Scholar]
- 88. Nguyen KD, Qiu Y, Cui X, Goh YPS, Mwangi J, David T, Mukundan L, Brombacher F, Locksley RM, Chawla A. Alternatively activated macrophages produce catecholamines to sustain adaptive thermogenesis. Nature 480: 104–108, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Nimah M, Zhao B, Denenberg AG, Bueno O, Molkentin J, Wong HR, Shanley TP. Contribution of MKP-1 regulation of p38 to endotoxin tolerance. Shock 23: 80–87, 2005 [DOI] [PubMed] [Google Scholar]
- 90. Nishimura S, Manabe I, Nagasaki M, Hosoya Y, Yamashita H, Fujita H, Ohsugi M, Tobe K, Kadowaki T, Nagai R, Sugiura S. Adipogenesis in obesity requires close interplay between differentiating adipocytes, stromal cells, and blood vessels. Diabetes 56: 1517–1526, 2007 [DOI] [PubMed] [Google Scholar]
- 91. Nixon M, Wake DJ, Livingstone DE, Stimson RH, Esteves CL, Seckl JR, Chapman KE, Andrew R, Walker BR. Salicylate downregulates 11β-HSD1 expression in adipose tissue in obese mice and in humans, mediating insulin sensitization. Diabetes 61: 790–796, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, Eagle AR, Vats D, Brombacher F, Ferrante AW, Chawla A. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature 447: 1116–1120, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Ofei F, Hurel S, Newkirk J, Sopwith M, Taylor R. Effects of an engineered human anti-TNF-alpha antibody (CDP571) on insulin sensitivity and glycemic control in patients with NIDDM. Diabetes 45: 881–885, 1996 [DOI] [PubMed] [Google Scholar]
- 94. Ogata A, Morishima A, Hirano T, Hishitani Y, Hagihara K, Shima Y, Narazaki M, Tanaka T. Improvement of HbA1c during treatment with humanised anti-interleukin 6 receptor antibody, tocilizumab. Ann Rheum Dis 70: 1164–1165, 2011 [DOI] [PubMed] [Google Scholar]
- 95. Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol 72: 219–246, 2010 [DOI] [PubMed] [Google Scholar]
- 96. Osto M, Zini E, Franchini M, Wolfrum C, Guscetti F, Hafner M, Ackermann M, Reusch CE, Lutz TA. Subacute endotoxemia induces adipose inflammation and changes in lipid and lipoprotein metabolism in cats. Endocrinology 152: 804–815, 2011 [DOI] [PubMed] [Google Scholar]
- 97. Ota K, Wildmann J, Ota T, Besedovsky HO, Del Rey A. Interleukin-1β and insulin elicit different neuroendocrine responses to hypoglycemia. Ann NY Acad Sci 1153: 82–88, 2009 [DOI] [PubMed] [Google Scholar]
- 98. Ouchi N, Kihara S, Arita Y, Okamoto Y, Maeda K, Kuriyama H, Hotta K, Nishida M, Takahashi M, Muraguchi M, Ohmoto Y, Nakamura T, Yamashita S, Funahashi T, Matsuzawa Y. Adiponectin, an adipocyte-derived plasma protein, inhibits endothelial NF-kappaB signaling through a cAMP-dependent pathway. Circulation 102: 1296–1301, 2000 [DOI] [PubMed] [Google Scholar]
- 99. Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Gorgun C, Glimcher LH, Hotamisligil GS. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306: 457–461, 2004 [DOI] [PubMed] [Google Scholar]
- 100. Pamir N, McMillen TS, Kaiyala KJ, Schwartz MW, LeBoeuf RC. Receptors for tumor necrosis factor-(alpha) play a protective role against obesity and alter adipose tissue macrophage status. Endocrinology 150: 4124–4134, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Pang C, Gao Z, Yin J, Zhang J, Jia W, Ye J. Macrophage infiltration into adipose tissue may promote angiogenesis for adipose tissue remodeling in obesity. Am J Physiol Endocrinol Metab 295: E313–E322, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Paquot N, Castillo MJ, Lefebvre PJ, Scheen AJ. No increased insulin sensitivity after a single intravenous administration of a recombinant human tumor necrosis factor receptor: Fc fusion protein in obese insulin-resistant patients. J Clin Endocrinol Metab 85: 1316–1319, 2000 [DOI] [PubMed] [Google Scholar]
- 103. Park PH, Huang H, McMullen MR, Bryan K, Nagy LE. Activation of cyclic-AMP response element binding protein contributes to adiponectin-stimulated interleukin-10 expression in RAW 264.7 macrophages. J Leukoc Biol 83: 1258–1266, 2008 [DOI] [PubMed] [Google Scholar]
- 104. Pasarica M, Sereda OR, Redman LM, Albarado DC, Hymel DT, Roan LE, Rood JC, Burk DH, Smith SR. Reduced adipose tissue oxygenation in human obesity: evidence for rarefaction, macrophage chemotaxis, and inflammation without an angiogenic response. Diabetes 58: 718–725, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Pedersen BK. IL-6 signalling in exercise and disease. Biochem Soc Trans 35: 1295–1297, 2007 [DOI] [PubMed] [Google Scholar]
- 106. Pedersen BK, Åkerström TCA, Nielsen AR, Fischer CP. Role of myokines in exercise and metabolism. J Appl Physiol 103: 1093–1098, 2007 [DOI] [PubMed] [Google Scholar]
- 107. Petit F, Jarrous A, Dickinson RD, Molina PE, Abumrad NN, Lang CH. Contribution of central and peripheral adrenergic stimulation to IL-1α-mediated glucoregulation. Am J Physiol Endocrinol Metab 267: E49–E56, 1994 [DOI] [PubMed] [Google Scholar]
- 108. Preiss D, Sattar N. Statins and the risk of new-onset diabetes: a review of recent evidence. Curr Opin Lipidol 22: 460–466, 2011 [DOI] [PubMed] [Google Scholar]
- 109. Qiao L, Kinney B, Schaack J, Shao J. Adiponectin inhibits lipolysis in mouse adipocytes. Diabetes 60: 1519–1527, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Qiu P, Cui X, Barochia A, Li Y, Natanson C, Eichacker PQ. The evolving experience with therapeutic TNF inhibition in sepsis: considering the potential influence of risk of death. Exp Opin Invest Drugs 20: 1555–1564, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Ranieri VM, Thompson BT, Barie PS, Dhainaut JF, Douglas IS, Finfer S, Gårdlund B, Marshall JC, Rhodes A, Artigas A, Payen D, Tenhunen J, Al-Khalidi HR, Thompson V, Janes J, Macias WL, Vangerow B, Williams MD. Drotrecogin alfa (activated) in adults with septic shock. N Engl J Med 366: 2055–2064, 2012 [DOI] [PubMed] [Google Scholar]
- 112. Reinhart K, Karzai W. Anti-tumor necrosis factor therapy in sepsis: update on clinical trials and lessons learned. Crit Care Med 29: S121–S125, 2001 [DOI] [PubMed] [Google Scholar]
- 113. Rohl M, Pasparakis M, Baudler S, Baumgartl J, Gautam D, Huth M, De Lorenzi R, Krone W, Rajewsky K, Bruning JC. Conditional disruption of I(kappa)B kinase 2 fails to prevent obesity-induced insulin resistance. J Clin Invest 113: 474–481, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Rosenvinge A, Krogh-Madsen R, Baslund B, Pedersen BK. Insulin resistance in patients with rheumatoid arthritis: effect of anti-TNFalpha therapy. Scand J Rheumatol 36: 91–96, 2007 [DOI] [PubMed] [Google Scholar]
- 115. Rui L, Aguirre V, Kim JK, Shulman GI, Lee A, Corbould A, Dunaif A, White MF. Insulin/IGF-1 and TNF-alpha stimulate phosphorylation of IRS-1 at inhibitory Ser307 via distinct pathways. J Clin Invest 107: 181–189, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Sabater M, Moreno-Navarrete JM, Ortega FJ, Pardo G, Salvador J, Ricart W, Fruhbeck G, Fernandez-Real JM. Circulating pigment epithelium-derived factor levels are associated with insulin resistance and decrease after weight loss. J Clin Endocrinol Metab 95: 4720–4728, 2010 [DOI] [PubMed] [Google Scholar]
- 117. Sabio G, Cavanagh-Kyros J, Ko HJ, Jung DY, Gray S, Jun JY, Barrett T, Mora A, Kim JK, Davis RJ. Prevention of steatosis by hepatic JNK1. Cell Metab 10: 491–498, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Sabio G, Davis RJ. cJun NH(2)-terminal kinase 1 (JNK1): roles in metabolic regulation of insulin resistance. Trends Biochem Sci 35: 490–496, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Sarraf P, Frederich RC, Turner EM, Ma G, Jaskowiak NT, Rivet DJ, 3rd, Flier JS, Lowell BB, Fraker DL, Alexander HR. Multiple cytokines and acute inflammation raise mouse leptin levels: potential role in inflammatory anorexia. J Exp Med 185: 171–175, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Schreyer SA, Chua SC, Jr, LeBoeuf RC. Obesity and diabetes in TNF-alpha receptor-deficient mice. J Clin Invest 102: 402–411, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Schultz O, Oberhauser F, Saech J, Rubbert-Roth A, Hahn M, Krone W, Laudes M. Effects of inhibition of interleukin-6 signalling on insulin sensitivity and lipoprotein (a) levels in human subjects with rheumatoid diseases. PloS one 5: e14328, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest 116: 1793–1801, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Siegmund B, Lehr HA, Fantuzzi G. Leptin: a pivotal mediator of intestinal inflammation in mice. Gastroenterology 122: 2011–2025, 2002 [DOI] [PubMed] [Google Scholar]
- 124. Somm E, Cettour-Rose P, Asensio C, Charollais A, Klein M, Theander-Carrillo C, Juge-Aubry CE, Dayer JM, Nicklin MJ, Meda P, Rohner-Jeanrenaud F, Meier CA. Interleukin-1 receptor antagonist is upregulated during diet-induced obesity and regulates insulin sensitivity in rodents. Diabetologia 49: 387–393, 2006 [DOI] [PubMed] [Google Scholar]
- 125. Somm E, Henrichot E, Pernin A, Juge-Aubry CE, Muzzin P, Dayer JM, Nicklin MJ, Meier CA. Decreased fat mass in interleukin-1 receptor antagonist-deficient mice: impact on adipogenesis, food intake, and energy expenditure. Diabetes 54: 3503–3509, 2005 [DOI] [PubMed] [Google Scholar]
- 126. Spitzer JA, Nelson KM, Fish RE. Time course of changes in gluconeogenesis from various precursors in chronically endotoxemic rats. Metabolism 34: 842–849, 1985 [DOI] [PubMed] [Google Scholar]
- 127. Stanley TL, Zanni MV, Johnsen S, Rasheed S, Makimura H, Lee H, Khor VK, Ahima RS, Grinspoon SK. TNF-alpha antagonism with etanercept decreases glucose and increases the proportion of high molecular weight adiponectin in obese subjects with features of the metabolic syndrome. J Clin Endocrinol Metab 96: E146–E150, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Sun K, Asterholm IW, Kusminski CM, Bueno AC, Wang ZV, Pollard JW, Brekken RA, Scherer PE. Dichotomous effects of VEGF-A on adipose tissue dysfunction. Proc Natl Acad Sci USA 109: 5874–5879, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Sun K, Kusminski CM, Scherer PE. Adipose tissue remodeling and obesity. J Clin Invest 121: 2094–2101, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Tack CJ, Stienstra R, Joosten LAB, Netea MG. Inflammation links excess fat to insulin resistance: the role of the interleukin-1 family. Immunol Rev 249: 239–252, 2012 [DOI] [PubMed] [Google Scholar]
- 131. Talukdar S, Oh DY, Bandyopadhyay G, Li D, Xu J, McNelis J, Lu M, Li P, Yan Q, Zhu Y, Ofrecio J, Lin M, Brenner MB, Olefsky JM. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat Med 18: 1407–1412, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Tang T, Zhang J, Yin J, Staszkiewicz J, Gawronska-Kozak B, Mynatt R, Martin RJ, Keenan M, Gao Z, Ye J. Uncoupling of inflammation and insulin resistance by NF-kB in transgenic mice through induction of energy expenditure. J Biol Chem 285: 4637–4644, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Thaler JP, Choi SJ, Schwartz MW, Wisse BE. Hypothalamic inflammation and energy homeostasis: Resolving the paradox. Front Neuroendocrinol 31: 79–84, 2010 [DOI] [PubMed] [Google Scholar]
- 134. Trayhurn P, Wang B, Wood IS. Hypoxia in adipose tissue: a basis for the dysregulation of tissue function in obesity? Br J Nutr 100: 227–235, 2008 [DOI] [PubMed] [Google Scholar]
- 135. Tweedell A, Mulligan KX, Martel JE, Chueh FY, Santomango T, McGuinness OP. Metabolic response to endotoxin in vivo in the conscious mouse: role of interleukin-6. Metab Clin Exper 60: 92–98, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Vaisse C, Halaas JL, Horvath CM, Darnell JE, Jr, Stoffel M, Friedman JM. Leptin activation of Stat3 in the hypothalamus of wild-type and ob/ob mice but not db/db mice. Nat Genet 14: 95–97, 1996 [DOI] [PubMed] [Google Scholar]
- 137. Wallenius K, Wallenius V, Sunter D, Dickson SL, Jansson JO. Intracerebroventricular interleukin-6 treatment decreases body fat in rats. Biochem Biophys Res Commun 293: 560–565, 2002 [DOI] [PubMed] [Google Scholar]
- 138. Wogensen LD, Kolb-Bachofen V, Christensen P, Dinarello CA, Mandrup-Poulsen T, Martin S, Nerup J. Functional and morphological effects of interleukin-1 beta on the perfused rat pancreas. Diabetologia 33: 15–23, 1990 [DOI] [PubMed] [Google Scholar]
- 139. Wu JJ, Bennett AM. Essential role for mitogen-activated protein (MAP) kinase phosphatase-1 in stress-responsive MAP kinase and cell survival signaling. J Biol Chem 280: 16461–16466, 2005 [DOI] [PubMed] [Google Scholar]
- 140. Wu JJ, Roth RJ, Anderson EJ, Hong EG, Lee MK, Choi CS, Neufer PD, Shulman GI, Kim JK, Bennett AM. Mice lacking MAP kinase phosphatase-1 have enhanced MAP kinase activity and resistance to diet-induced obesity. Cell Metab 4: 61–73, 2006 [DOI] [PubMed] [Google Scholar]
- 141. Xu AW, Kaelin CB, Takeda K, Akira S, Schwartz MW, Barsh GS. PI3K integrates the action of insulin and leptin on hypothalamic neurons. J Clin Invest 115: 951–958, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Xu H, Hirosumi J, Uysal KT, Guler AD, Hotamisligil GS. Exclusive action of transmembrane TNF alpha in adipose tissue leads to reduced adipose mass and local but not systemic insulin resistance. Endocrinology 143: 1502–1511, 2002 [DOI] [PubMed] [Google Scholar]
- 143. Yamato M, Shiba T, Ide T, Seri N, Kudo W, Ando M, Yamada Ki Kinugawa S, Tsutsui H. High-fat diet-induced obesity and insulin resistance were ameliorated via enhanced fecal bile acid excretion in tumor necrosis factor-alpha receptor knockout mice. Mol Cell Biochem 359: 161–167, 2012 [DOI] [PubMed] [Google Scholar]
- 144. Yan J, Gao Z, Yu G, He Q, Weng J, Ye J. Nuclear corepressor is required for inhibition of phosphoenolpyruvate carboxykinase expression by tumor necrosis factor-(alpha). Mol Endocrinol 21: 1630–1641, 2007 [DOI] [PubMed] [Google Scholar]
- 145. Ye J. Adipose tissue vascularization: its role in chronic inflammation. Curr Diabet Reports 11: 203–210, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Ye J. Emerging role of adipose tissue hypoxia in obesity and insulin resistance. Int J Obes 33: 54–66, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Ye J, Gao Z, Yin J, He H. Hypoxia is a potential risk factor for chronic inflammation and adiponectin reduction in adipose tissue of ob/ob and dietary obese mice. Am J Physiol Endocrinol Metab 293: E1118–E1128, 2007 [DOI] [PubMed] [Google Scholar]
- 148. Ye J, Keller J. Regulation of energy metabolism by inflammation: a feedback response in obesity and calorie restriction. Aging 2: 361–368, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Yuan M, Konstantopoulos N, Lee J, Hansen L, Li ZW, Karin M, Shoelson SE. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science 293: 1673–1677, 2001 [DOI] [PubMed] [Google Scholar]
- 150. Zhang J, Gao Z, Yin J, Quon MJ, Ye J. S6K directly phosphorylates IRS-1 on Ser270 to promote insulin resistance in response to TNF-α signaling through IKK2. J Biol Chem 283: 35375–35382, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Zhang J, Henagan TM, Gao Z, Ye J. Inhibition of glyceroneogenesis by histone deacetylase 3 contributes to lipodystrophy in mice with adipose tissue inflammation. Endocrinology 152 1829–1838, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Zhao Q, Shepherd EG, Manson ME, Nelin LD, Sorokin A, Liu Y. The role of mitogen-activated protein kinase phosphatase-1 in the response of alveolar macrophages to lipopolysaccharide: attenuation of proinflammatory cytokine biosynthesis via feedback control of p38. J Biol Chem 280: 8101–8108, 2005 [DOI] [PubMed] [Google Scholar]
- 153. Zhou Y, Lee J, Reno CM, Sun C, Park SW, Chung J, Fisher SJ, White MF, Biddinger SB, Ozcan U. Regulation of glucose homeostasis through a XBP-1-FoxO1 interaction. Nat Med 17: 356–365, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Zilverschoon GRC, Tack CJ, Joosten LAB, Kullberg BJ, van der Meer JWM, Netea MG. Interleukin-18 resistance in patients with obesity and type 2 diabetes mellitus. Int J Obes 32: 1407–1414, 2008 [DOI] [PubMed] [Google Scholar]
- 155. Zumbach MS, Boehme MWJ, Wahl P, Stremmel W, Ziegler R, Nawroth PP. Tumor necrosis factor increases serum leptin levels in humans. J Clin Endocrinol Metab 82: 4080–4082, 1997 [DOI] [PubMed] [Google Scholar]