

Abstract
Biliary pancreatitis is the most common etiology for acute pancreatitis, yet its pathophysiological mechanism remains unclear. Ca2+ signals generated within the pancreatic acinar cell initiate the early phase of pancreatitis, and bile acids can elicit anomalous acinar cell intracellular Ca2+ release. We previously demonstrated that Ca2+ released via the intracellular Ca2+ channel, the ryanodine receptor (RyR), contributes to the aberrant Ca2+ signal. In this study, we examined whether RyR inhibition protects against pathological Ca2+ signals, acinar cell injury, and pancreatitis from bile acid exposure. The bile acid tauro-lithocholic acid-3-sulfate (TLCS) induced intracellular Ca2+ oscillations at 50 μM and a peak-plateau signal at 500 μM, and only the latter induced acinar cell injury, as determined by lactate dehydrogenase (LDH) leakage. Pretreatment with the RyR inhibitors dantrolene or ryanodine converted the peak-plateau signal to a mostly oscillatory pattern (P < 0.05). They also reduced acinar cell LDH leakage, basolateral blebbing, and propidium iodide uptake (P < 0.05). In vivo, a single dose of dantrolene (5 mg/kg), given either 1 h before or 2 h after intraductal TLCS infusion, reduced the severity of pancreatitis down to the level of the control (P < 0.05). These results suggest that the severity of biliary pancreatitis may be ameliorated by the clinical use of RyR inhibitors.
Keywords: calcium channels, dantrolene, ryanodine, tauro-lithocholic acid-3-sulfate, acinar cell injury
acute pancreatitis is a painful, inflammatory disorder of the pancreas for which there are few targeted therapies (16). In both children and adults, the most common etiology is biliary pancreatitis (4, 31), which accounts for ∼30–60% of cases. Since the early twentieth century, it has been suggested that biliary pancreatitis results from the obstruction of gallstones or sludge in the distal common bile duct (1, 34). For this reason, the main treatment includes removal of the stone by endoscopic retrograde cholangiopancreatography (ERCP) (49). Despite this, there is an 8% risk of pancreatitis even after ERCP (10), and current medical therapies are not particularly effective. Thus, there is a need to better understand the molecular pathophysiology of biliary pancreatitis to develop targeted therapies for this disease.
In humans, the primary bile acids are cholic acid and chenodeoxycholic acid, whereas secondary bile acids include the dehydroxylated, less hydrophilic forms lithocholic acid and deoxycholic acid (14, 52). Although bile acids can cause some change in the permeability of the pancreatic duct mucosal barrier (42), their major effects appear to occur on the pancreatic acinar cell, the initiating site for pancreatitis. One of the earliest pathological changes observed in the acinar cell is the emergence of high-amplitude, globalized cytosolic Ca2+ signals (40). These Ca2+ signals are critical to the progression of pancreatitis (27, 41, 43). Bile acids have been demonstrated to induce Ca2+ signals in hepatocytes (3, 9). Tauro-lithocholic acid induces Ca2+ release from intracellular stores in saponin-permealized human platelets and neuronal cell lines (11). Although in initial reports deoxycholate did not induce Ca2+ release in pancreatic acinar cells (47), more recent studies have demonstrated that the taurine-conjugated bile acids induce acinar cell Ca2+ signals even at micromolar concentrations (50). Preventing cytosolic Ca2+ rise by using the cytosolic Ca2+ chelator 1,2-bis(o-aminophenoxy) ethane-N,N,N′,N′-tetra-acetic acid mitigated vacuolization (24), mitochondrial depolarization (51), acinar cell injury (24), and death (7).
There are several known mechanisms by which bile acids could induce acinar cell Ca2+ signals. First, the sulfated monohydroxy bile acid tauro-lithocholic acid-3-sulfate (TLCS) at micromolar concentrations has been shown to induce apical to basolateral Ca2+ waves in acinar cells, a pattern that is typical for Ca2+ release from intracellular pools (50). Second, bile acids induce Ca2+ signals even in the absence of external Ca2+ (50). Third, acinar cells appear to express plasma membrane bile acid transporters (24) and receptors (38) that provide a mechanism for bile acid entry into the cell or receptor binding to transduce Ca2+ signals. Genetic deletion of the luminal bile acid receptor, Gpbar1, prevented TLCS-induced acinar cell injury and pancreatitis (38).
Bile acids can modulate the acinar cell Ca2+ signal by affecting multiple aspects of the Ca2+ machinery. Bile acids were demonstrated to inhibit the sarcoendoplasmic reticulum ATPase, leading to endoplasmic reticulum (ER) Ca2+ depletion and activation of capacitative Ca2+ entry (13, 24). In the intact acinar cell, bile acids were shown to potentiate Ca2+ release from the ER and vesicular Ca2+ stores through opening of the two major ER Ca2+ channels, the inositol 1,4,5-trisphosphate receptor (IP3R) and the ryanodine receptor (RyR) (18, 50). TLCS-induced Ca2+ transients were reduced by the IP3R inhibitor caffeine (20 mM) (50) and in two-photon permeabilized acinar cells by the RyR inhibitor ruthenium red (18).
The latter result is of particular interest because RyR-dependent Ca2+ release in the pancreatic acinar cell is a potential target for blocking aberrant Ca2+ signals. The RyR is a selective Ca2+ channel that forms a large homotetramer of 2.3 megadaltons and localizes to the basolateral region (28). We have previously reported that the RyR mediates pancreatitis in a secretagogue hyperstimulation model of pancreatitis (22, 35). RyR inhibitors reduced intra-acinar protease activation due to secretagogue hyperstimulation without affecting physiological enzyme secretion from the acinar cell. We then demonstrated in an in vivo caerulein hyperstimulation pancreatitis model that the RyR mediated early pancreatitis events such as protease activation and acinar cell injury, as well as the histological severity of pancreatitis. In the current study, we hypothesized that bile acids similarly caused acinar cell injury and pancreatitis through activation of RyR channel function. We, therefore, examined whether RyR inhibition would protect against pathological Ca2+ signals and acinar cell injury from bile acid exposure in vitro, as well as pancreatitis due to bile acid infusion in vivo, and found substantial protection against surrogate markers for the development of pancreatitis.
MATERIALS AND METHODS
Reagents and animals.
All reagents were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise stated. Male C57BL/6 mice weighing 20–25 g (Harlan Laboratories, Boston, MA) were fed standard laboratory chow, given free access to water, and randomly assigned to control or experimental groups. TLCS was dissolved in either 20 mM HEPES buffer (for Ca2+ imaging experiments) or Dulbecco's modified Eagle medium (DMEM)-F-12 buffer (for kinetic enzyme and cell injury assays), both at pH 7.4, and diluted to a final concentration of 50–500 μM for incubation. Dantrolene powder was first dissolved in dimethyl sulfoxide (DMSO), and then a 1 mg/ml stock was diluted to a final DMSO concentration of 5%. For in vivo use, dantrolene stocks were filtered through a 0.2-um polyvinylidene difluoride filter to remove any residual particulate matter. Ryanodine was dissolved in water. All animal treatments and euthanasia protocols were approved by the Animal Care and Use Committee.
Preparation of pancreatic acini for Ca2+ imaging.
Groups of pancreatic acinar cells were isolated as previously described (22), with minor modifications. Briefly, the pancreas was removed and then minced for 5 min in buffer containing 20 mM HEPES (pH 7.4), 95 mM NaCl, 4.7 mM KCl, 0.6 mM MgCl2, 1.3 mM CaCl2, 10 mM glucose, and 2 mM glutamine, plus 1% BSA, 1× minimum essential medium nonessential amino acids (GIBCO-BRL), 200 U/ml type 4 collagenase (Worthington, Freehold, NJ), and 1 mg/ml soybean trypsin inhibitor. The tissue was incubated for 30 min at 37°C with shaking at 90 rpm. The digest was transferred to a 15-ml conical tube and washed with collagenase-free buffer. The suspension was vigorously shaken for 15–20 s to separate the cells into smaller clusters.
Detection and analysis of cellular Ca2+ signals.
Acinar cells were loaded at room temperature with the low-affinity Ca2+-sensing dye Fluo-5F (Kd = 2.3 μM; Invitrogen). Cells were then pretreated with or without one of the following RyR inhibitors for 30 min: dantrolene (100 μM) or ryanodine (500 μM). Acinar cells were plated on acid-washed glass cover slips and then mounted on a perifusion chamber. Thereupon, they were stimulated at room temperature with the bile acid TLCS at the concentrations indicated. A Zeiss LSM510 laser scanning confocal microscope was used with a ×40, 1.4 numerical aperture objective. The dye was excited at 488 nm wavelength, and emission signals of >515 nm were collected every 2.5 s. Fluorescence from individual acinar cells was recorded. Analysis of recordings was performed using Image J software (NIH, Bethesda, MD), and mean fluorescence over time in each region was graphed. Transients were characterized as either peak-plateau, oscillations, or no response.
Preparation of pancreatic acini for enzyme kinetics/secretion assays.
Groups of pancreatic acinar cells were isolated as previously described (36) with minor modifications. Briefly, the pancreas was removed and then minced for 5 min in DMEM-F-12 1× buffer without phenol red (Invitrogen, Carlsbad, CA) containing 15 mM HEPES (pH 7.4), 120.6 mM NaCl, 4.16 mM KCl, 0.301 mM MgCl2, 1.05 mM CaCl2, 17.51 mM dextrose, 0.05 M HCl, 2 mM sodium pyruvate, and 2.5 mM glutamine, plus 0.1% BSA and 2 mg/ml type 4 collagenase (Worthington). The suspension was briefly incubated with oxygen for 5 min at 37°C with shaking at 90 rpm. The buffer was removed and replaced with new collagenase buffer, again briefly oxygenated, and then incubated for 35 min. The suspension was filtered through a 300-μm mesh (Sefar American, Depew, NY) and then washed three times with collagenase-free buffer. Acinar cells were allowed to equilibrate for 5 min at 37°C before use.
Morphological assessment of pancreatic acini.
Acinar cells were examined under light microscopy (model IX70; Olympus, Tokyo, Japan). Blebbing was defined as distinct protrusions of the basolateral membrane seen by light microscopy. Blebs were quantified from 12 to 25 fields at ×200 magnification and expressed as the number of blebs per 100 cells.
Cell injury assays.
Acinar cell injury was measured using a cytotoxicity assay for lactate dehydrogenase (LDH) leakage (Promega, Madison, WI). Absorbance was measured at 490 nm, 15 min after stopping the enzyme reaction. Results were expressed as percent LDH released in the media. For propidium iodide (PI) uptake, acinar cells were stained with 50 μg/ml of PI (Sigma) for 30 min as described by Saluja et al. (43). Fluorescence was measured using a fluorescent plate reader (Synergy H1; BioTek) at 536 nm excitation and 617 nm emission wavelengths.
Intraductal bile acid infusion model of pancreatitis.
Pancreatitis was induced by retrograde infusion of the bile acid TLCS (3 mM) or taurocholic acid (TC, 37 mM) in the distal common bile duct and pancreatic duct, as recently described (39). Briefly, C57BL/6 mice between 8 and 12 wk (20–25 g) were anesthetized with a ketamine (120 mg/kg)-xylazine (12 mg/kg) mixture (Butler Schein, Chicago, IL). A ventral incision was made to reveal the abdominal cavity. The duodenum was flipped to reveal its distal side and held in place by ligatures. The bile duct was identified, and a 30-gauge needle was inserted through the contralateral aspect of the duodenum to cannulate the biliopancreatic duct. TLCS or TC was infused at 10 μl/min for 5 min using a P33 perfusion pump (Harvard Apparatus, Holliston, MA). The exterior wound was closed using 7-mm wound clips, and a single injection of buprenorphine (0.075 mg/kg) was given immediately after the surgery. Normal saline-infused animals served as shams. Animals were allowed to recover on a heating pad for 90 min after the procedure. Mice were killed 6 or 24 h after induction.
Tissue preparation and histological grading.
Pancreas, duodenum, and spleen were fixed at room temperature for 24 h in 10% formalin solution and transferred to 70% ethanol. Paraffin-embedded sections were stained with hematoxylin and eosin and graded using a ×20 objective over 10 separate fields in a blinded fashion. Pancreas tissue was graded for edema, acinar cell vacuole formation, inflammation, and necrosis, as described by Wildi et al. (53).
PCR for cytokine expression.
RNA was isolated from pancreas tissue using TRIzol Reagent (Life Technologies, Grand Island, NY) according to the manufacturer's instructions. RNA was further purified using the RNeasy kit (Qiagen, Valencia, CA), with genomic DNA elimination columns and subsequent on-column DNase treatment to eliminate genomic DNA contamination. Total RNA (2 μg) was reverse transcribed using the Transcriptor First Strand cDNA Synthesis Kit (Roche, Mannheim, Germany). The resultant cDNA was amplified using PCR SuperMix (Invitrogen, San Diego, CA) with the following set of mouse specific primers: interleukin (IL)-6 5′-AGTTGCCTTCTTGGGACTGA-3′ and 5′-TCCACGATTTCCCAGAGAAC-3′; interferon (IFN)-γ 5′-ACTGAAGCCAGCTCTCTCTTCCTC-3′ and 5′-TTCCTTCTTGGGGTCAGCACAGAC-3′; and β-actin 5′-TTCTACAATGAGCTGCGTGTGGC-3′ and 5′-CTCATAGCTCTTCTCCAGGGAGGA-3′. PCR for each cytokine was conducted with the optimal numbers of cycles consisting of 80°C for 10 min, 94°C for 15 s, optimal annealing temperature 60°C for 1 min, 72°C for 1 min followed by incubation at 72°C for 10 min. The amplified product was fractioned on a 1.5% agarose gel and visualized by ethidium bromide staining. The band intensities were measured using Image J software. Data were normalized to β-actin and represented as fold vs. maximal.
Statistical analysis.
Data were expressed as means ± SE unless otherwise stated. Statistical analysis was performed using a Student's t-test or two-way ANOVA. Statistical significance was defined as a P value <0.05.
RESULTS
To examine the effects of bile acids on acinar cell Ca2+ signaling, we used TLCS because 1) it is the least hydrophilic and, therefore, most potent of the naturally occurring bile acids, and 2) it induces Ca2+ signals at low micromolar concentrations that are below the critical micellar concentration (21).
To examine Ca2+ signals, acini were loaded with the low-affinity Ca2+ dye Fluo-5F (Fig. 1A). Using time lapse laser scanning confocal microscopy we found that perfusing 50 μM TLCS caused Ca2+ oscillations in over half of the cells (55%). This low concentration did not cause acinar cell injury (Fig. 2B). Only 5% of the cells demonstrated a peak-plateau Ca2+ signal, defined as a plateauing of the Ca2+ fluorescence after an initial peak along with an absence of oscillations. Ca2+ oscillations are generally associated with a physiological stimulus, whereas peak-plateau signals are associated with insults that lead to acinar cell injury and pancreatitis (27, 41). At the end of each experiment, the muscarinic agonist carbachol (CCh, 1 mM) was given as a positive control to confirm that the cell could mobilize intracellular Ca2+. Of the CCh-inducible cells, 40% failed to elicit a Ca2+ signal with 50 μM TLCS administration. By contrast, all of the cells manifested a Ca2+ signal with 500 μM TLCS (Fig. 1C), a concentration that induced acinar cell injury (Fig. 2). Furthermore, most of the cells (64%) had a peak-plateau response. A little over a third of cells (36%) had Ca2+ oscillations.
Fig. 1.
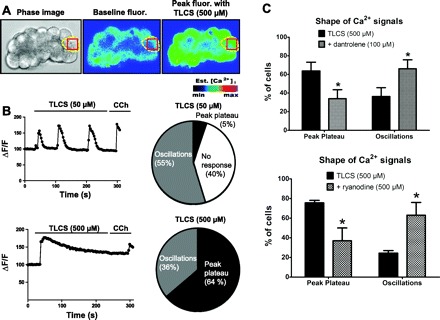
Ryanodine receptor (RyR) inhibition shifts the pattern of Ca2+ in individual acinar cells from pathological to physiological signals. A: acinar cells were stimulated with tauro-lithocholic acid-3-sulfate (TLCS, 500 μM); phase-contrast image of an acinus (left), fluorescent image of cells loaded with fluo-5F at baseline (middle), or peak fluorescence with TLCS perifusion (right). B: representative Ca2+ signals observed in cells stimulated with 50 μM (top) or 500 μM (bottom) TLCS are depicted on the left. The column on the right displays in pie chart of the proportion of cells with each type of response. Cells not responding to TLCS were counted as “no response” as long as they exhibited a Ca2+ signal to a positive control [carbachol (CCh), 1 mM] given at the end of each experiment; CCh nonresponders were not counted. C: profile of Ca2+ signals induced by TLCS (500 μM) with or without dantrolene (100 μM) (top) or ryanodine (500 μM) (bottom) (n = 50–60 cells/condition). *P < 0.05 compared with TLCS alone.
Fig. 2.
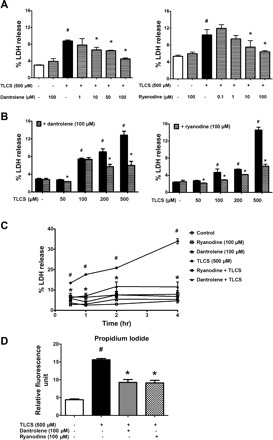
RyR inhibition mitigates TLCS-induced acinar cell injury. In isolated acinar cells, %lactate dehydrogenase (LDH) release or propidium iodide (PI) uptake was used as a biochemical indicator of injury. A: acinar cells were pretreated with increasing concentrations of dantrolene (left) or ryanodine (right) for 30 min before a 2-h incubation with TLCS. B: effects of the RyR inhibitors on varying concentrations of TLCS. C: pretreatment with the RyR inhibitors reduced cell injury as early as 30 min after TLCS administration and up to a 4-h period of observation. D: RyR inhibition reduces PI uptake after 2 h of TLCS stimulation (n = 3). P < 0.05 compared with the control (#) and TLCS alone (*).
To determine whether this aberrant Ca2+ signal is dependent on RyR opening, isolated acini were pretreated with the RyR inhibitors dantrolene (100 μM) or ryanodine (500 μM). Almost twice as many cells (63–66% of cells; P < 0.05) demonstrated Ca2+ oscillations with 500 μM TLCS in the presence of the RyR inhibitors. Conversely, the peak-plateau response was reduced by half, suggesting a shift toward a more physiological response (34–37% of cells; P < 0.05). We used 500 μM ryanodine because, although it is a prototypic RyR inhibitor, it can have a partial agonist effect at low micromolar concentrations.
Cell injury can be biochemically gauged in isolated acini by measuring the leakage of LDH in the media or by assessing PI uptake. To determine whether RyR-dependent Ca2+ transients are necessary for bile acid injury, dantrolene or ryanodine was administered at varying concentrations (Fig. 2A). Pretreatment for 30 min with either RyR inhibitor caused a concentration-dependent reduction in cell injury due to TLCS (500 μM) given over 2 h, with a nearly complete prevention of injury at the highest concentration of either inhibitor used. TLCS caused a concentration-dependent increase in cell injury that was blocked by the RyR inhibitors (Fig. 2B). Greater concentrations of TLCS (i.e., mM) are known to induce detergent injury on the cell (21). As expected, neither RyR inhibitor reduced cell injury with millimolar applications of TLCS. By contrast, the RyR inhibitors reduced acinar cell injury as early as 30 min from the time of TLCS (500 μM) administration, and this was sustained over a 4-h incubation (Fig. 2C). Similarly, pretreatment with dantrolene (100 μM) or ryanodine (100 μM) significantly reduced PI staining by 56 and 58%, respectively (P < 0.05, Fig. 2D).
Cell blebbing can be a harbinger of cell injury in the acinar cell. Within minutes of administering supraphysiological concentrations of cholecystokinin, one notes the emergence of these blebs as protrusions from the basolateral cell membrane (2, 8, 33, 44, 48). We found that TLCS (500 μM) also causes basolateral blebbing (Fig. 3), which is markedly reduced by both dantrolene and ryanodine. It would be of therapeutic relevance to know whether the RyR inhibitors reduce cell injury when given concurrently with TLCS or, more importantly, post-TLCS treatment. As shown in Fig. 4, pretreatment was most effective, but both coadministration of the RyR inhibitors with TLCS as well as post-TLCS administration also significantly reduced cell injury.
Fig. 3.
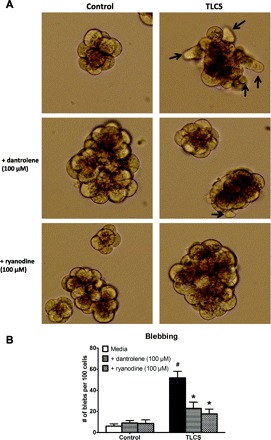
RyR inhibition mitigates TLCS-induced acinar cell blebbing. Acinar cells were pretreated with dantrolene (100 μM) or ryanodine (100 μM) for 30 min before a 2-h incubation with TLCS (500 μM). A: in each of the representative images, blebbing is denoted by arrows. B: quantification of blebbing in 6–10 fields. P < 0.05 compared with the control (#) and TLCS alone (*).
Fig. 4.
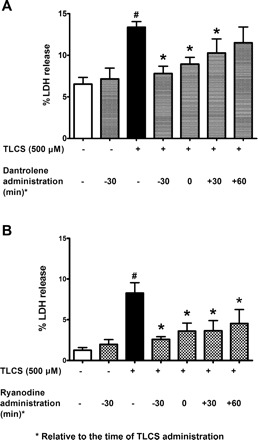
Both pretreatment and posttreatment with RyR inhibitors attenuates TLCS-induced acinar cell injury. Dantrolene (100 μM, A) or ryanodine (100 μM, B) was administered at the times indicated relative to the time of TLCS. RyR inhibition reduced cell injury in both pre- or post-TLCS treatment schemes, although the reduction was greater with pretreatment (n = 6). P < 0.05, compared with the control (#) and TLCS alone (*).
Recently, IP3Rs and store-operated Ca2+ entry (SOCE) have been shown to contribute to bile acid-induced acinar cell pathology (18, 19, 24, 25). To determine the relative effects of these Ca2+ release or influx mechanisms on bile acid-induced acinar cell injury, we used the IP3R inhibitors, 2-APB and xestospongin C, and the SOCE inhibitor BTP-2, which reduced LDH leakage by 50, 65, and 93% down to control levels, respectively (P < 0.05, Fig. 5). It should be noted that 2-APB is also an inhibitor of SOCE. To evaluate whether our findings with RyR inhibitors on TLCS responses in isolated acinar cells are relevant in vivo, we employed our recently published pancreatitis model in which TLCS is briefly infused in the PD of anesthetized mice (39). This results in inflammation and necrosis of the pancreatic head that is maximal after 24 h (Fig. 6). To examine the role of the RyR, we used dantrolene because it can be administered in vivo and is clinically used to treat the RyR channelopathy Malignant Hyperthermia (5).
Fig. 5.
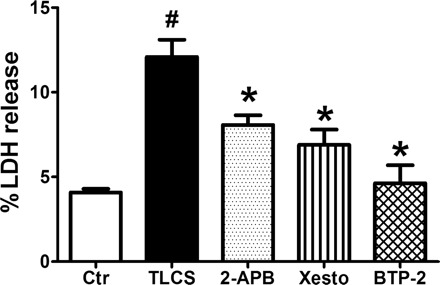
Inhibition of inositol 1,4,5-trisphosphate receptors (IP3Rs) or store-operated Ca2+ entry (SOCE) mitigates TLCS-induced acinar cell injury. Acinar cells were pretreated with the IP3R inhibitors 2-APB (10 μM) and xestospongin C (1 μM), or the SOCE inhibitor BTP-2 (10 μM) for 30 min before a 2-h incubation with TLCS (500 μM) (n = 3). P < 0.05 compared with the control (#) and TLCS alone (*).
Fig. 6.
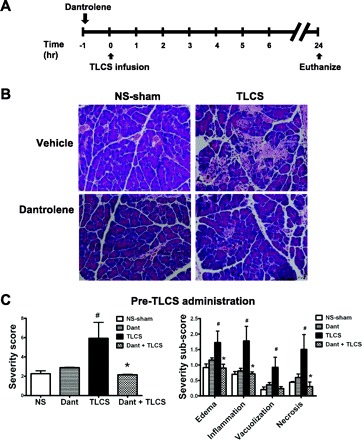
Dantrolene pretreatment reduces the histological severity of pancreatitis due to TLCS infusion. A: schema for in vivo RyR inhibition with a single ip injection of dantrolene (5 mg/kg) and pancreatitis induction using an intraductal infusion of TLCS. Mice were killed 24 h later. B: the row on top shows hematoxylin and eosin sections of the pancreatic head from normal saline sham (NS-sham; left) and TLCS-infused (right) mice that received treatment with vehicle only. The row on the bottom represents dantrolene pretreatment. C: overall severity score (left) along with subscores (right) (n = 4 for the NS-sham, 4 for dantrolene alone, 6 for TLCS alone, and 7 for dantrolene + TLCS). P < 0.05 compared with the NS-sham (#) and TLCS alone (*).
Dantrolene was given as a single intraperitoneal dose (5 mg/kg) 1 h before TLCS infusion (Fig. 6). This pretreatment regimen markedly reduced the histological severity of pancreatitis down to the level of the sham-operated controls. Notably, each of the severity criteria was reduced by dantrolene, including edema, inflammation, vacuolization, and necrosis.
The severity of pancreatitis also correlates with early expression of certain cytokines, particularly IL-6 (6). Of the various cytokines examined by semiquantitative PCR, only pancreatic IL-6 and IFN-γ were elevated above the normal saline-sham 2 h after TLCS infusion. The lack of difference in elevation among the other cytokines may be due to a generalized cytokine stress response from manipulating the PD even in the normal saline-sham animals. Nevertheless, dantrolene pretreatment prevented the expression of both IL-6 and IFN-γ observed with TLCS, as demonstrated by semiquantitative PCR (Fig. 7). In limited experiments, we also tested the role of RyR inhibition as a post hoc treatment for TLCS pancreatitis in vivo. A single dose of dantrolene (5 mg/kg) was administered 2 h after TLCS infusion and improved pancreatic (Fig. 8) histology 24 h after the TLCS.
Fig. 7.
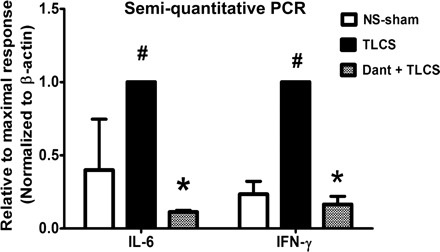
Dantrolene pretreatment reduces TLCS-induced expression of pancreatic interleukin (IL)-6 and interferon (IFN)-γ. cDNA from whole pancreas was amplified by semiquantitative PCR. Expression of IL-6 and IFN-γ from the pancreatic head was obtained 6 h after TLCS infusion (n = 2–3 animals/group). Densitometric measurements were normalized to β-actin and shown relative to the maximal response (i.e., TLCS alone). P < 0.05 compared with the NS-sham (#) and TLCS alone (*).
Fig. 8.
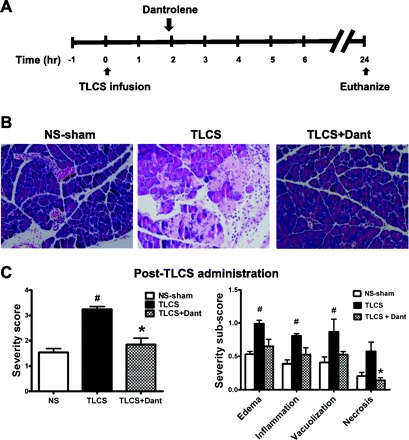
Dantrolene given after TLCS infusion reduces the severity of pancreatitis. A: schema for in vivo RyR inhibition with a single ip injection of dantrolene (5 mg/kg) and pancreatitis induction using an intraductal infusion of TLCS. B: hematoxylin and eosin sections of the pancreatic head. C: overall severity score (left) along with subscores (right) (n = 3 animals/group). P < 0.05 compared with the NS-sham (#) and TLCS alone (*).
To determine whether the more common physiological bile acids are able to trigger RyR-mediated acinar cell injury and pancreatitis, we used the bile acids sodium TC (10 mM), taurochenodeoxycholic acid (TCDC; 1 mM), and taurodeoxycholic acid (TDC; 1 mM). Pretreatment with dantrolene or ryanodine reduced acinar cell injury resulting from each of the bile acids (P < 0.05, Fig. 9). We also tested the role of RyR inhibition as prophylaxis for TC-induced pancreatitis in vivo, using a protocol we have previously published in mice (39). A single intraperitoneal dose of dantrolene (5 mg/kg) was given 1 h before TC (37 mM) infusion (Fig. 10). TC induced elevations in serum amylase and LDH, which were reduced by dantrolene pretreatment. In addition, TC infusion caused marked histological damage in pancreas after 24 h. Dantrolene improved the histological score in both tissues (P < 0.05). Overall, the in vivo data point to both a protective as well as therapeutic role for dantrolene and lend support to the hypothesis that pathological activation of the RyR significantly contributes to the development and progression of bile-infusion pancreatitis.
Fig. 9.
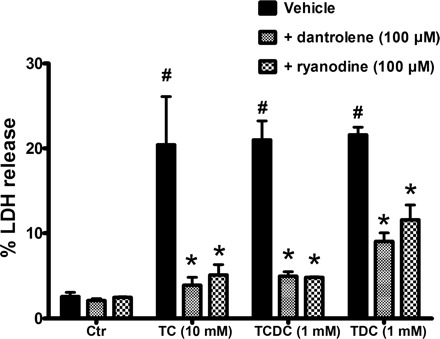
RyR inhibition attenuates acinar cell injury due to taurocholic acid (TC), taurochenodeoxycholic acid (TCDC), and taurodeoxycholic acid (TDC). Isolated acinar cells were pretreated with either dantrolene (100 μM) or ryanodine (100 μM) for 30 min before a 2-h incubation with the bile acids (n = 3). P < 0.05 compared with the control (#) and each corresponding bile acid (*).
Fig. 10.
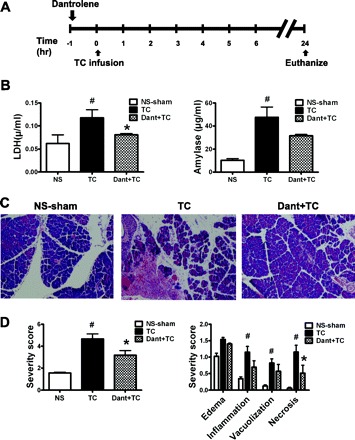
Dantrolene pretreatment reduces the severity of pancreatitis due to TC infusion. A: schema for in vivo RyR inhibition with a single ip injection of dantrolene (5 mg/kg) and pancreatitis induction using an intraductal infusion of TC. B: serum lactate dehydrogenase (LDH, left) and amylase (right) levels. C and D: hematoxylin and eosin sections of the pancreatic head along with corresponding severity scores (n = 3–4 animals/group). P < 0.05 compared with the NS-sham (#) and TLCS alone (*).
DISCUSSION
The key findings in this study were that, in isolated acinar cells, the RyR inhibitors dantrolene and ryanodine changed the pattern of the acinar cell Ca2+ signal from a pathological peak-plateau to physiological oscillations, attenuated acinar cell injury, and basolateral cell blebbing. In vivo, administration of dantrolene blocked pancreatic injury when given before bile acid infusion, and it also ameliorated pancreatitis when given as therapy after bile infusion. In previous work, we and others have shown that basolateral Ca2+ amplitude and Ca2+ wave speed with supraphysiological secretagogue stimulation were dependent on the RyR (22, 29, 32, 36, 45). In our current work, we find that RyR inhibition reversed the pattern of the acinar cell Ca2+ signals (in most acinar cells) induced by pathological concentrations of bile acids from a peak-plateau to that of Ca2+ oscillations. Another novel feature of the current study is the demonstration that RyR inhibition blocks cell injury.
Bile acids cause release of Ca2+ through the opening of both IP3Rs and RyRs (18). Furthermore, the intracellular Ca2+ release can trigger opening of Ca2+ influx channels via SOCE (25). Thus we cannot exclude the contribution of IP3R-Ca2+ release or SOCE in mediating Ca2+-dependent processes in pancreatitis. Indeed, IP3R inhibition or its genetic deletion protects acinar cells against alcohol-induced trypsin activation (19, 20). Furthermore, acini from mice with a genetic deletion of the SOCE channel TRPC3 have reduced pancreatic pERK phosphorylation and LC3 conversion due to sodium taurocholate (25). We demonstrate in the current study that the IP3R inhibitors 2-APB and xestospongin C or the SOCE inhibitor BTP-2 each markedly reduced bile acid-mediated cell injury. Taken together, the data suggest that each of the Ca2+ channels is necessary to generate the Ca2+ signals that cause acinar cell injury and that their roles are interrelated.
It is notable though that IP3R-Ca2+ release is critical to the physiological secretion of pancreatic enzymes from the apical lumen of the acinar cell (17). Thus, blocking IP3R-Ca2+ release might actually be deleterious to the overall health of the acinar cell. The RyR, on the other hand, is primarily localized to the basolateral region (15, 22, 30, 45), and RyR-Ca2+ does not appear to mediate pancreatic enzyme secretion (22). For this reason, RyR-Ca2+ channel blockers hold promise as a viable target for pancreatitis therapy.
The mechanism by which RyR blockade converts pathological Ca2+ signals to physiological oscillations is not entirely clear. A likely reason is that RyR blockade attenuates the large-scale emptying of intracellular Ca2+ stores induced by high micromolar concentrations of TLCS. This then prevents the opening of store-operated Ca2+ channels and the subsequent sustained cytosolic Ca2+ signal, manifested as a peak-plateau response. Instead, with reduced RyR opening, the levels of Ca2+ released mimic the physiological state and thus allow for the sequential regenerative discharges of stored Ca2+, or oscillations (12).
In the in vivo model of intraductal TLCS infusion, it is notable that a single dose of dantrolene given 1 h before bile acid infusion, or as treatment given 2 h after infusion, reduced pancreatic necrosis and inflammation 24 h after the insult. Dantrolene is a hydantoin derivative that is clinically approved for the treatment of Malignant Hyperthermia, a life-threatening skeletal muscle disorder caused by a surge of SR Ca2+ release and Ca2+ entry upon exposure of mutant RyR1 to halothane or other volatile anesthetics (23). Dantrolene binds to a specific domain on RyR1 and has been shown to stabilize protein-protein interactions supported by this domain (26, 37). However, it is unclear whether dantrolene directly inhibits RyR1 opening (54). Although dantrolene suppresses RyR1-stimulated Ca2+ transients, it has no effect on RyR1 single channel activity in a lipid bilayer (46). Taken together, the data suggest that dantrolene suppresses RyR1-coupled Ca2+ mechanisms, but not necessarily by acting as a classical channel blocker.
To provide support for our findings using dantrolene, we also used ryanodine, which is a prototypic RyR channel inhibitor at micromolar concentrations (28), and demonstrated that it had similar effects on Ca2+ signals and injury in isolated cells. The effects of ryanodine could not be examined in vivo because of its skeletal muscle and cardiotoxicity. However, the data in isolated acinar cells with post-TLCS treatment regimens using both inhibitors, and the in vivo findings with dantrolene given as a treatment dose, suggest that RyR-coupled Ca2+ mechanisms not only trigger events leading to pancreatic injury but that ongoing RyR-Ca2+ propagates the injury. As is the case for Malignant Hyperthermia, our findings suggest that dantrolene may also be useful as a treatment for pancreatitis.
A novel clinical implication of this study is the potential for the use of RyR modulators as a targeted therapy for bile-induced pancreatitis. In addition, the RyR mutations that cause a gain of function in diseases such as Malignant Hyperthermia (28) allow us to speculate whether gain of function polymorphisms in the RyR could predispose the 4–8% of patients with gallstones or sludge to develop pancreatitis.
In summary, we have demonstrated that the bile acid TLCS causes pathological Ca2+ signals that are dependent upon the RyR. Inhibition of the RyR in isolated acinar cells mitigates acinar cell injury due to bile acids, including basolateral blebbing. In vivo, dantrolene can be used to both prevent and treat pancreatitis due to the intraductal infusion of bile acids. The findings suggest that RyR modulators may provide a therapeutic mechanism for biliary pancreatitis.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) Grants DK-083327, DK-093491, DK-34989 (Yale Liver Center), a Children's Digestive Health and Nutrition Young Investigator Award (to S. Z. Husain), and NIDDK Grants DK-091327 to Michael L. Steer (Tufts University) and DK-092460 (to V. P. Singh).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: S.Z.H., M.A., V.B., and G.P. conception and design of research; S.Z.H., A.I.O., K.A.M., Y.L., S.S., S.M.M., D.W., and R.C.-W. analyzed data; S.Z.H., A.I.O., K.A.M., Y.L., S.S., S.M.M., V.P.S., J.P., M.A., V.B., and G.P. interpreted results of experiments; S.Z.H. drafted manuscript; S.Z.H., A.I.O., K.A.M., V.P.S., J.P., M.A., V.B., and G.P. edited and revised manuscript; S.Z.H., A.I.O., K.A.M., Y.L., S.S., S.M.M., D.W., R.C.-W., V.P.S., J.P., M.A., V.B., and G.P. approved final version of manuscript; A.I.O., K.A.M., Y.L., S.S., S.M.M., D.W., R.C.-W., and G.P. performed experiments; A.I.O., K.A.M., Y.L., S.S., and S.M.M. prepared figures.
ACKNOWLEDGMENTS
We thank Drs. M. Nathanson and F. Gorelick for helpful discussion throughout the study and Mateus Guerra for technical expertise.
REFERENCES
- 1.Acosta JM, Ledesma CL. Gallstone migration as a cause of acute pancreatitis. N Engl J Med 290: 484–487, 1974 [DOI] [PubMed] [Google Scholar]
- 2.Adler G, Kern HF, Pan GZ, Gardner JD. Secretagogue-induced membrane alterations in dispersed acini from rat pancreas. Eur J Cell Biol 33: 234–241, 1984 [PubMed] [Google Scholar]
- 3.Anwer MS, Engelking LR, Nolan K, Sullivan D, Zimniak P, Lester R. Hepatotoxic bile acids increase cytosolic Ca++ activity of isolated rat hepatocytes. Hepatology 8: 887–891, 1988 [DOI] [PubMed] [Google Scholar]
- 4.Bai HX, Lowe ME, Husain SZ. What have we learned about acute pancreatitis in children? J Pediatr Gastroenterol Nutr 52: 262–270, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Betzenhauser MJ, Marks AR. Ryanodine receptor channelopathies. Pflugers Arch 460: 467–480, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bhatia M. Inflammatory response on the pancreatic acinar cell injury. Scand J Surg 94: 97–102, 2005 [DOI] [PubMed] [Google Scholar]
- 7.Booth DM, Murphy JA, Mukherjee R, Awais M, Neoptolemos JP, Gerasimenko OV, Tepikin AV, Petersen OH, Sutton R, Criddle DN. Reactive oxygen species induced by bile acid induce apoptosis and protect against necrosis in pancreatic acinar cells. Gastroenterology 140: 2116–2125, 2011 [DOI] [PubMed] [Google Scholar]
- 8.Burnham DB, Williams JA. Effects of high concentrations of secretagogues on the morphology and secretory activity of the pancreas: a role for microfilaments. Cell Tissue Res 222: 201–212, 1982 [DOI] [PubMed] [Google Scholar]
- 9.Combettes L, Dumont M, Berthon B, Erlinger S, Claret M. Release of calcium from the endoplasmic reticulum by bile acids in rat liver cells. J Biol Chem 263: 2299–2303, 1988 [PubMed] [Google Scholar]
- 10.Cooper ST, Slivka A. Incidence, risk factors, and prevention of post-ERCP pancreatitis. Gastroenterol Clin North Am 36: 259–276, 2007 [DOI] [PubMed] [Google Scholar]
- 11.Coquil JF, Berthon B, Chomiki N, Combettes L, Jourdon P, Schteingart C, Erlinger S, Claret M. Effects of taurolithocholate, a Ca2(+)-mobilizing agent, on cell Ca2(+) in rat hepatocytes, human platelets and neuroblastoma NG108–15 cell line. Biochem J 273: 153–160, 1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dupont G, Combettes L, Bird GS, Putney JW. Calcium oscillations. Cold Spring Harb Perspect Biol 3: 45–57, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fischer L, Gukovskaya AS, Penninger JM, Mareninova OA, Friess H, Gukovsky I, Pandol SJ. Phosphatidylinositol 3-kinase facilitates bile acid-induced Ca2+ responses in pancreatic acinar cells. Am J Physiol Gastrointest Liver Physiol 292: G875–G886, 2007 [DOI] [PubMed] [Google Scholar]
- 14.Fisher MM, Yousef IM. Sex differences in the bile acid composition of human bile: studies in patients with and without gallstones. Can Med Assoc J 109: 190–193, 1973 [PMC free article] [PubMed] [Google Scholar]
- 15.Fitzsimmons TJ, Gukovsky I, McRoberts JA, Rodriguez E, Lai FA, Pandol SJ. Multiple isoforms of the ryanodine receptor are expressed in rat pancreatic acinar cells. Biochem J 351: 265–271, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frossard JL, Steer ML, Pastor CM. Acute pancreatitis. The Lancet 371: 143–152, 2008 [DOI] [PubMed] [Google Scholar]
- 17.Futatsugi A, Nakamura T, Yamada MK, Ebisui E, Nakamura K, Uchida K, Kitaguchi T, Takahashi-Iwanaga H, Noda T, Aruga J, Mikoshiba K. IP3 receptor types 2 and 3 mediate exocrine secretion underlying energy metabolism. Science 309: 2232–2234, 2005 [DOI] [PubMed] [Google Scholar]
- 18.Gerasimenko JV, Flowerdew SE, Voronina SG, Sukhomlin TK, Tepikin AV, Petersen OH, Gerasimenko OV. Bile acids induce Ca2+ release from both the endoplasmic reticulum and acidic intracellular calcium stores through activation of inositol trisphosphate receptors and ryanodine receptors. J Biol Chem 281: 40154–40163, 2006 [DOI] [PubMed] [Google Scholar]
- 19.Gerasimenko JV, Lur G, Ferdek P, Sherwood MW, Ebisui E, Tepikin AV, Mikoshiba K, Petersen OH, Gerasimenko OV. Calmodulin protects against alcohol-induced pancreatic trypsinogen activation elicited via Ca2+ release through IP3 receptors. Proc Natl Acad Sci USA 108: 5873–5878, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gerasimenko JV, Lur G, Sherwood MW, Ebisui E, Tepikin AV, Mikoshiba K, Gerasimenko OV, Petersen OH. Pancreatic protease activation by alcohol metabolite depends on Ca2+ release via acid store IP3 receptors. Proc Natl Acad Sci USA 106: 10758–10763, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hofmann AF, Roda A. Physicochemical properties of bile acids and their relationship to biological properties: an overview of the problem. J Lipid Res 25: 1477–1489, 1984 [PubMed] [Google Scholar]
- 22.Husain SZ, Prasad P, Grant WM, Kolodecik TR, Nathanson MH, Gorelick FS. The ryanodine receptor mediates early zymogen activation in pancreatitis. Proc Natl Acad Sci USA 102: 14386–14391, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Inan S, Wei H. The cytoprotective effects of dantrolene: a ryanodine receptor antagonist. Anesth Analg 111: 1400–1410, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim JY, Kim KH, Lee JA, Namkung W, Sun AQ, Ananthanarayanan M, Suchy FJ, Shin DM, Muallem S, Lee MG. Transporter-mediated bile acid uptake causes Ca2+-dependent cell death in rat pancreatic acinar cells. Gastroenterology 122: 1941–1953, 2002 [DOI] [PubMed] [Google Scholar]
- 25.Kim MS, Hong JH, Li Q, Shin DM, Abramowitz J, Birnbaumer L, Muallem S. Deletion of TRPC3 in mice reduces store-operated Ca2+ influx and the severity of acute pancreatitis. Gastroenterology 137: 1509–1517, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kobayashi S, Bannister ML, Gangopadhyay JP, Hamada T, Parness J, Ikemoto N. Dantrolene stabilizes domain interactions within the Ryanodine receptor. J Biol Chem 280: 6580–6587, 2005 [DOI] [PubMed] [Google Scholar]
- 27.Kruger B, Albrecht E, Lerch MM. The role of intracellular calcium signaling in premature protease activation and the onset of pancreatitis. Am J Pathol 157: 43–50, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lanner JT, Georgiou DK, Joshi AD, Hamilton SL. Ryanodine receptors: structure, expression, molecular details, and function in calcium release (Abstract). Cold Spring Harb Perspect Biol 2: a003996, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leite MF, Burgstahler AD, Nathanson MH. Ca2+ waves require sequential activation of inositol trisphosphate receptors and ryanodine receptors in pancreatic acini. Gastroenterology 122: 415–427, 2002 [DOI] [PubMed] [Google Scholar]
- 30.Leite MF, Dranoff JA, Gao L, Nathanson MH. Expression and subcellular localization of the ryanodine receptor in rat pancreatic acinar cells. Biochem J 337: 305–309, 1999 [PMC free article] [PubMed] [Google Scholar]
- 31.Lowenfels AB, Sullivan T, Fiorianti J, Maisonneuve P. The epidemiology and impact of pancreatic diseases in the United States. Curr Gastroenterol Rep 7: 90–95, 2005 [DOI] [PubMed] [Google Scholar]
- 32.Nathanson MH, Padfield PJ, O'Sullivan AJ, Burgstahler AD, Jamieson JD. Mechanism of Ca2+ wave propagation in pancreatic acinar cells. J Biol Chem 267: 18118–18121, 1992 [PubMed] [Google Scholar]
- 33.O'Konski MS, Pandol SJ. Effects of caerulein on the apical cytoskeleton of the pancreatic acinar cell. J Clin Invest 86: 1649–1657, 1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Opie EL. The relation of cholelithiasis to disease of the pancreas and to fat-necrosis. Johns Hopkins Hospital Bulletin 12: 19–21, 1901 [Google Scholar]
- 35.Orabi AI, Shah AU, Ahmad MU, Choo-Wing R, Parness J, Jain D, Bhandari V, Husain SZ. Dantrolene mitigates caerulein-induced pancreatitis in vivo in mice. Am J Physiol Gastrointest Liver Physiol 299: G196–G204, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Orabi AI, Shah AU, Muili K, Luo Y, Mahmood SM, Ahmad A, Reed A, Husain SZ. Ethanol enhances carbachol-induced protease activation and accelerates Ca2+ waves in isolated rat pancreatic acini. J Biol Chem 286: 14090–14097, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paul-Pletzer K, Yamamoto T, Bhat MB, Ma J, Ikemoto N, Jimenez LS, Morimoto H, Williams PG, Parness J. Identification of a dantrolene-binding sequence on the skeletal muscle ryanodine receptor. J Biol Chem 277: 34918–34923, 2002 [DOI] [PubMed] [Google Scholar]
- 38.Perides G, Laukkarinen JM, Vassileva G, Steer ML. Biliary acute pancreatitis in mice is mediated by the G protein-coupled cell surface bile acid receptor Gpbar1. Gastroenterology 138: 715–725, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Perides G, van Acker GJD, Laukkarinen JM, Steer ML. Experimental acute biliary pancreatitis induced by retrograde infusion of bile acids into the mouse pancreatic duct. Nat Protocols 5: 335–341, 2010 [DOI] [PubMed] [Google Scholar]
- 40.Petersen OH, Tepikin AV. Polarized calcium signaling in exocrine gland cells. Annu Rev Physiol 70: 273–299, 2008 [DOI] [PubMed] [Google Scholar]
- 41.Raraty M, Ward J, Erdemli G, Vaillant C, Neoptolemos JP, Sutton R, Petersen OH. Calcium-dependent enzyme activation and vacuole formation in the apical granular region of pancreatic acinar cells. Proc Natl Acad Sci USA 97: 13126–13131, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reber HA, Mosley JG. The effect of bile salts on the pancreatic duct mucosal barrier. Br J Surg 67: 59–62, 1980 [DOI] [PubMed] [Google Scholar]
- 43.Saluja AK, Bhagat L, Lee HS, Bhatia M, Frossard JL, Steer ML. Secretagogue-induced digestive enzyme activation and cell injury in rat pancreatic acini. Am J Physiol Gastrointest Liver Physiol 276: G835–G842, 1999 [DOI] [PubMed] [Google Scholar]
- 44.Singh VP, McNiven MA. Src-mediated cortactin phosphorylation regulates actin localization and injurious blebbing in acinar cells. Mol Biol Cell 19: 2339–2347, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Straub SV, Giovannucci DR, Yule DI. Calcium wave propagation in pancreatic acinar cells: functional interaction of inositol 1,4,5-trisphosphate receptors, ryanodine receptors, and mitochondria. J Gen Physiol 116: 547–560, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Szentesi P, Collet C, Sarkozi S, Szegedi C, Jona I, Jacquemond V, Kovacs L, Csernoch L. Effects of dantrolene on steps of excitation-contraction coupling in mammalian skeletal muscle fibers. J Gen Physiol 118: 355–375, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Takeyama Y, Nakanishi H, Ohyanagi H, Saitoh Y, Kaibuchi K, Takai Y. Enhancement of secretagogue-induced phosphoinositide turnover and amylase secretion by bile acids in isolated rat pancreatic acini. J Clin Invest 78: 1604–1611, 1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Torgerson RR, McNiven MA. The actin-myosin cytoskeleton mediates reversible agonist-induced membrane blebbing. J Cell Sci 111: 2911–2922, 1998 [DOI] [PubMed] [Google Scholar]
- 49.van Geenen EJ, van der Peet DL, Bhagirath P, Mulder CJ, Bruno MJ. Etiology and diagnosis of acute biliary pancreatitis. Nat Rev Gastroenterol Hepatol 7: 495–502, 2010 [DOI] [PubMed] [Google Scholar]
- 50.Voronina S, Longbottom R, Sutton R, Petersen OH, Tepikin A. Bile acids induce calcium signals in mouse pancreatic acinar cells: implications for bile-induced pancreatic pathology. J Physiol 540: 49–55, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Voronina SG, Barrow SL, Gerasimenko OV, Petersen OH, Tepikin AV. Effects of secretagogues and bile acids on mitochondrial membrane potential of pancreatic acinar cells: comparison of different modes of evaluating DeltaPsim. J Biol Chem 279: 27327–27338, 2004 [DOI] [PubMed] [Google Scholar]
- 52.Weinman SA, Sajid J. Bile secretion and cholestasis. In: Textbook of Gastroenterology, edited by Yamada T. Hoboken, NJ: Wiley-Blackwell, 2009, p. 401–428 [Google Scholar]
- 53.Wildi S, Kleeff J, Mayerle J, Zimmermann A, Bottinger EP, Wakefield L, Buchler MW, Friess H, Korc M. Suppression of transforming growth factor β signalling aborts caerulein induced pancreatitis and eliminates restricted stimulation at high caerulein concentrations. Gut 56: 685–692, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhao X, Weisleder N, Han X, Pan Z, Parness J, Brotto M, Ma J. Azumolene inhibits a component of store-operated calcium entry coupled to the skeletal muscle ryanodine receptor. J Biol Chem M602306200 281: 33477–33486, 2006 [DOI] [PubMed] [Google Scholar]